

Abstract
Purpose:
Acquired hematopoietic failure is commonly caused by therapeutic and accidental exposure to toxic agents to the bone marrow (BM). Efficient recovery from the BM failure is not only dictated by the intrinsic sensitivity and proliferation capacity of the hematopoietic stem and progenitor cells, but also nourished by the BM environment niche. Identification of genetic factors that improve the recovery from hematopoietic failure is essential. Vertebrate SETD4 is a poorly characterized and putatively non-histone methyltransferase. This study aims to identify the roles of SETD4 in BM recovery.
Methods and Materials:
An inducible Setd4 knock out mouse model (Setd4flox/flox;Rosa26-CreERT2+ ) was used. Adult sex-matched littermates were treated with tamoxifen to induce Setd4 deletion or Oil as the control. Tamoxifen treated Setd4wt/wt;Rosa26-CreERT2+mice were included as another control. Those mice were irradiated to induce hematopoietic syndrome and analyzed to identify the roles and mechanisms of Setd4 in of BM recovery.
Results:
Loss of Setd4 in adult mice improved the survival of whole-body irradiation induced BM failure. This was associated with improved recoveries of long-term and short-term hematopoietic stem cells (HSC), and early progenitor cells. BM transplantation analyses surprisingly showed that the improved recovery was not due to a radiation resistance of the Setd4 deficient HSC, but that Setd4 deficient HSC were actually more sensitive to radiation. However, the Setd4 deficient mice were better recipients for allogeneic HSC transplantation. Furthermore, there was an enhanced splenic erythropoiesis in Setd4 deficient mice.
Conclusion:
These findings not only revealed a previously unrecognized role of the Setd4 as a unique modulator of hematopoiesis, but also underscored the critical role of the BM niche in the recovery of hematopoietic failure. Our study also implicated Setd4 as a potential target for therapeutic inhibition to improve the conditioning of the BM niche prior to allogeneic transplantation.
Keywords: SETD4, radiation-induced bone marrow failure, hematopoiesis, hematopoietic stem cell (HSC), bone marrow niche
Summary:
Deletion of Setd4 in adult mice improved the survival from radiation-induced hematopoietic failure. Setd4 deficiency sensitized HSCs to radiation but improved bone marrow environment niche. Our study suggests that SETD4 as a potential inhibitory target to improve the niche function for recovery of BM failure.
Introduction
The mammalian hematopoietic system is subject to a consistent renewal during the life span of an individual. Inherited and acquired hematopoietic failure can be caused by diseases such as myelodysplastic syndrome, acute myeloid leukemia, and Fanconi anemia(1–3), as well as a consequence of chemotherapy treatment or accidental over-dose exposure to radiation and other toxic agents(4–7). Depleting the host bone marrow (BM) hematopoietic cells before allogeneic hematopoietic stem cell (HSC) transplantation is commonly used to treat a wide array of malignancies including multiple myeloma, leukemias, lymphomas, and some solid tumors(8–10). Understanding the molecular factors that modulate hematopoietic recovery is essential to improve the outcomes of these diseases and treatments.
Lysine methylation plays an important role in diverse cellular processes to regulate chromatin structures, genome stability, and fate determination of stem cells, etc. (11–19). Many lysine methyltransferases harbor a Su(var)3-9-Enhancer of zeste–Trithorax (SET) domain, which is responsible for transferring a methyl group from S-adenosylmethionine to lysine residues. The human genome encodes an estimated of 58 SET domain methyltransferases(20). Although most of the SET domain proteins have documented histone modifying activity, the SETD6 group of SET proteins (including SETD6, SETD3 and SETD4) represents a distinct class of non-histone methyltransferases(20), and they share a similar substrate-binding domain resembling that of the plant Rubisco methyltransferase(21–27). SETD6 methylates p65 (RelA)(24,28), and other non-histone proteins(29–31). SETD3 was initially reported to methylate histones(32,33), but recently found to a histidine residue in β-actin(25,26,34). However, little is known about the function of mammalian SETD4.
The brine shrimp Artemia Parthenogenetica SETD4 was initially suggested to play a role in methylating histone H4K20 and H3K79(21), and the human SETD4 protein may have the same activity (35). However, another report failed to identify these sites as SETD4 substrates, and instead suggested H3K4 as the substrate of SETD4 in macrophages (36). Emerging evidence suggests that SETD4 may have an oncogenic activity(37–39). According to the TCGA database, oncogenic fusion of SETD4 was found to be with TMPRSS2 in prostate cancer, with FTCD in invasive ductal carcinoma, with KIAA1958 in serous ovarian cancer, and with B4Galt6 in lung squamous cancer. SETD4 was highly expressed in the quiescent breast cancer stem cell sub-population of MCF7 cells(35), and an induced Setd4 deletion in adult mice delayed the radiation-induced T-lymphoma development in mice(40).
In this study, we found that induced Setd4 knockout in adult mice improved the hematopoietic recovery after total body irradiation (TBI). Interestingly, the improved recovery was not associated with a resistance of the Setd4 deficient HSCs to radiation, rather the Setd4 deficient HSC were actually more sensitive to radiation than wild type HSC in vivo. However, Setd4 deficient mice were more receptive to HSC transplantation from wild type donors, and that Setd4 deficient HSCs had a repopulating advantage in recipient mice after transplantation. These results underscore the unique mode by which Setd4 regulates hematopoiesis and implied that Setd4 inhibition may not only improve the competence of donor HSCs but also the ability of the recipient BM to receive the transplantation.
Materials and Methods
Detailed materials and methods can be found in the Supplemental Materials and Methods.
Mouse lines, tamoxifen (Tam) treatment, and TBI
The sources and properties of three mouse lines (Setd4flox/flox;Rosa26-CreERT2/+;Tp53flox/flox) used in this study are summarized in Table S1. The construction, verification, and genotyping strategies of a mouse line with an allele of floxed exon-6 of Setd6, designated Setd4flox/flox, were described in details (40). Crossing of the Setd4flox/flox with Rosa26-CreERT2 (B6.129, Jackson Laboratory Stock No 008463) produced the Setd4flox/flox;Rosa26-CreERT2+ mice. Adult Setd4flox/flox;Rosa26-CreERT2+ mice were i.p. injected with Tam (or corn oil as the control) to induce Setd4 deletion(40). Mice were temporarily restrained in the ventilated pie-shaped chambers of the RadDisk cage (a circular cage with 12 pie-shaped chambers and a rotating lid that covers 11 of the chambers). Up to 11 mice were separately placed into the individual chamber of the RadDisk cage, and then the cage was placed inside the chamber of the Gammacell 40 Extractor (MDS Nordion) 137Cs γ-irradiator. The dose rate of of the radiation source was 86 cGy/min. The specific radiation doses are given when describing the experiments.
