

Abstract
Aims/hypothesis
Recent histological analysis of pancreata obtained from long-standing type 1 diabetes patients identified chronic islet inflammation as well as limited evidence suggestive of beta cell replication. Studies in rodent models also suggest beta cell replication can be induced by certain inflammatory cytokines as well as with gastrin. We therefore tested the hypothesis that beta cell replication would be observed in non-autoimmune human pancreatic disorders in which localized inflammation or elevation in gastrin were present.
Methods
Resected operative pancreatic specimens were obtained from patients diagnosed with primary adenocarcinoma (with or without chronic severe pancreatitis) or gastrinoma. Additional pancreatic tissue was obtained from autopsy control patients. Immunohistochemistry was used to assess fractional insulin area, beta cell numbers and replication rates, and differentiation factors relevant to beta cell development.
Results
Fractional insulin area was similar among groups. Patients with pancreatic adenocarcinoma and localized chronic severe pancreatitis displayed significant increases in beta cell number as single cells, as well as increased beta cell replication rates and levels of neurogenic differentiation 1 in islets. Patients with gastrinoma demonstrated significant increases in single beta cell numbers, yet beta cell replication rates and islet differentiation factor levels were similar to control groups.
Conclusions/interpretation
These findings indicate that chronic severe pancreatic inflammation can be associated with significant effects on beta cell numbers or replication rates, depending on their cellular distribution. This information may prove useful for attempts seeking to design therapies aimed at inducing beta cell replication as a means to reverse diabetes.
Keywords: Pancreatic adenocarcinoma, Beta cells, Beta cell replication, Islets of Langerhans, Chronic pancreatitis, Gastrinoma
Introduction
A longstanding dogma regarding the pathogenesis of type 1 diabetes portends that in the months to years following the initiation of insulin therapy, beta cell mass is irrevocably lost; with the pancreas becoming an organ devoid of insulin producing cells. This notion has, however, recently come under scrutiny, with interest having evolved for the concept of “beta cell” regeneration [1]. Evidence supporting such a notion finds its basis in many fields including those of incretin pharmacology, stem cell biology, obesity, pregnancy, and type 2 diabetes. However, a recent study involving the detection of insulin producing cells within pancreata from subjects with type 1 diabetes of intermediate to longstanding duration (i.e., 5 to 59 years) has been used by some, to bolster the case for beta cell regeneration [2]. Specifically, this investigation indicated that pancreata from nearly 90 percent of longstanding type 1 diabetes patients contained insulin positive cells and that the presence of such cells was unrelated to duration of disease.
At least three distinct mechanisms have been put forward to explain the proposed process of beta cell regeneration, including replication, neogenesis, and transdifferentiation [3–5]. Based on studies of mice, some have proposed that beta cell growth is entirely mediated by replication of existing beta cells [6]. Other investigations involving pancreata from type 1 diabetes patients support the concept of regeneration, although if true, the beta cell regenerative capacity appears to be lower in humans than rodents [2]. Additional studies suggest that beta cells bud off from the pancreatic ducts, suggesting this might be a process by which beta cells are derived post-pancreatectomy [7]. While partial pancreatectomy in rodents rapidly induces beta cell regeneration, evidence for beta regeneration was not found in patients with pancreatic resections involving more than 50% of the initial pancreatic mass [8]. Evidence for beta cell transdifferentiation has been observed both in vitro as well as in vivo [9, 10], yet the physiologic validity remains controversial in humans [11].
A potential role for gastrin in beta cell regeneration was suggested by studies that observed increased beta cell mass in mice treated with pharmacological administration of this factor, especially when co-administered with epidermal growth factor (EGF) [12]. Additionally, treatment of non-obese diabetic (NOD) mice with a combination of gastrin and EGF increased insulin content eight-fold [13]. In humans, hypergastrinaemia is observed in settings of primary gastrinoma (“Zollinger-Ellison syndrome”) or secondary to pharmacological induced chronic acid suppression [14]. However, a recent study analyzing surgical samples from four gastrinoma patients showed no evidence for increased beta cell replication [15]. A second setting in which increased beta cell replication might be observed has been noted in rodent studies including those involving over expression of γ-interferon, transforming growth factor-β, IL-1β, and others [16–19]. This notion has also found recent support in humans where pancreatic specimens obtained from patients with chronic pancreatitis showed evidence of increased numbers of insulin-positive cells in ducts, as well as cells expressing transcriptional markers of endocrine origin (i.e., pancreatic and duodenal homeobox 1 (PDX1))[20]. This study was, however, somewhat restricted in that the control group consisted of autopsy specimens from children and young adults and the extent or nature of chronic pancreatitis was not provided.
With this continued uncertainty about conditions in which beta cell regeneration might occur in the human, we elected to examine the uninvolved regions of pancreata surgically removed from patients with pancreatic adenocarcinoma. In these samples, the presence or absence of localized inflammation might give an indication of whether inflammation could stimulate changes in beta cell mass. To also allow for inter-hospital comparisons to existing related studies [20, 21], we also included autopsy-based specimens from patients without clinical evidence of pancreatic disease as normal controls. Histopathological assessment of the severity of chronic pancreatitis was performed, as was assessment of morphological markers of beta cell replication and cellular lineage. These latter efforts included studies charactering the levels of PDX1, neurogenic differentiation 1 (NEUROD1), and neurogenin 3 (NEUROG3) as these transcription factors are intimately involved in the differentiation of precursor endocrine cells to beta cells [22, 23]. Since insulin positive cells were not restricted solely to islets (i.e., they were also observed within small clusters or as single cells), we sought to quantify the numbers and replication rates of insulin positive cells in all three forms of cellular distribution.
