

Abstract
Neural tube defects (NTDs) represent a failure of the neural plate to complete the developmental transition to a neural tube. NTDs are the most common birth anomaly of central nervous system. Following mandatory folic acid fortification of dietary grains, a dramatic reduction in the incidence of NTDs was observed in areas where the policy was implemented, yet the genetic drivers of NTDs in humans, and the mechanisms by which folic acid prevents disease remain disputed. Here we discuss current understanding of human NTD genetics, recent advances regarding potential mechanisms by which folic acid might modify risk through effects on the epigenome and transcriptome, and new approaches to study refined phenotypes for a greater appreciation of the developmental and genetic causes of NTDs.
Keywords: Anencephaly, folate, folic acid, myelomeningocele, neural tube defect, spina bifida
Neural tube defect is a genetically complex disease
Neural tube defects (NTDs; see Glossary) are the most common birth anomaly of the central nervous system (CNS), encompassing a range of defects of varying severity. NTDs can occur anywhere along the neural axis and present with a range of clinical severity, and their subtypes are named based upon the anatomic site and severity of the defect. The most severe forms of NTD are anencephaly or craniorachischisis, in which either the forebrain or the entire CNS, respectively, fails to transition from a neural plate to a neural tube. The least severe forms show near complete closure of the neural tube, in conditions such as spinal lipoma or spina bifida occulta, occurring in 4–6% of the general population, identified incidentally during spine imaging in many cases [1]. NTDs account for approximately 88,000 deaths/year worldwide (disease burden and mortality estimates of World Health Organization: https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html) [2]. In the US, NTDs are one of the conditions tracked by the Centers for Disease Control and Prevention (CDC) longitudinally across the country, to define disease incidence, and identify populations at risk. Most patients with symptomatic disease remain wheelchair bound throughout life, with urinary or fecal incontinence and learning difficulties [3]. Moreover, it is likely many more fetuses with NTD are spontaneously or electively terminated. Despite its frequency, the genetic basis of NTDs in humans remains mostly unknown, and therefore molecular characterization and effective quantification of risk remains unachievable.
Up to 70% of the NTD risk is predicted to be attributable to genetic factors, although these estimates were made over 40 years ago, prior to dietary folic acid (FA) supplementation, so these bear repeating [4]. There are multiple lines of evidence supporting NTD as a genetic disease, but the lack of mendelian inheritance in most cases suggests it is a genetically complex disease (Fig. 1). Most evidence is from twin studies, where higher concordance was observed in same-sex twins (which includes monozygotic and dizygotic twins) than in opposite-sex twins (1.93 vs 1.00 per 1,000) [5, 6], which was further validated in a more recent study [7]. However, the concordance rate varies with NTD subtypes, implying that there might be multiple genetic mechanisms or environmental factors influencing the genetic architecture. In familial cases, NTDs phenotypes tend to ‘breed true’ within families; in other words, if the proband has spina bifida then the recurrence tends to have spina bifida rather than a different NTD [8]. This observation suggests that not all NTDs are equivalent and that different genetic factors may determine the location and severity of the NTD.
Figure 1. Neural tube defects (NTDs) are likely to be impacted by many types of genetic variants.
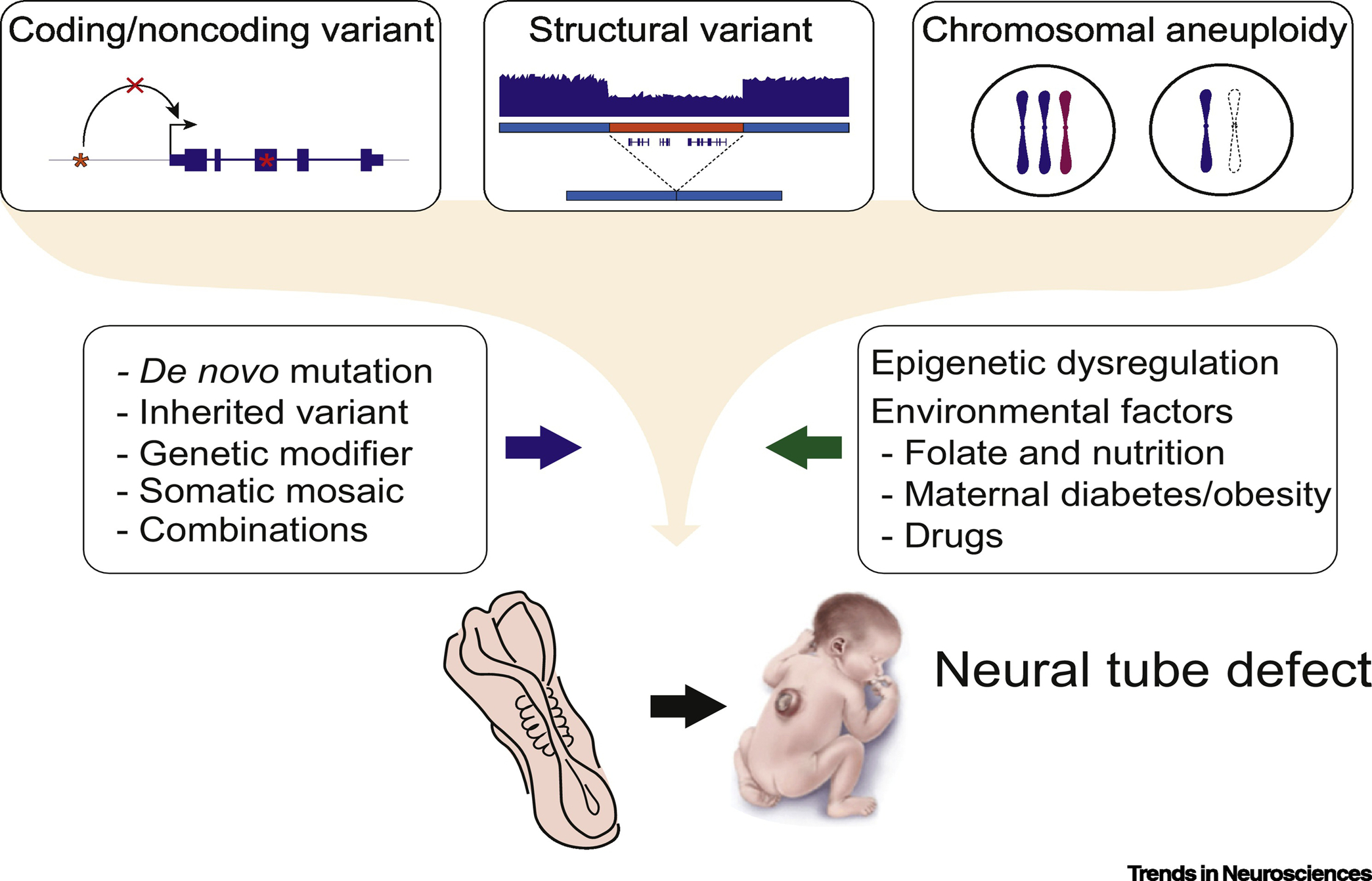
These variants include coding and noncoding ones, structural ones, and chromosomal aneuploidy. Sources of variants include de novo, inherited, modifiers, somatic and combinations of these. Genetic variants are influenced by epigenetic factors and the environment to establish the NTD risk. Newborn image adopted from the CDC Public Library of Images.
Can an understanding of genetic architecture of NTDs be achieved using similar approaches to those used for conditions like congenital heart disease and autism spectrum disorder (ASD)? These conditions share features with NTDs in that: 1) they show complex inheritance; 2) most cases are sporadic; 3) in the severe case of the disease, most affecteds are unlikely to bear children, and thus mutations are likely under strong purifying selection [9, 10]. What was most insightful for these conditions was the realization that a large collection of phenotypically diverse patients could capture both rare and common variants contributing to risk and that simplex cases could take the place of large pedigrees that were used for traditional linkage associations. Current trends are to apply similar approaches to what was successful for congenital heart disease and ASD, by recruiting large and phenotypically-diverse cohorts of NTD simplex cases, stratified based upon FA exposure. The goal of this review is to discuss the current understanding of human NTD genetic architecture, NTD risk factors, and potential NTD candidate genes. We also address potential ways in which FA works to reduce risk of NTDs through transcriptional and epigenetic mechanisms, which has potential implications on the genetic architecture of disease.