Analysis of BM and peripheral blood cells
BM single-cell suspensions were counted to determine the total number of cells. Then, aliquots were stained for surface markers to determine the relative abundance of each cell types(41). The sources of antibodies are listed in Tables S2 and S3. For BM transplantation, a given number of sorted donor Lin- cells were transplanted into lethally irradiated recipient mice. Blood was collected to monitor the success of the transplantation based on the donor CD45.1 or CD45.2 congenic markers.
Statistics
Statistical analyses were performed using Prism GraphPad, including Student’s t-test and Log-rank (Mantel-Cox) test.
Results
Deletion of Setd4 protects mice from ionizing radiation (IR) induced BM failure
To avoid any potential impact of a constitutive Setd4 deletion on the embryonic and early postnatal development of mice, the Tam-inducible knockout mouse model (Setd4flox/flox;Rosa26-CreERT2+) (40) was used in this study. Whenever possible, sex-matched littermates were treated with Tam (Setd4Δ) or Oil (Setd4flox), We challenged these mice with TBI of 8 or 9 Gy that cause approximately 50% or 90% death due to hematopoietic failure for this strain of mice. Surprisingly, we found that Setd4Δ/Δ mice survived significantly better than the mice treated with Oil when exposed to 8 Gy TBI (Fig. 1A). When the mice were exposed to a higher dose of 9 Gy (Fig. 1B), the Setd4Δ/Δ mice survived significantly better than the control mice, but the Setd4Δ/wt mice had similar survival rate as the control mice. Tam-treatment of wild type mice (including Setd4flox/flox;Rosa26CreERT2−/−, Setd4flox/wt;Rosa26CreERT2−/−, and Setd4wt/wt;Rosa26-CreERT2+) did not alter the overall survival of the mice (Fig. 1C). It is interesting that the improved survival of the Setd4Δ/Δ mice was similar as the level of p53Δ/Δ mice, but co-deletion of p53 and Setd4 had an additive effect and better protected the mice from irradiation (Fig. S1). In Figs. 1A–1C, roughly equal number of males and females were used in each group (see figure insert for specific number of males and females). We did not observe differences of survivals between males and females.
Figure 1. Sensitivity of Setd4 deficient mice to TBI.

(A&B): Tam or control treated sex-matched littermates of Setd4flox/flox;Rosa26-CreERT2+ or Setd4flox/wt;Rosa26-CreERT2+ were divided into three experiments groups: 1) Control: Oil-treated Setd4flox/flox;Rosa26-CreERT2+ or Setd4flox/wt;Rosa26-CreERT2+; 2) Setd4Δ/wt: Tam-treated Setd4flox/wt;Rosa26-CreERT2+; and 3) Setd4Δ/Δ: Tam-treated Setd4fox/flox;Rosa26-CreERT2+. Mice were treated with 8 Gy (A) or 9 Gy (B) of TBI, and monitored for survival. The P-values and number of mice in each group are shown. (C): Control mice treated with tamoxifen and Oil and then TBI as indicated. Oil: Oil-treated Setd4flox/flox;Rosa26CreERT2−/−, Setd4flox/wt;Rosa26CreERT2−/−, and Setd4wt/wt;Rosa26-CreERT2+ mice. For 8 Gy, 4, 3 and 2 mice of the above genotypes were included. For 9 Gy, 6, 2, and 2 mice were used. Tam: Tam-treated wt mice with the same genotypes above. For 8 Gy, 4, 4, and 4 mice with the same three genotypes were used. For 9 Gy, 6, 4, and 4 were included. The survivals of each group (panels A, B, and C) were obtained from multiple irradiation experiments of 1-5 pairs of mice per irradiation depending on the availability of matched littermates at the time of irradiation. Shown are the results of the combined number of mice. The total number of mice (n) and the number of males (M) and females (F) in each group are shown above the survival curves. (D): Repopulation of BM cells after TBI. At least 3 mice in each group were pathologically analyzed by hematoxylin-and-eosin (HE) staining. Shown are by representative images from the staining of cross sections of femur BM cavities from the indicated group of mice 14 days after 8 Gy of TBI. Hematopoietic cells clusters had dramatically repopulated the bone marrows of Tam-treated Setd4flox/flox;Rosa26CreERT2+ (Setd4Δ/Δ) mice, but not in Oil-treated Setd4flox/flox;Rosa26CreERT2+ (Setd4flox/flox) or the Tam-treated Setd4wt/wt;Rosa26-CreERT2+ (Tam-Setd4wt/wt). Upper panel, 40x; lower panel, 100x; scale bar = 100 μm. (E): The total number of BM mono-nuclear cell (BM-MNC) collected from one paired femur and tibia. Three independent experiments with an average of 3 mice per subset of time points were performed. The combined data are plotted.
It is well established in the radiation biology field that WBI-induced BM failure often results in death within 30 days and mostly between 2-3 weeks. The results in Fig. 1A–B suggested that Setd4 deficient mice had a decreased sensitivity to radiation induced BM failure. To verify this, we performed Hematoxylin-and-Eosin (HE) staining of the femur BM cavities 14 days after 8 Gy TBI. We designed our approaches with three animal groups: 1) Oil-treated Setd4flox/flox;Rosa26-CreERT2+ (Setd4flox/flox) littermate mice as a control; 2) Setd4Δ/Δ; and 3) Tam-treated Setd4wt/wt ;Rosa26-CreERT2+ (Tam-Setd4wt/wt) as another control. We found that hematopoietic cells clusters had dramatically repopulated the BM cavity of Setd4Δ/Δ mice but not those of Setd4flox/flox and Tam-Setd4wt/wt mice (Fig. 1D). Furthermore, we isolated the BM cells and determined the number of the BM mono-nuclear cells (BM-MNC) in the survived mice. Four days after irradiation, there were equal depletion of BM-MNC for knockout and the two control groups (Fig. 1E). However, by day 14 and 21, there was more BM-MNC in the Setd4Δ/Δ mice than control mice, suggesting an enhanced recovery of BM-MNC when Setd4 was deleted.
The mice that survived the first 30 days from the hematopoietic syndrome after 8 Gy of TBI were kept for long-term observation. No major differences in term of overall survival (Fig. S2), or in the incidences of tumors were observed. This result suggests that once the mice survived the initial hematopoietic failure, Setd4 loss did not significantly impact the overall survival or tumor development. Furthermore, we irradiated the littermates of Setd4Δ/Δ and Setd4flox/flox mice with 13 Gy TBI, and all mice died within 10 days post TBI (Fig. S3), which was likely due to TBI-induced GI syndrome. The similar sensitivity of these mice in both groups suggested that there was unlikely an effect of Setd4 loss in protecting the mice from TBI-induced GI syndrome.
