

Abstract
Ever since the pioneering discoveries in the mid nineteen hundreds, the hypothalamus was recognized as a crucial component of the neural system controlling appetite and energy balance. The new wave of neuron-specific research tools has confirmed this key role of the hypothalamus and has delineated many other brain areas to be part of an expanded neural system sub serving these crucial functions. However, despite significant progress in defining this complex neural circuitry, many questions remain. One of the key questions is why the sophisticated body weight regulatory system is unable to prevent the rampant obesity epidemic we are experiencing. Why are pathologically obese body weight levels defended, and what can we do about it? Here we try to find answers to these questions by 1) reminding the reader that the neural controls of ingestive behavior have evolved in a demanding, restrictive environment and encompass much of the brain’s major functions, far beyond the hypothalamus and brainstem, 2) hypothesizing that the current obesogenic environment impinges mainly on a critical pathway linking hypothalamic areas with the motivational and reward systems to produce uncompensated hyperphagia, and 3) proposing adequate strategies for prevention and treatment.
1. Introduction
In less than a century, obesity rates have drastically increased in Western as well as in rapidly developing countries. Given the strong association with diabetes, cardiovascular disease, and cancer, obesity has become one of the major health burdens with a huge economic impact. Clearly, the rapid rise of obesity is inconsistent with changes in the gene pool as a causative factor. More likely, the modern obesogenic environment interacts with individual genetic predisposition and in addition may cause epigenetic changes that are propagated to offspring [1]. For more in-depth discussions of the genetics of obesity the reader should consult relevant recent reviews [1–4]. Here, we focus mainly on the obesogenic environment causing obesity in individuals with a polygenetic obesity predisposition, also referred to as “common obesity”.
Whether body weight/adiposity is homeostatically regulated and how this regulation is accomplished has been the subject of much debate. Homeostatic regulation of body weight hypothesizes that body weight (or perhaps body fat) is actively defended owing to a coordinated response to any disturbance, similar to the regulation of body temperature in warm-blooded animals. The set-point model of homeostatic body weight regulation was mainly based on observations in laboratory rats and seasonal animals such as hamsters. When rats that have lost body weight after a period of food restriction are allowed unrestricted access to food, they overeat and body weight returns to exactly the same level of unrestricted rats within a few days or weeks, even taking into account an ascending body weight curve during growth [5, 6]. More impressively, after the same manipulation in Siberian hamsters during their seasonal weight loss phase, body weight returns to exactly the same low level of non-restricted hamsters, suggesting that “…the point of energy balance is continuously re-adjusted, reflecting an apparent sliding set point” [7–9]. Many more papers have focused on the dynamics of body weight/adiposity regulation, with evidence both for and against the concept of a body weight set point. The interested reader should consider the following reviews [10–18].
The goal of this present paper is to review the literature supporting the view that higher brain functions such as hedonic and cognitive processing are an integral part of the neural control of food intake and body weight/adiposity regulation, and in particular that the distinction between “homeostatic” and “hedonic” may no longer be useful. First, we will remind the reader that ingestive behavior is not simply the act of consumption, but is instead a complex behavior that depends on multiple neural systems. We will review the evidence for cross-talk between the “classical” homeostatic system and the reward, emotional, and cognitive systems in general, particularly focusing on recent insights into the physiology of basomedial hypothalamic agouti-related protein (AGRP) and pro-opio-melanocortin (POMC) neurons that are considered master nutrient sensors and motivational drivers. Second, we will discuss the hypothesis that one of the main mechanisms by which the obesogenic environment increases the level of defended body weight in genetically prone individuals is by impinging on this motivational drive. Third, we will briefly discuss evidence for some candidate mechanisms that might be responsible for the chronic elevation of the defended obese body weight, which makes weight loss so difficult. Finally, we will lay out some of the implications for the prevention and treatment of obesity prompted by our integrative neural model.
2. Multiple neural systems controlling food intake and body weight
Historical background
Some of the pioneers in ingestive behavior research had already included forebrain structures other than the hypothalamus in the overall neural system governing this crucial behavior. For example, Eliot Stellar included the thalamus, striatum, and cortical structures in his working model [19]. It may also not be appreciated that much of our understanding of higher brain functions such as learning & memory and decision making is based on experiments using food reward in food-restricted experimental animals. However, for a long time, the bulk of research on ingestive behavior was directed towards the brainstem and hypothalamus. The few exceptions were studies looking at ingestive behavior following early manipulations of dopamine projections and signaling from the midbrain to the nucleus accumbens and other forebrain area [20–24], or opioid stimulation of the ventral striatum [25]. Even during the first few years after the discovery of leptin in 1995, the focus on the hypothalamus was reinvigorated, with little attention to higher brain functions.
This focus on hypothalamus and brainstem started to change when two influential, anatomically-inspired review papers [26, 27] made the case for a much larger neural system underlying the control of ingestive behavior and ultimately the regulation of body weight. Looking at humans in the modern world and the typically housed laboratory rodent with food almost omnipresent, it might not be obvious that ingestive behavior requires much strategy and a sophisticated neural control system. However, throughout most of evolution, finding food and water in sufficient quantity and quality was a daily challenge and may well have contributed to the evolution of sophisticated higher brain functions such as learning and memory. Figure 1 shows the role of the brain in integrating external and internal information and orchestrating adaptive behavioral, autonomic, and endocrine responses necessary for body energy allostasis. Neural processing within cortico-limbic and external sensory brain areas and communication with the hypothalamus should be particularly important for human food intake, which is more and more guided by sensory, emotional and cognitive rather than metabolic aspects in the obesogenic environment of affluent societies. Watts provided the first comprehensive model of the workflow during the different phases of ingestive behaviors and its potential underlying neural substrate [28]. A modified diagram showing these phases in the greater context of the control of body weight is depicted in Fig. 2. The initiation and procurement phases can be particularly challenging in a restrictive natural environment, requiring a response plan and extensive foraging behavior, all depending on prior experience and the current environmental conditions.
Fig. 1.
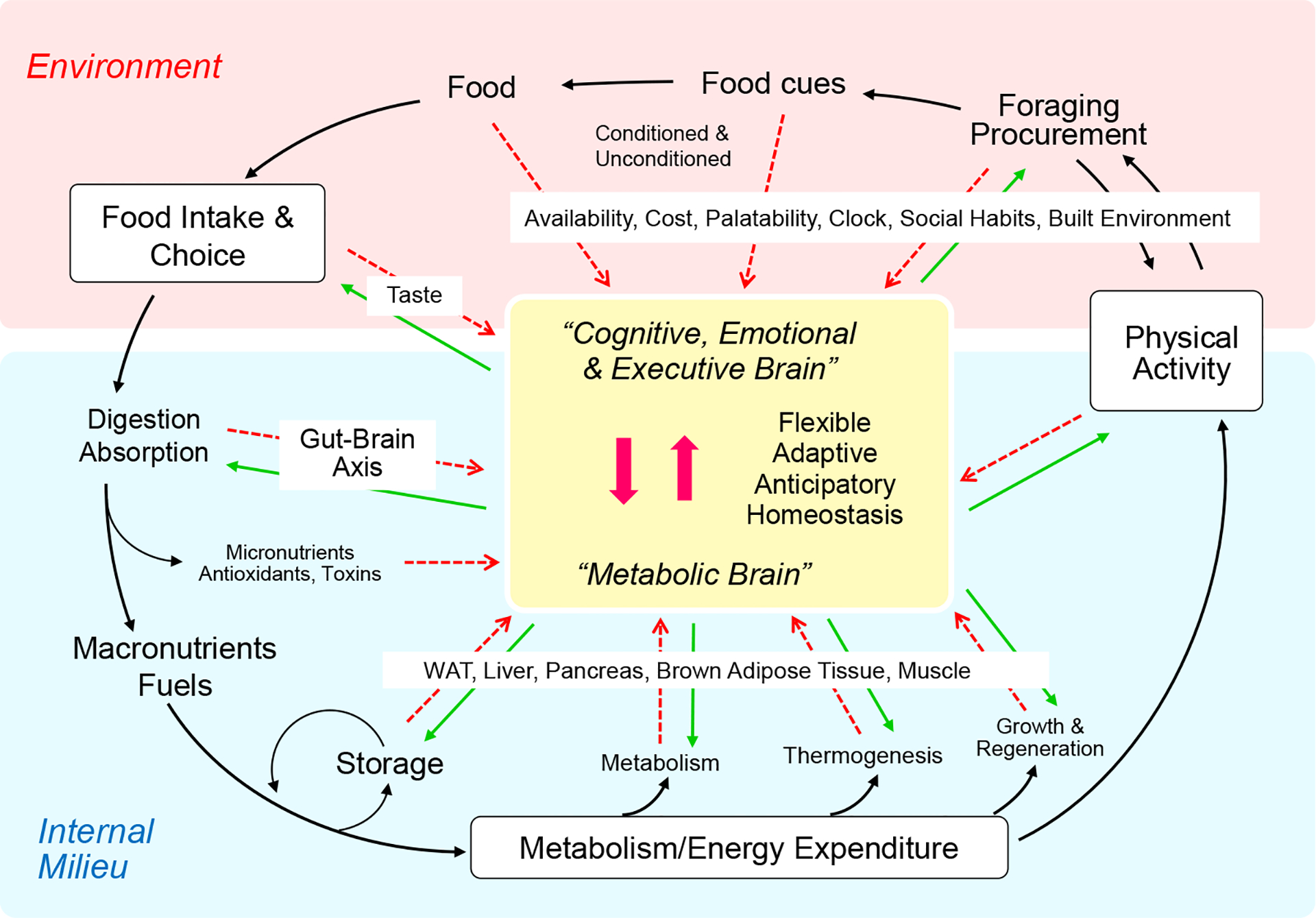
By integrating external (red) and internal (blue) information, the brain (yellow) can regulate long-term body weight/adiposity flexibly and adaptively, to accommodate special circumstances (allostasis). Red dashed arrows represent sensory information to the brain. Green arrows represent behavioral, autonomic, and endocrine motor outflow from the brain. Reproduced with permission from [32].