Mouse models of NTDs
To understand mechanism of NTDs, mouse has proven to be a tractable model, but of relatively limited impact on our understanding of human NTDs, mostly attributable to the polygenic nature of human NTDs. In mouse models, most results rely on recessive or dominant models, which are not common in humans. Over 300 NTD mouse models are reported (see Table S1 in the supplemental information online) [11], some of which show reduced penetrance or severity with FA supplementation (e.g. Pax3, Cart1), whereas others are FA resistant (e.g. Ct, Axd) [12]. One of the best known models is the FA-responsive crooked tail (Cd), a fully penetrant recessive NTD model linked to a damaging variant in LRP6 encoding Low-density lipoprotein receptor-related protein 6, a component of the Wnt-Fzd-LRP5-LRP6 complex that triggers beta-catenin signaling [13]. Several NTD patients with damaging variants in LRP6 were described [14], suggesting an evolutionarily conserved role, but few such examples exist. Multiple reasons could possibly account for the fundamental differences between humans and mice NTDs. Some possibilities are that (1) NTDs have different developmental mechanism in rodents; (2) body size differences impart differential risk; and (3) the human orthologs of mouse NTD genes predispose to risk in humans in ways that are not yet fully understood.
Genetic studies of human NTDs
Early linkage analysis within multiplex NTD families suggested candidate loci on chromosomes 2, 7, 10, and X [15–17]. However, logarithm of odds (LOD) scores were below genome-wide significance, and fine mapping was not successful. Moreover, there are no published large-scale genome-wide association studies (GWASs) of NTDs to date. Instead, most published NTD genetics studies have focused on candidate genes derived from the folate metabolic pathway (e.g. MTHFR), or mouse NTD models (see Table S2 in the supplemental information online). With the advent of next-generation sequencing (NGS), and the availability of approaches like whole exome (WES) and whole genome sequencing (WGS), the numbers of genes tested as candidates has increased. A number of consortia have started to emerge to aggregate cohorts and sequencing data, such as the Spina Bifida Sequencing Consortium (https://sbseqconsortium.org) which we took part in initiating, given the need for multi-site and multi-ethnic aggregation for comprehensively studying disease architecture.
De novo mutations
De novo mutations (DNMs) can increase risk of both rare and common disease, and are the most likely to be under purifying selection [9, 10]. Given the success of DNM discovery in ASD and congenital heart disease [9, 10], investigating DNMs in NTDs would seem like an obvious strategy, further evidenced by positive results from small scale studies [18]. In a cohort of 43 NTD trios (parents and affected child) of mixed clinical presentation, about 10% carried loss-of- function (LoF) DNMs [18], some in genes associated with mouse NTD models. For instance, SHROOM3, mutated in the mouse NTD model Shrm [19], showed LoF DNMs in two offspring, one with myelomeningocele and one with anencephaly. SHROOM3 is a strong NTD candidate, but since even healthy offspring show LoF DNMs at a basal rate, genes identified with this approach require validation, and a statistical framework. Such a framework would not be difficult to develop. Assuming 100 different genes that can lead to NTDs with a LoF DNM, and with a 50% excess burden of such haploinsufficient LoF DNM in cases vs. controls, and a false discovery rate of <10%, we calculate about 1000 trios would need to be sequenced in order to identify 10 recurrently mutated genes (Fig. 2).
Figure 2. Estimation of cohort size for causative variants discovery.
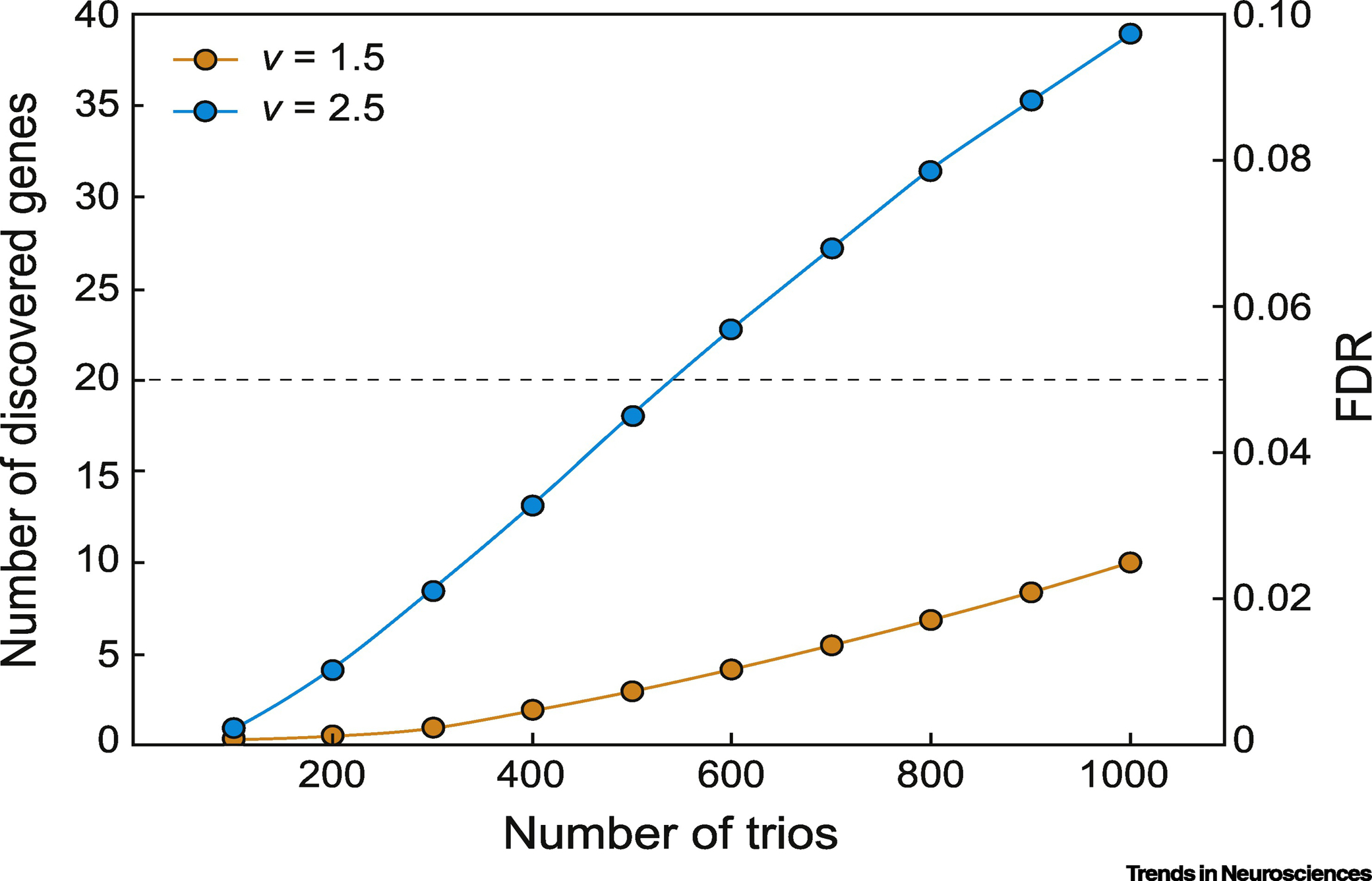
Power calculation curve indicating estimated number of trios necessary to discover a given numbers of genes as recurrently mutated, assuming that there are 100 ‘discoverable’ genes for haploinsufficient neural tube defects (NTDs) in humans. The variable ‘v’ is defined as the ratio of the number of de novo loss-of-function (LoF) mutations in affected individuals divided by the number of de novo LoF mutations in unaffected individuals across the population. For these calculations, we estimate v as 1.5 or 2.5, meaning that subjects with NTD will have 1.5 or 2.5 as many de novo LoF mutations as controls. Assuming v=2.5, then a cohort of 1000 trios would discover 40 recurrently mutated genes with a false discovery rate of 10% (blue line). If on the other hand v = 1.5 then 1000 trios would only discover 10 recurrently mutated genes. These calculations are based upon the basal de novo mutation rate, and assumes a total of 20,000 human genes that can be mutated. Abbreviation: FDR, false discovery rate.
Beyond the success with coding DNM, some NTD risk alleles are likely to reside in the noncoding genome. The noncoding genome contains (among other elements) regulatory elements that determine gene expression specificity, and thus extending discovery of rare genetic variants of high effect to the noncoding genome is essential to gain high-resolution insight into the spatiotemporal and cell type specific dynamics of the biology of NTD. WGS analysis holds the potential to detect the most noncoding genome variation, but a major challenge is a lack of a priori evidence for association testing, relying instead on an ever-increasing number of annotations describing noncoding genome functionality [20]. Such high noncoding genome dimensionality will require consideration of multiple comparisons problem, requiring again a statistical framework to test associations. Recently, category-wide association study (CWAS) has been developed to test multiple hypotheses for noncoding variants from WGS data [21]. From the WGS of 1,902 ASD families, CWAS has successfully identified noncoding DNMs that contribute to ASD risk, with the strongest contributors in conserved promoter regions [22]. These combined approaches have the potential to paint a complete picture of risk associated with noncoding variation, but will require enormous cohort sizes.