Setd4 deletion promotes BM recovery
Since the deletion of Setd4 protected mice from the fatality of radiation induced hematopoietic syndrome, we hypothesized that Setd4 may modulate BM recovery from radiation damage. To test this hypothesis, we quantified the number of different cell types in the hierarchy of the BM regeneration with well-established biomarkers and methods(41–44) (detailed in Fig S4 and Supplemental Materials and Methods). Representative flow cytometry profiles for a pair of littermates of Setd4Δ/Δ and Setd4flox/flox are shown in Fig. S4B.
The lineage relationships of these cell populations are simplified in Fig. 2A. Among the LSK population of cells (Fig. 2B–F), which represent the stem and early progenitor cells, there were no significant difference of total LSK, LT-HSC, ST-HSC, MPP, and LMPP populations between Setd4 knockout mice and control mice without irradiation. Shortly after irradiation, all cell populations were equally depleted at 4 days. However, by 2 weeks after irradiation, the Setd4 knockout mice had an enhanced recovery of LT-HSC, ST-HSC, and MPP, but not LMPP populations. Among the LS−K and the LSlowKlow populations, which contains the committed progenitor cell populations (Fig. 2G–J), there were enhanced recoveries of the CMP, MEP, and the GMP populations by 2 weeks, but not the CLP population. By 3 weeks post TBI, the difference among these groups were largely diminished, which is expected because only the mice that survived were available for this analysis and these mice had survived past the critical time period of hematopoietic failure and had likely succeeded in replenishing damaged BM at 3 weeks post TBI. Together, these data indicate that Setd4 inactivation leads to an improved recovery of IR-induced BM injuries by more efficient expansion of early stem and progenitor cells, such as LT-HSC, ST-HSC, and the myeloid progenitors, but not the LMPP and CLP which ultimately repopulate the peripheral lymphocyte pools.
Figure 2. Cell counts of hematopoietic stem cells (HSCs) and progenitor cells in the BM after 8 Gy of irradiation.
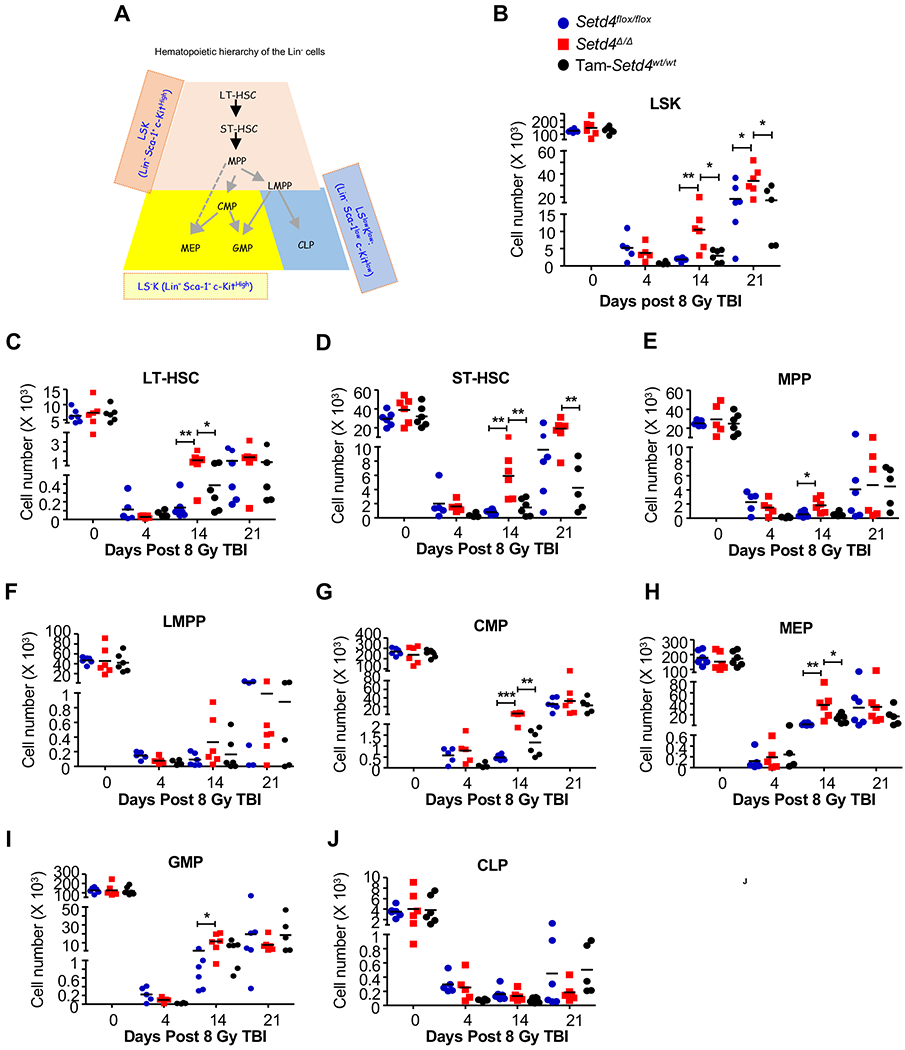
(A): A simplified illustration of the lineage hierarchy of the Lin− cells. Based on Sca-1 and c-Kit expression, the Lin- cells were divided into three populations shown with different colors. In each population, the specific cell types can be identified by additional cell markers (see Supplemental Materials and Methods, and Fig. S4 for details). LSK: Sca-1+ and c-Kit+; LS− K: Sca-1− and c-Kit+; LSlowKlow: Sca-1low c-Kitlow; LT-HSC: long-term HSC; ST-HSC: short-term HSC; MPP: multipotent progenitor; LMPP: lymphoid-primed MPPs; CMP: common myeloid progenitors; GMP: granulocyte-macrophage progenitors; MEP: megakaryocyte-erythroid progenitors. CLP: common lymphoid progenitors.
(B-J): The numbers of total LSK, LT-HSC, ST-HSC, MPP, LMPP, CMP, MEP, GMP, and CLP cells at different the indicated times after 8 Gy of TBI. The BM-MNC (Fig. 1E) were then used to sort out the Lin- cells, and to determine the relative percentage of each of the cell types in the hematopoietic hierarchy as labeled above each panel. The designation of animal genotypes is shown in Panel B. For each tested mouse, a pair of femur and tibia were crushed to collect all BM cells at 4, 14, and 21 days after 8Gy of TBI. The mouse genotypes were: 1) Oil-treated Setd4flox/flox;Rosa26-CreERT2+ (Seid4flox/flox, blue filled cycle); 2) Tamoxifen-treated Setd4flox/flox;Rosa26-CreERT2+ (Setd4Δ/Δ red square); and 3) tamoxifen-treated Setd4wt/wt;Rosa26-CreERT2+ (Tam-Setd4wt/wt, filled black cycles). The mice in groups 1 and 2 were male littermates. In all panels, each dot represents the data from an independent mouse, and none-irradiated mice are shown as day 0. *: p<0.05, **: P<0.01. Shown are combined results from individual mice from 2-3 independent experiments with an average of 3 mice per time point in each experiment.