Fig. 2.
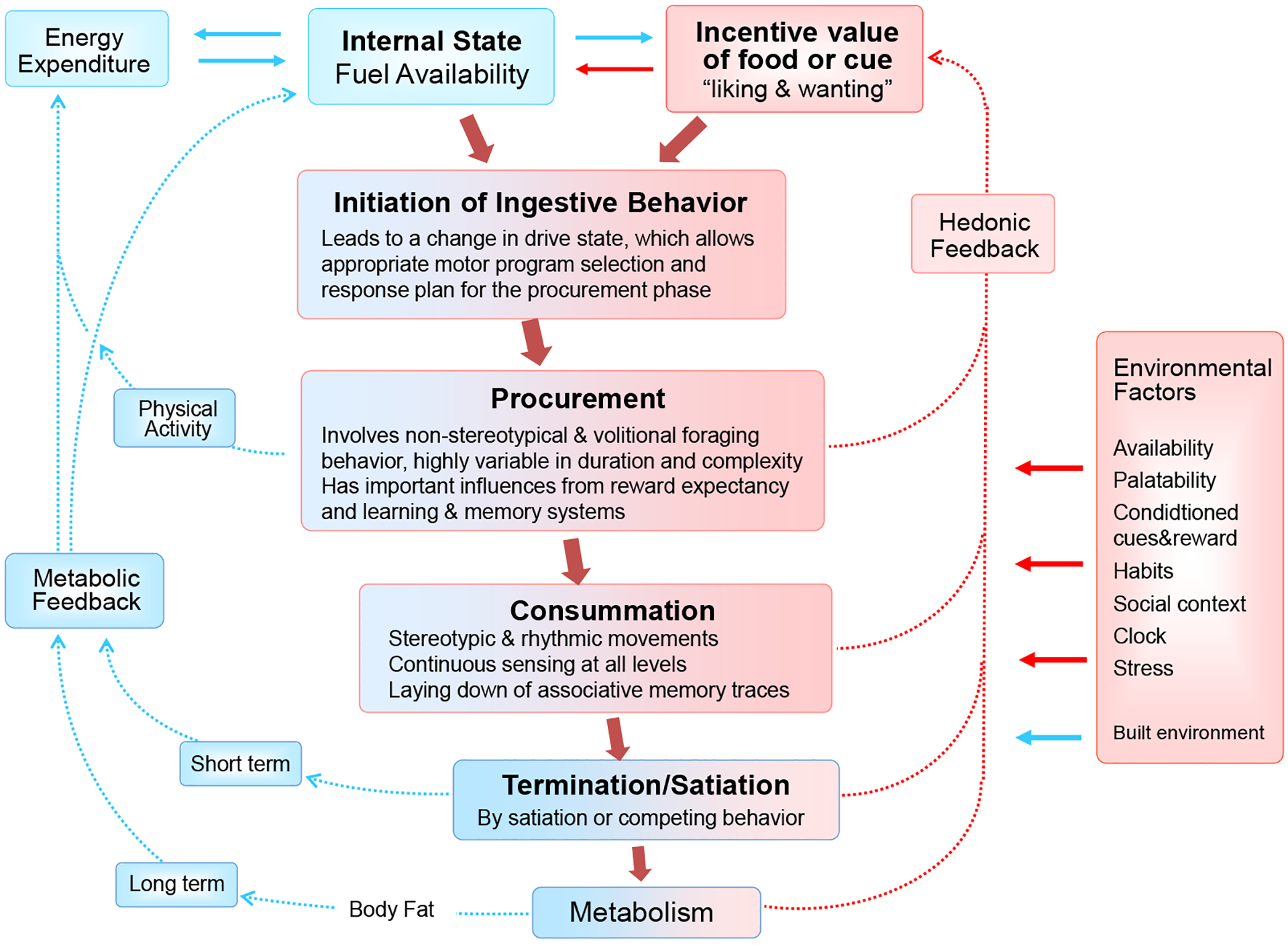
The phases of ingestive behavior and its logistical tasks. Internal state and environmental signals interact to initiate ingestive behavior. In a natural restrictive environment, the procurement phase is the most complex and typically relies heavily on previous experience. It can involve extensive foraging, requiring considerable physical activity and energy, and it generates new memories for guiding future foraging behavior. The consumatory phase is typically less demanding when not contested. Besides rhythmic movements and autonomic support for ingestion and digestion, associative memories of the sensory qualities detected at all levels are formed, a process that continues after termination of the ingestive bout during digestion and absorption. The procurement, consummation, and termination phases are influenced by environmental factors. Short and long-term metabolic feedback signals lead to satiation and satiety. Hedonic feedback signals are derived from both sensory and postabsorptive consequences of food. Parts of the diagram are adapted from [28]. Blue boxes and arrows depicts metabolic, red boxes and arrows depict hedonic processes and signals.
Since then, many studies demonstrated such top-down modulation of homeostatic hypothalamic controls by cortico-limbic systems, as well as bottom-up modulation of higher brain functions by interoceptive signals of nutrient availability. The new genetics-driven neuroscience tool set was instrumental in this endeavor. We have regularly summarized such observations during the last 15 years [29–36] and suggested that hedonics act in unison with the classical homeostatic system in the neural control of appetite and regulation of body weight. A schematic diagram of identified and potential neural pathways accomplishing such integration and coordination is shown in Fig. 3. Here we further discuss these well-known neural pathways and mechanisms in the light of some more recent studies.
Fig. 3.
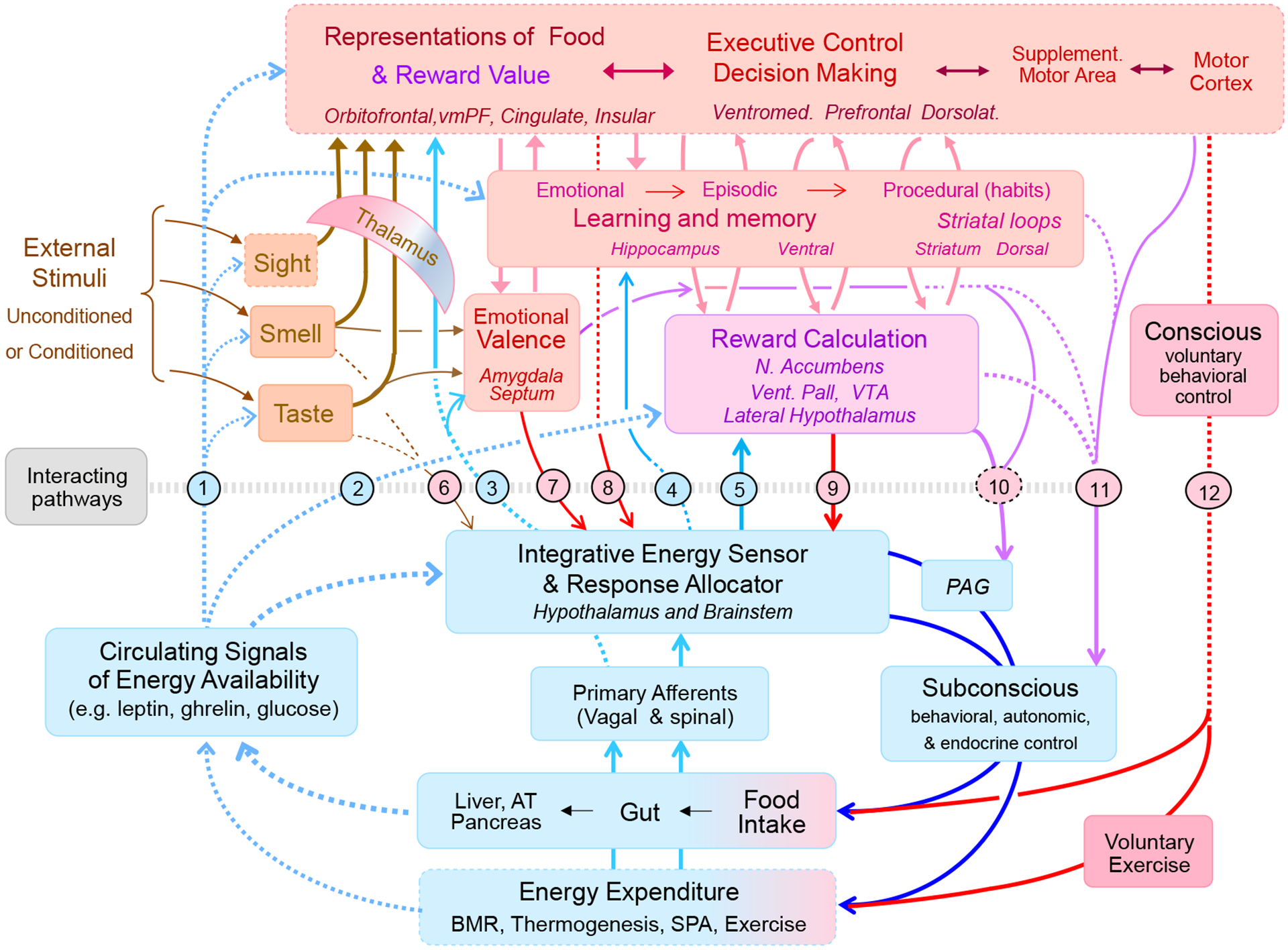
Schematic diagram showing the intimate relationship between the classical homeostatic (light blue) and the hedonic (pink/red) neural systems controlling appetite, energy balance, and body weight. The classical homeostatic system (integrative energy sensor and response allocator) in the brainstem and hypothalamus is capable of sensing the internal milieu through circulating (broken light blue lines) and neural (solid light blue lines) signals and control energy intake and expenditure subconsciously (dark blue lines). The hedonic system senses signals from the environment, calculates emotional valence and reward value of potential goal objects through learning and memory, and can influence energy intake and expenditure through both conscious voluntary (red arrows) and unconscious (purple arrows) actions. Known interacting pathways are numbered, with blue numbers/pathways representing bottom-up modulation and red numbers/pathways representing top-down modulation. Bottom-up modulation: Circulating interoceptive signals have been shown to modulate external sensory inputs, various cortical areas, and other areas involved in learning and memory (pathway 1), as well as reward processing areas (pathway 2). Interoceptive signals mainly carried by vagal afferents reach vast cortical and subcortical areas, including the insular cortex (pathway 3). In addition, interoceptive information processed in the hypothalamus and brainstem reaches thalamus, hippocampus, and the reward system (pathways 3, 4, and 5). Specifically, there are orexin projections from the LH to the paraventricular nucleus of the hypothalamus and in turn to the nucleus accumbens (pathway 3), and to the ventral tegmental area (pathway 5). Top-down modulation: Taste, olfactory, and visual information can directly reach parts of the hypothalamus (pathway 6). Amygdala to lateral hypothalamic pathways mediate conditioned food intake (pathway 7). The hypothalamus receives massive inputs from many cortical and subcortical areas (pathway 8). The LH receives direct GABA-ergic input from the ventral striatum (pathway 9). Subconscious motor actions including autonomic nervous system outflow can originate from the striatum, the central amygdala (emotional motor system), and certain cortical areas, some of them passing through the periaqueductal gray (Pathways 10 and 11). Finally conscious willful motor actions can affect both food intake, food choice, and energy expenditure. Abbreviations: PAG, periaqueductal gray; VTA, ventral tegmental area; Vent. Pall, ventral pallidum; vmPFC, ventromedial prefrontal cortex.