Rare inherited coding variants
The plethora of individual genetic factors contributing to NTDs in mice, and the genes involved in folate metabolism, have provided ample fodder for candidate gene testing in NTD singletons. Efforts to detect rare coding variants using genome-wide approaches have been pursued with WES mainly on singleton patients [23, 24]. These early efforts generally relied on publicly available cohorts like the Genome Aggregation Database (gnomAD) to establish baseline minor allele frequencies [25], and as such may suffer errors from population stratification. One effort applied WGS analysis from ethnically diverse NTD cases, revealing enrichment of rare LoF variants in cases [23], and suggesting that NTDs may be caused by the sum of effects of multiple genes, similar to the oligogenic model. A study of a cohort of 51 NTD families showed cases had a greater burden of damaging variants in MYO1E, encoding an unconventional nonmuscle class I myosin [24]. These approaches, while promising, will require confirmation and genotype-phenotype correlations in larger cohorts.
Functional validation of human NTD genetic studies
Functional validation is a critical step that should follow the discovery of candidate genes or variants. A hurdle is overcoming the potential complexity of human NTDs, including zygosity, modifiers, and environmental factors. The lack of simple genetically faithful and highly penetrant model of human NTD is a potential hurdle, yet still important information can be gained by studying the impact of FA on existing or new animal models. Higher throughput than achievable in mouse can be realized in frog or zebrafish, but a caveat is that neural tube closure mechanisms differ across species [26]. Although in vivo vertebrate animal models are the most robust validation approach currently, other tools such as stem cell models, neural tube organoids, or 3-dimensional bioprinting have advantages, for instance in incorporating human-specific variants, and may prove fruitful in modeling human NTD [27].
Genes and pathways implicated in NTD
Folate metabolism pathway
Since folate was implicated in NTD risk, folate metabolic pathway genes have been studied as candidates in NTD. MTHFR encodes methylenetetrahydrofolate reductase, required for the conversion of folate into its active form, L-methylfolate. The MTHFR c.677C>T SNP (rs1801133) p.V222A reduces enzyme activity in cells by 35% when heterozygous and 70% when homozygous [28], and is the most reproduced single marker of NTD risk, conferring an odds ratio between 1.34–1.44 when present in the mother or the child, but not the father [29]. However, there are still some discrepancies in its effect on NTD risk, as the effect does not replicate in some populations [29, 30]. Similarly, the MTHFR promoter SNP (rs3737965) regulates MTHFR expression and contributes to spina bifida risk [31, 32], hinting at the kinds of non-coding variants likely to function in NTDs. There are other MTHFR SNPs studied but since they may be in linkage disequilibrium with other SNPs, it is difficult to separate their individual effects. SNPs in other folate-related genes such as MTHFD1, MTRR, MTR, TYMS, SLC19A1 or LRP2 have been associated with only a moderate effect on NTD risk [24, 33–36], and similarly the mitochondrial folate metabolism glycine cleavage system genes AMT, GCSH, and GLDC are attracting attention as NTD risk factors [37, 38].
Planar cell polarity (PCP) and canonical Wnt pathways
PCP has been appreciated as a critical pathway for neural tube closure, ever since rare coding variants in VANGL1 were found enriched in human NTDs [39], and a missense variant in Vangl2 was identified as the cause for the partially-penetrant Loop-tail (Lp) NTD mouse [40, 41]. The PCP pathway determines cellular orientation necessary for shape transitions from a neural plate to a tube. Moreover, PCP is a key driver of ‘convergent extension’, the process in which cellular movements converge (narrow) along one axis and extend (elongate) along the perpendicular axis. A PCP defect should thus result in an abnormally wide neural plate, hampering proper neural tube closure [41]. Since convergent extension of the embryo precedes neural tube closure, the idea is that early defects could lead to mis-patterning of the embryo that could increase NTD risk indirectly [41], and that defects in early developmental processes preceding neural tube closure could potentially impact NTD risk. PCP pathway gene mutations have been identified in 1–20% of NTD cases, depending upon the study, including genes SCRIB, DACT1, CELSR1, FUZ, FZD6, PRICKLE1, VANGL1 and VANGL2 [42], but the relative importance of these genes remains unclear. The canonical Wnt pathway has been also implicated in NTD risk. For example, LRP6, a Wnt co-receptor mutated in a mouse NTD model, was assessed for polymorphisms in 190 individuals with NTD, identifying four individuals with predicted deleterious variants, some of which impacted Wnt signaling (a key pathway regulating PCP) [14].
Sonic Hedgehog (Shh) pathway
While Shh loss-of-function mouse mutants often show gross patterning defects, it is hyperactivation of Shh signaling that leads to NTDs [43], and thus disruption of negative regulators of Shh pathway often leads to NTDs [44]. For instance, mutations in the Shh negative regulator GPR161 were enriched in 384 human spina bifida patients [45]. The Shh pathway repressor GLI3 was also linked to NTD risk in a WES study in a French cohort of 23 individuals [35]. Other Shh pathway genes, such as DISP1 and PTCH1, have also been implicated in NTD pathogenesis [46, 47]. PRKACB is important for the formation of the GLI3 repressor and has been implicated in human NTD risk [48]. NTD phenotypes resulting from Shh pathway mutations may share important components with ciliary pathway genes and PCP pathway mutants, since these pathways are known to interact [49].
Cytoskeleton and cilia genes
Genes controlling the cytoskeleton are strong candidates for NTD risk. However, it remains to be clarified in which cell populations these genes function, since there are many cell populations interacting during neural tube closure. These cell populations include neural cells, paraxial mesoderm, overlying fibroblasts, bone progenitors, etc. To date, only a handful of cytoskeletal genes are associated with human NTDs. The aforementioned SHROOM3 is a PDZ-domain containing actin regulator (sometimes included in the PCP pathway) and was implicated as an NTD risk gene in two separate studies with de novo and gene burden analyses [18, 24]. ITGB1, a signal transduction protein functioning between the cytoskeleton and extracellular matrix, and DLC1, a GTPase-activating protein important for cytoskeletal remodeling and cell migration/proliferation [18, 24], were also implicated. A number of ciliary genes lead to NTD when mutated, such as Tulp3 [50], because the graded response to Shh depends upon cilia architecture [51]. Sequencing of 281 candidate NTD genes in 373 NTD patients and 222 healthy controls revealed eight rare mutations in the cilia-related gene DNAAF1, which encodes a protein for preassembly of the dynein-arm complex within the cilium [52].
Inositol metabolism genes
NTDs were identified in mice lacking Itpk1, a regulator of inositol hexakisphosphate [53], and subsequently polymorphisms in ITPK1 were identified in human NTD patients [54]. Inositol is required for neural tube closure, and inositol supplementation can dramatically reduce curly tail NTD penetrance from ~70% to ~3–7% [55]. This led to the model that deficiency in inositol may impart risk of NTD, particularly in mothers found to be FA resistant. Currently there is a clinical trial of inositol as an add-on to FA supplementation in NTD prevention [56].
Chromosomal abnormality and structural variation
Given the complex genetic architecture of NTDs, it is not surprising that chromosomal abnormalities and structural variations (SVs) contribute to NTD risk. Large chromosomal abnormalities such as trisomy 13 or 18, among others, have been reported as risk factors for various NTDs [57, 58], accompanied by multiple congenital anomalies.
Structural variations
SVs such as chromosomal microdeletions or microduplications have not been studied much because most of the genetic assays used were unable to detect SVs, and therefore, effects of SV on NTD risk is not well established. For instance, NTD cases with a 2q35–36.2 deletion containing PAX3 have been reported in patients showing NTD with or without Waardenburg syndrome, which is an autosomal dominant PAX3 haploinsufficiency characterized by pigment abnormalities and sensorineural deafness [59–61]. Deletions in a few genes associated with proteoglycans or cilia also have been implicated in the NTD risk [62, 63].
DiGeorge/Velocardiofacial 22q11.2 deletion syndrome (22q11.2DS) is relatively common (1 per 3,000–6,000 live births), since the locus harbors four low copy number repeat sequence blocks (LCRs) which predispose to unequal crossing-over, leading to de novo deletions (or duplications) [64]. Although a few NTD cases with 22q11.2DS have been reported, it is still unclear which, if any, gene(s) among the ~90 genes in the interval confer NTD risk [64–66]. Likewise, SOX3 duplication (Xq27.1) has been reported in three female myelomeningocele patients [67], and duplication of 15q24.2-q26.2 containing ~100 genes was also reported in a single case of anencephaly and NTD [68]. Presumably, these SVs may contain either gene(s) of which decreased dosage increases risk NTDs, or noncoding regulatory elements including the ones impacting long-range chromatin effects, although it is not possible to distinguish them without refinement of the minimal interval or animal modeling.