Enhanced splenic erythropoiesis in Setd4 deficient mice post TBI
In adult rodents, erythropoiesis in peripheral lymphoid organs occurs to support the animals when BM repopulation is impaired(45). During the course of the study, we noticed that Setd4Δ/Δ mice had enlarged spleens more frequently than control mice (Fig. 3A–B). This prompted us to investigate whether there was any effect of Setd4 loss on splenic erythropoiesis. Based on the stages of erythroid maturation (Fig. 3C), we analyzed cell surface markers CD71 and Ter119, which are commonly used to define the maturation stages of erythroid progenitor cells(46). We observed significantly more cells in the S3 (CD71hi/Ter119hi) stage and correspondingly reduced numbers of cells in the immature S1 (CD71−/Ter119−) stage at 2 weeks post TBI (Fig. 3D–E). There were significantly more late-stage cells (S2-S5) in the spleens of Setd4Δ/Δ than in Setd4flox/flox and Setd4wt/wt mice. In addition, the percentage of erythroid cell population (S2-S5), relative to lymphoid and myeloid cell populations, were significantly increased in Setd4Δ/Δ mice compared to the other groups in the spleens (Fig. 3F). These data indicate that loss of Setd4 enhanced splenic erythropoiesis after TBI.
Figure 3. Enhanced splenic erythropoiesis in Setd4 deficient mice after 8 Gy of TBI.

The spleens were collected to analyze their cellular components.
(A): Representative gross features of spleens, and spleen weight at 2 weeks post 8 Gy TBI from mice with the indicated genotypes.
(B): Representative H&E staining of spleens at 2 weeks post 8 Gy TBI, scale bar: 100 μm. A denser population of cells were observed in the Setd4 deficient mice than the control mice.
(C): A schematic representation of developmental sequence of erythroid cells from Stage 1 (SI) to Stage 5 (S5). HSC: hematopoietic stem cell; BFU-E: burst-forming unit-erythroid; CFU-E: colony-forming unit-erythroid; Pro-E: pro erythroblast; Chromo-E: chromatophilic erythroblasts; RBC: reticulocyte and erythrocyte.
(D): Representative CD71 and TER119 staining profiling for spleen erythroid populations for the indicated mouse genotypes. The gating scheme to identify the different erythroid cell populations is shown on the right.
(E): The numbers of total splenic erythroid cell populations (S2-S5) at 2 weeks post 8 Gy. Shown are combined quantification from two independent experiments.
(F): Percentage distribution of B cells, T cells, granulocytes, and erythroid cells in the spleens at 2 weeks post 8 Gy for the indicated mouse genotypes.
Setd4Δ/Δ reconstituted BM and engrafted Setd4Δ/Δ HSCs were more sensitive to IR than wild type counterparts
It is generally believed that both the fitness of the stem and progenitor cells in the hematopoiesis hierarchy, and the regulatory function of the BM microenvironment niche contribute to the recovery from the hematopoietic failure(7,47). To understand the mechanism(s) by which Setd4 deficiency conferred an enhanced BM recovery, we first investigated whether Setd4 deletion confers radiation resistance to the stem and progenitor cells in the hematopoietic hierarchy using two independent approaches.
First, we measured the role of Setd4 in the sensitivity of hematopoietic stem and progenitor cells (Fig. 4A). Bone marrows of the wild type B6.CD45.1 recipient mice were depleted by 2 doses of 6.5 Gy TBI separated by 4 hours, which is known to completely depletes BM and peripheral hematopoietic cells while sparing the BM niche(48). Within 24 hours, the recipient mice were transplanted with 1 million of Lin- cells isolated from the male littermates of Setd4Δ/Δ or Setd4flox/flox, which express the CD45.2 congenic surface marker. Both the CD45 staining of peripheral blood cell from transplanted recipients and the genotyping of DNA extracted from ear clips 8 weeks after transplantation (Fig. 4B and 4C) confirmed that the donor HSCs had successfully implanted in the recipient mice and were functioning as the source of hematopoiesis. Thus, these mice were chimeric mice where the HSC-hierarchy was contributed by the donor HSCs (Setd4flox/flox or Setd4Δ/Δ), but the BM niche and other stromal cells were derived from the recipient wild type mice.
Figure 4. Radiation sensitivity of wild type and Setd4 deficient BM chimeric mice.

(A): Schema outlining the non-competitive BM reconstitution assay. B6.CD45.1 recipient mice (B6 mice expressing the congenic CD45.1 marker) were exposed to 2 split doses of 6.5 Gy (total of 13 Gy) separated by 4 hours to deplete their native BM. Then one million Lin– BM cells from sex-matched littermates of either Setd4flox/flox or Setd4Δ/Δ mice (with CD45.2 marker) were transplanted into recipient mice. Eight weeks after the transplantation, peripheral blood and ear clips were sampled to confirm the successful implantation of the donor BM. Sixteen weeks after the transplantation, the mice were exposed to 8 Gy of TBI to induce hematopoietic syndrome and to address whether there is any difference of sensitivity caused by the transplanted donor BM.
(B): Representative flow cytometric profiles of peripheral blood cells from the B6.CD45.1 recipients reconstituted with CD45.2 Setd4flox/flox or CD45.2 Setd4Δ/Δ BM cells. The peripheral blood cells were sampled 8 weeks after BM transplantation, and then stained antibodies to differentiate the transplanted cells (CD45.2+) from the recipient cells (CD45.1+). The small percentage (6-7%) of the remaining CD45.1 host cells are likely radio-resistant that were not killed by the irradiation.
(C): Conformation of the genotyping of the BM chimeric mice. Genomic DNA were extracted from ear snips (containing blood) of recipients mice that were reconstituted with Setd4Δ/Δ BM cells (lanes 1, 2, 3, 4, and 6) or with Setd4flox/flox BM cells (lanes 5, 7, 8). Multiplex PCR genotyping was performed to simultaneously identify the wild type, floxed exon 6, and ΔExon6 alleles using three mixed primers: A (5′-TCCTGGGCTCTGCCATCCATG), B (5′-CTGTTGCAATGGAAATGCCAG), and C (5′-CTAAAGCTCTGCCCTAAGGTC). Pairing between primers A and B can amplify a 234bp wt allele and a 318bp flox-Exon6 allele but cannot amplify the ΔExon6 allele because region of primer B is deleted in the ΔExon6 allele. Paring between primers A and C can amplify a 369bp ΔExon6 allele. (Details of the PCR genotype strategies can be found in the Supplement Information of a previous report (40).