Recent evidence for crosstalk between the classical hypothalamic homeostatic regulator and reward pathways
With the ability to selectively record neural activity from behaving animals, as well as to stimulate, inhibit, or permanently silence the activity of neuronal populations with specific molecular fingerprints, a new era of functional systems neuroscience has begun. The critical roles of AGRP/NPY and POMC/CART neurons in the arcuate nucleus were discovered by demonstrating complete cessation of food intake after selective deletion of AGRP neurons in adult mice [37, 38] and powerful stimulation of food intake and gain of body weight during 4 days of chemogenetic activation of AGRP neurons [39]. Conversely, POMC deficiency causes severe hyperphagia and early onset obesity in mice and humans [40–42], and optogenetic activation over longer periods of time decreases food intake [43].
Selective optogenetic activation of AGRP neurons in satiated mice evokes the complete behavioral responses necessary to find and eat food, abandoning any competing motivational drives [43–45]. The amount eaten strictly depends on the duration of stimulation, independent of any delay between stimulation and access to food for up to an hour, suggesting that “it transmits a hunger signal that accumulates in a downstream circuit even after AGRP neurons have been silenced” [44]. AGRP neuron stimulation produces conditioned and motivated eating, as mice will lever press for food reward on a progressive ratio schedule [39, 44]. Thus, the relatively small group of AGRP neurons in the basomedial hypothalamus appears to be at the center of food intake control and regulation of energy balance, and understanding its outputs and inputs should provide a comprehensive picture of how this regulation works.
Among the downstream targets of AGRP neurons are the lateral hypothalamus (LH) and the paraventricular nucleus of the hypothalamus (PVH), as optogenetic stimulation of AGRP neuron terminals in these brain areas elicited the same complete behavioral responses [44]. AGRP neuron projections to the LH likely activate diverse populations of LH neurons, including GABA, and orexin neurons known to project to the ventral tegmental area (VTA) [46–50], and in turn engage the mesolimbic dopamine reward system including the nucleus accumbens [48] (interactive pathway # 5 in Fig. 3). The connections to the mesolimbic dopamine system are likely responsible for eliciting the “wanting” of food upon AGRP neuron activation and the inputs to the PVH for engaging brainstem motor programs as well as the autonomic support necessary for ingestive behavior. Each of these subsystems has its own complexity, and the reader should consult [51, 52] for in-depth discussions.
Most relevant to this discussion are the connections between the hypothalamus and other forebrain structures such as the cortex, amygdala, and hippocampus, and the way nutritionally important information is integrated by these structures. With their capacity to integrate large amounts of information [53], medium spiny neurons in the nucleus accumbens are key for selecting appropriate behavioral action [54]. Besides dopaminergic input from the VTA, they notably receive glutamatergic inputs from the hippocampus and prefrontal cortex. The hippocampus is a brain region that integrates feeding-relevant internal and external signals with learning and memory processes [55–60]. Ghrelin signaling in the ventral hippocampus increased meal size by reducing the effectiveness of a number of interoceptive satiety signals such as GLP-1, cholecystokinin, and amylin through a pathway involving lateral hypothalamic orexin neurons and their brainstem projections [61]. Lesions of parts of the hippocampus [62] and selective optogenetic inhibition of glutamatergic pyramidal neurons in the dorsal or ventral hippocampus ([63] during the postmeal period increases future food intake. Glutamatergic inputs to medium spiny neurons in the nucleus accumbens from the prefrontal cortex are thought to modulate eating behavior in the context of other important drives and executive decisions. The orbitofrontal cortex, which is a major hub in reward processing contains specific populations of neurons differentially responsive to aspects of food intake and social environment ([64–67]. Thus, the downstream targets of AGRP neurons are in a position to orchestrate appropriate behavioral, autonomic and endocrine actions necessary for the display of complete appetitive and consumatory behavior.
Regarding inputs to AGRP and POMC neurons, they primarily respond to interoceptive signals of nutrient availability, either through relevant receptors directly expressed by these neurons, or indirectly via neural inputs. In ad libitum fed mice, when baseline activity is low, the hunger-inducing gut hormone ghrelin increased activity of AGRP and decreased activity of POMC neurons [44, 68]. In fasted mice, when baseline activity is high, re-feeding, as well as systemic administration of leptin, glucose, fat, PYY, CCK, and GLP-1 decreased activity of AGRP neurons [68, 69]. These effects are consistent with AGRP and POMC mRNA expression levels. Notably, AGRP mRNA expression is about 2-fold higher in 24 hour food-deprived and chronically food-restricted mice compared with ad libitum fed mice [70, 71] and returns to normal levels after 24 but not 6 hours re-feeding [71], suggesting that AGRP mRNA expression is a convenient readout for “hungriness”.
Interestingly, AGRP and POMC neuron activity changed before the hungry mice ingested any food. Simply seeing caged food or smelling inaccessible food was sufficient to produce a transient and small reduction in AGRP and increase in POMC neuron activity that rapidly reversed when consumption did not follow [68, 72]. This learned anticipatory response seems logical given the observation that elevated activity of AGRP neurons is acting as a negative valence teaching signal [73]. Importantly however, it indicates the existence of neural inputs from the environment via external sensory channels. While the exact nature of neural inputs to AGRP and POMC neurons has not been fully explored, they include direct and multisynaptic inputs originating in other parts of the hypothalamus [74], and other brain areas. Using the Cre-recombinase-enabled, cell-specific monosynaptic neuron mapping technique, strong excitatory direct inputs from thyrotropin-releasing hormone (TRH) and pituitary adenylate cyclase-activating polypeptide (PACAP) expressing neurons in the paraventricular nucleus of the hypothalamus, as well as from the dorsomedial nucleus of the hypothalamus, while the supraoptic nucleus, medial preoptic area, and lateral hypothalamus provided much weaker inputs to AGRP neurons [74]. These direct hypothalamic inputs were confirmed in another study that also found direct inputs from a large number of brain areas including septum, striatum, amygdala, pallidum, thalamus, hippocampus, midbrain, pons, and hindbrain, although most of them quite sparse [75]. A much earlier study, using virally-assisted multi-synaptic retrograde tracing found AGRP neuron inputs from amygdala, cortical areas and other brain regions [76].
In summary, the ability to manipulate specific neuron populations in behaving animals tremendously enriched our understanding of the classical hypothalamic appetite control system. Although we have discussed here only a selection of relevant recent publications, some fundamental new insights emerge. First, AGRP neurons are not only responsive to interoceptive signals of nutrient availability, but also to environmental signals via the external sensory pathways and cortico-limbic areas of the brain. Second, AGRP neurons are uniquely capable of driving and coordinating the complete behavioral sequence underlying natural food intake behavior, specifically they engage the mesolimbic dopamine reward system to generate the intense “wanting” of food in a hungry animal. Third, a number of other cortico-limbic brain areas with the ability to integrate exteroceptive and interoceptive information are anatomically and functionally connected to the “classical homeostatic” areas of the hypothalamus and brainstem, including AGRP and POMC neurons. Further, we hypothesize that environmental influences on food intake and energy balance regulation gain access to “classical homeostatic” brain areas mainly via these cortico-limbic structures.
3. The obesogenic environment likely impinges on the powerful motivational system that is inextricably linked to the extended homeostatic regulatory system
Although a number of potential causes for the recent (last 50 years or so) steep increase in the prevalence of obesity have been mentioned, it seems clear that the obesogenic environment is the main driver. The most convincing fact is that we can reproduce this scenario over and over again, at least in animals. The key experiments were done a while ago by feeding rats a “Supermarket diet” and controlling their level of exercise. Compared to regular chow diet, the supermarket diet led to overeating and obesity within a short time and the less the rats had the opportunity to move, the more they became obese [77]. This experiment has been replicated hundreds of times in rats and mice with other diets high in fat and sugar and relatively empty of micronutrients and fibers - the typical supermarket and fast food diet. It can be shown in most species, including Drosophila [78], cats, and even elephants [79]. And this experiment is going on in the huge urban centers of rapidly developing and industrializing countries such as China [80]. Besides the diet, driving factors are the built environment, mechanization and automatization, which greatly reduce the opportunity for physical activity. Advertisement and pricing of palatable but poor quality foods are other important drivers. This obesogenic environment, originally generated in the spirit of re-building the economy after World War II, continues to be dominated by non-sustainable economic models, leading to propagation of obesity for profit. Given the dominance of profit as a motive and an almost complete lack of political will to regulate, the obesogenic environment is not likely to be reversed or even halted soon.