Monogenic syndromes potentially increasing risk of NTDs
Several human syndromes where mutant gene function is well established display an increased risk of NTDs, which can provide insight into the pathogenesis of NTDs. The risk of NTD appears to be increased in individuals with Waardenburg syndrome, caused by haploinsufficiency of PAX3. PAX3 is a member of the paired box transcription factor family expressed by multipotent neural crest and somitic mesoderm precursors, implicating these cell types in NTD. In the literature there are reports of six of 617 Waardenburg syndrome patients with NTDs [61], i.e. risk of roughly 1:100, which is many times higher than the baseline population risk. This is supported by the splotch mouse Pax3 recessive mutation, that results in NTDs in 8–56% of offspring, depending upon the background and folate status [69]. TBXT aka brachyury, encoding the T-box transcription factor T, was identified in a human monogenic NTD with sacral agenesis, abnormal ossification of the vertebral bodies and persistent notochordal canal, as well as in isolated chordoma [46, 70]. Focal dermal hypoplasia, or Goltz syndrome, is an X-linked dominant disease caused by mutation of PORCN [71], encoding a homologue of porcupine, an O-acyltransferase involved in Wnt protein processing. Although only a few cases are reported, this condition has been associated with NTDs such as spina bifida occulta or myelomeningocele and Chiari 2 malformation [72]. A case study reported recessive variants of APAF1 and CASP9, which are key apoptotic genes, as potential causative genes for FA resistant NTD such as spina bifida, holoanencephaly, or spinal rachischisis in patients with multiple anomalies [73].
Gene-environment and gene-gene interactions
The effect of folate on NTD risk offers a striking example of a gene-environment interaction. Evidence for gene-environment interaction by FA include: 1) FA alters the sex ratio in human NTDs (a male/female ratio: from 0.48–0.77 to about 1), 2) FA may reduce NTD severity in human [74, 75], and 3) FA reduces the incidence and severity of mouse NTD models, such as splotch (Pax3 mutant) or Lrp2null [76, 77]. However, despite the impact of folate in reducing NTD incidence, genes and pathways regulated by folate remain mostly unknown [78]. Other environmental factors influencing NTD risk are maternal hyperglycemia, obesity, elevated body temperature, disadvantaged socio-economic status, and drugs such as valproic acid, opioids or potentially the antiretroviral dolutegravir [79–85].
There are now over 20 genes implicated in digenic pathogenesis of mouse NTD [86–88]. Gene-gene interaction examples in mouse include digenic interaction between PCP genes (e.g. Vangl1 and Vangl2) [87]. Digenic inheritance was proposed in human NTD, especially for SNPs near folate metabolism genes [89, 90], and between PCP genes. As an example, 1% of more than 500 patients were double heterozygous for PCP mutations, meaning they carried damaging variants in two different genes [91], but establishing the specific combinations of these gene mutations in determining risk requires studies in additional patients.
While risk for NTDs is generally assumed to depend upon the genetic constitution of the offspring, it is conceivable that some risk might trace back to the parents’ genetic makeup or risk factors, which could interact with the fetal genetic constitution. One obvious place to look would be the mother’s genotype for folate metabolism genes. Multiple meta-analyses have shown the association of MTHFR rs1801133 with NTD maternal risk, although they are limited to candidate SNP genotyping [92]. Perhaps the greatest maternal factors will turn out to be genetic risk of diabetes, which in turn imparts risk of fetal NTDs. Supporting this model, interaction between maternal hyperglycemia and insulin resistance genes with fetal glucose homeostasis genes increases risk of NTDs from a cohort of 737 NTD trios [89]. Paternal risk is generally not considered, but an important contribution to the number of DNMs in an offspring is paternal age at the time of conception. Indeed, paternal age correlates with the risk of NTDs, with an odds ratio of 1.35 for 40-year old vs. 20-year old father, about the same risk attributed to the MTHFR rs1801133 [93].
Epigenetic regulation in NTD
Given that FA is a methyl donor, researchers have long hypothesized that FA can impact genes expression, through FA-dependent DNA and histone methylation, which are major components of ‘epigenome’ (Fig. 3) [36, 94, 95]. Since most or all DNA methylation marks are removed post-fertilization, and then undergo re-marking [96], there is probably a substantial demand for methyl groups, which folate provides. In one example, genetic risk of birth defects (including NTDs) in mice was attributed to the grandparents genotype, specifically the enzyme methionine synthase reductase (Mtrr), necessary for utilization of methyl groups from the folate cycle [97]. Moreover, findings from Fbp1 (folate binding protein 1, partially responsible for folate uptake) knockout mice suggest a requirement in gene transcription for G-proteins, transcription factors, growth factors, methyltransferases, and cell cycle mRNAs [98], but assessment in the neural tube remains to be explored. Mouse studies also support the potential for other epigenetic regulation pathways (in addition to folate-related ones) in NTDs. For instance, exencephaly is observed in several murine chromatin regulator mutations, including DNA methylases, histone methylases, acetylases and deacetylases [94]. Moreover, maternal exposure to valproate, which is an histone deacetylase inhibitor, is a risk factor for NTDs in humans [83].
Figure 3. Schematic of folate as a methyl group donor.
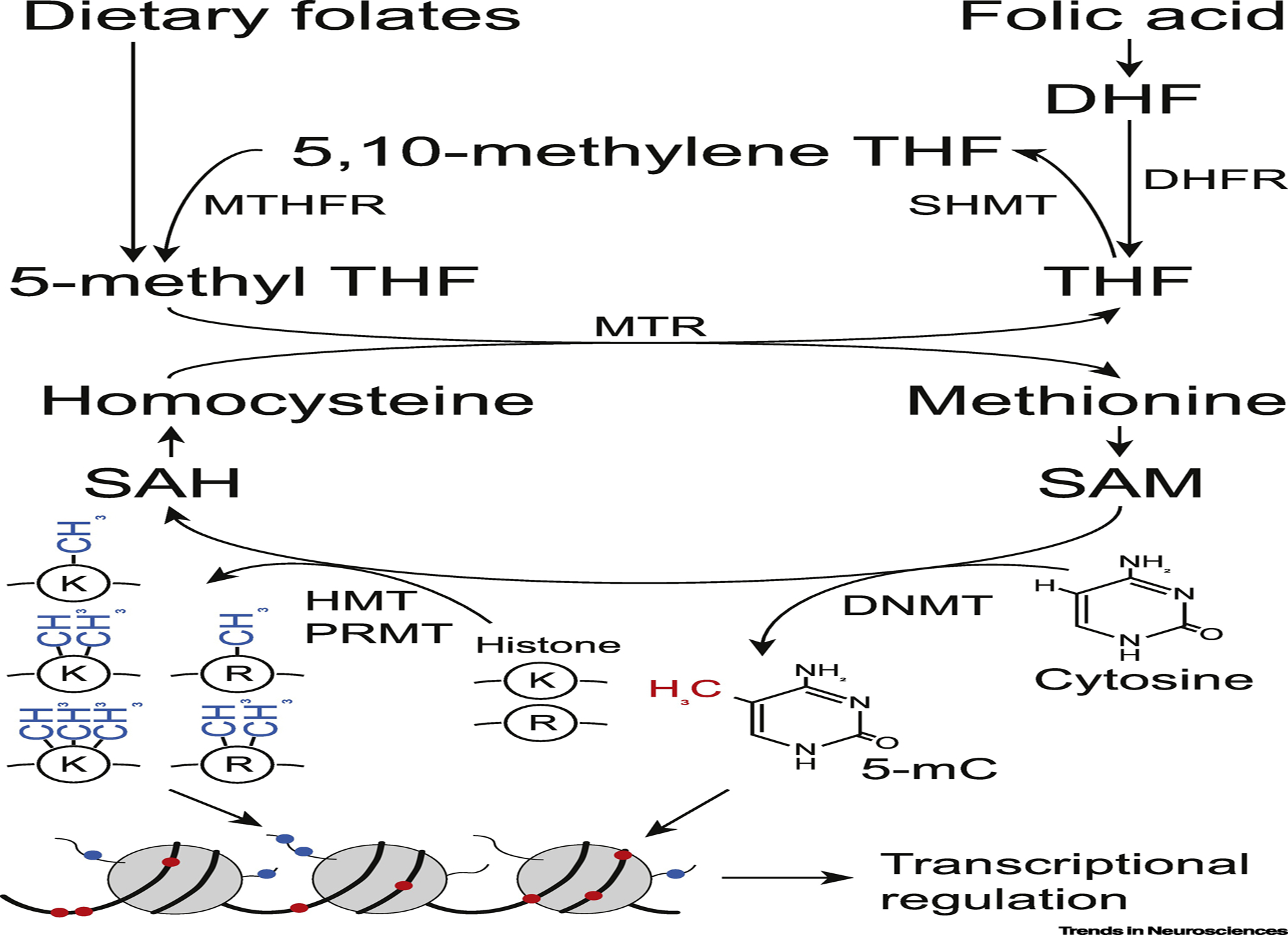
Either dietary folates or synthetic folic acid are incorporated into folate one-carbon metabolism pathway and serve as methyl group donors for various substrates including protein, DNA, RNA and lipids. Epigenetic regulation by DNA and histone methylation plays crucial roles in transcriptional regulation and embryonic development. DNMT, DNA methyltransferase; DHFR, dihydrofolate reductase; HMT, histone methyltransferase; K, lysine; mC, methylcytosine; MTHFR, methylenetetrahydrofolate reductase; MTR, methionine synthase; PRMT: protein arginine methyltransferase; R, arginine; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate.