(D): Kaplan-Meier survival curves of BM chimeric mice after γ-radiation. Sixteen weeks after transplantation, the B6.CD45.1 recipients were treated with 8 Gy of TBI, and animal survival from the expected hemopoietic failure was monitored. Shown are the survival curves of the mice transplanted with Setd4Δ/Δ (n=13) or Setd4flox/flox (n=11). p = 0.0049.
To allow the transplanted LT-HSCs to replenish the entire hematopoiesis hierarchy(49), the recipient chimeric mice were kept for 16 weeks, and then treated with TBI (8Gy). Surprisingly, the mice transplanted with Setd4Δ/Δ HSCs actually were more sensitive than the mice transplanted with the Setd4flox/flox HSCs (Fig. 4D). This finding is contradictory to our initial prediction that the Setd4 deletion may confer a resistance of the cells in the HSC hierarchy, and suggested that Sedt4 deletion actually made the cells in the hematopoietic hierarchy more sensitive than Setd4flox/flox mice, which did not explain why the Setd4Δ/Δ mice were more resistant to radiation induced hematopoietic syndrome than the Setd4flox/flox mice.
To verify the above findings, we carried out a competitive transplantation assays as outlined in Fig. 5A. First, we depleted the BM of wild-type recipient mice (CD45.1) as described previously(48). Then, Lin- BM cells of Setd4Δ/Δ (CD45.2) and Setd4wt/wt (CD45.1/2) were mixed at approximately 1:1 ratio and transplanted into the recipients (CD45.1). The relative contribution of each type of donor cells to hematopoiesis was assessed by determining the ratio of CD45.2 over CD45.1/2 in peripheral blood cells with flow cytometric analysis at 4-week intervals, during the first 8-16 week after transplantation. As demonstrated in Fig. S5A, the donor Setd4Δ/Δ (CD45.2) and competitor wild type (CD45.1/2) cells successfully implanted and there were few recipient (CD45.1) cells in the peripheral blood.
Figure 5. Radiation sensitivity of engrafted HSCs from Setd4 deficient mice.

(A): Shema detailing the competitive repopulation assay. Lin– BM cells (>95% Lin–) from Setd4Δ/Δ mice (CD45.2) were mixed, at a ratio of 1:1, with WT competitor Lin–cells from B6.CD45.1/2 mice, and 1 × 106 total cells were injected i.v. into lethally irradiated B6.CD45.1 recipients. The contribution of HSCs to reconstitute and subsequent refurbishment of the peripheral hematopoietic cells was monitored by flow cytometric analysis every 4 weeks. Twenty weeks after the transplantation, 6 of 11 reconstituted mice were exposed to sublethal irradiation (6.5 Gy) (IR: n=6; non-IR: n=5), and peripheral blood was monitored for 12 weeks, until final sacrifice for BM analysis at 18 weeks after the irradiation (or 48 weeks after the transplantation).
(B): Peripheral blood from lethally irradiated transplanted mice were monitored for the relative contribution of Setd4Δ/Δ donor (CD45.2) and wild type competitor (CD45.1/2) cells every 4 weeks, starting 8 weeks post-transplantation. Shown are the ratio of CD45.2 cells over CD45.1/2 competitor cells in the total peripheral blood cells and the granulocytes. Data show mean ± SEM among the mice, and are representative of two identical experiments. See Table S4 for the number of animals in each group. The presented data were the combination of two independent experiments.
(C): The relative contribution of irradiated Setd4Δ/Δ (donor) and WT (competitor) cells in all cells (left) or granulocytes (right) was plotted at 16, 24, 28 and 32 wk post transplantation. *, p < 0.05 by t-test.
(D): The relative contribution of Setd4Δ/Δ and Setd4wt/wt BM HSCs and HPCs at 48 weeks after the transplantation. Left: irradiated mice (3 mice per group). Right: non-irradiated mice (2 mice per group). Mean and SEM are also shown. See Fig. S5 for representative flowcytometry profiles.
We reasoned that if the radiation sensitivities of Setd4Δ/Δ (CD45.2) and Setd4wt/wt (CD45.1/2) BM cells are similar, then the relative percentage of these populations should remain consistent in the mice after irradiation. The reconstituted mice were kept for 16 weeks and then exposed to 6.5 Gy of gamma-radiation 20 weeks after the initial transplantation. We then measured the relative population sizes of the Setd4Δ/Δ (CD45.2) and Setd4wt/wt (CD45.1/2) cells in peripheral blood every 4 weeks after the irradiation (i.e. 24, 28, and 32 weeks after the initial transplantation). Fig. 5B shows the relative population sizes of the irradiated (top panel) and non-irradiated (bottom panel) mice. When comparing the donor ratio at 16 weeks (4 weeks before irradiation) with that at 24 weeks (4 weeks after irradiation), a remarkable reduction in the CD45.2 (Setd4Δ/Δ) cells was observed (Fig. 5B, S5B), while there was an increase in these cells in non-irradiated mice (Fig. 5B & S5B). The total ratio of Setd4Δ/Δ cells to wild type competitor cells dropped from 1.5:1 to 0.5:1 by 4 weeks after irradiation and never recovered at later time points (Fig. 5C). This indicated that upon irradiation the Setd4Δ/Δ hematopoietic cells had reduced competence in the recipient mice when compared with the Setd4 proficient cells. These results were in agreement with results observed above (Fig. 4) and confirmed that the Setd4 deficient hematopoietic cells were more sensitive to radiation. Another important finding from this experiment is that, in the absences of irradiation, the Setd4 deficient hematopoietic cells may have had an advantage in re-populating the BM over the Setd4wt/wt hematopoietic cells (see bottom panels of Fig. 5B, S5B).
To assess the long-term fitness of the BM cells, the above mice were sacrificed 28 weeks after irradiation (or 48 weeks after the initial transplantation) and lineage analysis was performed on their BM cells as described in Fig. 2 and S4. As shown in Fig. 5D and S6 the ratio of the Setd4Δ/Δ to Setd4wt/wt cells in the BM were significantly reduced at this late stage after the irradiation. These data again suggested that the Setd4 deficient LT-HSCs and progeny were more sensitive to radiation. However, among the two available pairs of non-irradiated mice, there were significantly more early progenitors in Setd4Δ/Δ than Setd4wt/wt, although the advantage of Setd4Δ/Δ LSK, LT-HSC and ST-HSC was marginal. These data again implied that the Setd4Δ/Δ cells had an advantage in repopulating the BM when not stressed by irradiation.