Why and how does the obesogenic environment bypass or overpower the homeostatic regulatory system? The answer is likely not a simple one, as energy balance depends on both energy intake and energy expenditure, and the very purpose of the homeostatic regulator is to respond to an insult on one side of the equation with compensatory adjustments on the other side. So we would expect that reduced physical activity caused by the obesogenic environment is compensated by a commensurate decrease in energy intake. Conversely, the homeostatic regulator should increase energy expenditure in the face of overeating. Perhaps the simple fact that the obesogenic environment unfavorably affects both arms of the energy balance is key. It could be that the increased energy intake caused by the obesogenic environment is not compensated by commensurate changes in energy expenditure simply because the drastic changes in the built environment prevent rather than stimulate physical activity-induced energy expenditure.
We hypothesize that the obesogenic environment impinges on a neural node that is most crucial for the defense of body weight, thereby resetting the level of defended body weight in the obese. Arcuate AGRP and POMC neurons, and their reciprocal connections with the reward system, potentially represent this crucial central node. The obesogenic environment somehow sensitizes this AGRP-dopamine motivational system so that it is hyperactive when metabolically hungry and remains active even when metabolically replete. The recent demonstration that AGRP and dopamine signaling are interdependent, meaning that changes in one automatically leads to changes in the other [69], could be one mechanism by which signals from the obesogenic environment drive overeating (Fig. 4). It is conceivable that, similarly to drugs of abuse, signals from the obesogenic environment not only activate the dopamine system, but also AGRP neurons, leading to hyperphagia. The fact that simply seeing or smelling food without ingestion produces small transient decreases, not increases, in AGRP activity [68, 69], seems at odds with such a hypothesis. However, it should be noted that the rapid decrease of AGRP neuronal activity is anticipatory and short-lived if not immediately followed by eating. Also, these experiments were carried out in food-deprived mice with high basal AGRP neuron activity, while one of the hallmarks of hedonic eating is that it mostly occurs in the absence of metabolic hunger, when baseline activity is low. It will be interesting to model rodent studies more closely to the human situation and investigate the effect of environmental stimuli on AGRP neuron activity under non-food deprivation/restriction conditions.
Fig. 4.
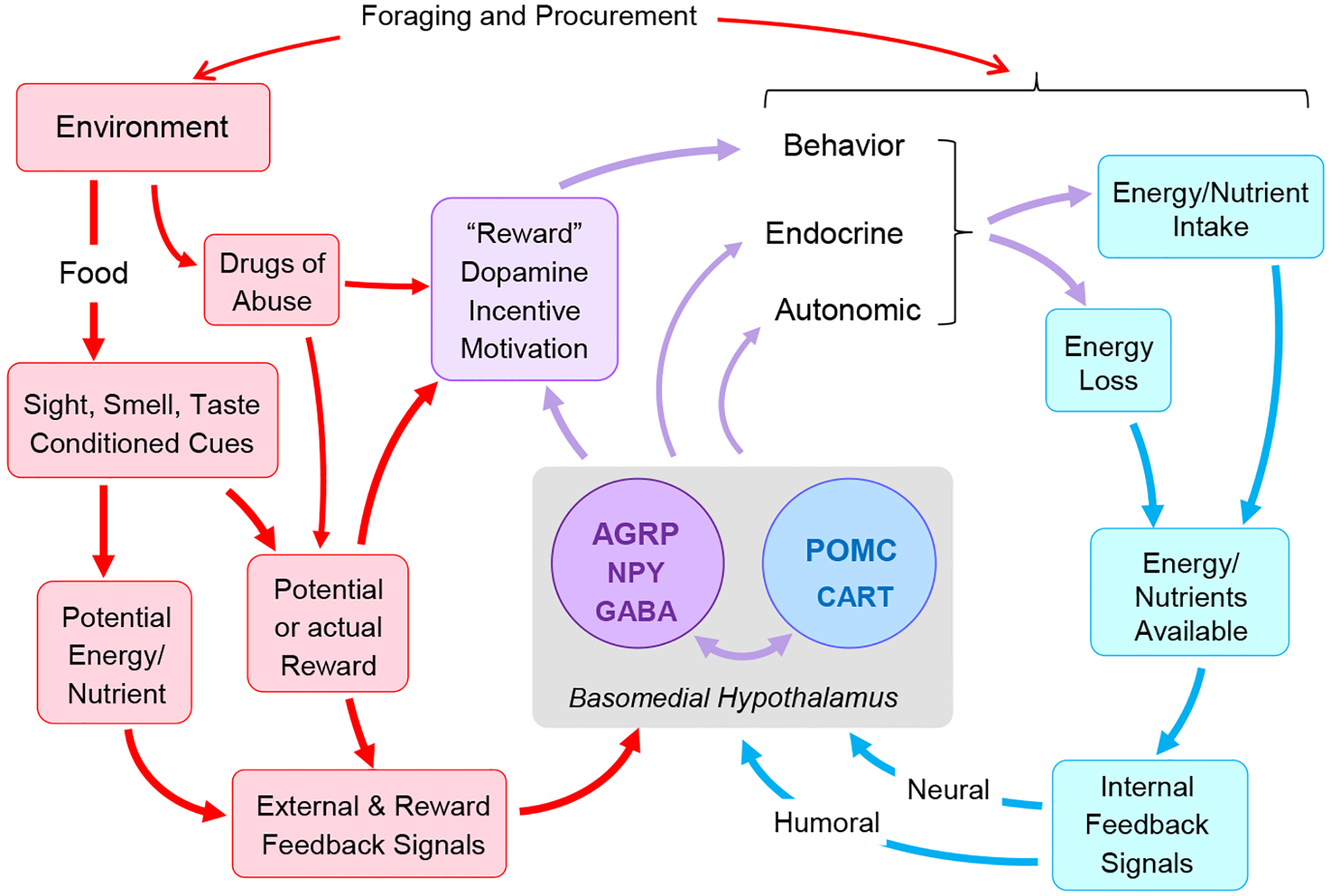
Potential role of the basomedial hypothalamus and mesolimbic dopamine system in integrating classical homeostatic and hedonic controls of food intake and energy balance. AGRP and POMC neurons are key in sensing the nutritional state (blue arrows and boxes) and orchestrating adaptive anabolic and catabolic responses through behavioral, autonomic and endocrine actions (purple arrows). Notably, nutrient deficiency drives AGRP neuron activity and in turn the mesolimbic dopamine reward system to produce a sustained “wanting” of food. Environmental factors gain access to both AGRP neurons and the mesolimbic dopamine system through sensory channels and conditioned reward and energy expectancies (red boxes and arrows). We hypothesize that environmental pressure enhanced by easy availability, palatability, conditioned reward expectancy, and other factors further stimulate the AGRP-dopamine pathway, so that it is active even in the absence of a metabolic deficit.
Changes in AGRP neuron activity may not be required for the obesogenic environment to drive hyperphagia. This was demonstrated in mice with neonatal AGRP neuron ablation, which in contrast to adult mice, does not lead to cessation of eating and starvation [38]. While under normal low-fat diet conditions, ghrelin can no longer elicit a feeding response, high-fat diet still leads to hyperphagia, and restores the feeding response to ghrelin in these mice by acting on the dopamine system in the ventral tegmental area [81]. These findings suggest that AGRP and dopamine neurons, both critical for driving food intake, are able to interact and partially substitute for each other.
In summary, we propose that exteroceptive stimuli emanating from the obesogenic environment gain access to the powerful motivational mechanisms underlying the natural drive to eat and produce a hyperphagic bias. The exact neural pathways and mechanisms of this bias are not known, but recently identified external sensory inputs to basomedial hypothalamic AGRP neurons and the bi-directional interdependent modulation of AGRP and mesolimbic dopamine neurons may play a role. However, whether the environmental pressure leads to a permanent resetting of defended body weight/adiposity, and the underlying mechanisms mediating the reset remain largely speculative. We will next discuss some of the molecular and neural mechanisms proposed to propagate the obese state and prevent its reversal.
4. Does the obese state promote further resistance to body weight defense mechanisms: Evidence for a vicious circle?
Most rodent studies using inbred C57BL6 mice or outbred Sprague Dawley rats find that high-fat diet-induced obesity lasting up to 20 weeks is largely reversible after switching back to normal chow diet [82]. Although others find incomplete reversal, there is always a large and sustained weight loss after the switch back to normal chow [83]. It is also clear that when weight loss is induced through caloric restriction in rodents on a high-fat diet, they regain all the lost weight when ad libitum high-fat conditions are reinstated. These findings are consistent with the idea that in rodents the defended level of body weight/adiposity is strictly a function of diet and likely other environmental factors. The first type of study, namely completely removing the obesogenic diet and environment, has not been done in a controlled fashion over extended periods of time in humans, but the outcome would likely be similar to rodent studies. Although observations in prisoners and during war times tend to support such a conclusion, none of them looked at obese subjects and truly obesogenic environments (for review see [84]).