How does folic acid reduce NTD incidence?
What can we learn from the decades of FA supplementation? Incidence of NTDs has dropped by ~15–70% in every region where FA supplementation policy has been implemented, with a strong female sex bias. NTDs previously showed a female preponderance, but this bias has disappeared with FA supplementation [6, 74, 99]. In addition to the sex-specific bias, differences are also evident between NTD subtypes. Specifically, FA supplementation had a greater impact on reducing anencephaly than other types of NTD [6, 74, 99]. Although further studies are needed, this drop seems to have coincided with a decrease in severity of NTDs. There are two possible explanations for this observation (although a combination of the two is feasible as well): 1) FA has prevented NTDs only in those that would have had the most severe phenotypes. 2) FA has reduced the severity of NTD across the board, essentially rescuing those at the mild end of the spectrum, and shifting those from the severe end to the mild end (Fig. 4). The latter explanation seems more plausible, and is supported by observations in mice [13, 76, 77].
Figure 4. A. Model of impact of folic acid (FA) supplementation on neural tube defect (NTD) incidence and severity.
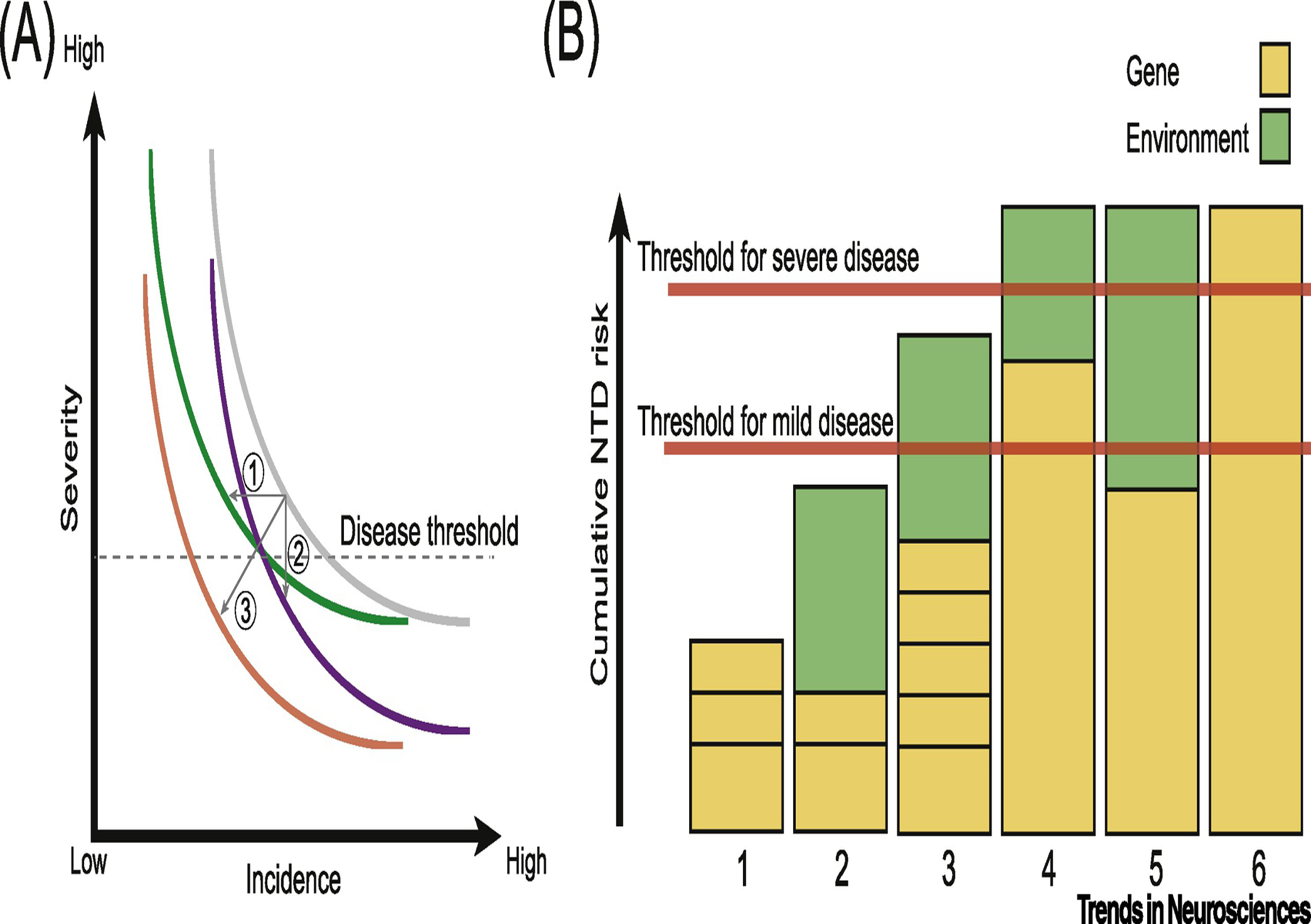
In the absence of FA, a hypothetical gray curve represents the relationship between incidence and severity. FA supplementation may decrease NTD incidence (arrow 1, curve moves to left) or severity (arrow 2, curves moves down), or may impact both (arrow 3, curve moves both left and down). B. Cumulative NTD risk along y-axis and individual example risk profiles numbered at bottom. Yellow: genetic risk; green: environment risk attributed to FA deficiency or other factors. Individuals 1 and 2 have risk below the threshold. Individual 3 has cumulative risk above threshold for mild disease, but if exposed to folate, would fall below threshold for mild disease. Individuals 4 and 5 have cumulative risk above threshold for severe disease, but if exposed to folate, 4 would fall into mild disease and 5 would fall below mild disease threshold. Individual 6 has genetic risk above threshold for severe disease that is not modified by folate.
This phenomenon is somewhat reminiscent of ‘female protective effect’ in ASD. The female protective effect was proposed to explain the observation of a ~5:1 sex bias of ASD toward males, together with a 2-fold enrichment of de novo protein-truncating variants in female ASD individuals [100]. In other words, females require a greater genetic insult to manifest ASD compared to males. Many hypotheses have been put forward to explain the female protective effect, but there is still lack of clarity regarding the mechanisms involved.
The ‘female protective effect’ in ASD, where a greater mutation burden seems to be required for disease in females, is reminiscent of the effect of FA supplementation in the protection from NTDs. Consider the potential outcomes of a hypothetical experiment comparing DNMs in NTD cases conceived in folate-deficient areas vs. folate-replete areas (or in cases born before vs. after FA implementation). If the effect of FA was to reduce the burden of DNMs as a means of protecting from NTDs, then we would expect to observe a drop in DNM burden after FA fortification. As a specific example, since the entire US population was subjected to FA supplementation since 1998, we would expect to observe fewer DNMs in NTD cases conceived after fortification compared with those conceived before fortification, and since the entire population was subject to FA fortification, we would expect this drop in DNM to extend to the generation population. While it is possible that FA supplementation coincided with reduced DNM rate in humans, no such observation has been reported in the literature. In fact, the DNM rate in offspring seems to be increasing in recent years, probably correlating with advanced parental age [101]. If the effect of FA was independent of DNMs, then we would expect the DNM rate to be constant before and after fortification. However, if the effect of FA was to increase the threshold of mutations required for NTD, much like females with ASD, then we would expect to observe an increased burden of DNMs after FA supplementation in cases of the same NTD severity. In other words, the same clinical severity now requires a greater burden of DNMs, and thus FA preferentially rescued cases that had milder DNM burden (Fig. 4). To our knowledge, such a test has not yet been performed.