Methods
Patients
A total of twenty-five patients were included in this study (Table 1). Patients were first selected by a retrospective search of the clinical database in the University of Florida and Shands hospital during the last 20 years for patients treated by pancreatic resection with a diagnosis of primary pancreatic adenocarcinoma or pancreatic gastrin-secreting tumor (gastrinoma). Patients with a clinical history of symptomatic pancreatitis were excluded from selection, since the goal of this study was to examine pancreatic tissue damaged by localized mass lesions, rather than pancreatitis arising in the setting of systemic conditions (e.g., alcoholism). Gastrinoma patients had no other clinical diagnosis than a primary pancreatic tumor. From an initial search yielding thirty three cases, twenty five patients were subsequently found to have one or more uninvolved tissue blocks available for further study. The uninvolved tissue blocks were selected through review of the final pathology findings for an ancillary pathological diagnosis of no pancreatitis or chronic severe pancreatitis. Of the nine cases listed as having no pancreatitis on the pathology report, four were subsequently deemed to have incidental chronic focal, mild pancreatitis following histopathology review and hereafter were called mild pancreatitis patients. Mild chronic pancreatitis was assigned to samples having <5% of acinar tissue infiltrated with mononuclear cells and replaced by fibrosis, while severe chronic pancreatitis was assigned to samples with >20% of the acinar area infiltrated with mononuclear cells and fibrosis. Ultimately fourteen patients with pancreatic adenocarcinoma were selected and were subsequently stratified based on the absence (N=5) or presence of chronic mild (N=4) or severe (N=5) pancreatitis in the uninvolved tissue block (hereafter referred to as no, mild, and severe pancreatitis groups). Six patients with preoperative and final pathological diagnosis of intrapancreatic gastrinomas, confirmed by gastrin immunohistochemistry, had uninvolved sample blocks available for further evaluation. In addition, tissue samples were collected from five autopsy patients (hereafter referred to as “autopsy control”) who had no clinical history of diabetes or pancreatic disease, and whose pancreata were normal by histological examination. Exclusion criteria for all sample block selections were: >5% tumor infiltration in the uninvolved sample, acute pancreatitis, or >5% acinar autolysis. The study protocol for retrospective pathology report review and analyses of beta cells was approved by the University of Florida Institutional Review Board as an exempt protocol with waiver of informed consent.
Table 1.
Study patients and demographics
| Clinical diagnosis | Histopathology | Gendera | Age (years) | Ethnicityb |
|---|---|---|---|---|
| Autopsy control | No pancreatitis | F | 20 | C |
| Autopsy control | No pancreatitis | F | 27 | C |
| Autopsy control | No pancreatitis | F | 57 | C |
| Autopsy control | No pancreatitis | F | 71 | C |
| Autopsy control | No pancreatitis | M | 55 | C |
| Pancreatic adenocarcinoma | No pancreatitis | F | 36 | B |
| Pancreatic adenocarcinoma | No pancreatitis | F | 47 | C |
| Pancreatic adenocarcinoma | No pancreatitis | F | 69 | C |
| Pancreatic adenocarcinoma | No pancreatitis | M | 58 | - |
| Pancreatic adenocarcinoma | No pancreatitis | M | 77 | C |
| Pancreatic adenocarcinoma | No pancreatitis | M | 82 | - |
| Pancreatic adenocarcinoma | Mild pancreatitis | F | 24 | B |
| Pancreatic adenocarcinoma | Mild pancreatitis | F | 48 | C |
| Pancreatic adenocarcinoma | Mild pancreatitis | F | 48 | C |
| Pancreatic adenocarcinoma | Mild pancreatitis | F | 62 | C |
| Pancreatic adenocarcinoma | Severe pancreatitis | F | 76 | - |
| Pancreatic adenocarcinoma | Severe pancreatitis | M | 43 | C |
| Pancreatic adenocarcinoma | Severe pancreatitis | M | 54 | C |
| Pancreatic adenocarcinoma | Severe pancreatitis | M | 55 | H |
| Hypergastrinaemia | No pancreatitis | F | 21 | C |
| Hypergastrinaemia | No pancreatitis | F | 34 | C |
| Hypergastrinaemia | No pancreatitis | F | 48 | C |
| Hypergastrinaemia | No pancreatitis | F | 53 | C |
| Hypergastrinaemia | No pancreatitis | F | 70 | C |
| Hypergastrinaemia | No pancreatitis | M | 79 | C |
F, Female; M, Male
B, African American; H, Hispanic; C, Caucasian; -, not available.
Pancreatic histology
Paraffin blocks were obtained from the Shands hospital archive. The standard histological procedure was fixation in 10% zinc formalin followed by paraffin embedding. Paraffin blocks were serially sectioned (4 µm) and sections were stained with aldehyde fuschin and hematoxylin and eosin (AF-H&E) followed by immunolocalization studies. AF-H&E stained slides were reviewed by two investigators to confirm histopathological findings and exclusion criteria.
