

Abstract
Adulteration remains an issue in the dietary supplement industry, including botanical supplements. While it is common to employ a targeted analysis to detect known adulterants, this is difficult when little is known about the sample set. With this study, untargeted metabolomics using liquid chromatography coupled to ultraviolet-visible spectroscopy (LC-UV) or high-resolution mass spectrometry (LC-MS) was employed to detect adulteration in botanical dietary supplements. A training set was prepared by combining Hydrastis canadensis L. with a known adulterant, Coptis chinensis Franch., in ratios ranging from 5% to 95% adulteration. The metabolomics datasets were analyzed using both unsupervised (principal component analysis and composite score) and supervised (SIMCA) techniques. Palmatine, a known H. canadensis metabolite, was quantified as a targeted analysis comparison. While the targeted analysis was the most sensitive method tested in detecting adulteration, statistical analyses of the untargeted metabolomics datasets detected adulteration of the goldenseal samples, with SIMCA providing the greatest discriminating potential.
Keywords: metabolomics, goldenseal, Hydrastis canadensis, mass spectrometry, principal component analysis, dietary supplements
1. Introduction
The 2017 Council for Responsible Nutrition survey found that botanicals make up 39% of the total dietary supplement usage in the United States, the overall use of which has increased by 8% since 2015 [1]
Botanical dietary supplements encompass a wide range of over-the-counter products including capsules, tea, tinctures, and loose powders prepared from plant material. The Dietary Supplement Health and Education Act (DSHEA) of 1994 assigns the Federal Drug Administration (FDA) regulatory oversight of dietary supplements; however, regulation and quality control of these products is challenging due to their inherent complexity and variability, and because the landscape of companies is vast and constantly changing [2]. These regulatory and analytical challenges constitute a problem since contaminated or adulterated product may put the consumer at risk of adverse interactions [3].
A botanical product is considered adulterated when the composition reported on the label does not match the actual material being sold [4]. This problem can occur due to limited availability of the natural product (either from cultivation or ethical and legal wildcrafting), economic incentives to substitute other natural products or introduce other compounds, or poor quality control during production [5, 6]. While most adulteration and quality control are monitored by targeted analytical methods [7, 8], untargeted metabolomics methodologies have been employed to detect unknown adulteration in botanical dietary supplements [9–12].
One botanical product for which there are known issues with contamination and adulteration is Hydrastis canadensis L. (Ranunculaceae), commonly known as goldenseal [4, 5]. While the benzylisoquinoline alkaloid berberine is present in goldenseal and frequently attributed as the main bioactive principle, it is common across a wide variety of plants including Berberis vulgaris L. (Berberidaceae), Mahonia aquifolium (Pursh) Nutt. (Berberidaceae), and Coptis chinensis Franch. (Ranunculaceae) [13, 14]. However, beyond berberine, these other species possess distinct secondary metabolite profiles from that of goldenseal; two defining secondary metabolites found in goldenseal are hydrastine and canadine, which are absent in other berberine-containing plants [4, 9, 15], while B. vulgaris (barberry), M. aquifolium (Oregon grape), and C. chinensis (Chinese goldthread) all have additional alkaloids (e.g.,coptisine, dihydrocoptisine, palmatine, and jatrorrhizine) that are not present in goldenseal [16–18]. The presence of these marker compounds are signs of possible adulteration; however, trace amounts of contamination may or may not be detectable and it is difficult to detect adulteration when the identity of the adulterants are unknown.
Both targeted and untargeted methods are used to interpret the data obtained from mass spectrometric analysis of mixtures. For a targeted analysis, the analyst chooses a series of analytes a priori and analyzes the dataset to determine whether these analytes are present. Targeted analyses have the advantage of higher sensitivity and specificity compared to untargeted methodologies but requires knowledge about the sample and the potential adulterant prior to analysis.
Untargeted metabolomics methods have the advantage of comparing multiple complex products without any a priori knowledge of their composition or identification of major metabolites [19]. While it is not possible to measure the entirety of small molecules produced by an organism due to analytical limitations, by detecting as many of these small molecules as possible, untargeted metabolomics approaches enable a holistic analysis in comparing complex samples [20, 21]. Metabolomics has been utilized in a wide variety of applications in the natural product industry including natural product drug discovery [22], dietary supplement adulteration [9], and botanical products (e.g., green tea, goldenseal, Ginkgo biloba, black cohosh, and ginseng) for authenticity and possible adulteration/contamination [9, 11, 21, 23–29]. In analyzing for potential adulteration, the variations in metabolite profiles can represent alterations in the chemical composition, which could be attributed to naturally occurring biological or genetic variability in the source material [26, 27]. This variance is typically visualized using unsupervised statistical analysis (principal component analysis, PCA) and followed up with a supervised statistical analysis (soft independent modelling of class analogy, SIMCA) for a more quantitative assessment of outliers. A 95% confidence interval, calculated using Hotelling’s T2 [30, 31] or Q statistic [32, 33], may be applied to unsupervised statistical analysis to give a mathematical representation of outliers in addition to a visual interpretation [32].
Several studies have been conducted to assess the authenticity of goldenseal supplements, including targeted quantitative analysis [28], untargeted Fourier-transform near-infrared spectroscopy (FT-NIR) analysis [10], and untargeted ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS) metabolomics [9, 12]. Some of these methods employed targeted analysis of known metabolites of goldenseal using HPLC-UV and GC-MS [8, 10, 15], and compounds from adulterating species were found in several of the commercial products [15].
An untargeted metabolomics study using FT-NIR analysis assessed a sample set comprised of goldenseal and common adulterants [34]. In this study, goldenseal adulteration was simulated computationally for four different adulterant species, yellow dock (Rumex crispus L., Polygonaceae), yellow root (Xanthorhiza simplicissima Marshall, Ranunculaceae), goldenthread (Coptis chinensis Franch., Ranunculaceae), and Oregon grape (Mahonia aquifolium (Pursh) Nutt., Berberidaceae) [34]. Employing two supervised statistical analyses, SIMCA and PLS (partial least squares), a 5% adulteration level (i.e., 95% goldenseal, 5% adulterant) was identified as a statistical outlier [34].
In the current study, goldenseal reference materials were physically (rather than computationally as in the study by Liu et al., 2018) blended with C. chinensis plant material to form a series of intentionally adulterated products. These products were analyzed using a metabolomics approach designed to detect adulteration in goldenseal products [9], while changing several analytical and statistical variables to compare approaches. Data were acquired from two different platforms: an LC-MS system featuring a hybrid quadrupole-Orbitrap mass analyzer and an LC-UV system. For the data analytics aspect of the study, multiple statistical procedures were employed to compare analysis of the resulting datasets. Composite score analysis, PCA, and SIMCA were contrasted to compare unsupervised versus supervised analysis. In addition, quantitative analysis of palmatine, a common adulterant of goldenseal, was performed to serve as a targeted analysis comparison against the untargeted methods. The goal of this study was to compare the sensitivity of outlier detection with different analytical platforms and statistical approaches.