It remains unclear whether FA reduces all NTD subtypes globally or certain subtypes more specifically; it also remains unclear whether FA reduces NTD incidence across the board or decreases disease severity in terms of the spinal level of the defect and the extent of open neural tissue [74, 75, 102, 103]. Large scale studies so far indicate that the answer to these questions may depends on sex, ethnicity, geographic region, and background nutritional status [36, 74, 75, 103]. For example, for females in NTD prevalent areas such as high-elevation Argentina and Northern China, FA supplementation tends to have more impact on anencephaly prevention, while many other populations show comparable rates on both anencephaly and spina bifida [74, 75]. Here again, genetic factors may play a role of modifying the severity of NTD. Studies with detailed phenotyping, including defect level and extent would be necessary to answer these questions.
Unaccounted-for variables in persistent cases post-FA supplementation include genetic and/or environmental factors regulating maternal folate levels. For instance, there may be some pregnant women with very low levels of circulating folate despite their being exposed to supplementation. So far, a few small-scaled SNP genotyping genetic studies in humans have uncovered variants in folate metabolic genes in mothers contributing to risk [33], although further studies are needed. It is interesting that among rare essential B vitamins like B1, B2, B9 and B12, only folate (B9) shows no evidence of genetic regulation, meaning that genotypes do not seem to determine folate levels [104]. What then determines folate levels? Obviously dietary intake is one driver, and population level studies demonstrate that this significantly increases serum folate levels [105]. But a largely unexplored area is the role of the gut microbiome in this context. Specifically, it has been noted that the gut microbiome can produce folate, which is subsequently absorbed by the host [106]. It remains to be tested whether alterations in this process are associated with NTD risk.
Concluding Remarks and Future Perspectives
Understanding the genetic architecture of human NTDs and the role of folate is crucial, not only to achieve a mechanistic understanding of this most common CNS structural defect, but also for effective clinical screening or further prevention and treatment. However, the heterogeneous nature of NTDs in terms of phenotypes and genetic architecture, together with the recognized contribution of environmental factors, have hindered progress. The other consequence of this complexity is a lack of large-scale genetic studies stratified by NTD subtypes, which is partially responsible for low yield of the expected insight by prior genetic studies applying approaches like GWAS and candidate gene testing. Yet, noticeable advances have been made, including the discovery of an association between NTDs and folate metabolism, PCP, and environmental risk factors. As sequencing costs decrease, new NGS studies are likely to reveal proposed and novel candidate genes, enabling the detection of rare alleles including SNVs, indels, short tandem repeats and SVs using exome- or genome-wide sequencing applied to trios or quads (trio + unaffected sibling), as have been performed for ASD and congenital heart disease.
Large scale sequencing of cohorts from diverse ethnic groups is needed to disentangle the population-level genetic architecture of NTDs (see Outstanding Questions). To solve this problem, researchers from all over the world are organizing consortia to aggregate NTD samples, including preexisting cases that were conceived prior to FA supplementation, or from parts of the world yet to include FA fortification. With the gradual replacement of “phenotype-driven” syndrome delineation by “genotype-driven” by virtue of high-throughput sequencing and extensive collaborative efforts, not only the size of a cohort but also its quality (in terms of refined phenotyping and detailed description of environmental factors) will be crucial for future research [107].
Supplementary Material
Outstanding Questions.
- Has folic acid (FA) supplementation changed the landscape of de novo mutations in terms of their rate and severity?
- Is the genetic contribution to neural tube defect (NTD) risk different before and after FA supplementation? In other words, are the mutations found in NTD patients severer or milder after FA was introduced into the diet?
- Are there mutated genes or affected pathways in that are specific to affected individuals that were conceived after FA supplementation that are ‘folate-nonresponsive’?
- Could residual cases of NTD in the US or other parts of the world already supplementing be prevented if the dose of FA was higher?
- Are there other dietary supplements that could added to further reduce NTD rates, such as inositol supplementation?
- Why has the rate of NTDs dropped more significantly following FA supplementation in geographies with higher baseline NTD rates?
- How much do noncoding variants contribute to NTD risk and what is their pathogenic mechanisms?
- Are there subtypes- (e.g. anencephaly or myelomeningocele) specific or level- (e.g. thoracic, lumbar, or sacral) specific genetic factors?
- Are there population-specific genetic risk factors?
- Can the risk of NTDs be predictable by polygenic risk score?
Highlights.
- Neural tube defects (NTDs) are the most common birth anomaly of the central nervous system (CNS). They result from a failure of the neural plate to complete the developmental transition to a neural tube.
- Mandatory folic acid (FA) supplementation of dietary grains led to a significant decrease in the incidence of NTDs, but the mechanisms involved remain disputed.
- NTD is a relatively common phenotype in mouse knockouts, showing both simple and complex inheritance, as well as responses to folate, yet few of the genes identified in mouse models have been established as causes in human NTDs.
- Candidate gene approaches in human studies support a role for Wnt/planar cell polarity, cilia, Sonic Hedgehog (Shh), bone morphogenetic protein (BMP) signaling factors in NTD risk.
- Whole genome sequencing, as opposed to SNP genotyping or candidate gene resequencing, allows for detection of most coding, noncoding, and structural variants, including rare, common, de novo, and somatic variants.
- Recently, there have been collaborative efforts to assemble larger cohorts of phenotypically diverse patients from diverse ethnic groups, both prior to and after FA fortification, for comprehensive risk assessment.
Acknowledgements
The authors wish to acknowledge the UCSD CTRI UL1TR001442 of CTSA funding, support from the Rady Children’s Institute for Genomic Medicine, and Gabriella Miller Kids First research program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors wish to thank Kathleen Fisch and Sara Brin Rosenthal of the Center for Computational Biology & Bioinformatics of University of California, San Diego for power calculation (Fig. 2), Aakash Patel for literature review, and Allison Elizabeth Ashley-Koch, José F. Cordero, Osvaldo Mutchinick, Andrew J. Copp, Lee Niswander, Kit Sing Au, Joon-Yong An, members of the Spina Bifida Sequencing Consortium, and members of the Gleeson lab for useful feedback.
Glossary
- Anencephaly
a condition of absent skull and major portion of brain due to developmental abnormality
- Craniorachischisis
a condition whereby the neural tube is not closed along both of the brain and spinal cord
- Epigenome
sum of any modifications on DNA or histones without alterations in the DNA sequence itself
- Folate
a generic term referring to any kinds of natural food folate (in tetrahydrofolate form). Folate in fortified foods and most of dietary supplements is in the form of folic acid, which is artificially synthesized as a pure chemical compound
- Folic acid (FA) fortification
many countries mandate manufacturers to add FA to various dietary grains based on the previous clinical trials showing FA’s effect on neural tube defect prevention. Concentration of fortified FA varies from country to country, but is typically in the range 140–220 μg/100 g of flour
- Genetic architecture
understanding of the genetic factors responsible for heritable phenotypic variability, which also includes their interactions with each other (genetic interaction) and with the environment (gene-environment interaction)
- Genome-wide association study (GWAS)
an approach to associate genetic variations with particular diseases
- Haploinsufficient
a single copy of a certain gene that does not produce sufficient protein product for normal phenotypes
- Linkage association (linkage analysis)
an approach to find co-segregated genetic segments within family
- Loss-of-function (LoF) mutation
mutations (usually coding ones) that result in the gene product having no function, including stopgain, frameshift, or splicing mutations
- Mendelian inheritance
inheritance patterns of single gene (monogenic) diseases
- Microdeletion/microduplication
relatively small chromosomal deletion/duplication that usually cannot be detected by conventional cytogenetic method under light microscope
- Multiple comparisons problem
statistical problem whereby an increase in simultaneous tests increases the likelihood of an erroneous result
- Myelomeningocele
a condition of protrusion of meninges and spinal cord through vertebra and skin defects. Also known as ‘spina bifida’
- Neural tube defect
a group of developmental disorders that is caused by failed neural tube closure during development
- Oligogenic model
an inheritance model involving several genes
- Population stratification
a systematic difference in allele frequencies of genetic variants among subpopulations due to different ancestry
- Purifying selection
an evolutionary tendency to eliminate protein-altering or -damaging gene variants
- Regulatory elements
genetic regions that control the transcription of nearby genes
- Spina bifida
see myelomeningocele
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict statement: Neither author has any conflicts to report.