Immunohistochemistry and immunofluorescence
Immunolocalization was performed with primary and secondary antibodies (ESM Table 1) using detection methods as previously described [24]. In brief, deparaffinized sections were incubated in 3% hydrogen peroxide in methanol for 10 min to block endogenous peroxidase activity. Antigen retrieval was performed, when required, by heating samples at 95˚C for 25 min in citrate buffer, pH 6.0 (Dako, Carpentina, CA, USA). Sections were incubated in 10% goat or rabbit serum and blocked for endogenous avidin-biotin binding (Vector Labs, Burlingame, CA, USA) when required. The primary antibody was incubated for 1 hr at room temperature or overnight at 4˚C. Following extensive washing, the secondary antibody was applied and the antibody complex detected using a Vectastain ABC kit with diaminobenzidine (DAB) chromagen (Vector Labs, Burlingame, CA, USA) or the MACH2 polymer system used with Vulcan Fast Red chromagen (Biocare Medical, Concord, CA, USA). Double staining was performed using sequential application and detection of each primary antibody. Sections were lightly counterstained in hematoxylin and mounted. Slides were photographed with a Zeiss Axioskop and Axiocam HR color camera or scanned with a ScanScope CS imaging system (Aperio, Vista, CA, USA).
For double staining immunofluorescence, Ki-67 was detected using Alexa Fluor 488-conjugated secondary antibodies (Invitrogen Molecular Probes, Carlsbad, CA, USA) followed by insulin detection using an Alexa Fluor 594-conjugated secondary antibody. Nuclei were stained with 4,6-diamidio-2-phenylindole (DAPI) (Vectashield, Vector Labs, USA). Sections were photographed using the Zeiss Axioskop with a dual FITC/Texas Red filter for visualization of both fluorochromes using Axiovision 4.0 software. The same exposure settings were used for all samples.
Incubation conditions for primary antibodies were optimized using control human tissue samples and testing several enzymatic and heat-based antigen retrieval methods. A positive and negative control sample was included in each assay run to allow inter-assay comparisons. Negative controls included substitution of the primary with immunoglobulin (IgG) from the host species used to generate the primary antibody and by incubation with primary antibody dilution buffer alone.
Morphometric analysis
Fractional insulin area was calculated as the ratio of insulin immunoreactive area/total pancreatic area per field of view (5–10 non-overlapping acinar regions; 20x objective) using Metamorph 6.0 software (Molecular Devices Corporation, Sunnyvale, CA, USA) for immunofluorescence. Insulin positive cells were selected by color using their red fluorescence followed by selection of all colors for total acinar tissue. Fractional insulin area was also determined on whole slide scans using the positive pixel count algorithm in ImageScope 9.0 (Aperio, Vista, CA, USA). The pen tool was used to exclude regions of fibrosis in pancreatic tumors samples.
To quantify beta cells with Ki-67 co-localization, 5–10 non-overlapping fields of view (20x objective) were photographed using a dual FITC/Texas Red filter set. Insulin-positive cells in each field of view were manually counted using the image zoom function on a 21” video monitor to aid discrimination. Ki-67-positive beta cell nuclei were counted and distribution recorded in islets, clusters (2–6 positive cells), or as single cells. Total beta cell number per field of view was calculated from a summation of islet, cluster, and single cell counts. Criteria for a Ki-67-positive beta cell required a Ki-67-positive nucleus of similar size to other beta cell nuclei within an insulin-positive cytoplasm. Analyses were performed by staff blinded to patient tumor group.
Immunohistochemistry scoring
PDX1, NEUROD1, and NEUROG3 immunopositivity was scored in nuclei of cells in islets, ductular epithelium, and acinar regions. Each patient had at least five medium power field of views (40x objective) randomly selected that contained islets and ducts each with a minimum of 25 cells. Percentage of stained nuclei was estimated. For acinar cells, five high power fields of view (60x objective) were randomly selected and an overall percentage of nuclear staining estimated.
Statistical analysis
Data are shown as mean ± SEM with N = number of patients. Comparisons between patient groups were carried out by single factor ANOVA and post-hoc t-test (two-tailed). A p value < 0.05 was considered statistically significant. A two sample test using average values for single beta cell numbers was performed using the statistical power calculator (Researcher’s tool kit, DSS Research, Fort Worth, TX).
Results
Routine histopathological examination of pancreata showed no significant findings in normal autopsy controls or patients with gastrinoma, other than mild focal fibrosis in the latter (Fig. 1). Samples of pancreata from patients with adenocarcinoma and chronic pancreatitis had varying degrees of perilobular fibrosis, ductular proliferation or hyperplasia, and acinar atrophy that was heterogeneous between lobules (Fig. 1). Islet inflammation (insulitis) was not observed in any sample as assessed by the lack of several mononuclear cells surrounding or within the islet by AF-H&E staining. Insulin staining by acinar region was variable within patients and was recognized in islets of varying sizes, small clusters, and single cells (Fig. 1, 2). Using cytokeratin 7 immunostaining to localize duct epithelium, insulin-positive cells were infrequently observed within the ductular epithelium in all groups (Fig. 2).
Fig. 1.

Pancreatic histopathology in uninvolved regions from patients with primary adenocarcinoma. Paraffin sections were stained with AF-H&E to delineate insulin-positive beta cells (dark purple). a Section from a patient with adenocarcinoma and no pancreatitis (1.25x). Red dye at lower margin delineates orientation towards the adenocarcinoma. Mild intralobular adipose tissue was present. b Focal ductular proliferation in a patient with no pancreatitis. Cross sections of ducts are visible (arrows) in addition to a large group of cells that are negative by AF-H&E staining. Elastic fibers in a large blood vessel stain dark purple with AF-H&E. c Pancreatic morphology in a patient with chronic mild pancreatitis. Islets of varying sizes are observed in different lobules. Focal ductular hyperplasia (arrow) is observed in a large duct with multifocal mild adipose tissue (clear spaces) in the acinar regions. d Pancreatic morphology in a patient with chronic severe pancreatitis. Focal, mild acinar atrophy was observed (white star) with focal, moderate ductular proliferation and hyperplasia (arrows).