Setd4 deletion confers an improved BM microenvironment to support HSC transplantation
Collectively, data from the BM transplantation assays (Fig. 4 and 5) implied that Setd4 deletion makes the hematopoietic cells acutely more sensitive to radiation, while conferring an advantage to the Setd4Δ/Δ hematopoietic progenitors in the absences of irradiation. Nevertheless, these findings suggested that the improved recovery of Setd4Δ/Δ mice from acute BM failure wasn’t due to a radiation resistance of the Setd4Δ/Δ HSC or HPC at the time of exposure. This led us to form an alternative hypothesis that the enhanced recovery of Setd4Δ/Δ mice from acute hematopoietic syndrome may be attributed to a more favorable Sedt4 deficient BM niche. To test this, we performed HSC dilution survival assays (Fig. 6A). Male Setd4flox/flox:Rosa26-CreERT2 littermates were injected with Tam (Setd4Δ/Δ) or Oil (Setd4flox/flox). Seven days later, the mice were subjected to lethal doses of radiation (2 doses of 6.5 Gy separated by 4 hours) to deplete the BM, and then transplanted with titrated numbers of wild-type Lin− BM cells from CD45.1 mice (Table S5). In this assay, the survival of recipients depends on whether the damaged BM niche was able to support transplanted HSCs after limiting numbers are transplanted. As shown in the Fig. 6B, Setd4Δ/Δ had a significantly improved survival than Setd4flox/flox when transplanted with the same number of wild type Lin- cells. Based on a survival calculation, the number of cells required for 63% survival (or 37% fatality rate) was 43.8 x 103 for Setd4Δ/Δ mice and 69 x103 for Setd4flox/flox mice, a reduction of 1.6-fold. This indicates that Setd4Δ/Δ mice have a more favorable BM environment to host the HSC transplantation. Thus, the radiation-resistance of Setd4 deficient mice was likely due to an improved BM niche that nourishes the damaged hematopoietic cells. This highlights the importance of the BM niche in the recovery of the hematopoiesis in contrast to the intrinsic radiation sensitivity of the hematopoietic cells themselves.
Figure 6. Survival of lethally irradiated Setd4 deficient and proficient mice after transplantation of wild type Lin- BM donor cells.
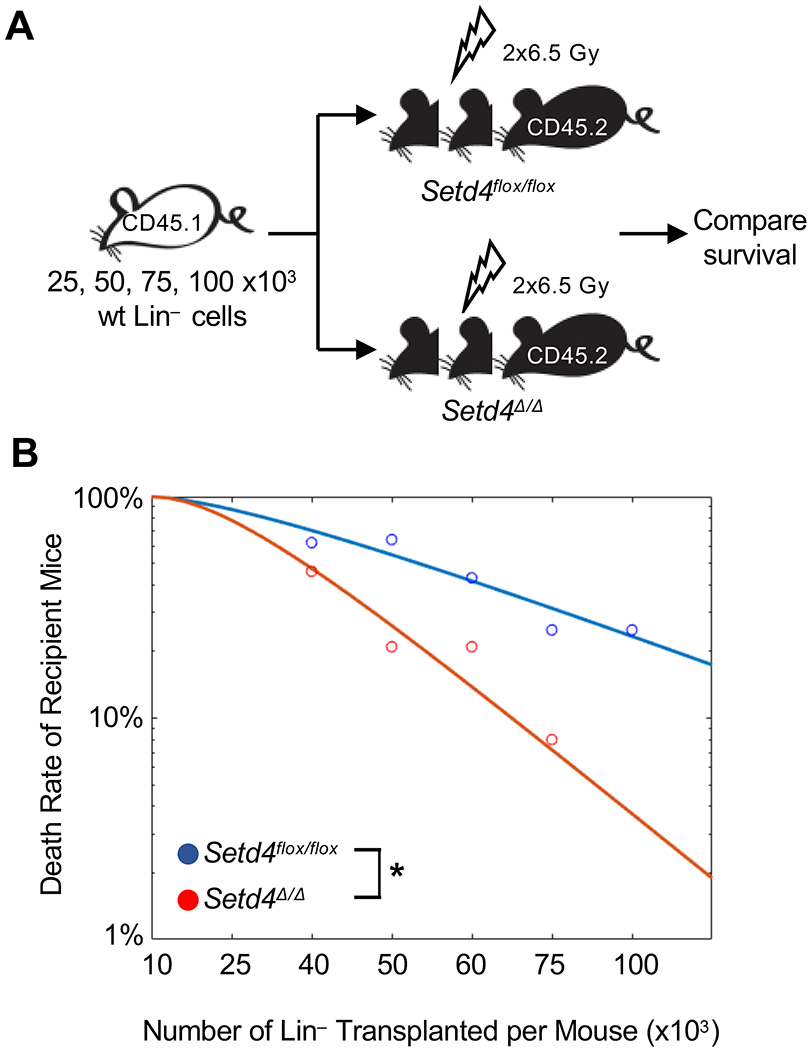
(A): Shema detailing HSC transplantation to rescue the lethally irradiated recipient mice. Serially diluted donor Lin- BM cells (10, 25, 40, 50, 60, 75, and 100x103) from B6.CD45.1 mice were transferred into Setd4Δ/Δ or Setd4flox/flox recipients that were irradiated with to 2 doses of 6.5 Gy separated by 4 hours (total of 13 Gy). Survival was monitored for 30 days.
(B): Death rate of the transplanted recipient mice as a function of the numbers of transplanted Lin− cells per mouse. For each experimental point, 6-14 recipients were used. The overall death rates were based on combined results from multiple independent transplantation experiments (see Table S5 for the specific number of mice used in each time point). The curves were fit by nonlinear regression. The calculated number of cells for a 63% of survival rate is 69.0x103 for Setd4flox/flox mice, and 43.8x103 for Setd4Δ/Δ mice. P=0.029 by two-way Anova test.
Discussion
In this study, we use an inducible Setd4-knockout mouse model to directly identify the function of SETD4 in mammalians, and surprisingly found that Setd4-deficient mice were more resistant to radiation-induced hematopoietic syndrome. Interestingly, Setd4 deletion conferred a radio-sensitivity to the HSCs in vivo, but the non-irradiated Setd4 deficient HSCs had an advantage over wild type HSCs in recipient mice, and the BM niches of the Setd4-deficient recipient mice had an enhanced ability to engraft transplanted HSCs. Furthermore, there was an enhanced splenic erythropoiesis in Setd4 deficient mice. These novel findings suggest a unique mode of action by which Setd4 modulates the recovery from BM failure and underscores the importance of the BM niche in the recovery from hematopoietic failure.