References
- 1.Brown E, et al. (1994) Prevalence of incidental intraspinal lipoma of the lumbosacral spine as determined by MRI. Spine (Phila Pa 1976) 19, 833–836 [DOI] [PubMed] [Google Scholar]
- 2.Zaganjor I, et al. (2016) Describing the Prevalence of Neural Tube Defects Worldwide: A Systematic Literature Review. PLoS One 11, e0151586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rocque BG, et al. (2015) Assessing health-related quality of life in children with spina bifida. J Neurosurg Pediatr 15, 144–149 [DOI] [PubMed] [Google Scholar]
- 4.Leck I (1974) Causation of neural tube defects: clues from epidemiology. Br Med Bull 30, 158–163 [DOI] [PubMed] [Google Scholar]
- 5.Windham GC, et al. (1982) The association of twinning and neural tube defects: studies in Los Angeles, California, and Norway. Acta Genet Med Gemellol (Roma) 31, 165–172 [DOI] [PubMed] [Google Scholar]
- 6.Windham GC and Sever LE (1982) Neural tube defects among twin births. Am J Hum Genet 34, 988–998 [PMC free article] [PubMed] [Google Scholar]
- 7.Deak KL, et al. (2008) Further evidence for a maternal genetic effect and a sex-influenced effect contributing to risk for human neural tube defects. Birth Defects Res A Clin Mol Teratol 82, 662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drainer E, et al. (1991) Do familial neural tube defects breed true? J Med Genet 28, 605–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iossifov I, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin SC, et al. (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49, 1593–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bult CJ, et al. (2019) Mouse Genome Database (MGD) 2019. Nucleic Acids Res 47, D801–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juriloff DM and Harris MJ (2000) Mouse models for neural tube closure defects. Hum Mol Genet 9, 993–1000 [DOI] [PubMed] [Google Scholar]
- 13.Carter M, et al. (1999) Crooked tail (Cd) models human folate-responsive neural tube defects. Hum Mol Genet 8, 2199–2204 [DOI] [PubMed] [Google Scholar]
- 14.Lei Y, et al. (2015) Rare LRP6 variants identified in spina bifida patients. Hum Mutat 36, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rampersaud E, et al. (2005) Whole genomewide linkage screen for neural tube defects reveals regions of interest on chromosomes 7 and 10. J Med Genet 42, 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamm DS, et al. (2006) High-density single nucleotide polymorphism screen in a large multiplex neural tube defect family refines linkage to loci at 7p21.1-pter and 2q33.1-q35. Birth Defects Res A Clin Mol Teratol 76, 499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newton R, et al. (1994) Linkage analysis of 62 X-chromosomal loci excludes the X chromosome in an Icelandic family showing apparent X-linked recessive inheritance of neural tube defects. Clin Genet 45, 241–249 [DOI] [PubMed] [Google Scholar]
- 18.Lemay P, et al. (2015) Loss-of-function de novo mutations play an important role in severe human neural tube defects. J Med Genet 52, 493–497 [DOI] [PubMed] [Google Scholar]
- 19.Zohn IE, et al. (2005) Using genomewide mutagenesis screens to identify the genes required for neural tube closure in the mouse. Birth Defects Res A Clin Mol Teratol 73, 583–590 [DOI] [PubMed] [Google Scholar]
- 20.Sanders SJ, et al. (2017) Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nat Neurosci 20, 1661–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werling DM, et al. (2018) An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet 50, 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An JY, et al. (2018) Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z, et al. (2018) Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Res 28, 1039–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemay P, et al. (2019) Whole exome sequencing identifies novel predisposing genes in neural tube defects. Mol Genet Genomic Med 7, e00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karczewski KJ, et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv, 531210 [Google Scholar]
- 26.Gont LK, et al. (1993) Tail formation as a continuation of gastrulation: the multiple cell populations of the Xenopus tailbud derive from the late blastopore lip. Development 119, 991–1004 [DOI] [PubMed] [Google Scholar]
- 27.Cao X, et al. (2020) Loss of RAD9B impairs early neural development and contributes to the risk for human spina bifida. Hum Mutat 41, 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Put NM, et al. (1995) Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet 346, 1070–1071 [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, et al. (2015) Association between MTHFR C677T polymorphism and neural tube defect risks: A comprehensive evaluation in three groups of NTD patients, mothers, and fathers. Birth Defects Res A Clin Mol Teratol 103, 488–500 [DOI] [PubMed] [Google Scholar]
- 30.Amorim MR, et al. (2007) Non-Latin European descent could be a requirement for association of NTDs and MTHFR variant 677C > T: a meta-analysis. Am J Med Genet A 143A, 1726–1732 [DOI] [PubMed] [Google Scholar]
- 31.Martinez CA, et al. (2009) Genetic association study of putative functional single nucleotide polymorphisms of genes in folate metabolism and spina bifida. Am J Obstet Gynecol 201, 394 e391–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Consortium GT (2013) The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai CQ, et al. (2019) Association of neural tube defects with maternal alterations and genetic polymorphisms in one-carbon metabolic pathway. Ital J Pediatr 45, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng J, et al. (2015) Association between MTHFD1 polymorphisms and neural tube defect susceptibility. J Neurol Sci 348, 188–194 [DOI] [PubMed] [Google Scholar]
- 35.Renard E, et al. (2019) Exome sequencing of cases with neural tube defects identifies candidate genes involved in one-carbon/vitamin B12 metabolisms and Sonic Hedgehog pathway. Hum Genet 138, 703–713 [DOI] [PubMed] [Google Scholar]
- 36.Au KS, et al. (2017) Finding the genetic mechanisms of folate deficiency and neural tube defects-Leaving no stone unturned. Am J Med Genet A 173, 3042–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung KY, et al. (2017) Partitioning of One-Carbon Units in Folate and Methionine Metabolism Is Essential for Neural Tube Closure. Cell Rep 21, 1795–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narisawa A, et al. (2012) Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum Mol Genet 21, 1496–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kibar Z, et al. (2007) Mutations in VANGL1 associated with neural-tube defects. N Engl J Med 356, 1432–1437 [DOI] [PubMed] [Google Scholar]
- 40.Kibar Z, et al. (2001) Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet 28, 251–255 [DOI] [PubMed] [Google Scholar]
- 41.Torban E, et al. (2004) Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family. J Biol Chem 279, 52703–52713 [DOI] [PubMed] [Google Scholar]
- 42.Juriloff DM and Harris MJ (2012) A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol 94, 824–840 [DOI] [PubMed] [Google Scholar]
- 43.Murdoch JN and Copp AJ (2010) The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol 88, 633–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiang C, et al. (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413 [DOI] [PubMed] [Google Scholar]
- 45.Kim SE, et al. (2019) Dominant negative GPR161 rare variants are risk factors of human spina bifida. Hum Mol Genet 28, 200–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beaumont M, et al. (2019) Targeted panel sequencing establishes the implication of planar cell polarity pathway and involves new candidate genes in neural tube defect disorders. Hum Genet 138, 363–374 [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, et al. (2013) Association between PTCH1 polymorphisms and risk of neural tube defects in a Chinese population. Birth Defects Res A Clin Mol Teratol 97, 409–415 [DOI] [PubMed] [Google Scholar]
- 48.Wu J, et al. (2013) Association between PKA gene polymorphism and NTDs in high risk Chinese population in Shanxi. Int J Clin Exp Pathol 6, 2968–2974 [PMC free article] [PubMed] [Google Scholar]
- 49.Onishi K and Zou Y (2017) Sonic Hedgehog switches on Wnt/planar cell polarity signaling in commissural axon growth cones by reducing levels of Shisa2. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cameron DA, et al. (2009) Tulp3 is a critical repressor of mouse hedgehog signaling. Dev Dyn 238, 1140–1149 [DOI] [PubMed] [Google Scholar]
- 51.Caspary T, et al. (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 12, 767–778 [DOI] [PubMed] [Google Scholar]
- 52.Miao C, et al. (2016) Mutations in the Motile Cilia Gene DNAAF1 Are Associated with Neural Tube Defects in Humans. G3 (Bethesda) 6, 3307–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson MP, et al. (2009) Neural tube defects in mice with reduced levels of inositol 1,3,4-trisphosphate 5/6-kinase. Proc Natl Acad Sci U S A 106, 9831–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guan Z, et al. (2014) The maternal ITPK1 gene polymorphism is associated with neural tube defects in a high-risk Chinese population. PLoS One 9, e86145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cockroft DL, et al. (1992) Inositol deficiency increases the susceptibility to neural tube defects of genetically predisposed (curly tail) mouse embryos in vitro. Teratology 45, 223–232 [DOI] [PubMed] [Google Scholar]