Fig. 2.

Pancreatic immunohistochemistry for pancreatic ductular epithelium and insulin. Pancreatic sections were double stained for cytokeratin 7 (CK7, brown) and insulin (red) as described in Methods. CK7 immunopositivity was found in ducts and centroacinar cells (a-c, black arrows) adjacent to unstained acinar cells. Insulin positive cells were identified in close approximation to or within ductular epithelium in all patients groups. Insulin positive cells were found as single cells (a, d, white arrows) or clusters (a-d) and were only rarely observed in ducts with hyperplasia (d).
As previous studies have suggested that beta cell mass may be influenced by a variety of pathological conditions including those providing the focus for this report, we considered it important to address the issue of fractional insulin area. Fractional insulin area provides a macro-like assessment of the contribution of all insulin-positive cells as an assessment of beta cell mass [21, 25–27]. Using this well accepted method of analysis, very limited differences in fractional insulin area were noted amongst the five study groups using immunohistochemistry ( 1.4%±0.1% autopsy control; 1.1%±0.1% no pancreatitis; 2.0±0.2% mild pancreatitis; 1.8±0.7% severe pancreatitis; 1.5%±0.2% gastrinoma). Indeed, the only comparison within the five study groups reaching statistical significance was in comparing patients with no pancreatitis to those with severe pancreatitis using the immunofluorescence field of view measurements, where a significantly increased fractional area was noted in the latter subjects (3.2± 0.5% versus 5.9±1.4%, respectively, (p<0.05)). Fractional insulin area in the samples from autopsy patients was 4.1% ± 0.6 which was slightly lower than that from patients with mild pancreatitis (5.2%± 0.9%) or hypergastrinaemia (5.3%±1.0%).
The numbers of insulin positive cells in islets, clusters, or as single cells were quantified and the total number of insulin positive cells was calculated as a summation of these three distributions (Fig. 3a). These data represent a total of 20,693 beta cell counts (i.e., 4451 autopsy control; 3407 no pancreatitis; 3731 mild pancreatitis; 4122 severe pancreatitis; and 4982 gastrinoma). Similar to the fractional insulin area data, total beta cell numbers were highest in the severe pancreatitis group, yet these data were also the most variable (209±89, Fig. 3a). The no pancreatitis group had the lowest total beta cell number (101±13) compared to autopsy controls (178±35, p=0.053), mild pancreatitis (173±26, p=0.027), and gastrinoma groups (200±45, p=0.046) (Fig. 3a). Islet beta cell numbers were constant between all groups except for the no pancreatitis group which was significantly lower than the mild pancreatitis group (92±13 versus 157±20, respectively, p=0.020) (Fig. 3b). These two groups showed the lowest variability in beta cell counts. Beta cell numbers in clusters were similar among autopsy controls (15±4) and patients with no (6±1) or mild pancreatitis (10±5) (Fig. 3c). These three groups were lower than those with severe pancreatitis (45±15, p=0.051 compared to autopsy controls, p=0.008 compared to no pancreatitis, p=0.057 compared to mild pancreatitis). Furthermore, gastrinoma patients revealed significantly increased beta cell numbers in clusters when compared to the no pancreatitis group (27±6, p=0.005) as for the severe pancreatitis group. Strikingly, the largest increase in beta cell counts was observed in the single beta cell compartment, and indeed, both severe pancreatitis (21.3±2.2) and gastrinoma (19.6±5.1) groups were significantly higher (p<0.005) when compared to controls (3.6±0.9) and no (2.4±0.4) or mild (5.2±2.5) pancreatitis study groups (statistical power analysis = 99%, α=0.05, β=0.80) (Fig. 3d).
Fig. 3.

Beta cell numbers and distribution within islets or clusters, or as single cells. Beta cells were identified by anti-insulin immunofluorescence, images acquired using a 20x objective, and positive cells counted per field of view. a Mild pancreatitis and gastrinoma subjects had significantly higher total beta cell numbers compared to patients with no pancreatitis. b Beta cell numbers within islets were lowest in the no pancreatitis group when compared to patients with mild pancreatitis (*p=0.02). c Beta cell numbers within clusters were significantly higher in severe pancreatitis and gastrinoma patients compared to no pancreatitis patients (*p<0.01). d Patients with severe pancreatitis and gastrinoma had significantly higher beta cell counts as single cells compared to controls and both no and mild pancreatitis groups (***p<0.005).
For assessment of beta cell replication, Ki-67 nuclear staining in insulin positive cells was used as an index of the cellular proliferation rate. Of the aforementioned 20,693 beta cells analyzed, eighty-six beta cells had Ki-67-positive nuclei (overall 0.42%). Beta cell proliferation rates, expressed as either the %Ki-67 positive cells for total or islet beta cells, were significantly increased (p<0.005) in the severe pancreatitis group compared to all other groups (Fig. 4). Beta cell proliferation rates within clusters were similar between groups (overall 0.2%±0.1%) (Fig.4). Single beta cells showed highly variable proliferation rates ith no significant differences between groups, however, the severe pancreatitis group showed the highest single beta cell proliferation rate (3.4%±1.7%) (Fig.4). As age has been reported to influence beta cell replication rates [21], patient group ages were analyzed for statistical differences and none were found (autopsy control, 43±8 (N=5); pancreatic carcinoma- no pancreatitis, 57±7 (N=5); pancreatic carcinoma- mild pancreatitis, 46±8 (N=4); pancreatic carcinoma- severe pancreatitis, 62±7 (N=5); gastrinoma-no pancreatitis, 44±7, (N=6)).