2. Materials and Methods
2.1. Solvents and Samples
All solvents and chemicals used were of reagent or spectroscopic grade, as required, and obtained from ThermoFisher Scientific (Waltham, MA, USA) or Cayman Chemical (Ann Arbor, MI, USA). A palmatine chloride standard was purchased from Chromadex (Irvine, CA, USA) and was found to have a purity of 98% determined by UPLC-UV (data not shown).
2.1.A. Sample Selection and Reference Materials
Ten commercial goldenseal products were selected based on their popularity in online consumer sales reports [35]. All products were capsules and derived from root/rhizome of Hydrastis canadensis. Each sample was randomly coded with an internal reference number (beginning with the letters “GS”) to maintain manufacturer anonymity (see Electronic Supplementary Material (ESM), Table S1).
Botanical reference samples for Hydrastis canadensis root (GS-13) and Coptis chinensis root (GS-14) were obtained from Chromadex (Irvine, CA). Both reference materials were obtained as dried powders and extracted using the same methods applied for the goldenseal samples.
2.1.B. Sample Adulteration
Samples were intentionally adulterated in house by combining goldenseal and C. chinensis in different ratios. A representative and verified goldenseal commercial product (verified through prior LC-MS analysis), GS-4, was combined in with the C. chinensis reference material, GS-14, to achieve a range of ratios ranging from 5% to 95% adulteration (ESM, Table S2). Samples were extracted as described below.
2.1.C. Sample Extraction
Samples were weighed into scintillation vials (200 mg of material per sample) and 20.0 mL methanol were added. Extractions were performed in triplicate to provide process replicates for analysis. Samples were shaken for 24 h, decanted into clean, weighed vials, and dried under N2 gas. Samples were stored at room temperature prior to analysis.
2.1.D. Compound identification
Variables, unique m/z value and retention time (m/z-RT) pairs, present in the loadings plot were used to confirm and explain the variance in the corresponding scores plot. These ions were identified by using exact mass (< 5 ppm) and retention time. The compounds berberine (1), canadine (2), hydrastine (3), coptisine (4), palmatine (5), jatrorrhizine (6), and dihydrocoptisine (7) are all known constituents of the botanicals under investigation (ESM, Fig. S1).
2.2. Sample Analysis
Liquid chromatography-mass spectrometry (LC-MS) data were acquired utilizing a Q Exactive Plus quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific) with a heated electrospray ionization (HESI) source coupled to an Acquity UPLC system (Waters, Milford, MA, USA). Samples were resuspended in CH3OH to a concentration of 0.1 mg/mL. Injections of 3 μL were performed on an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm, Waters) with a flow rate of 0.3 mL/min using the following binary solvent gradient of H2O (0.1% formic acid added) and CH3CN (0.1% formic acid added): initial isocratic composition of 95:5 (H2O:CH3CN) for 1.0 min, increasing linearly to 0:100 over 7 minutes, followed by an isocratic hold at 0:100 for 1 min, gradient returned to starting conditions of 95:5 and held isocratic again for 2 min. The positive ionization mode was utilized over a full scan of m/z 150–900 with the following settings: capillary voltage, 5 V; capillary temperature, 300 °C; tube lens offset, 35 V; spray voltage, 3.80 kV; sheath gas flow and auxiliary gas flow, 35 and 20 units, respectively. Each sample was injected in triplicate to provide analytical replicates for analysis. Extracted ion chromatographs were obtained from the XCalibur software (ThermoFisher Scientific).
2.2.A. Quantitative Analysis
Targeted analysis was performed using a palmatine standard purchased from ChromaDex (Los Angeles, California). A range of concentrations were prepped using serial dilutions in optima grade methanol (ESM, Table S2). Extracts were prepared at a concentration of 0.1 mg/mL (mass extract per volume of solvent) for analysis. The same parameters and LC method were utilized on LC-UV and LC-MS platforms. On the mass spectrometer platform, a selected ion monitoring (SIM) scan was performed from a m/z range of 350.1549–354.1549. The LC-UV data were collected in a range of 150–600 nm, but for processing purposes a range of 346.3–346.4 nm, the wavelength at which palmatine absorbs, was selected.
The limit of detection (LOD) for each approach was calculated using the equation LOD = 3s ÷ m, where s is the standard deviation of the lowest point in the linear range and m is the slope of the regression line. The limit of quantitation (LOQ) was determined as the lowest concentration of standard in the calibration curve that provided a residual of less than 15%, as described previously [38]. The limit of detection was expressed in two different forms, ppm palmatine in the plant and w/w % of Coptis chinensis adulterant, for comparison to the untargeted methodologies. The quantity of palmatine in the plant was calculated using the initial plant mass (199.10 mg) and extract mass (45.78 mg) of the C. chinensis reference material. The w/w % of C. chinensis adulterant was calculated using the quantity of palmatine in the plant and the amount of palmatine in the C. chinensis reference material (1.24 mg of palmatine per gram of plant material).
2.2.B. LC-UV Analyses
LC-UV data were collected in the same run on the Q Exactive Plus mass spectrometer, using the photodiode array detector (PDA) on the Waters Acquity UPLC across a range of 189 – 600 nm. The retention time and peak area for each sample were exported from Xcalibur into Excel for analysis. A data matrix was created of all the samples with retention time and peak area. This was analyzed in Sirius to produce the principal component analysis (PCA) scores and loadings plots.
2.3. Data Treatments
The LC-MS data were analyzed, aligned, and filtered using MZmine 2.28 software (http://mzmine.github.io/) with a slightly modified version of a previously reported method [9]. The following parameters were used for peak detection of the data acquired from the Q Exactive Plus: noise level (absolute value), 1×105 counts; minimum peak duration 0.5 min; tolerance for m/z intensity variation, 20%. Peak list filtering and retention time alignment algorithms were performed to refine peak detection. The join algorithm was used to integrate all the chromatograms into a single data matrix using the following parameters: the balance between m/z and retention time was set at 10.0 each, m/z tolerance was set at 0.001 or 5 ppm, and retention time tolerance was defined as 0.5 min. The peak areas for individual ions detected in the process replicates and analytical replicates were exported from the data matrix for further analysis.
Relative standard deviation (RSD) filtering was utilized for all datasets. Analytical replicates would be expected to have comparable profiles. Ions detected within the analytical replicates with for which peak area differed by more than 25% [36] were assigned as artefacts of the instrument and excluded from the metabolomics analysis. The peak area of any feature (m/z and retention time pair) with an RSD value above 25% was replaced with a 0. Principal component analysis (PCA) was performed using Sirius version 10.0 (Pattern Recognition Systems AS, Bergen, Norway). Data transformation was carried out by a fourth root transform of peak area to reduce heteroscedasticity. The 95% confidence interval was calculated using Hotelling’s T2 with the R package ‘car’ [37].