Given that BM injury is a major adverse side effect of anticancer therapy and excessive exposure to radiation, it is possible that inhibition of Setd4 during these circumstances may be beneficial to the recovery of the BM. Based on our findings, it is foreseeable that inhibition of Setd4 would: 1) sensitize cells of the HSC lineage in the host to BM depletion, 2) make the host BM niche more amenable to receive allogeneic transplantation, and 3) improve the competence of the donor HSCs. Furthermore, it has been shown that Setd4 deletion delayed the development of radiation-induced T-lymphoma(40). Thus, Setd4 would potentially be an attractive target for inhibition to help condition the BM before allogeneic transplantation, a strategy used to treat widely disseminated malignancies and some solid tumors(8–10).
It is well known that the overall function of the hematopoietic system is both intrinsically regulated by the HSCs and their progenies, as well as by the BM niche nurturing the HSCs. One striking finding of our study is that the Setd4 deficient HSCs are actually more sensitive to irradiation, but the BM niche of the same Setd4 deficient mice is more receptive to the engraftment of HSC upon transplantation. However, the overall consequence of these confounding effects was an improved recovery from radiation induced hematopoietic failure. This finding underscores the importance of the BM niche in the overall function of the hematopoietic system. It is generally believed that the BM niche is supported by osteoblasts, mesenchymal stromal cells, endothelial cells, and likely additional cellular components. Thus, an important aspect of future studies would be determining whether and how Setd4 regulates the function(s) of these BM niche cells. Because the mice received transplantation of Setd4 HSCs were more sensitive to radiation (Fig. 4), more quantitative approaches, such as CFU-S assay, may be adapted in the future to quantify the role of Setd4 in HSC sensitivity to radiation damage and to identify the associated molecular mechanisms.
It should be noted that our results also suggested that the improved recovery of Setd4 deficient mice is independent of p53 deletion, and Setd4 loss further enhanced the survival of p53-deficient mice post IR (Fig. S1). Pharmacological inhibition of p53 has been proposed to protect individuals from acute BM injury and attenuate the development of radiation-related blood cancer(50). Similarly, it is foreseeable that targeting Setd4 may represent another strategy to protect BM from radiation and radiation-induced lymphomagenesis.
The mammalian SETD4 is largely an orphan methyltransferase since no physiological substrate of mammalian SETD4 has been reported. Several preliminary reports have suggested a potentially oncogenic activity of mammalian SETD4(37–39), and oncogenic fusions of SETD4 had been reported in TCGA database. The conditional Setd4 deletion did not significantly change spontaneous tumor formation (40), which is in an agreement with these preliminary reports. Clearly, our study has only just begun to reveal the function of mammalian SETD4, and additional studies would need to be performed to further understand SETD4 activities in mammalians.
In summary, our studies revealed a critical role of Setd4 in maintaining hematopoiesis, suggesting that Setd4 inhibition might be an attractive strategy to mitigate radiation-induced BM injury, and improve the outcome of allogeneic HSC transplantation after TBI for patients with widely disseminated malignancies. Future studies will be needed to identify physiological SETD4 substrates and to elucidate the regulatory mechanisms of SETD4 expression.
Supplementary Material
Acknowledgments:
The authors thank Ms. Neta Schneider for assistance with some of the mouse work, Mr. Louis Osorio (Rutgers Child Health Institute of New Jersey) for generous technical assistance with performing the mouse BM transplantation studies; and Dr. Arthur Roberts (Rutgers Cancer Institute of New Jersey) for cell sorting; Dr. Ping Xie and Dr. Sam Bunting (Rutgers the State University of New Jersey) for suggestions throughout the study.
Funding Statement: This research was supported by NIH R01CA156706 and R01CA195612 grants to ZS and R21AI105747 to LKD, by support from Robert Wood Johnson Foundation to CINJ, and by the Biospecimen Repository and Histopathology Service, Genome Editing, and Flow Cytometry/Cell Sorting shared resources of The Rutgers Cancer Institute of New Jersey (P30CA072720).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement: The authors declare that there were no competing interests.
References
- 1.Pronk E, Raaijmakers MH. The mesenchymal niche in mds. Blood 2019;133:1031–1038. [DOI] [PubMed] [Google Scholar]
- 2.Sieff CA. Introduction to acquired and inherited bone marrow failure. Hematology/Oncology Clinics 2018;32:569–580. [DOI] [PubMed] [Google Scholar]
- 3.Bogliolo M, Surralles J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Current opinion in genetics & development 2015;33:32–40. [DOI] [PubMed] [Google Scholar]
- 4.Shimoni A, Nagler A. Radioimmunotherapy and stem-cell transplantation in the treatment of aggressive b-cell lymphoma. Leukemia & lymphoma 2007;48:2110–2120. [DOI] [PubMed] [Google Scholar]
- 5.Calado RT. Immunologic aspects of hypoplastic myelodysplastic syndrome Seminars in oncology. Elsevier; 2011. pp. 667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blumenfeld Z. Chemotherapy and fertility. Best practice & research Clinical obstetrics & gynaecology 2012;26:379–390. [DOI] [PubMed] [Google Scholar]
- 7.Shao L, Wang Y, Chang J, et al. Hematopoietic stem cell senescence and cancer therapy-induced long-term bone marrow injury. Translational cancer research 2013;2:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong JYC, Filippi AR, Dabaja BS, et al. Total body irradiation: Guidelines from the international lymphoma radiation oncology group (ilrog). International journal of radiation oncology, biology, physics 2018;101:521–529. [DOI] [PubMed] [Google Scholar]
- 9.Quast U. Whole body radiotherapy: A tbi-guideline. Journal of medical physics 2006;31:5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paix A, Antoni D, Waissi W, et al. Total body irradiation in allogeneic bone marrow transplantation conditioning regimens: A review. Critical reviews in oncology/hematology 2018;123:138–148. [DOI] [PubMed] [Google Scholar]
- 11.De Haro LP, Wray J, Williamson EA, et al. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res 2010;38:5681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradhan S, Chin HG, Estève P-O, et al. Set7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 2009;4:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi MH, Palanichamy Kala M, Ow JR, et al. Glp inhibits heterochromatin clustering and myogenic differentiation by repressing mecp2. Journal of molecular cell biology 2018;10:161–174. [DOI] [PubMed] [Google Scholar]
- 14.Wang GG, Cai L, Pasillas MP, et al. Nup98–nsd1 links h3k36 methylation to hox-a gene activation and leukaemogenesis. Nature cell biology 2007;9:804. [DOI] [PubMed] [Google Scholar]