Fig. 4.

Beta cell proliferation rates determined by Ki-67 levels. Proportion of Ki-67–positive beta cells are shown for each group expressed as total cells or beta cells within islets, clusters, and single cells. Despite variability, the severe pancreatitis group showed significantly higher proportions of total Ki-67-positive beta cells (***p<0.005) compared to all other groups. The average count of Ki-67-positive beta cells in islets was also significantly higher in the severe pancreatitis patients compared to all other groups (***p<0.005). Ki-67 positivity was most variable within single beta cells. Legend: Control (white); No pancreatitis (stippled); Mild pancreatitis (light grey); Severe pancreatitis (dark grey); Gastrinoma (black).
Pancreata samples from all study groups were examined for levels of three transcription factors (i.e., PDX1, NEUROD1, and NEUROG3) associated with the differentiation of pancreatic endocrine precursors into adult beta cells. Surprisingly, transcription factor levels in islets from the autopsy control samples obtained at autopsy were the lowest compared to all other groups (Fig. 5d). These data suggested that pancreatic sample harvesting post-mortem could alter levels for these transcription factors.
Fig. 5.
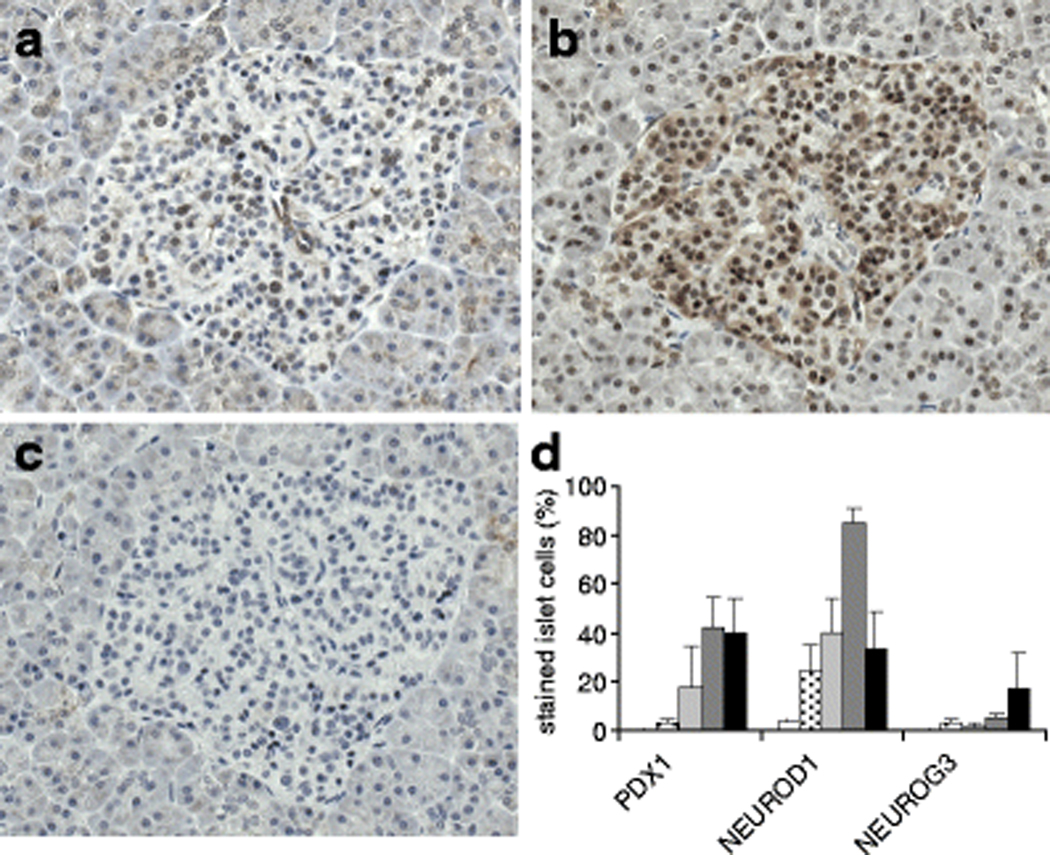
PDX1, NEUROD1, and NEUROG3 levels in pancreatic islets. Representative islet images showing immunostaining for PDX1 (a), NEUROD1 (b), and NEUROG3 (c) in serial sections from a patient with mild pancreatitis. d Semi-quantitative analysis of percentage nuclear staining for PDX1, NEUROD1, and NEUROG3 staining in islets. The level of PDX1 was significantly higher in severe pancreatitis and gastrinoma patients compared to both autopsy control and no pancreatitis patients (**p<0.03). NEUROD1 was significantly higher in severe pancreatitis patients compared to all other groups (***p<0.02). Subjects with either mild pancreatitis or gastrinoma also showed intermediate NEUROD1 levels when compared to controls (*p<0.05). NEUROG3 levels were lower than PDX1 or NEUROD1 and no significant differences were observed between groups. Legend: Control (white); No pancreatitis (stippled); Mild pancreatitis (light grey); Severe pancreatitis (dark grey); Gastrinoma (black).