2.3.A. Composite Score Analysis
Composite score analysis was performed using a custom R script (available from https://github.com/jjkellogg/Composite-score). Principal component analysis was conducted on the main dataset, and the model was cross-validated using the Kaiser-Guttman rule, Jolliffe’s modification of the Kaiser-Guttman rule, and Broken stick criterion to determine the optimal number of principal components. The optimal number of components to include in the model was determined to be four principal components, which were used to calculate the pair-wise similarity metric, the composite score, as previously reported [25, 39]. This matrix was exported to Cytoscape 3.6.1 (Seattle, WA) for network visualization, displayed as a network of nodes, with edges described by the composite score value. A sub-network was generated by defining a minimum significant similarity score delineating similar samples, either 0.1 or 0.3, as applicable.
2.3.B. Supervised Statistical Analysis
Data were analyzed by SIMCA, a supervised method of analysis, using Solo (Eigenvector Research Inc., Wenatchee, WA, USA). SIMCA fits a PCA model to each pre-specified class of samples and then compares the models to determine the similarity (or difference) of the classes. However, for authentication, or detection of adulterants, only one class of samples needs to be identified, the authentic or reference samples. Hence, one-class modeling is a subset of SIMCA.
Analytical data were imported to Solo from Excel (Microsoft, WA, USA) as a 2 dimensional matrix; 19 samples (12 authentic samples and 7 adulterated samples) versus averaged counts for 1462 masses (variables). The data were pre-processed by dividing each variable by the square root of the average count, normalizing each sample by the sum of the squares of the counts (a unit vector), and mean centering each variable.
The one-class PCA modeling produced scores and loadings based on the characteristics of the authentic samples, in this case the authentic goldenseal. The loadings also produced scores for the unknown samples. Unknown samples were compared to the authentic samples using the Q statistic. The Q statistic describes the distance of the sample from the model and is a more accurate indicator of adulteration than the Hotelling T2 statistic. In general, the variance of the Q residual is proportional to the degree of adulteration.
3. Results
3.1. Adulteration of Goldenseal Samples
C. chinensis contains characteristic marker compounds: magnofluroine ([M]+ 342.1700), coptisine ([M]+ 320.0918), dihydrocoptisine ([M]+ 322.1075), palmatine ([M]+ 352.1542), and jatrorrhizine ([M]+ 338.1392) [18]. As expected, these compounds were found to increase in abundance, corresponding to increases in the C. chinensis ratio (Fig. 1). In addition, Hydrastine ([M+H]+ 384.1435) and canadine ([M+H]+ 340.1538) are unique to goldenseal and absent in other berberine-containing species [15, 40], and the relative intensity of these alkaloids also decreased as the percentage of goldenseal decreased in the adulterated samples. Clear differences within the base peak chromatograms are visible at 5% adulteration (coptisine is visible), however, a distinct shift in the ratio between hydrastine and berberine was observed visually at 25% adulteration (Fig. 1).
Fig. 1:
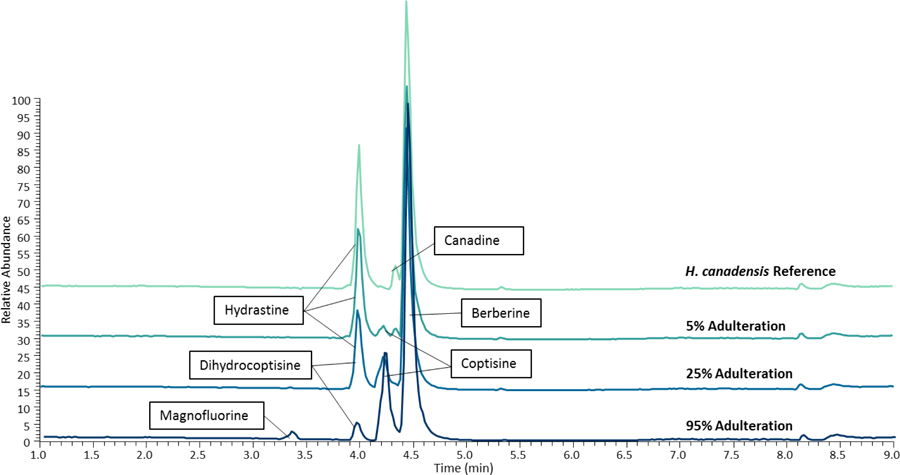
Extracted ion chromatograms of three adulterated samples and the H. canadensis reference material on the LC-MS system. Each set includes chromatograms of 5, 25, and 95% adulteration (percentage of Coptis chinensis present, remaining material is goldenseal). Marker compounds were identified by accurate mass and retention time, <5 ppm. Hydrastine and canadine are marker compounds in Hydrastis canadensis, while magnofluroine, coptisine, dihydrocoptisine, and palmatine are unique to C. chinensis. As the percentage of C. chinensis in the samples increases, the presence of hydrastine and canadine decreases and is replaced by an increase in coptisine, palmatine, magnofluroine, and dihydrocoptisine. Chromatograms were normalized to the same relative intensity for comparison.
3.2. Unsupervised Statistical Analysis
Unsupervised analysis of the untargeted metabolomics data was performed using principal component analysis (PCA) on both datasets to determine at which percentage adulteration could be detected. PCA is used to reduce the dimensions of a large data set into a series of orthogonal variables of decreasing variance that capture the patterns of the data. Thus, a PCA scores plot shows the relationship between different samples, where each data point is representative of that sample’s chemical profile (as described by features detected and associated peak area). The PCA data for the mass spectrometry platform (Fig. 2a) evidenced a trend in percentage of adulteration; i.e., the higher the adulteration, the further that the adulterated sample (orange squares) was spatially from the cluster of unadulterated goldenseal samples (blue diamonds). The purple diamond and red triangle represent the goldenseal and C. chinensis reference materials, respectively. The goldenseal reference material clustered with the group of commercial supplements, while the C. chinensis reference was observed to lie further away from the commercial supplements, closely aligned with the with 95% C. chinensis / 5% goldenseal sample. This suggested that the 95% adulterated supplement can be distinguished as not pure C. chinensis, rather, it still contains some constituents found in goldenseal. Visually, it was clear that at 5% adulteration, the sample no longer clustered with the main goldenseal sample cluster. This would raise suspicion regarding the product identity. The same trend was observed on the LC-UV platform.
Fig. 2:

Principal component analysis (PCA) scores plot (a), PCA loadings plot (b), and Hotelling’s T2 95% confidence interval (c) from the LC-MS data. The points in the scores plot are labeled to indicate the percentage of adulterant (Coptis chinensis) added to the Hydrastis canadensis plant material (A-5 is 5% C. chinensis, 95% H. canadensis, A-10 is 10% C. chinensis, etc.). Similar variance was showcased with a PC 1 (53.5%) versus PC 2 (9.3%) comparison of the LC-MS data for a total of 62.8% variance. The red triangle denotes the reference material for C. chinensis, while the purple diamond represents the reference material for H. canadensis. In the loadings (b) each data point represents a feature (a unique m/z value-retention time pair) detected in the sample. Variables in red are associated with Coptis chinensis, while those in green are associated with Hydrastis canadensis. The 95% confidence interval (c) was applied to PCA analysis of the goldenseal samples with only a single adulterated sample included in the dataset. The inclusion of just one outlier improves the ability to detect outliers as compared to datasets where multiple outliers were included (ESM, Fig. S3). At 5% adulteration the sample was not an outlier however, at 10% adulteration the sample was beyond the confidence interval and became a visible outlier.