Most human studies with obese subjects can achieve meaningful weight loss for up to 2 years with the combination of caloric restriction and lifestyle modification [85]. However, the majority of these subjects re-gains the lost weight within a year or two of ending therapy [86], This weight regain is epitomized by the scientific follow-up of the “Biggest Loser” television contest, showing that a significant reduction of metabolic energy expenditure and increased hunger were the drivers of weight regain [87]. These counter-regulatory responses are consistent with the view that in most humans the obese body weight is actively defended, representing one of the biggest hurdles in the treatment of obesity [87]. It should be noted, however, that after stopping the intervention, the subjects typically go right back to the same obesogenic environment that caused their obesity in first place. Therefore, the conclusion should be modified, stating that most obese humans defend their obese body weight if they remain in an obesogenic environment. This is distinct from the reversal of obesity in rodents that are returned to regular (low-fat) chow.
A number of potential mechanisms have been proposed through the years that may explain the development of obesity and the resistance to reverse it. One proposed mechanism has been functional leptin-resistance, based on the observation that “common” obesity is typically associated with high circulating leptin levels that are unable to produce a catabolic response profile and counteract obesity induced by an obesogenic environment [88, 89]. The exact molecular and neural mechanisms leading to this inability of high circulating leptin levels to prevent or reverse obesity are not well understood. A limited number of experimental involuntary overfeeding studies lasting 2–3 weeks and producing mild obesity and increased circulating leptin levels in rats have provided some answers. One study demonstrated that intragastric overfeeding increased POMC mRNA expression in the arcuate nucleus, and that blockade of the melanocortin-3/4 receptor with intraventricular SHU9119 during the post-gastric infusion period completely rescued the suppressed food intake of control rats with intraventricular saline infusion [90]. Furthermore, while intragastrically overfed control Zucker rats spontaneously reduced their food intake for 6 days in the immediate post-overfeeding period, obese Zucker rats with impaired leptin-signaling, failed to exhibit this persistent hypophagia, with intake returning to near baseline in only one day [91]. Together, these studies suggest that in these reversible rodent obesity models, increased leptin does contribute to the compensatory reduction in food intake, mainly by stimulating POMC neurons and MC3/4 signaling. Therefore, resistance to these particular actions of leptin and melanocortins could be responsible for the irreversible “common” obesity in humans. However, it is important to note that the obesity induced in these models was relatively mild and that the diet used for overfeeding was low-fat and bypassed the oral cavity and its ability to activate cephalic phase responses. Also, other studies found that preventing hyperleptinemia and leptin resistance in high-fat-diet induced obese mice has no effect on high fat diet induced weight gain and suggests that high fat diet drives weight gain and insulin resistance independent of leptin resistance [92].
Perhaps the most influential idea is that consumption of unhealthy foods which are high in saturated fats and sugars but low in fibers and micronutrients leads to inflammation in the hypothalamus [93, 94] that corrupts the proper function of arcuate AGRP and POMC neurons. However, recent studies [95, 96] suggest that high-fat diet-induced inflammation in the hypothalamus and perhaps other brain areas [97], is not the primary cause of obesity, but may contribute to its irreversible effects.
Another idea that has gathered considerable momentum is the diet- or obesity-induced weakening of satiety mechanisms both in the periphery at the level of vagal afferents, and centrally at the level of the hippocampus. Gastric vagal mechanosensitivity is persistently reduced in chronic high-fat diet-induced obese mice for 12 weeks following the return to normal chow [98], impairing satiation signaling to the brain [99] and possibly mediated by elevated nitric oxide signaling [100]. As discussed above, the hippocampus is a brain region that integrates feeding-relevant internal and external signals with learning and memory processes and has the ability of interoceptive states such as satiety to inhibit responding to previously rewarded cues [60]. Western diets high in fat and sugar are weakening this mechanism both in rats [101] and humans [102], leading to overeating and propagating obesity.
Another potential mechanism that may explain the development of obesity and the resistance to its reversal is reward-deficiency. This hypothesis suggests that obesogenic diets and the obese state erode the ability of the mesolimbic dopamine pathways to generate reward, and that subjects with obesity further increase consumption of high-energy foods in an attempt to self-medicate to maintain food reward [103, 104].
To summarize, in distinction to animal models of obesity, our understanding of human obesity and its reversibility is complicated by the fact that the obesogenic environment (the main cause of obesity in first place) cannot be easily and completely removed. This suggests that all the proposed reasons for why the obese body weight is defended ultimately hinge on this inability to remove the obesogenic environment. There is no convincing evidence in animal models that a vicious circle propagates an irreversible obesity that persists once the obesogenic environment is completely removed. In contrast, there is lots of evidence that obesity is irreversible only when the obesogenic environment persists. As we see next, this concept has major implications for the prevention and treatment of obesity.
5. Implications for prevention and treatment
The most effective measure to prevent or reverse obesity would be to remove the root cause – the obesogenic environment. Of course, the most optimistic mind realizes that a complete reversal of the modern environment is simply not possible, but small changes could go a long way. For this to happen, the public, scientists, health care professionals, food and insurance companies, and governments need to be on the same page and put aside self-interests. However, under the current political and corporate climate which is downplaying environmental concerns, it is unrealistic to expect much progress in reversing the obesogenic environment in the near future.
Unable to significantly reverse the obesogenic environment, the next best approach is to change people’s relationship with the obesogenic environment. Such a strategy may not be useful for adults with moderate to severe obesity, as the obese body weight is staunchly defended and habits are extremely difficult to break. However, this strategy is particularly indicated for children and adolescents that have a high probability to develop obesity later in life but are not yet obese. In this population, we need to instill immunity to the toxic effects of the obesogenic environment. Multi-component strategies that target the child, family, school, social network, and community, as well as diet, physical activity, sleep quality, and behavior/cognitive change, are likely to be most successful [105–108], particularly, if they can be combined with, at least, small changes to the food and built environment. In addition, behavioral phenotyping of children before they are overweight or obese will allow treatments that are tailored to children’s individual predispositions and will be more effective [109]. A number of behavioral phenotypes have been identified, such as 1) the ability to calorically compensate for a preload (also called selfregulation), 2) the ability to resist high energy-dense snack foods when not metabolically hungry (eating in the absence of hunger), 3) the willingness to work for food reward (relative reinforcing value of food), 4) ability to resist the drive to palatable food items (reward sensitivity), and 5) the rate of eating [109]. While phenotype-specific treatment seems straight forward for some of these behavioral subtypes such as high eating rate and eating snacks in the absence of hunger, it is currently unknown how to modify self-regulation, the relative reinforcing value of food, and reward sensitivity. Small changes in the environment such as banning television advertisements for food to children may go a long way. Also, we do not have a good understanding of the underlying neural mechanisms for each of the behavioral subtypes, except that some of them (e.g. self-regulation) depend more on impaired interoceptive signaling and others more on impaired exteroceptive signals and reward expectations. Therefore, much needs to be done to understand the underlying neural mechanisms and develop adequate treatment methods for each behavioral subtype.
In children, adolescents, and young adults that are already obese, the initial use of low-calorie diets may be necessary to achieve meaningful weight loss. In clinical “behavioral and lifestyle modification” studies in adults, intensive behavioral therapy is typically accomplished with regular face-to-face or electronic coaching sessions elastically spaced over a year, with or without low-calorie meal replacements for the first few weeks or months [85, 110]. Typical weight loss at 1 year in subjects with obesity is about 5–10%, with one study reporting an impressive 17.9% [111]. However, in almost all studies the intervention is stopped at 1 year, and most of the weight is regained. Changing habits, which is really the goal of this therapy, is extremely difficult for most humans, and if there is no continued pressure, old habits take over again [112–118]. The use of electronic media to do the bulk of counseling is promising because more people can be reached for longer times, and the cost for long-term treatment can be kept minimal [119, 120].
Pharmacotherapy or surgery should always be the last resort for treatment of obesity and only used in patients with a heavy genetic predisposition for obesity before they become obese, or in morbidly obese patients that failed to respond to behavioral modification. For in-depth discussions of obesity pharmacotherapy and surgery, the reader is directed to two recent reviews [121, 122]. The critical question for this discussion is whether pharmacological and surgical treatments are purely symptomatic or whether they are able to reset the elevated and defended body weight associated with obesity and suppress the powerful counter-regulatory adaptive responses, namely increased hunger and hypo-metabolism.
Four of the five currently available drugs approved for long-term treatment of obesity in the USA act on the brain [121]. Lorcaserin is a 5-HT2c receptor agonist. Liraglutide and Semaglutide are GLP-1 receptor agonists. Phentermine + Topiramate is a combination of a norepinephrine-releasing/uptake inhibitor and a drug (Topiramate) with a number of actions (L-type sodium channel blocker, AMPA/kainite glutamate receptor antagonist, enhancer of GABA-mediated chloride fluxes, and carbonic anhydrase inhibitor). Finally, Naltrexone + Bupropion is a combination of an opioid receptor antagonist and an atypical antidepressant with norepinephrine and dopamine reuptake inhibitory as well as nicotinic receptor antagonist activities.
The modest effectiveness of the 5-HT2c receptor agonist Lorcaserin as an anti-obesity drug appears to be based on its actions on at least 3 key pathways, i) hypothalamic [123] and ii) brainstem [124] POMC neurons, and iii) ventral tegmental dopamine neurons [125, 126]. The GLP-1 receptor agonists Liraglutide, and the newer, slow-release Semaglutide, are the most effective anti-obesity drugs to date, with Semaglutide (1 mg once weekly for 30 weeks) resulting in 5.8% weight loss and a meaningful reduction of HbA1c by 1.7 percentage points [127]. These effects are likely due to both, peripheral actions on insulin secretion and gastric emptying, and central actions on hypothalamic and extrahypothalamic systems involved in the control of food intake and body weight regulation [128–131].