- 15.Rea S, Eisenhaber F, O’carroll D, et al. Regulation of chromatin structure by site-specific histone h3 methyltransferases. Nature 2000;406:593. [DOI] [PubMed] [Google Scholar]
- 16.Peters AH, O’Carroll D, Scherthan H, et al. Loss of the suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001;107:323–337. [DOI] [PubMed] [Google Scholar]
- 17.Lehnertz B, Ueda Y, Derijck AA, et al. Suv39h-mediated histone h3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Current Biology 2003;13:1192–1200. [DOI] [PubMed] [Google Scholar]
- 18.Shen X, Liu Y, Hsu Y-J, et al. Ezh1 mediates methylation on histone h3 lysine 27 and complements ezh2 in maintaining stem cell identity and executing pluripotency. Molecular cell 2008;32:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett RL, Swaroop A, Troche C, et al. The role of nuclear receptor-binding set domain family histone lysine methyltransferases in cancer. Cold Spring Harbor perspectives in medicine 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrossian TC, Clarke SG. Uncovering the human methyltransferasome. Molecular & Cellular Proteomics 2011;10:M110. 000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai L, Ye S, Li HW, et al. Setd4 regulates cell quiescence and catalyzes the trimethylation of h4k20 during diapause formation in artemia. Molecular and cellular biology 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trievel RC, Flynn EM, Houtz RL, et al. Mechanism of multiple lysine methylation by the set domain enzyme rubisco lsmt. Nature Structural Biology 2003;10:545. [DOI] [PubMed] [Google Scholar]
- 23.Trievel RC, Beach BM, Dirk LMA, et al. Structure and catalytic mechanism of a set domain protein methyltransferase. Cell 2002;111:91–103. [DOI] [PubMed] [Google Scholar]
- 24.Chang Y, Levy D, Horton JR, et al. Structural basis of setd6-mediated regulation of the nf-kb network via methyl-lysine signaling. Nucleic acids research 2011;39:6380–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson AW, Diep J, Dai S, et al. Setd3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 2019;565:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Q, Liao S, Kwiatkowski S, et al. Structural insights into setd3-mediated histidine methylation on beta-actin. eLife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abaev-Schneiderman E, Admoni-Elisha L, Levy D. Setd3 is a positive regulator of DNA-damage-induced apoptosis. Cell Death Dis 2019;10:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy D, Kuo AJ, Chang Y, et al. Lysine methylation of the nf-κb subunit rela by setd6 couples activity of the histone methyltransferase glp at chromatin to tonic repression of nf-κb signaling. Nature immunology 2011;12:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feldman M, Vershinin Z, Goliand I, et al. The methyltransferase setd6 regulates mitotic progression through plk1 methylation. Proceedings of the National Academy of Sciences of the United States of America 2019;116:1235–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vershinin Z, Feldman M, Chen A, et al. Pak4 methylation by setd6 promotes the activation of the wnt/beta-catenin pathway. The Journal of biological chemistry 2016;291:6786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy D, Liu CL, Yang Z, et al. A proteomic approach for the identification of novel lysine methyltransferase substrates. Epigenetics & chromatin 2011;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DW, Kim KB, Kim JY, et al. Characterization of a novel histone h3k36 methyltransferase setd3 in zebrafish. Biosci Biotechnol Biochem 2011;75:289–94. [DOI] [PubMed] [Google Scholar]
- 33.Eom GH, Kim KB, Kim JH, et al. Histone methyltransferase setd3 regulates muscle differentiation. The Journal of biological chemistry 2011;286:34733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwiatkowski S, Seliga AK, Vertommen D, et al. Setd3 protein is the actin-specific histidine n-methyltransferase. eLife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye S, Ding YF, Jia WH, et al. Set domain-containing protein 4 epigenetically controls breast cancer stem cell quiescence. Cancer Res 2019;79:4729–4743. [DOI] [PubMed] [Google Scholar]
- 36.Zhong Y, Ye P, Mei Z, et al. The novel methyltransferase setd4 regulates tlr agonist-induced expression of cytokines through methylation of lysine 4 at histone 3 in macrophages. Molecular immunology 2019;114:179–188. [DOI] [PubMed] [Google Scholar]
- 37.Faria JAQA, Corrêa NCR, de Andrade C, et al. Set domain-containing protein 4 (setd4) is a newly identified cytosolic and nuclear lysine methyltransferase involved in breast cancer cell proliferation. Journal of cancer science & therapy 2013;5:58–65. [PMC free article] [PubMed] [Google Scholar]
- 38.Li G-M, Wang Y-G, Pan Q, et al. Rnai screening with shrnas against histone methylation-related genes reveals determinants of sorafenib sensitivity in hepatocellular carcinoma cells. International journal of clinical and experimental pathology 2014;7:1085. [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu S, Xu Y, Song M, et al. Prdm16 is associated with evasion of apoptosis by prostatic cancer cells according to rna interference screening. Molecular medicine reports 2016;14:3357–3361. [DOI] [PubMed] [Google Scholar]
- 40.Feng X, Lu H, Yue J, et al. Loss of Setd4 delays radiation-induced thymic lymphoma in mice. DNA Repair (Amst) 2020;86:102754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amrani YM, Gill J, Matevossian A, et al. The paf oncogene is essential for hematopoietic stem cell function and development. Journal of Experimental Medicine 2011:jem. 20102170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nimmo RA, May GE, Enver T. Primed and ready: Understanding lineage commitment through single cell analysis. Trends in cell biology 2015;25:459–67. [DOI] [PubMed] [Google Scholar]
- 43.Adolfsson J, Borge OJ, Bryder D, et al. Upregulation of flt3 expression within the bone marrow lin(−)sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 2001;15:659–69. [DOI] [PubMed] [Google Scholar]
- 44.Woolthuis CM, Park CY. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood 2016;127:1242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim CH. Homeostatic and pathogenic extramedullary hematopoiesis. Journal of blood medicine 2010;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siatecka M, Bieker JJ. The multifunctional role of eklf/klf1 during erythropoiesis. Blood 2011;118:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine 2014;20:833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui Y, Hisha H, Yang G, et al. Optimal protocol for total body irradiation for allogeneic bone marrow transplantation in mice. Bone marrow transplantation 2002;30:843. [DOI] [PubMed] [Google Scholar]
- 49.Dykstra B, Kent D, Bowie M, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell stem cell 2007;1:218–229. [DOI] [PubMed] [Google Scholar]
- 50.Yu H, Shen H, Yuan Y, et al. Deletion of puma protects hematopoietic stem cells and confers long-term survival in response to high-dose γ-irradiation. Blood 2010;115:3472–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.