- 56.Greene ND, et al. (2016) Inositol for the prevention of neural tube defects: a pilot randomised controlled trial. Br J Nutr 115, 974–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen CP (2007) Chromosomal abnormalities associated with neural tube defects (I): full aneuploidy. Taiwan J Obstet Gynecol 46, 325–335 [DOI] [PubMed] [Google Scholar]
- 58.Chen CP (2007) Chromosomal abnormalities associated with neural tube defects (II): partial aneuploidy. Taiwan J Obstet Gynecol 46, 336–351 [DOI] [PubMed] [Google Scholar]
- 59.Goumy C, et al. (2014) De novo 2q36.1q36.3 interstitial deletion involving the PAX3 and EPHA4 genes in a fetus with spina bifida and cleft palate. Birth Defects Res A Clin Mol Teratol 100, 507–511 [DOI] [PubMed] [Google Scholar]
- 60.Drozniewska M and Haus O (2014) PAX3 gene deletion detected by microarray analysis in a girl with hearing loss. Mol Cytogenet 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hart J and Miriyala K (2017) Neural tube defects in Waardenburg syndrome: A case report and review of the literature. Am J Med Genet A 173, 2472–2477 [DOI] [PubMed] [Google Scholar]
- 62.Bassuk AG, et al. (2013) Copy number variation analysis implicates the cell polarity gene glypican 5 as a human spina bifida candidate gene. Hum Mol Genet 22, 1097–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, et al. (2013) Detection of copy number variants reveals association of cilia genes with neural tube defects. PLoS One 8, e54492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDonald-McGinn DM, et al. (2015) 22q11.2 deletion syndrome. Nat Rev Dis Primers 1, 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leoni C, et al. (2014) Neural tube defects and atypical deletion on 22q11.2. Am J Med Genet A 164A, 2701–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canda MT, et al. (2012) Prenatal diagnosis of a 22q11 deletion in a second-trimester fetus with conotruncal anomaly, absent thymus and meningomyelocele: Kousseff syndrome. J Obstet Gynaecol Res 38, 737–740 [DOI] [PubMed] [Google Scholar]
- 67.Hureaux M, et al. (2019) SOX3 duplication: A genetic cause to investigate in fetuses with neural tube defects. Prenat Diagn 39, 1026–1034 [DOI] [PubMed] [Google Scholar]
- 68.Chen CP, et al. (2017) Molecular cytogenetic characterization of a duplication of 15q24.2-q26.2 associated with anencephaly and neural tube defect. Taiwan J Obstet Gynecol 56, 550–553 [DOI] [PubMed] [Google Scholar]
- 69.Epstein DJ, et al. (1991) Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell 67, 767–774 [DOI] [PubMed] [Google Scholar]
- 70.Shaheen R, et al. (2015) T (brachyury) is linked to a Mendelian form of neural tube defects in humans. Hum Genet 134, 1139–1141 [DOI] [PubMed] [Google Scholar]
- 71.Grzeschik KH, et al. (2007) Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet 39, 833–835 [DOI] [PubMed] [Google Scholar]
- 72.Peters T, et al. (2014) Focal dermal hypoplasia: report of a case with myelomeningocele, Arnold-Chiari malformation and hydrocephalus with a review of neurologic manifestations of Goltz syndrome. Pediatr Dermatol 31, 220–224 [DOI] [PubMed] [Google Scholar]
- 73.Spellicy CJ, et al. (2018) Key apoptotic genes APAF1 and CASP9 implicated in recurrent folate-resistant neural tube defects. Eur J Hum Genet 26, 420–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu J, et al. (2018) Sex differences in the prevalence of neural tube defects and preventive effects of folic acid (FA) supplementation among five counties in northern China: results from a population-based birth defect surveillance programme. BMJ Open 8, e022565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poletta FA, et al. (2018) Neural tube defects: Sex ratio changes after fortification with folic acid. PLoS One 13, e0193127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burren KA, et al. (2008) Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum Mol Genet 17, 3675–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sabatino JA, et al. (2017) Prevention of neural tube defects in Lrp2 mutant mouse embryos by folic acid supplementation. Birth Defects Res 109, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krauss RS and Hong M (2016) Gene-Environment Interactions and the Etiology of Birth Defects. Curr Top Dev Biol 116, 569–580 [DOI] [PubMed] [Google Scholar]
- 79.McMahon DM, et al. (2013) Maternal obesity, folate intake, and neural tube defects in offspring. Birth Defects Res A Clin Mol Teratol 97, 115–122 [DOI] [PubMed] [Google Scholar]
- 80.Jia S, et al. (2019) Maternal, paternal, and neonatal risk factors for neural tube defects: A systematic review and meta-analysis. Int J Dev Neurosci 78, 227–235 [DOI] [PubMed] [Google Scholar]
- 81.Kerr SM, et al. (2017) Periconceptional maternal fever, folic acid intake, and the risk for neural tube defects. Ann Epidemiol 27, 777–782 e771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grewal J, et al. (2009) Neural tube defects: an analysis of neighbourhood- and individual-level socio-economic characteristics. Paediatr Perinat Epidemiol 23, 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nau H (1994) Valproic acid-induced neural tube defects. Ciba Found Symp 181, 144–152; discussion 152–160 [DOI] [PubMed] [Google Scholar]
- 84.Yazdy MM, et al. (2013) Periconceptional use of opioids and the risk of neural tube defects. Obstet Gynecol 122, 838–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zash R, et al. (2019) Neural-Tube Defects and Antiretroviral Treatment Regimens in Botswana. N Engl J Med 381, 827–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murdoch JN, et al. (2014) Genetic interactions between planar cell polarity genes cause diverse neural tube defects in mice. Dis Model Mech 7, 1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Torban E, et al. (2008) Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc Natl Acad Sci U S A 105, 3449–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Juriloff DM and Harris MJ (2018) Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. J Dev Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lupo PJ, et al. (2014) Maternal-fetal metabolic gene-gene interactions and risk of neural tube defects. Mol Genet Metab 111, 46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dutta HK, et al. (2017) Evidence of gene-gene interactions between MTHFD1 and MTHFR in relation to anterior encephalocele susceptibility in Northeast India. Birth Defects Res 109, 432–444 [DOI] [PubMed] [Google Scholar]
- 91.Wang L, et al. (2018) Digenic variants of planar cell polarity genes in human neural tube defect patients. Mol Genet Metab 124, 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Molloy AM, et al. (2017) Genetic Risk Factors for Folate-Responsive Neural Tube Defects. Annu Rev Nutr 37, 269–291 [DOI] [PubMed] [Google Scholar]
- 93.Green RF, et al. (2010) Association of paternal age and risk for major congenital anomalies from the National Birth Defects Prevention Study, 1997 to 2004. Ann Epidemiol 20, 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wilde JJ, et al. (2014) Genetic, epigenetic, and environmental contributions to neural tube closure. Annu Rev Genet 48, 583–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H and Niswander L (2018) Does DNA methylation provide a link between folate and neural tube closure? Epigenomics 10, 1263–1265 [DOI] [PubMed] [Google Scholar]
- 96.Sasaki H and Matsui Y (2008) Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 9, 129–140 [DOI] [PubMed] [Google Scholar]
- 97.Padmanabhan N, et al. (2013) Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 155, 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spiegelstein O, et al. (2004) Folate-regulated changes in gene expression in the anterior neural tube of folate binding protein-1 (Folbp1)-deficient murine embryos. Neurochem Res 29, 1105–1112 [DOI] [PubMed] [Google Scholar]
- 99.Rittler M, et al. (2004) Sex ratio and associated risk factors for 50 congenital anomaly types: clues for causal heterogeneity. Birth Defects Res A Clin Mol Teratol 70, 13–19 [DOI] [PubMed] [Google Scholar]
- 100.Satterstrom FK, et al. (2019) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. bioRxiv, 484113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jonsson H, et al. (2017) Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 [DOI] [PubMed] [Google Scholar]
- 102.Santos LM, et al. (2016) Prevention of neural tube defects by the fortification of flour with folic acid: a population-based retrospective study in Brazil. Bull World Health Organ 94, 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams LJ, et al. (2005) Decline in the prevalence of spina bifida and anencephaly by race/ethnicity: 1995–2002. Pediatrics 116, 580–586 [DOI] [PubMed] [Google Scholar]
- 104.Tanaka T, et al. (2009) Genome-wide association study of vitamin B6, vitamin B12, folate, and homocysteine blood concentrations. Am J Hum Genet 84, 477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abdollahi Z, et al. (2011) Efficacy of flour fortification with folic acid in women of childbearing age in Iran. Ann Nutr Metab 58, 188–196 [DOI] [PubMed] [Google Scholar]
- 106.Engevik MA, et al. (2019) Microbial Metabolic Capacity for Intestinal Folate Production and Modulation of Host Folate Receptors. Front Microbiol 10, 2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bamshad MJ, et al. (2019) Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet 105, 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.