The loadings plot (Fig. 2b) provides a plot of the features (m/z-retention time-pairs) in which their positioning indicates their influence on the spatial distribution of the samples observed in the scores plot (Fig. 2a), thus, the loadings can be used to qualitatively correlate the features to the representative samples, and highlights the compounds that are different between the plant species. For the sample set, the green markers signified compounds unique to goldenseal, while red markers represented metabolites unique to C. chinensis. Hydrastine, canadine, as well as the 13C isotope of hydrastine were located in the upper left region, which corresponded to the position of the goldenseal supplements in the scores plot (Fig. 2a). Palmatine, coptisine, dihydrocoptisine, and the 13C isotopes of palmatine and coptisine were visible in the lower right region of the plot, which corresponded to the position of the C. chinensis reference material (Fig. 2a and b). This supports the distinction between groupings of samples observed in the scores plot.
PCA is one of the most common methods employed as an unsupervised method to visualize metabolomics datasets and glean initial information on the relationship of samples without any a priori assumptions having been made about the dataset. PCA modeling and interpretation has often been employed to detect outliers from a dataset using the Hotelling’s T2 95% confidence ellipse [30, 31]. As such, the 95% confidence interval was used as a first method to detect outliers within the PCA dataset. While visual inspection of the PCA scores plot (Fig. 2a) suggested samples with adulteration at the 5% were differentiated from the main goldenseal sample set, the 95% confidence interval was employed to provide a more quantitative analysis using the standard deviation of the sample set. This was used to determine the percentage of adulterant needed to be labelled as an outlier within this approach. This calculation can be applied to multivariate data to assess similarity among samples- samples that fall within the confidence interval are believed to be similar with 95% certainty, while samples that lie beyond the confidence ellipse are considered statistically distinct. In the case where all adulterated and unadulterated samples were included in the sample set (ESM, Table S1), the application of the Hotelling’s T2 test enabled several outliers (the adulterated samples containing 75% - 95% C. chinensis) to be distinguished from the rest of the samples (ESM, Fig. S2).
It was hypothesized that the inclusion of so many adulterated samples within the dataset extends the confidence interval and limits the discriminatory power of the method; as the number of outliers increases, or as they are spatially located further from the norm, the standard deviation within the dataset increases and the resulting confidence interval becomes too broad to be an accurate determiner of outliers. As the original dataset included a range of adulteration percentages, the variability between the adulterated samples was large, and contributed an inability to distinguish adulterated and unadulterated samples. To heighten sensitivity of the confidence interval, one could increase the number of unadulterated goldenseal samples, which would reduce the goldenseal sample variability, or reduce the number (and thus variation) of suspected adulterated samples, which would increase the inter-category variance, and result in higher sensitivity and greater applicability for untargeted situations. A semi-supervised approach was adopted, including only one adulterated sample at a time with the unadulterated goldenseal samples, which resulted in a decrease in variance for the adulterated samples and the overall discriminatory power of the confidence interval heightened (Fig. 2c). This iterative method enabled a substantial reduction in the level of adulteration that was determined to be an outlier (50% adulteration versus 10% adulteration via the LC-MS system) as compared to the dataset that included all adulterated samples.
3.3. LC-UV Metabolomics
The data obtained using liquid chromatography separation with an ultraviolet (UV) photodiode array (PDA) detector were also utilized for metabolomics analysis. Using UV or PDA data as an input source has appeal, as these spectroscopy instruments are more cost-efficient for entities that might not have access to mass spectrometry equipment. With UV/Vis data, the independent variable was retention time, and peak intensity was used in place of peak area for PCA analysis (Fig. 3).
Fig. 3:

LC-UV metabolomics PCA scores and loadings plots. The scores plot (a) yielded a similar trend in adulteration percentage as seen with mass spectrometry data (Fig. 2). The trend was not as exaggerated and followed a more linear trend between adulterated samples (orange squares) and goldenseal samples (blue diamonds). The C. chinensis and H. canadensis reference materials were spatially located with the appropriate clusters. The loadings plot (b) showed the directionally correlated retention time with the scores plot. Loops were observed due to the continuous nature of the UV/Vis signal as a peak eluted. The retention time can be compared against standards for a targeted analysis and achieve tentative peak identification. The dominant discriminating features were similar to the mass spectrometry analysis; however, dihydrocoptisine and jatrorrhizine were absent from the C. chinensis region of the plot. Canadine was also absent from the H. canadensis ions. In calculating the outlier determination (c) an adulterated sample of 25% falls on the 95% confidence interval line (ESM Fig. S3). This could be deemed an outlier, while a sample with 50% adulteration (c) was a clear outlier, plotting far outside the 95% confidence interval.
The same trend in composition was observed with analysis of the LC-UV data (Fig. 3) as with the mass spectrometry-based metabolomics (Fig. 2). The variance of the adulterated samples was proportional to the peak intensity of the unique metabolites in C. chinensis, as the peak area increases so does the weight of that variable within the statistical analysis. The PC 1 versus PC 2 scores plot encompassed 97.7% of the variance in the dataset. UV absorbance was generally not as sensitive as mass spectrometry; however, this did not seem to impact the PCA scores plot (Fig. 3a). One main disadvantage of utilizing UV absorbance data for metabolomics analysis approach was the reduction in useful information gleaned from the loadings plot. With no discrete m/z values as input data, the loadings plot is a near-continuous plot of retention time (Fig. 3b). Thus, the loops visible in the loadings plot corresponded to the gradual increase in intensity associated with peak elution over time. Loops were observed because the variance grew as the peak elutes at a certain retention time and then receded. The resulting retention time values can be coupled with a targeted mass spectrometry-based analysis to achieve tentative peak identification. However, using only UV absorbance data yielded little additional information to discern responsible metabolites underpinning the visible trends. Mass spectrometry provides additional information, specifically m/z values, to improve identification of unknown compounds and determine which metabolites were responsible for the variation observed in the samples.
The advantage of collecting mass spectrometry data, as opposed to UV data, was apparent in the additional information garnered about the metabolites, chemical profile, and the ability to relate chemical composition to the variation of the samples. Pairing m/z value with retention time allowed for putative identification of secondary metabolites, which could suggest an identification of the possible adulteration source, in this case C. chinensis. However, the LC-UV metabolomics approach is a more cost-effective analytical input to gauge sample relationships and authenticity and could be improved if more data was known about the sample set. Ultimately, both dataset sources were successful in detecting adulterated samples via untargeted methodologies; however, the data obtained with the LC-UV did not provide as sensitive a level of distinction between adulterated and non-adulterated samples in this application. The underlying reasons for this difference are likely multifaceted, as there are many differences in the instrumentation and methods used for data acquisition and analysis on the different platforms.