PDX1 immunoreactivity was found predominantly in islet cells (Fig. 5a, d) with low levels in pancreatic ducts and acinar cells. PDX1 islet levels were significantly higher in both severe pancreatitis patients (42%±13%, p=0.009) and subjects with gastrinoma (40%±14%, p=0.02) compared to patients with no pancreatitis (3%±2%) (Fig. 5d). As indicated, autopsy control patients had virtually absent PDX1 levels (0.2%±0%) (Fig. 5d). Interestingly, patients with mild pancreatitis had intermediate yet highly variable PDX1 levels (18%±10%).
In contrast to the largely restricted islet levels for PDX1, NEUROD1 was observed in islets (Fig. 5b) as well as ducts and acinar cells. The proportion of NEUROD1-positive islets cells was significantly higher in patients with severe pancreatitis (85%±6%, p< 0.02) compared to all other groups (Fig. 5d). Subjects with either mild pancreatitis (40%±14%) or gastrinoma (34%±14%) showed moderately higher islet NEUROD1 levels compared to patients without pancreatitis (24%±11%) (Fig. 5d). NEUROD1 in ducts and acinar cells was higher in both severe pancreatitis patients and gastrinoma patients compared to patients with no or mild pancreatitis, yet variability precluded demonstration of significant differences (data not shown). As expected for adult tissues, NEUROG3 levels were lower than those for PDX1 and NEUROD1 (Fig. 5c, d). In addition, while no single study group demonstrated statistically significant differences in NEUROG3 islet level, it was observed that patients with gastrinoma had the highest levels of all study groups (18%±14%) (Fig. 5d
Discussion
Studies on the regulation of beta cell mass show a remarkable degree of plasticity exists for these cells in response to a variety of experimental and physiological conditions ranging from pregnancy to obesity. As noted earlier, additional information would suggest that pancreatic disorders associated with elevations in localized cytokine production could increase beta cell mass. In theory, cytokines released from the inflammatory cells or the tumor itself could induce beta cell replication [19]. Intriguing new data show that immune cell infiltration in islets also triggers beta cell replication [28]. Gastrin is a potent growth factor for the entire gastrointestinal tract and is expressed in fetal islet cells [29, 30]. Excessive serum gastrin levels could produce profound growth effects on cell types expressing gastrin receptors including pancreatic ductular epithelium and acinar cells [25, 31]. To address our hypothesis that beta cell replication would be observed in non-autoimmune human pancreatic disorders in which localized inflammation or elevation in gastrin were present in subjects with chronic pancreatitis, we performed a series of studies assessing the fractional insulin area as a marker of beta cell mass, followed by detailed analysis of beta cell numbers and replication rates within three different cellular compartments.
Marked heterogeneity in the fractional insulin area of autopsy subjects has been noted, with variance of up to 80-fold in the same patient [32]. Our study reiterates variability in fractional insulin area while also showing the trend for patients with no pancreatitis to show the lowest fractional insulin area. Fractional insulin area was mirrored by enumeration of total and islet beta counts. Thus, our data, despite a relatively small sample set, are consistent with others who report that patients with pancreatic adenocarcinoma can show decreased beta cell numbers in islets [33, 34]. Since these differences were not significantly different from autopsy controls, these patients would not be expected to have alterations in blood glucose though we were not able to evaluate this last question, owing to the nature of our Institutional review board approval. Of note, the presence of pancreatitis, whether chronic mild or severe, resulted in fractional insulin area and total and islet beta cell counts at levels comparable to autopsy controls and gastrinoma patients.
Importantly, our studies suggest that analysis of beta cell distribution rate could prove more sensitive than fractional insulin area for accurately accounting for the dynamics of beta cell homeostasis. Stated differently, summation of all the beta cells, within well-formed islets or appearing as isolated cells or clusters, is necessary to account for total beta cell mass. Indeed, our data indicate that beta cells appearing as single cells show significant plasticity in severe pancreatitis patients as well as in subjects with gastrinoma patients. While exocrine tissue area can be expected to be reduced with severe pancreatitis, these data showing that both severe pancreatitis and gastrinoma patients had increased numbers of single beta cells, would tend to argue that single beta cells were not more discernable in severe pancreatitis samples.
The prevailing thought holds that beta cell replication in adult pancreas is rare. Indeed, carefully sorted human beta cells from islet preparations failed to replicate in vitro in marked contrast to beta cells isolated from rat islets [35]. However, recent reports do suggest that the capacity for life-long beta cell replication exists in humans [21, 28, 32, 36]. While such data in humans are limited, autopsy-based samples showed that 0.04% of human beta cells express Ki67 [32]. Furthermore, low beta cell replication rates using Ki-67 co-localization (0.13% ± 0.08%) are reported in autopsy control subjects [36]. Similar to those reports, we observed consistent low proportions of total Ki-67-positive beta cells in islets across all study groups (~0.3%) except when severe pancreatitis (1.5%±0.2) was present. Although significantly different from other groups, this low proportion of beta cells undergoing proliferation was not expected to alter fractional insulin area, as indeed was found.
While the current study is limited due to the small case numbers and expected variability associated with cell counting methodologies, our study supports a recent case report that showed increased islet beta cell replication rates in a 89-year old patient with recent onset type 1 diabetes and pancreatic adenocarcinoma [32]. Taken together, these studies show the potential for beta cell replication to continue during adulthood.