Bariatric surgery, in particular gastric bypass surgery and ventral sleeve gastrectomy, are by far the most effective obesity treatments, with sustained weight loss of 15–25% for up to 20 years. For patients with severe obesity and diabetes, the long-term benefits of a higher life expectancy and quality clearly outweigh the disadvantages of the surgery being invasive and irreversible. Given the remarkable anti-obesity effects of the GLP-1 receptor agonists Liraglutide and Semaglutide discussed above and the drastic increases in circulating GLP-1 after both gastric bypass and vertical sleeve gastrectomy [132, 133], central GLP-1 action seemed to be the key mechanism by which these bariatric surgeries suppress food intake. However, a role for GLP-1 could not be confirmed in GLP-1 receptor-deficient mouse models of gastric bypass [134] and vertical sleeve gastrectomy [135]. One explanation for this conundrum may be that unlike the stable analogs Liraglutide and Semaglutide, even high circulating levels of endogenous peripheral GLP-1 may not reach these central receptors in sufficient amounts.
Interestingly, gastric bypass surgery does not indiscriminately reduce appetite and lower body weight through a restrictive mechanism, but rather appears to change the defended body weight or body weight set point. Unlike weight loss induced by food restriction, which results in increased hunger and hypometabolism, weight loss induced by gastric bypass surgery appears to suppress these counter-regulatory adaptations. Consistent with this conclusion, we recently demonstrated that AGRP mRNA expression in mice, unlike after starvation-induced weight loss, is not increased after gastric bypass-induced weight loss [70], suggesting that a new set point has been established. The observation that gastric bypass surgery is less effective in MC4R-deficient compared with wildtype mice is consistent with this interpretation [136]. At present, the mechanisms through which bariatric surgery acts on the brain to establish this new body weight set point are unclear. Identification of this mechanism might be key for developing pharmacological tools that can eventually replace the invasive surgery.
Newly available experimental neuroscience techniques open up new ways to further explore the mechanisms of obesity drugs and surgery. Interesting experiments include in vivo recording of AGRP and POMC neuron activity to test whether these drugs and surgeries are able to suppress hunger or increase satiety at the key central node.
6. Summary and Conclusions
The sobering conclusion is that in the absence of political will to halt harmful environmental changes propagated by for-profit thinking, the prevention and treatment of obesity remains largely symptomatic and thus relatively ineffective. The first line of defense is thus relegated to improving behavioral modification therapies, particularly in children and adolescents, even before they develop overweight or obesity. Realizing that the obese body weight is defended by strong adaptive biological responses to weight loss, makes prevention the number one priority. Much can be gained by developing all aspects of behavioral modification therapy in children, such as the involvement of the entire community, behavioral phenotyping leading towards personalized approaches, and the use of electronic techniques to reach more subjects for longer periods. The goal should be no less than making children immune to the toxic effects of the obesogenic environment. In subjects that are already obese and have not or have poorly responded to behavioral modification therapy, pharmacotherapy and surgery are valid treatment options. A more complete understanding of how the obesogenic environment corrupts normal functions of the neural systems governing appetitive and consumatory behaviors will be instrumental in improving behavioral modification efforts, in making drugs more selective and efficient, and in revealing the mechanisms which make bariatric surgeries so effective.
Highlights.
Despite presence of physiological mechanisms for the regulation of body weight, the obesity epidemic continues unabated.
Here we critically discuss potential reasons for this conundrum and identify the obesogenic environment as primary driver of obesity.
We hypothesize that the obesogenic environment impinges on critical neural circuitry in the hypothalamus and limbic system to raise the defended body weight.
Implications for prevention and treatment of obesity are discussed
Funding:
Research by the authors was partially funded by National Institutes of Health grants DK 047348 and OT2OD023864 (HRB), R01DK105032 (CDM), R01DK092587 and OT2OD23864(HM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare no conflicts of interest
Literature
- [1].Fall T, Mendelson M, Speliotes EK Recent Advances in Human Genetics and Epigenetics of Adiposity: Pathway to Precision Medicine? Gastroenterology. 2017,152:1695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Loos RJF, Janssens A Predicting Polygenic Obesity Using Genetic Information. Cell Metab. 2017,25:535–43. [DOI] [PubMed] [Google Scholar]
- [3].van der Klaauw AA, Farooqi IS The hunger genes: pathways to obesity. Cell. 2015,161:119–32. [DOI] [PubMed] [Google Scholar]
- [4].Farooqi IS, O’Rahilly S The Genetics of Obesity in Humans In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al. , eds. Endotext. South Dartmouth (MA)2000. [Google Scholar]
- [5].Keesey RE, Powley TL Hypothalamic regulation of body weight. Am. Sci 1975,63:558–65. [PubMed] [Google Scholar]
- [6].Harris RB, Kasser TR, Martin RJ Dynamics of recovery of body composition after overfeeding, food restriction or starvation of mature female rats. J. Nutr 1986,116:2536–46. [DOI] [PubMed] [Google Scholar]
- [7].Steinlechner S, Heldmaier G, Becker H The seasonal cycle of body weight in the Djungarian hamster: photoperiodic control and the influence of starvation and melatonin. Oecologia. 1983,60:401–5. [DOI] [PubMed] [Google Scholar]
- [8].Morgan PJ, Ross AW, Mercer JG, Barrett P What can we learn from seasonal animals about the regulation of energy balance? Prog. Brain Res 2006,153:325–37. [DOI] [PubMed] [Google Scholar]
- [9].Morgan PJ, Mercer JG The regulation of body weight: lessons from the seasonal animal. Proc. Nutr. Soc 2001,60:127–34. [DOI] [PubMed] [Google Scholar]
- [10].Keesey RE, Corbett SW Metabolic defense of the body weight set-point. Res. Publ. Assoc. Res. Nerv. Ment. Dis 1984,62:87–96. [PubMed] [Google Scholar]
- [11].Keesey RE, Powley TL The regulation of body weight. Annu. Rev. Psychol 1986,37:109–33. [DOI] [PubMed] [Google Scholar]
- [12].Keesey RE, Hirvonen MD Body weight set-points: determination and adjustment. J. Nutr 1997,127:1875S–83S. [DOI] [PubMed] [Google Scholar]
- [13].Boyle PC, Storlein LH, Keesey RE Increased efficiency of food utilization following weight loss. Physiol. Behav 1978,21:261–4. [DOI] [PubMed] [Google Scholar]
- [14].Levitsky DA, Raea Limb JE, Wilkinson L, Sewall A, Zhong Y, Olabi A, et al. Lack of negative autocorrelations of daily food intake on successive days challenges the concept of the regulation of body weight in humans. Appetite. 2017,116:277–83. [DOI] [PubMed] [Google Scholar]
- [15].Levitsky DA The non-regulation of food intake in humans: hope for reversing the epidemic of obesity. Physiol. Behav 2005,86:623–32. [DOI] [PubMed] [Google Scholar]
- [16].Geary N Control-theory models of body-weight regulation and body-weight-regulatory appetite. Appetite. 2020,144:104440. [DOI] [PubMed] [Google Scholar]
- [17].Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR Energy balance and its components: implications for body weight regulation. Am. J. Clin. Nutr 2012,95:989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Speakman JR, Levitsky DA, Allison DB, Bray MS, de Castro JM, Clegg DJ, et al. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech. 2011,4:733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stellar E The physiology of motivation. Psychol. Rev 1954,61:5–22. [DOI] [PubMed] [Google Scholar]
- [20].Koob GF, Riley SJ, Smith SC, Robbins TW Effects of 6-hydroxydopamine lesions of the nucleus accumbens septi and olfactory tubercle on feeding, locomotor activity, and amphetamine anorexia in the rat. J. Comp. Physiol. Psychol 1978,92:917–27. [DOI] [PubMed] [Google Scholar]
- [21].Rowland N, Marshall JF, Antelman SM, Edwards DJ Hypothalamic hyperphagia prevented by damage to brain dopamine-containing neurons. Physiol. Behav 1979,22:635–40. [DOI] [PubMed] [Google Scholar]
- [22].Antelman SM, Szechtman H Tail pinch induces eating in sated rats which appears to depend on nigrostriatal dopamine. Science. 1975,189:731–3. [DOI] [PubMed] [Google Scholar]
- [23].Cador M, Kelley AE, Le Moal M, Stinus L Ventral tegmental area infusion of substance P, neurotensin and enkephalin: differential effects on feeding behavior. Neuroscience. 1986,18:659–69. [DOI] [PubMed] [Google Scholar]
- [24].Schneider LH, Gibbs J, Smith GP D-2 selective receptor antagonists suppress sucrose sham feeding in the rat. Brain Res. Bull 1986,17:605–11. [DOI] [PubMed] [Google Scholar]
- [25].Bakshi VP, Kelley AE Feeding induced by opioid stimulation of the ventral striatum: role of opiate receptor subtypes. J. Pharmacol. Exp. Ther 1993,265:1253–60. [PubMed] [Google Scholar]
- [26].Saper CB, Chou TC, Elmquist JK The need to feed: homeostatic and hedonic control of eating. Neuron. 2002,36:199–211. [DOI] [PubMed] [Google Scholar]
- [27].Berthoud HR Multiple neural systems controlling food intake and body weight. Neurosci. Biobehav. Rev 2002,26:393–428. [DOI] [PubMed] [Google Scholar]
- [28].Watts AG Understanding the neural control of ingestive behaviors: helping to separate cause from effect with dehydration-associated anorexia. Horm. Behav 2000,37:261–83. [DOI] [PubMed] [Google Scholar]
- [29].Berthoud HR Neural control of appetite: cross-talk between homeostatic and non-homeostatic systems. Appetite. 2004,43:315–7. [DOI] [PubMed] [Google Scholar]