3.4. Composite Score Analysis
Composite score analysis was performed using the PCA model from data acquired on the LC-MS system. In the previous approaches, the PCA data was limited to just two principal components (i.e., PC1 and PC2). However, there is additional variation in the dataset that is not encapsulated within only two principal components. Expanding the analysis to include multiple principal components would allow for a more comprehensive analysis of the dataset.
From the multi-component PCA, a similarity score was calculated between every sample, a correlation coefficient that ranges from −1.0 to 1.0. The correlations can serve as the foundation for a network diagram, with nodes (individual samples) while connections are derived from the correlation to connect nodes. For the analysis of all samples (Fig. 4a) a similarity score threshold of >0.3 was set. From the composite analysis, there were two distinct clusters observed: the adulterated samples (orange) and the unadulterated goldenseal samples (blue) (ESM, Tables S1 and S2). More connections were observed in the composite score analysis plot with all positive connections (0–1.0) but the two groups were still distinct (ESM, Fig. S6).
Fig. 4:
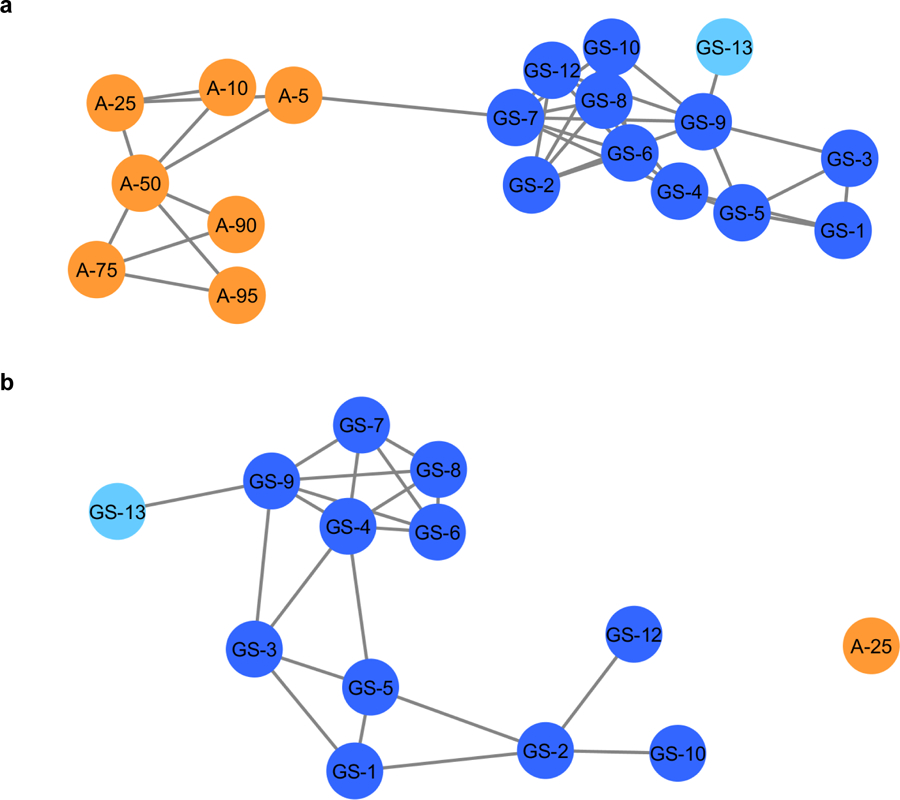
Composite score analysis of the entire sample set, a, (ESM, Tables S1 and S2) and composite score analysis with 25% adulterated product (b). The adulterated samples, in orange, are separated from the goldenseal commercial samples (in blue). The light blue node represents the goldenseal vouchered reference material. The connected lines represent a similarity score of >0.3, the highest score being 1.0. In the composite score analysis of the authentic supplements versus the 25% adulterated product (b) a similarity score range of 0.10–1.0 was applied. The 25% adulterated product (orange) is no longer connected to the other nodes (blue).
When calculating a composite score network comparing the goldenseal sample cluster against a single adulterated product (Fig. 4b), the analysis was not as sensitive as principal component analysis based upon the LC-MS untargeted metabolomics (Fig. 2a). This is due to the similarity between the plants; there should be some overlap in metabolite content given the plants belong to the same family (Ranunculaceae). In addition, the “adulterated” samples are still partially comprised of goldenseal so a level of connectivity should be expected. Using the composite score’s network diagram facilitates visual determination of potential outlier samples; the score serves as a quantitative measure to differentiate dissimilar samples. Restricting the connectivity threshold to 0.3, the distinction between the two groups was clear (Fig. 4b). While the cutoff point is relative and will vary among datasets and combination of samples, it provides an important metric for authentication. In this sample set, using a similarity score of >0.10, 25% Coptis chinensis was completely differentiable (no connection edges) from the goldenseal sample cluster. Again, ascribing a definitive score cutoff is a more restrictive way to use this model (compared to a holistic interpretation of the whole network) but was successful in this application in differentiating the two sample types (adulterated vs. unadulterated). Similar to the statistical methods described earlier, this approach could be made more sensitive if the same goldenseal product were being analyzed rather than an assortment, or if the two samples were vastly different botanicals. However, composite score analysis is a useful way to utilize and display principal component analysis data in a more quantitative and comprehensive way than a traditional 1x1 principal component comparison.
3.5. Supervised Statistical Analysis
While the adulterated samples were visually separated from the authentic samples (Fig. 2), with the application of the Hotelling’s T2 95% confidence interval as cut-off, it was not possible to fully resolve the adulterated samples at the lowest concentration analyzed (5% C. chinensis). This limitation was observed even using the semi-supervised approach, in which only one adulterated sample was included in the dataset at a time. Supervised statistical techniques such as SIMCA are better suited for distinguishing outliers in a dataset than unsupervised methods, although they require some a priori knowledge of the underlying groupings present in the dataset.
In this study, SIMCA analysis was conducted in which only authentic H. canadensis samples were identified and subjected to PCA, i.e., a one-class model. The loadings were used to compute Q statistic scores for both the authentic and unknown, adulterated samples. For detection of adulteration, the Q statistic (which provides the distance of the sample from the model) has been shown to take precedence over the Hotelling’s T2 statistic [32]. Plotting the samples versus the 95% confidence interval for the one-class model (Fig. 5a), all the reference samples were observed to fall below the 95% confidence limit (and thus be within the model’s limitations) while all the adulterated samples fall above the limit (fall outside the model). Thus, all the adulterated samples were correctly judged to be adulterated.
Fig. 5:

A one-class model (SIMCA) plot based on goldenseal products (a) and plot of Q residual versus adulterant concentration (10–95%) showing a linear relationship between the Q residual and concentration of adulteration (b). The Q residual (y-axis) is the distance of the sample from the model (a). The dotted line represents the 95% confidence limit. The confirmed goldenseal products (blue diamonds) clustered below or on the 95% confidence interval. The adulterated products (orange squares) fell above the confidence interval with the Q residual proportional to the level of adulteration. In (B) the blue diamond represents the mean Q residual value for the unadulterated botanicals and the horizontal dashed line provides the 95% confidence limit. As the concentration of adulteration increases, the sample is plotted further away from the model and thus moves further away from the 95% confidence interval. The outlier limitation determined using the linear detection methodology was 5% adulteration.