As suggested by many investigators, the absence of an increase in periductal islets or intraductal insulin-positive cells implies that beta cell regeneration in humans is accomplished predominately by beta cell replication [21]. Islet ductal complexes and isolated insulin positive cells in ducts have also been reported in pancreata obtained from autopsy specimens and organ donors [21, 37]. Similarly, we observed insulin positive cells in ducts from all patient groups. While the increase in single beta cell numbers in both the severe pancreatitis group and patients with gastrinoma was not associated with a simultaneous increase in insulin positive cells in ductal complexes or ducts, the exact origin of the single beta cells remains unknown.
We elected to characterize levels of beta cell differentiation factors within islets as well as ducts and acinar regions. During fetal pancreatic endocrine development, PDX1 specifies the early pancreatic epithelium, permitting its proliferation, branching, and subsequent differentiation [38]. In the current study, PDX1 levels were moderately increased in islets from both severe pancreatitis and gastrinoma samples yet were not found in ducts. Our data contrast those showing ductal levels of PDX1 in chronic pancreatitis [20]; however, since the primary antibodies were different, this and differences in staging of pancreatitis could account for this discrepancy. NEUROD1 and NEUROG3 are also critical regulators of endocrine cell fate specification in the pancreas [26, 39]. NEUROD1 levels were significantly increased in islets from patients with severe pancreatitis yet, as expected; NEUROG3 levels were low in adult cells.
In conclusion, beta cells in humans with chronic severe pancreatitis demonstrated increased single beta cell numbers, islet beta cell Ki-67 replication rates, and islet NEUROD1 levels. Despite these alterations, total beta cell numbers, fractional insulin area, and apoptosis rates remained similar to other study groups. We believe these data are important for ongoing efforts seeking to convey human beta cell regeneration in vivo. While these current studies certainly provide clues as to the nature of cytokine-induced beta cell proliferation, future studies would be aided by the inclusion of additional patients, freshly obtained specimens, and the inclusion of additional immunological (e.g., monitors of lymphoid and myeloid phenotype, cytokine expression, apoptosis, etc.) and developmental (e.g., nestin) markers. Indeed, if the specific factors imparting beta cell replication induced by chronic pancreatitis can be identified, these mediators could assist with the development of therapeutic strategies for restoring beta cell mass in settings of insulin deficiency.
Supplementary Material
Acknowledgements
These studies were funded by the National Institutes of Health (PO1 42288), Juvenile Diabetes Research Foundation, Sebastian and Keene Family Endowments, and Department of Pathology Clinical Research funds. We are grateful to Melissa Chen, Erkya Gayle, and Lauren Tenace (Department of Pathology, University of Florida) for providing excellent technical support.
Abbreviations
- AF-H&E
Aldehyde fuschin and hematoxylin and eosin
- CK7
Cytokeratin 7
- DAPI 4
6-diamidio-2-phenylindole
- DAB 3
3-diaminobenzidine
- EGF
Epidermal growth factor
- Ki-67
Ki-67 antigen
- NOD
Non-obese diabetic mice
- NEUROD1
Neurogenic differentiation 1
- NEUROG3
Neurogenin 3
- PDX1
Pancreatic and duodenal homeobox 1
Footnotes
Duality of interest
One author (JMC) serves on the Medical Advisory Board for the Aperio Corporation. The authors otherwise declare that there is no duality of interest associated with this manuscript.
References
- [1].Atkinson MA, Rhodes CJ (2005) Pancreatic regeneration in type 1 diabetes: dreams on a deserted islet? Diabetologia 48: 2200–2202 [DOI] [PubMed] [Google Scholar]
- [2].Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC (2005) Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48: 2221–2228 [DOI] [PubMed] [Google Scholar]
- [3].Bonner-Weir S, Weir GC (2005) New sources of pancreatic beta-cells. Nat Biotechnol 23: 857–861 [DOI] [PubMed] [Google Scholar]
- [4].Bouwens L (2006) Beta cell regeneration. Curr Diabetes Rev 2: 3–9 [DOI] [PubMed] [Google Scholar]
- [5].Lipsett M, Aikin R, Hanley S, Al-Maleek J, Laganiere S, Rosenburg L (2006) Islet neogenesis: a potential therapeutic tool in type 1 diabetes. Int J Biochem Cell Biol 38: 715–720 [DOI] [PubMed] [Google Scholar]
- [6].Nir T, Melton DA, Dor Y (2007) Recovery from diabetes in mice by beta cell regeneration. The Journal of clinical investigation 117: 2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bonner-Weir S, Sharma A (2006) Are there pancreatic progenitor cells from which new islets form after birth? Nature clinical practice 2: 240–241 [DOI] [PubMed] [Google Scholar]
- [8].Menge BA, Tannapfel A, Belyaev O, et al. (2008) Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 57: 142–149 [DOI] [PubMed] [Google Scholar]
- [9].Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L (2005) In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia 48: 49–57 [DOI] [PubMed] [Google Scholar]