- [30].Berthoud HR Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr. Opin. Neurobiol 2011,21:888–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Berthoud HR The neurobiology of food intake in an obesogenic environment. Proc. Nutr. Soc 2012,71:478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Berthoud HR, Munzberg H, Morrison CD Blaming the Brain for Obesity: Integration of Hedonic and Homeostatic Mechanisms. Gastroenterology. 2017,152:1728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Munzberg H, Qualls-Creekmore E, Yu S, Morrison CD, Berthoud HR Hedonics Act in Unison with the Homeostatic System to Unconsciously Control Body Weight. Front Nutr. 2016,3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shin AC, Zheng H, Berthoud HR An expanded view of energy homeostasis: neural integration of metabolic, cognitive, and emotional drives to eat. Physiol. Behav 2009,97:572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zheng H, Berthoud HR Eating for pleasure or calories. Curr Opin Pharmacol. 2007,7:607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zheng H, Berthoud HR Neural systems controlling the drive to eat: mind versus metabolism. Physiology (Bethesda). 2008,23:75–83. [DOI] [PubMed] [Google Scholar]
- [37].Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci 2005,8:1289–91. [DOI] [PubMed] [Google Scholar]
- [38].Luquet S, Perez FA, Hnasko TS, Palmiter RD NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005,310:683–5. [DOI] [PubMed] [Google Scholar]
- [39].Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest 2011,121:1424–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet 1998,19:155–7. [DOI] [PubMed] [Google Scholar]
- [41].Yaswen L, Diehl N, Brennan MB, Hochgeschwender U Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med 1999,5:1066–70. [DOI] [PubMed] [Google Scholar]
- [42].Kuhnen P, Clement K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. N. Engl. J. Med 2016,375:240–6. [DOI] [PubMed] [Google Scholar]
- [43].Aponte Y, Atasoy D, Sternson SM AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci 2011,14:351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Y, Lin YC, Zimmerman CA, Essner RA, Knight ZA Hunger neurons drive feeding through a sustained, positive reinforcement signal. Elife. 2016,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Burnett CJ, Li C, Webber E, Tsaousidou E, Xue SY, Bruning JC, et al. Hunger-Driven Motivational State Competition. Neuron. 2016,92:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell. 2015,160:516–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, et al. Decoding neural circuits that control compulsive sucrose seeking. Cell. 2015,160:528–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, et al. Inhibitory Input from the Lateral Hypothalamus to the Ventral Tegmental Area Disinhibits Dopamine Neurons and Promotes Behavioral Activation. Neuron. 2016,90:1286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Aston-Jones G, Smith RJ, Moorman DE, Richardson KA Role of lateral hypothalamic orexin neurons in reward processing and addiction. Neuropharmacology. 2009,56 Suppl 1:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Burdakov D How orexin signals bias action: Hypothalamic and accumbal circuits. Brain Res. 2018. [DOI] [PubMed] [Google Scholar]
- [51].Berridge KC, Ho CY, Richard JM, Difeliceantonio AG The tempted brain eats: Pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010,1350:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Berthoud H-R The caudal brainstem and the control of food intake and energy balance In: Stricker EM, Woods SC, eds. Handbook of Behavioral Neurobiology. New York: Plenum; 2004. p. 195–240. [Google Scholar]
- [53].Groenewegen HJ, Wright CI, Beijer AV, Voorn P Convergence and segregation of ventral striatal inputs and outputs. Ann. N. Y. Acad. Sci 1999,877:49–63. [DOI] [PubMed] [Google Scholar]
- [54].Nunes EJ, Randall PA, Podurgiel S, Correa M, Salamone JD Nucleus accumbens neurotransmission and effort-related choice behavior in food motivation: effects of drugs acting on dopamine, adenosine, and muscarinic acetylcholine receptors. Neurosci. Biobehav. Rev 2013,37:2015–25. [DOI] [PubMed] [Google Scholar]
- [55].Kanoski SE, Grill HJ Hippocampus Contributions to Food Intake Control: Mnemonic, Neuroanatomical, and Endocrine Mechanisms. Biol. Psychiatry 2017,81:748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hsu TM, Noble EE, Reiner DJ, Liu CM, Suarez AN, Konanur VR, et al. Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology. 2018,131:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Suarez AN, Hsu TM, Liu CM, Noble EE, Cortella AM, Nakamoto EM, et al. Gut vagal sensory signaling regulates hippocampus function through multi-order pathways. Nat Commun. 2018,9:2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hsu TM, Hahn JD, Konanur VR, Lam A, Kanoski SE Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacology. 2015,40:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ross A, Barnett N, Faulkner A, Hannapel R, Parent MB Sucrose ingestion induces glutamate AMPA receptor phosphorylation in dorsal hippocampal neurons: Increased sucrose experience prevents this effect. Behav. Brain Res 2019,359:792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hargrave SL, Jones S, Davidson TL The Outward Spiral: A vicious cycle model of obesity and cognitive dysfunction. Curr Opin Behav Sci. 2016,9:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Suarez AN, Liu CM, Cortella AM, Noble EE, Kanoski SE Ghrelin and Orexin Interact to Increase Meal Size Through a Descending Hippocampus to Hindbrain Signaling Pathway. Biol. Psychiatry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Davidson TL, Kanoski SE, Chan K, Clegg DJ, Benoit SC, Jarrard LE Hippocampal lesions impair retention of discriminative responding based on energy state cues. Behav. Neurosci 2010,124:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hannapel R, Ramesh J, Ross A, LaLumiere RT, Roseberry AG, Parent MB Postmeal Optogenetic Inhibition of Dorsal or Ventral Hippocampal Pyramidal Neurons Increases Future Intake. eNeuro. 2019,6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].O’Doherty JP, Deichmann R, Critchley HD, Dolan RJ Neural responses during anticipation of a primary taste reward. Neuron. 2002,33:815–26. [DOI] [PubMed] [Google Scholar]
- [65].Keiflin R, Reese RM, Woods CA, Janak PH The orbitofrontal cortex as part of a hierarchical neural system mediating choice between two good options. J. Neurosci 2013,33:15989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Watson KK, Platt ML Social signals in primate orbitofrontal cortex. Curr. Biol 2012,22:2268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jennings JH, Kim CK, Marshel JH, Raffiee M, Ye L, Quirin S, et al. Interacting neural ensembles in orbitofrontal cortex for social and feeding behaviour. Nature. 2019,565:645–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Y, Lin YC, Kuo TW, Knight ZA Sensory detection of food rapidly modulates arcuate feeding circuits. Cell. 2015,160:829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Alhadeff AL, Goldstein N, Park O, Klima ML, Vargas A, Betley JN Natural and Drug Rewards Engage Distinct Pathways that Converge on Coordinated Hypothalamic and Reward Circuits. Neuron. 2019,103:891–908 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Patkar PP, Hao Z, Mumphrey MB, Townsend RL, Berthoud HR, Shin AC Unlike calorie restriction, Roux-en-Y gastric bypass surgery does not increase hypothalamic AgRP and NPY in mice on a high-fat diet. Int J Obes (Lond). 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Swart I, Jahng JW, Overton JM, Houpt TA Hypothalamic NPY, AGRP, and POMC mRNA responses to leptin and refeeding in mice. Am J Physiol Regul Integr Comp Physiol. 2002,283:R1020–6. [DOI] [PubMed] [Google Scholar]
- [72].Garfield AS, Shah BP, Burgess CR, Li MM, Li C, Steger JS, et al. Dynamic GABAergic afferent modulation of AgRP neurons. Nat. Neurosci 2016,19:1628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Betley JN, Xu S, Cao ZFH, Gong R, Magnus CJ, Yu Y, et al. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature. 2015,521:180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014,507:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, et al. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Frontiers in neuroanatomy. 2015,9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, et al. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001,291:2608–13. [DOI] [PubMed] [Google Scholar]
- [77].Sclafani A, Springer D Dietary obesity in adult rats: similarities to hypothalamic and human obesity syndromes. Physiol. Behav 1976,17:461–71. [DOI] [PubMed] [Google Scholar]
- [78].Musselman LP, Kuhnlein RP Drosophila as a model to study obesity and metabolic disease. J. Exp. Biol 2018,221. [DOI] [PubMed] [Google Scholar]
- [79].Morfeld KA, Meehan CL, Hogan JN, Brown JL Assessment of Body Condition in African (Loxodonta africana) and Asian (Elephas maximus) Elephants in North American Zoos and Management Practices Associated with High Body Condition Scores. PLoS One. 2016,11:e0155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hu L, Huang X, You C, Li J, Hong K, Li P, et al. Prevalence of overweight, obesity, abdominal obesity and obesity-related risk factors in southern China. PLoS One. 2017,12:e0183934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Denis RG, Joly-Amado A, Webber E, Langlet F, Schaeffer M, Padilla SL, et al. Palatability Can Drive Feeding Independent of AgRP Neurons. Cell Metab. 2015,22:646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007,5:181–94. [DOI] [PubMed] [Google Scholar]
- [83].Koza RA, Nikonova L, Hogan J, Rim JS, Mendoza T, Faulk C, et al. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2006,2:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Johnstone AM Fasting - the ultimate diet? Obes Rev. 2007,8:211–22. [DOI] [PubMed] [Google Scholar]
- [85].Webb VL, Wadden TA Intensive Lifestyle Intervention for Obesity: Principles, Practices, and Results. Gastroenterology. 2017,152:1752–64. [DOI] [PubMed] [Google Scholar]
- [86].Nordmo M, Danielsen YS, Nordmo M The challenge of keeping it off, a descriptive systematic review of high-quality, follow-up studies of obesity treatments. Obes Rev 2020,21:e12949. [DOI] [PubMed] [Google Scholar]
- [87].Hall KD, Guo J Obesity Energetics: Body Weight Regulation and the Effects of Diet Composition. Gastroenterology. 2017,152:1718–27 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Myers MG Jr., Leibel RL, Seeley RJ, Schwartz MW Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010,21:643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pan WW, Myers MG Jr. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci. 2018,19:95–105. [DOI] [PubMed] [Google Scholar]