An alternate means of examining the Q residual values is to plot them as a function of the concentration of Coptis chinensis (Fig. 5b). Interestingly, in the case of adulteration with a single entity (C. chinensis), a linear plot is obtained despite the complexity of the spectra and variation at many masses. However, the variation at each mass is proportional and a linear relationship is obtained as a function of concentration. The linearity of Fig. 5b establishes confidence in the one-class model and the Q residual as a means of detecting adulteration and determining the LOD for the method, slightly less than 5%.
3.6. Targeted analysis for detection of adulterants
A targeted approach (specifically selecting for the known alkaloid palmatine, present in the C. chinensis adulterant) demonstrated a lower limit of detection on all three platforms (Table 1) than the untargeted methods. For the least sensitive of the two instrument platforms, the LC-UV system, the targeted analysis yielded a limit of detection for palmatine of 0.027 μM, corresponding to a palmatine concentration of 20 ppm in the sample, or 1.7% w/w C. chinensis adulterant. The mass spectrometric methods were even more sensitive, with the LC-MS system giving a calculated limit of 0.3% w/w C. chinensis (Table 1). These values are well below even the lowest cut-off (10% adulterant) observed with untargeted metabolomics using unsupervised data analysis (5% adulterant). Thus, for situations where the adulterant is of known identity, a targeted analysis will detect adulteration at much lower levels (33-fold in this case). However, it is worth noting that a disadvantage of targeted analysis is that it requires a priori knowledge of the identity of the adulterant. Analyzing a sample set where there was no suspicion or prior knowledge concerning adulteration, or if the identity of potential marker compounds was not known, it would not be possible to utilize a targeted analysis.
Table 1:
Limit of detection (LODa) and limit of quantitation (LOQb) of palmatine with targeted analysis on different instrument platforms, as well as the LOD in ppm palmatine in the plantc, and the % w/w of C. chinensis that would be detectable as an adulterantd.
| Method of analysis | Limit of detection (LOD) palmatine (µM)a | Limit of quantitation (LOQ) palmatine (µM)b | Limit of detection expressed as ppm palmatine in C. chinensis plantc | Minimum detectable Coptis chinensis (% w/w )d |
|---|---|---|---|---|
| LC with UV/VIS | 0.027 | 0.54 | 20 | 1.7 |
| LC-MS | 0.0047 | 0.12 | 3.8 | 0.30 |
Limit of detection was calculated using the following equation: LOD = 3s ÷ m where s is the standard deviation and m is the slope from the regression line.
Limit of quantitation was determined as the lowest concentration of standard in the calibration curve that provided a residual of less than 15% [38].
Calculated using the limit of detection and the original plant mass and extract mass of the Coptis chinensis reference material to give a value of ppm palmatine in the plant.
The w/w % of Coptis chinensis adulterant that would yield a concentration of palmatine corresponding to the limit of detection, calculated using the quantity of palmatine in the Coptis chinensis reference material (1.24 mg of palmatine per gram).
4. Discussion
Untargeted metabolomics analyses, employing both supervised and unsupervised statistical analysis, were compared against a targeted analysis. In a completely unsupervised approach (PCA), the Hotelling’s 95% confidence interval was used to estimate the limitations of the detection and observe potential outliers. However, as the variance between the dataset and potential outliers increased, the confidence interval expands and its discriminatory ability in detecting outliers decreased substantially (ESM, Fig. S2). Switching to a semi-supervised approach, in which a single adulterated sample was included in the PCA analysis sequentially, improved the power of the confidence interval to differentiate between authentic and adulterated samples. This approach made it possible to detect adulteration at the 10% m/m and 50% m/m levels for the LC-MS and LC-UV datasets, respectively. Composite score analysis combined four principal components to encompass a larger percentage of the variation in the dataset, as compared to the two principal component comparisons of traditional PCA and had a similar sensitivity in detecting adulterated samples.
As PCA was not originally designed to distinguish outliers from a large dataset, a supervised statistical analysis, SIMCA, was also included in the analysis. SIMCA was more effective for outlier detection than the unsupervised methods, yielding a detectable amount of 5% or less adulteration for the samples tested. The disadvantage of model-based statistics is the relationship between the samples is not further related to the variables. This technique has applications in scenarios where there is considerable prior knowledge and reference samples available, such as manufacturing quality assurance/control [12]. While each of the statistical techniques used in the study (PCA with 95% confidence interval, composite score, and SIMCA) can be successfully applied for authentication of goldenseal samples, the supervised, model-based approach (i.e., SIMCA) yielded a more sensitive quality control measure with the identification of reference samples to guide the model formation.
Of the methods compared herein, the targeted analysis was the most sensitive to detecting adulteration, with low limits of detection (0.0047 µM and 0.25 µM for the LC-MS and LC-UV respectively) on both platforms. These limits corresponded to a w/w % of 0.3% and 1.7% of C. chinensis adulterant. It is worth noting that a generic chromatographic method was used for analysis on both platforms. This facilitates comparison across platforms, but it is possible that the sensitivity could have been improved by optimizing the method for any system. However, using a more general method for this test case demonstrated that all platforms were viable for detection of botanical adulteration.
5. Conclusion
This methodology provided an untargeted process for ascertaining the authentication of supplements as well as a targeted methodology employing quantified marker compounds. Untargeted metabolomics can be used as a tool to identify adulterated samples and provide information about potentially unknown marker compounds that contribute to the differentiation, which is especially beneficial in situations when there is little prior knowledge of the composition or adulterant. Targeted analysis can be used for a direct and quantitative comparison in addition to verifying the level of adulteration present.
With the application of untargeted metabolomics, it was possible to discern authentic and adulterated goldenseal samples using data obtained from two different analytical platforms. The mass spectrometry platform allowed heightened sensitivity within the analysis as well as useful information (m/z-RT pairs) about the sample set. However, mass spectrometers are costly to purchase and maintain. LC-UV is a common tool utilized in the natural product community. Here it is clear that either platform is able to differentiate between an authentic and adulterated set of products. Thus, LC-UV can be used in place of mass spectrometry in order to detect adulteration via untargeted or targeted analysis, but with lower sensitivity (at least in the test case evaluated here).
In this study, different commercial products were used to provide a robust test case to challenge the analytical and statistical methods. In other settings, such as an industrial quality control environment, an increased number of authenticated products would increase the sensitivity of any of the statistical methodology, as more references or authenticated materials would tighten the variation among goldenseal samples and heighten the variance between the potential outliers and the goldenseal sample clusters. In situations where a mass spectrometer is not accessible, LC-UV metabolomics offers a more affordable but comparable option. Regardless of the analytical instrumentation, untargeted metabolomics with unsupervised or supervised data analysis for adulteration detection could be adapted and enhanced for implementation in various applications.