- [10].Yang L, Li S, Hatch H, et al. (2002) In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proceedings of the National Academy of Sciences of the United States of America 99: 8078–8083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Butler AE, Huang A, Rao PN, et al. (2007) Hematopoietic stem cells derived from adult donors are not a source of pancreatic beta-cells in adult nondiabetic humans. Diabetes 56: 18101816 [DOI] [PubMed] [Google Scholar]
- [12].Brand SJ, Tagerud S, Lambert P, et al. (2002) Pharmacological treatment of chronic diabetes by stimulating pancreatic beta-cell regeneration with systemic co-administration of EGF and gastrin. Pharmacol Toxicol 91: 414–420 [DOI] [PubMed] [Google Scholar]
- [13].Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A (2005) Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes 54: 2596–2601 [DOI] [PubMed] [Google Scholar]
- [14].Tomassetti P, Migliori M, Lalli S, Campana D, Tomassetti V, Corinaldesi R (2001) Epidemiology, clinical features and diagnosis of gastroenteropancreatic endocrine tumours. Ann Oncol 12 Suppl 2: S95–99 [DOI] [PubMed] [Google Scholar]
- [15].Meier JJ, Butler AE, Galasso R, Rizza RA, Butler PC (2006) Increased islet beta cell replication adjacent to intrapancreatic gastrinomas in humans. Diabetologia 49: 2689–2696 [DOI] [PubMed] [Google Scholar]
- [16].Jones EM, Sarvetnick N (1997) Islet regeneration in IFNgamma transgenic mice. Hormone and metabolic research Hormon- und Stoffwechselforschung 29: 308–310 [DOI] [PubMed] [Google Scholar]
- [17].Luo X, Yang H, Kim IS, et al. (2005) Systemic transforming growth factor-beta1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice. Transplantation 79: 1091–1096 [DOI] [PubMed] [Google Scholar]
- [18].Maedler K, Schumann DM, Sauter N, et al. (2006) Low concentration of interleukin1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 55: 2713–2722 [DOI] [PubMed] [Google Scholar]
- [19].Smith U (2007) Introduction: Symposium on diabetes, inflammation and cardiovascular disease. J Intern Med 262: 142–144 [DOI] [PubMed] [Google Scholar]
- [20].Phillips JM, O’Reilly L, Bland C, Foulis AK, Cooke A (2007) Patients with chronic pancreatitis have islet progenitor cells in their ducts, but reversal of overt diabetes in NOD mice by anti-CD3 shows no evidence for islet regeneration. Diabetes 56: 634–640 [DOI] [PubMed] [Google Scholar]
- [21].Meier JJ, Butler AE, Saisho Y, et al. (2008) Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57: 15841594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sumi S, Gu Y, Hiura A, Inoue K (2004) Stem cells and regenerative medicine for diabetes mellitus. Pancreas 29: e85–89 [DOI] [PubMed] [Google Scholar]
- [23].Zhang YQ, Sarvetnick N (2003) Development of cell markers for the identification and expansion of islet progenitor cells. Diabetes/metabolism research and reviews 19: 363–374 [DOI] [PubMed] [Google Scholar]
- [24].Zhang B, Lu Y, Campbell-Thompson M, et al. (2007) Alpha1-antitrypsin protects betacells from apoptosis. Diabetes 56: 1316–1323 [DOI] [PubMed] [Google Scholar]
- [25].Baggio LL, Drucker DJ (2006) Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med 57: 265–281 [DOI] [PubMed] [Google Scholar]
- [26].Bouwens L, Rooman I (2005) Regulation of pancreatic beta-cell mass. Physiol Rev 85: 1255–1270 [DOI] [PubMed] [Google Scholar]
- [27].Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110 [DOI] [PubMed] [Google Scholar]
- [28].In’t Veld P, Lievens D, De Grijse J, et al. (2007) Screening for Insulitis in Adult Autoantibody-Positive Organ Donors. Diabetes [DOI] [PubMed] [Google Scholar]
- [29].Greider MH, McGuigan JE (1971) Cellular localization of gastrin in the human pancreas. Diabetes 20: 387–396 [PubMed] [Google Scholar]
- [30].Rehfeld JF, Friis-Hansen L, Goetze JP, Hansen TV (2007) The biology of cholecystokinin and gastrin peptides. Current topics in medicinal chemistry 7: 1154–1165 [DOI] [PubMed] [Google Scholar]
- [31].Dockray G, Dimaline R, Varro A (2005) Gastrin: old hormone, new functions. Pflugers Arch 449: 344–355 [DOI] [PubMed] [Google Scholar]
- [32].Meier JJ, Lin JC, Butler AE, Galasso R, Martinez DS, Butler PC (2006) Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia [DOI] [PubMed] [Google Scholar]
- [33].Katsumichi I, Pour PM (2007) Diabetes mellitus in pancreatic cancer: is it a causal relationship? American journal of surgery 194: S71–75 [DOI] [PubMed] [Google Scholar]
- [34].Schmied BM, Ulrich AB, Friess H, Buchler MW, Pour PM (2001) The patterns of extrainsular endocrine cells in pancreatic cancer. Teratog Carcinog Mutagen 21: 69–81 [DOI] [PubMed] [Google Scholar]
- [35].Parnaud G, Bosco D, Berney T, et al. (2008) Proliferation of sorted human and rat beta cells. Diabetologia 51: 91–100 [DOI] [PubMed] [Google Scholar]
- [36].Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B (2000) Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49: 1325–1333 [DOI] [PubMed] [Google Scholar]
- [37].Gianani R, Putnam A, Still T, et al. (2006) Initial results of screening of nondiabetic organ donors for expression of islet autoantibodies. J Clin Endocrinol Metab 91: 1855–1861 [DOI] [PubMed] [Google Scholar]
- [38].Fellous TG, Guppy NJ, Brittan M, Alison MR (2007) Cellular pathways to beta-cell replacement. Diabetes/metabolism research and reviews 23: 87–99 [DOI] [PubMed] [Google Scholar]
- [39].Mellitzer G, Martin M, Sidhoum-Jenny M, et al. (2004) Pancreatic islet progenitor cells in neurogenin 3-yellow fluorescent protein knock-add-on mice. Mol Endocrinol 18: 2765–2776 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.