- [90].Hagan MM, Rushing PA, Schwartz MW, Yagaloff KA, Burn P, Woods SC, et al. Role of the CNS melanocortin system in the response to overfeeding. J. Neurosci 1999,19:2362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].White CL, Purpera MN, Ballard K, Morrison CD Decreased food intake following overfeeding involves leptin-dependent and leptin-independent mechanisms. Physiol. Behav 2010,100:408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Knight ZA, Hannan KS, Greenberg ML, Friedman JM Hyperleptinemia is required for the development of leptin resistance. PLoS One. 2010,5:e11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jais A, Bruning JC Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest 2017,127:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013,62:2629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang X, Ge A, Cheng M, Guo F, Zhao M, Zhou X, et al. Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp Diabetes Res. 2012,2012:847246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kocalis HE, Turney MK, Printz RL, Laryea GN, Muglia LJ, Davies SS, et al. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARdelta) in mice leads to increased susceptibility to diet-induced obesity. PLoS One. 2012,7:e42981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Guillemot-Legris O, Muccioli GG Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017,40:237–53. [DOI] [PubMed] [Google Scholar]
- [98].Kentish SJ, O’Donnell TA, Frisby CL, Li H, Wittert GA, Page AJ Altered gastric vagal mechanosensitivity in diet-induced obesity persists on return to normal chow and is accompanied by increased food intake. Int J Obes (Lond). 2014,38:636–42. [DOI] [PubMed] [Google Scholar]
- [99].Kentish SJ, Page AJ The role of gastrointestinal vagal afferent fibres in obesity. J Physiol. 2015,593:775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Yu Y, Park SJ, Beyak MJ Inducible nitric oxide synthase-derived nitric oxide reduces vagal satiety signalling in obese mice. J Physiol. 2019,597:1487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sample CH, Jones S, Hargrave SL, Jarrard LE, Davidson TL Western diet and the weakening of the interoceptive stimulus control of appetitive behavior. Behav. Brain Res 2016,312:219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Attuquayefio T, Stevenson RJ, Boakes RA, Oaten MJ, Yeomans MR, Mahmut M, et al. A high-fat high-sugar diet predicts poorer hippocampal-related memory and a reduced ability to suppress wanting under satiety. Journal of experimental psychology. Animal learning and cognition. 2016,42:415–28. [DOI] [PubMed] [Google Scholar]
- [103].Blum K, Bailey J, Gonzalez AM, Oscar-Berman M, Liu Y, Giordano J, et al. Neuro-Genetics of Reward Deficiency Syndrome (RDS) as the Root Cause of “Addiction Transfer”: A New Phenomenon Common after Bariatric Surgery. Journal of genetic syndromes & gene therapy. 2011,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Blum K, Thanos PK, Gold MS Dopamine and glucose, obesity, and reward deficiency syndrome. Frontiers in psychology. 2014,5:919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Mihrshahi S, Gow ML, Baur LA Contemporary approaches to the prevention and management of paediatric obesity: an Australian focus. Med. J. Aust 2018,209:267–74. [DOI] [PubMed] [Google Scholar]
- [106].Brown T, Moore TH, Hooper L, Gao Y, Zayegh A, Ijaz S, et al. Interventions for preventing obesity in children. The Cochrane database of systematic reviews. 2019,7:CD001871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Fernandez-Jimenez R, Al-Kazaz M, Jaslow R, Carvajal I, Fuster V Children Present a Window of Opportunity for Promoting Health: JACC Review Topic of the Week. J. Am. Coll. Cardiol 2018,72:3310–9. [DOI] [PubMed] [Google Scholar]
- [108].Strauss WJ, Nagaraja J, Landgraf AJ, Arteaga SS, Fawcett SB, Ritchie LD, et al. The longitudinal relationship between community programmes and policies to prevent childhood obesity and BMI in children: the Healthy Communities Study. Pediatr Obes. 2018,13 Suppl 1:82–92. [DOI] [PubMed] [Google Scholar]
- [109].Kral TVE, Moore RH, Chittams J, Jones E, O’Malley L, Fisher JO Identifying behavioral phenotypes for childhood obesity. Appetite. 2018,127:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wadden TA, Tsai AG, Tronieri JS A Protocol to Deliver Intensive Behavioral Therapy (IBT) for Obesity in Primary Care Settings: The MODEL-IBT Program. Obesity (Silver Spring). 2019,27:1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Winkler JK, Schultz JH, Woehning A, Piel D, Gartner L, Hildebrand M, et al. Effectiveness of a low-calorie weight loss program in moderately and severely obese patients. Obes Facts. 2013,6:469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Teixeira PJ, Carraca EV, Marques MM, Rutter H, Oppert JM, De Bourdeaudhuij I, et al. Successful behavior change in obesity interventions in adults: a systematic review of self-regulation mediators. BMC medicine. 2015,13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Samdal GB, Eide GE, Barth T, Williams G, Meland E Effective behaviour change techniques for physical activity and healthy eating in overweight and obese adults; systematic review and meta-regression analyses. The international journal of behavioral nutrition and physical activity. 2017,14:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tate DF, Lytle LA, Sherwood NE, Haire-Joshu D, Matheson D, Moore SM, et al. Deconstructing interventions: approaches to studying behavior change techniques across obesity interventions. Translational behavioral medicine. 2016,6:236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Demos KE, McCaffery JM, Thomas JG, Mailloux KA, Hare TA, Wing RR Identifying the mechanisms through which behavioral weight-loss treatment improves food decision-making in obesity. Appetite. 2017,114:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Martin J, Chater A, Lorencatto F Effective behaviour change techniques in the prevention and management of childhood obesity. Int J Obes (Lond). 2013,37:1287–94. [DOI] [PubMed] [Google Scholar]
- [117].Burgermaster M, Contento I, Koch P, Mamykina L Behavior change is not one size fits all: psychosocial phenotypes of childhood obesity prevention intervention participants. Translational behavioral medicine. 2018,8:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Romain AJ, Attalin V, Sultan A, Boegner C, Gernigon C, Avignon A Experiential or behavioral processes: which one is prominent in physical activity? Examining the processes of change 1 year after an intervention of therapeutic education among adults with obesity. Patient Educ. Couns 2014,97:261–8. [DOI] [PubMed] [Google Scholar]
- [119].Harricharan M, Gemen R, Celemin LF, Fletcher D, de Looy AE, Wills J, et al. Integrating mobile technology with routine dietetic practice: the case of myPace for weight management. Proc. Nutr. Soc 2015,74:125–9. [DOI] [PubMed] [Google Scholar]
- [120].Martin CK, Gilmore LA, Apolzan JW, Myers CA, Thomas DM, Redman LM Smartloss: A Personalized Mobile Health Intervention for Weight Management and Health Promotion. JMIR mHealth and uHealth. 2016,4:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Gadde KM, Apolzan JW, Berthoud HR Pharmacotherapy for Patients with Obesity. Clin. Chem 2018,64:118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Sharples AJ, Mahawar K Systematic Review and Meta-Analysis of Randomised Controlled Trials Comparing Long-Term Outcomes of Roux-En-Y Gastric Bypass and Sleeve Gastrectomy. Obes. Surg 2019. [DOI] [PubMed] [Google Scholar]
- [123].Lam DD, Przydzial MJ, Ridley SH, Yeo GS, Rochford JJ, O’Rahilly S, et al. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology. 2008,149:1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].D’Agostino G, Lyons D, Cristiano C, Lettieri M, Olarte-Sanchez C, Burke LK, et al. Nucleus of the Solitary Tract Serotonin 5-HT2C Receptors Modulate Food Intake. Cell Metab. 2018,28:619–30 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Valencia-Torres L, Olarte-Sanchez CM, Lyons DJ, Georgescu T, Greenwald-Yarnell M, Myers MG Jr., et al. Activation of Ventral Tegmental Area 5-HT2C Receptors Reduces Incentive Motivation. Neuropsychopharmacology. 2017,42:1511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Xu P, He Y, Cao X, Valencia-Torres L, Yan X, Saito K, et al. Activation of Serotonin 2C Receptors in Dopamine Neurons Inhibits Binge-like Eating in Mice. Biol. Psychiatry 2017,81:737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Capehorn MS, Catarig AM, Furberg JK, Janez A, Price HC, Tadayon S, et al. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1–3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. 2019:101117. [DOI] [PubMed] [Google Scholar]
- [128].Chao AM, Wadden TA, Walsh OA, Gruber KA, Alamuddin N, Berkowitz RI, et al. Effects of Liraglutide and Behavioral Weight Loss on Food Cravings, Eating Behaviors, and Eating Disorder Psychopathology. Obesity (Silver Spring). 2019,27:2005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Blundell J, Finlayson G, Axelsen M, Flint A, Gibbons C, Kvist T, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. 2017,19:1242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Terrill SJ, Holt MK, Maske CB, Abrams N, Reimann F, Trapp S, et al. Endogenous GLP-1 in lateral septum promotes satiety and suppresses motivation for food in mice. Physiol. Behav 2019,206:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Williams DL Neural integration of satiation and food reward: role of GLP-1 and orexin pathways. Physiol. Behav 2014,136:194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ Exaggerated glucagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis. 2007,3:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Peterli R, Steinert RE, Woelnerhanssen B, Peters T, Christoffel-Courtin C, Gass M, et al. Metabolic and hormonal changes after laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy: a randomized, prospective trial. Obes. Surg 2012,22:740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ye J, Hao Z, Mumphrey MB, Townsend RL, Patterson LM, Stylopoulos N, et al. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am J Physiol Regul Integr Comp Physiol. 2014,306:R352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wilson-Perez HE, Chambers AP, Ryan KK, Li B, Sandoval DA, Stoffers D, et al. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency. Diabetes. 2013,62:2380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hatoum IJ, Stylopoulos N, Vanhoose AM, Boyd KL, Yin DP, Ellacott KL, et al. Melanocortin-4 receptor signaling is required for weight loss after gastric bypass surgery. J. Clin. Endocrinol. Metab 2012,97:E1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]