Supplementary Material
Acknowledgements
This project was supported by the National Institutes of Health National Center for Complementary and Integrative Health (NIH NCCIH), specifically the Center of Excellence for Natural Product Drug Interaction Research (NaPDI) [grant number U54AT008909] and a Ruth L. Kirschstein Postdoctoral National Research Service Award [ grant number F32AT009816] to Joshua Kellogg. The authors would like to thank our collaborator Dr. Olav M. Kvalheim (orcid.org/0000-0001-9432-8776) for his valuable assistance in data analysis and feedback for the semi-supervised analysis approach. Mass spectrometry analyses were conducted in the Triad Mass Spectrometry Facility at the University of North Carolina at Greensboro (https://chem.uncg.edu/triadmslab/).
1. Funding
Funding was provided by the National Institutes of Health National Center for Complementary and Integrative Health (NIH NCCIH), specifically the Center of Excellence for Natural Product Drug Interaction Research (NaPDI) [grant number U54AT008909] and a Ruth L. Kirschstein Postdoctoral National Research Service Award [ grant number F32AT009816] to Joshua Kellogg.
Biography

E. Diane Wallace is a Mass Spectrometry Research Associate at the University of North Carolina at Chapel Hill (UNC-CH). She earned her Master’s of Science degree while working in Dr. Nadja Cech’s laboratory at the University of North Carolina at Greensboro. Diane’s work focuses on analytical chemistry, specifically mass spectrometry-based metabolomics. She continues to apply her knowledge in her new position at UNC-CH while expanding her mass spectrometry skillset.

Daniel A. Todd, PhD, is the Director of the Triad Mass Spectrometry Facility at UNC Greensboro. He specializes in small molecule analysis and has been working the last six years to aid in the development of chemometric tools for identifying therapeutically relevant small molecule natural products.

James Harnly, is an analytical chemist with expertise in atomic and molecular spectroscopy and authentication of food and botanical materials using chemometric methods. He has more than 40 years of experience in industry and government and serves as the Research Leader for the newly organized Methods and Applications Food Composition Lab. Dr. Harnly has authored more than 150 peer-reviewed papers, 18 technical reports and book chapters, and holds two patents.

Nadja Cech, is Patricia A. Sullivan Distinguished Professor of Chemistry at the University of North Carolina at Greensboro (UNCG). Dr. Cech leads a dynamic research group at UNCG, for which a major focus in the development of mass spectrometry metabolomics as a tool to understand synergy and complexity in biologically active botanical natural products. This work has been continuously funded by the National Institutes of Health for more than 15 years and was awarded the Jack L. Beal Award from the Journal of Natural Products in 2011.

Joshua J. Kellogg is an Assistant Professor in the Department of Veterinary and Biomedical Sciences at the Pennsylvania State University. Dr. Kellogg obtained his PhD in natural product chemistry and ethnobotanical nutraceuticals from North Carolina State University. His lab largely focuses on the development and application of metabolomic and bioinformatic approaches for complex mixture analysis, with special emphasis on the identification and characterization of chemical entities from natural sources to drive discovery of novel bioactive compounds.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Compliance with Ethical Standards
Conflicts of Interest
The authors declare no conflicts of interest.
Availability of Data and Material
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Original material also available upon request.
Code Availability
MzMine is an open source software and readily available to the public. Sirius is created by Pattern Recognition Systems and can be purchased here: http://www.prs.no/Sirius/Sirius.html. The code for the composite score analysis is available here: https://github.com/jjkellogg/Composite-score.
References
- 1.Vogtman H Dietary Supplement Usage Incrases, Says New Survey The Council for Responsible Nutrition: The Council for Responsible Nutrition; 2017. [updated October 19th, 2017 Available from: https://www.crnusa.org/newsroom/dietary-supplement-usage-increases-says-new-survey.
- 2.Dwyer JT, Coates PM, Smith MJ. Dietary Supplements: Regulatory Challenges and Research Resources. Nutrients. 2018;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Izzo AA, Hoon-Kim S, Radhakrishnan R, Williamson EM. A Critical Approach to Evaluating Clinical Efficacy, Adverse Events and Drug Interactions of Herbal Remedies. Phytother Res: PTR 2016;30(5):691–700. [DOI] [PubMed] [Google Scholar]
- 4.Tims M On Adulteration of Hydrastis canadensis root and rhizome. Botanical Adulterants Bulletin [Internet]. 2016.
- 5.McGraw JB, Sanders SM, Van der Voort M. Distribution and abundance of Hydrastis Canadensis L. (Ranunculaceae) and Panax quinquefolius L. (Araliaceae) in the central Appalachian region. J Torrey Bot Soc 2003;130(2):62–9. [Google Scholar]
- 6.Lee J Marketplace analysis demonstrates quality control standards needed for black raspberry dietary supplements. Plant Food Hum Nutr (Dordrecht, Netherlands). 2014;69(2):161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbas O, Zadravec M, Baeten V, Mikus T, Lesic T, Vulic A, Prpic J, Jemersic L, Pleadin J. Analytical methods used for the authentication of food of animal origin. Food Chem 2018;246:6–17. [DOI] [PubMed] [Google Scholar]
- 8.Avula B, Wang Y-H, Khan IA. Quantitative determination of alkaloids from roots of Hydrastis canadensis L. and dietary supplements using ultra-performance liquid chromatography with UV detection. J AOAC Int 2012;95(5):1398–405. [DOI] [PubMed] [Google Scholar]
- 9.Wallace ED, Oberlies NH, Cech NB, Kellogg JJ. Detection of adulteration in Hydrastis canadensis (goldenseal) dietary supplements via untargeted mass spectrometry-based metabolomics. Food Chem Toxicol 2018;120:439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown PN, Roman MC. Determination of hydrastine and berberine in goldenseal raw materials, extracts, and dietary supplements by high-performance liquid chromatography with UV: collaborative study. J AOAC Int 2008;91(4):694–701. [PMC free article] [PubMed] [Google Scholar]
- 11.Geng P, Harnly JM, Sun J, Zhang M, Chen P. Feruloyl dopamine-O-hexosides are efficient marker compounds as orthogonal validation for authentication of black cohosh (Actaea racemosa)-an UHPLC-HRAM-MS chemometrics study. Anal Bioanal Chem 2017;409(10):2591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harnly J, Chen P, Sun J, Huang H, Colson KL, Yuk J, McCoy J-AH, Reynaud DTH, Harrington PB, Fletcher EJ. Comparison of Flow Injection MS, NMR, and DNA Sequencing: Methods for Identification and Authentication of Black Cohosh (Actaea racemosa). Planta Med 2016;82(3):250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pengelly A, Bennett K, Spelman K, Tims M. An Appalachian Plant Monograph: Goldenseal Hydrastis canadensis L. Appalachian Center for Ethnobotanical Studies. 2012.
- 14.Cicero AFG, Baggioni A. Berberine and Its Role in Chronic Disease. Adv Exp Med Biol 2016;928:27–45. [DOI] [PubMed] [Google Scholar]
- 15.Weber HA, Zart MK, Hodges AE, Molloy HM, O’Brien BM, Moody LA, Clark AP, Harris RK, Overstreet JD, Smith CS. Chemical comparison of goldenseal (Hydrastis canadensis L.) root powder from three commercial suppliers. J Agri Food Chem 2003;51(25):7352–8. [DOI] [PubMed] [Google Scholar]
- 16.Ivanovska N, Philipov S. Study on the anti-inflammatory action of Berberis vulgaris root extract, alkaloid fractions and pure alkaloids. Int J Immunopharmaco 1996;18(10):553–61. [DOI] [PubMed] [Google Scholar]
- 17.Rackova L, Majekova M, Kost’alova D, Stefek M. Antiradical and antioxidant activities of alkaloids isolated from Mahonia aquifolium. Structural aspects. Bioorgan Med Chem 2004;12(17):4709–15. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Peng J, Li F, Liu X, Deng M, Wu H. Determination of Alkaloid Contents in Various Tissues of Coptis Chinensis Franch. by Reversed Phase-High Performance Liquid Chromatography and Ultraviolet Spectrophotometry. J Chromatogr Sci 2017;55(5):556–63. [DOI] [PubMed] [Google Scholar]
- 19.Fiehn O. Metabolomics - the link between genotypes and phenotypes. Plant Mol Biol 2002;48(1–2):155–71. [PubMed] [Google Scholar]
- 20.Hong E, Lee SY, Jeong JY, Park JM, Kim BH, Kwon K, Chun HS. Modern analytical methods for the detection of food fraud and adulteration by food category. J Sci Food Agric 2017;97(12):3877–96. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez SD, Rolandelli G, Buera MP. Detection of quinoa flour adulteration by means of FT-MIR spectroscopy combined with chemometric methods. Food Chem 2019;274:392–401. [DOI] [PubMed] [Google Scholar]
- 22.Britton ER, Kellogg JJ, Kvalheim OM, Cech NB. Biochemometrics to Identify Synergists and Additives from Botanical Medicines: A Case Study with Hydrastis canadensis (Goldenseal). J Nat Prod 2017. [DOI] [PMC free article] [PubMed]
- 23.Deconinck E, Sokeng Djiogo CA, Courselle P. Chemometrics and chromatographic fingerprints to classify plant food supplements according to the content of regulated plants. J Pharmaceut Biomed 2017;143:48–55. [DOI] [PubMed] [Google Scholar]
- 24.Karu N, Deng L, Slae M, Guo AC, Sajed T, Huynh H, Wine E, Wishart DS. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal Chim Acta 2018;1030:1–24. [DOI] [PubMed] [Google Scholar]
- 25.Kellogg JJ, Graf TN, Paine MF, McCune JS, Kvalheim OM, Oberlies NH, Cech NB. Comparison of Metabolomics Approaches for Evaluating the Variability of Complex Botanical Preparations: Green Tea (Camellia sinensis) as a Case Study. J Nat Prod 2017;80(5):1457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortesniemi M, Sinkkonen J, Yang B, Kallio H. NMR metabolomics demonstrates phenotypic plasticity of sea buckthorn (Hippophae rhamnoides) berries with respect to growth conditions in Finland and Canada. Food Chem 2017;219:139–47. [DOI] [PubMed] [Google Scholar]
- 27.McGeachie MJ, Dahlin A, Qiu W, Croteau-Chonka DC, Savage J, Wu AC, Wan ES, Sordillo JE, Al-Garawi A, Martinez FD, Strunk RC, Lemanske RF Jr., Liu AH, Raby BA, Weiss S, Clish CB, Lasky-Su JA. The metabolomics of asthma control: a promising link between genetics and disease. Immun Inflamm Dis 2015;3(3):224–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paiga P, Rodrigues MJE, Correia M, Amaral JS, Oliveira MBPP, Delerue-Matos C. Analysis of pharmaceutical adulterants in plant food supplements by UHPLC-MS/MS. Eur J Pharm Sci 2017;99:219–27. [DOI] [PubMed] [Google Scholar]
- 29.Pinasseau L, Vallverdu-Queralt A, Verbaere A, Roques M, Meudec E, Le Cunff L, Peros J-P, Ageorges A, Sommerer N, Boulet J-C, Terrier N, Cheynier V. Cultivar Diversity of Grape Skin Polyphenol Composition and Changes in Response to Drought Investigated by LC-MS Based Metabolomics. Front Plant Sci 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen X, Zhu Z-J. MetFlow: An interactive and integrated workflow for metabolomics data cleaning and differential metabolite discovery. Bioinformatics (Oxford, England). 2019. [DOI] [PubMed]
- 31.Wu H, Wang D, Meng J, Wang J, Feng F A plasma untargeted metabolomic study of Chinese medicine Zhi-Zi-Da-Huang decoction intervention to alchol-induced hepatic steatosis. Anal Methods. 2017;9(4):586–92. [Google Scholar]
- 32.Brerton RG. Chemometrics for Pattern Recognition. West, Sussex, UK: John Wiley and Sons, Ltd.; 2009. p. 233–86. [Google Scholar]
- 33.Harnly J, Bergana MM, Adams KM, Xie Z, Moore. Variance of Commercial Powdered Milks Analyzed by Proton Nuclear Magnetic Resonance and Impact on Detection of Adulterants. J Agri Food Chem 2018;66(32):8478–88. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Finley J, Betz JM, Brown PN. FT-NIR characterization with chemometric analyses to differentiate goldenseal from common adulterants. Fitoterapia 2018;127:81–8. [DOI] [PubMed] [Google Scholar]
- 35.Amazon.com. Amazon Best Sellers [Available from: https://www.amazon.com/Best-Sellers-Health-Personal-Care-Goldenseal-Herbal-Supplements/zgbs/hpc/3765771/ref=zg_bs_nav_hpc_3_3764461.
- 36.Todd DA, Zich DB, Ettefagh KA, Kavanaugh JS, Horswill AR, Cech NB. Hybrid Quadrupole-Orbitrap mass spectrometry for quantitative measurement of quorum sensing inhibition. J Microbiol Meth 2016;127:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caesar LK, Kvalheim OM, Cech NB. Hierarchical cluster analysis of technical replicates to identify interferents in untargeted mass spectrometry metabolomics. Anal Chim Acta 2018;1021:69–77.29681286 [Google Scholar]
- 38.Fox JWS. An R Companion to Applied Regression. 2nd ed. Thousand Oaks, CA: Sage; 2011. [Google Scholar]
- 39.Kellogg JJ, Kvalheim Olav M., Cech Nadja B. Composite score analysis for unsupervised comparison and network visualization of metabolomics data. Anal Chim Acta 2019. [DOI] [PMC free article] [PubMed]
- 40.Le PM, McCooeye M, Windust A. Characterization of the alkaloids in goldenseal (Hydrastis canadensis) root by high resolution Orbitrap LC-MS(n). Anal Bioanl Chem 2013;405(13):4487–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.