

Summary
Changes in the actin cytoskeleton are a primary mechanism mediating the morphological and functional plasticity that underlies learning and memory. The synaptic RhoGEFs Kalirin and Trio have emerged as central regulators of actin dynamics at the synapse. The increased attention surrounding Kalirin and Trio stems from the growing evidence for their roles in the etiology of a wide range of neurodevelopmental and neurodegenerative disorders. In this review, we discuss recent findings revealing the unique and diverse functions of these paralog proteins in neurodevelopment, excitatory synaptic transmission, and plasticity. We additionally survey the growing literature implicating these proteins in various neurological disorders.
Keywords: Kalirin, Trio, RhoGEF, Synaptic Plasticity, Neurotransmission, Autism, Schizophrenia, Neurodegeneration, Actin
The Dbl RhoGEFs Kalirin and Trio
GTPases are enzymes that bind GTP, hydrolyzing it to GDP. Different conformational states act to limit GTPase activity [1]. These “molecular switches” function as signal transducers, regulating numerous cellular processes [2]. The Ras superfamily of small GTPases comprises more than 150 members functioning as monomeric G proteins [2]. The Ras homologous (Rho) family of small GTPases are a major branch of the Ras superfamily. Rho GTPases are regulators of actin dynamics, with Ras-related C3 botulinum toxin substrate 1 (Rac1), Ras homolog family member A (RhoA), and cell division control protein 42 homolog (Cdc42) being the most well-known [3, 4]. Structural changes in the actin cytoskeleton associated with synaptic plasticity involve Rho family GTPases through their regulation of actin polymerization, resulting in changes in spine morphology [5, 6].
GTPases are, in part, regulated by guanine nucleotide exchange factors (GEFs) that facilitate the exchange of GDP for GTP, opposing the action of GTPase-activating proteins (GAPs) [7]. RhoGEFs were discovered in the context of diffuse B-cell lymphoma (Dbl) [8]. A distinct feature of the Dbl family of RhoGEFs is the presence of a Dbl homology (DH) domain, which is recurrently followed by a regulatory C-terminal pleckstrin homology (PH) domain, together comprising the enzymatic GEF domain [9]. GEF domains are conserved across Dbl family members; however, the non-GEF sequences flanking the DH-PH domains are more divergent and act as additional regulators of intrinsic GEF function, both structurally and through facilitating protein-protein and protein-lipid interactions. This sequence diversity is a critical means by which various Dbl RhoGEF proteins display unique functions, through different subcellular targeting and sequestration, resulting in differing spatial-temporal activation of GTPases.
The paralog Dbl RhoGEFs Kalirin and Trio, the only RhoGEFs with tandem GEF domains, have been broadly investigated and have emerged as significant regulators of synaptic development, plasticity, and neurotransmission. Recent work has reinforced and expanded on these roles and has further positioned Kalirin and Trio as critical molecules in neurodevelopmental and neurodegenerative disorders. This review covers the recent advancements in understanding the relationship between Kalirin and Trio and highlights the current developments in their functions in synaptic physiology and, importantly, in disease.
Evolutionary Conservation and Proteomic Diversity
Kalirin and Trio are distinctive in that they are paralogous proteins evolving from a mutual ancestral gene via duplication. Orthologs are observed in Drosophila (dTrio) and in C. elegans (unc-73) (Figure 1). Kalirin and Trio are similarly divergent to both dTrio and to unc-73 [10]. Recent domain and phylogenetic analyses have shown that the duplication event occurred in Urbilateria, with the last common ancestor existing in Prebilateria [11]. The predicted core domain structure of the Kalirin/Trio-related protein in Prebilateria is thought to contain 2 spectrin repeats and dual GEF domains. The working model posits the single Trio/Kalrn ancestor acquired a kinase domain, resulting in the ancestral Kalrn, which was subsequently duplicated in Urbilateria to yield distinct Kalrn and Trio genes. Interestingly, the phylogenetic analysis found retention and loss events were unpredictable in invertebrates, with only a few invertebrate taxa having retained both proteins [11]. In vertebrates, Kalirin and Trio were found to contain multiple protein variants when the duplicate was present, suggesting an adaptability for novel protein function [11, 12]. This is consistent with understandings of gene duplication retention, in which gene copies are retained when they serve complementary functions (so-called subfunctionalization) [13]. Indeed, many Kalirin and Trio isoforms have been identified and the proteomic diversity of Kalirin and Trio mediates their specific functions, both in developmental and tissue-specific manners (Figure 1, Box 1). For instance, C-terminal variations in Kalirin affect its trafficking and regulate its function at excitatory synapses via the inclusion of a PDZ-ligand, which is not present in any Trio isoform [14, 15]. The emergence of a variety of Kalirin and Trio isoforms may indicate a degree of functional separation, which may account for the evolutionary retention of complementary Kalirin and Trio isoforms that mediated functions once served by the ancestral Kalirin/Trio.
Figure 1. Major Trio and Kalirin isoforms.
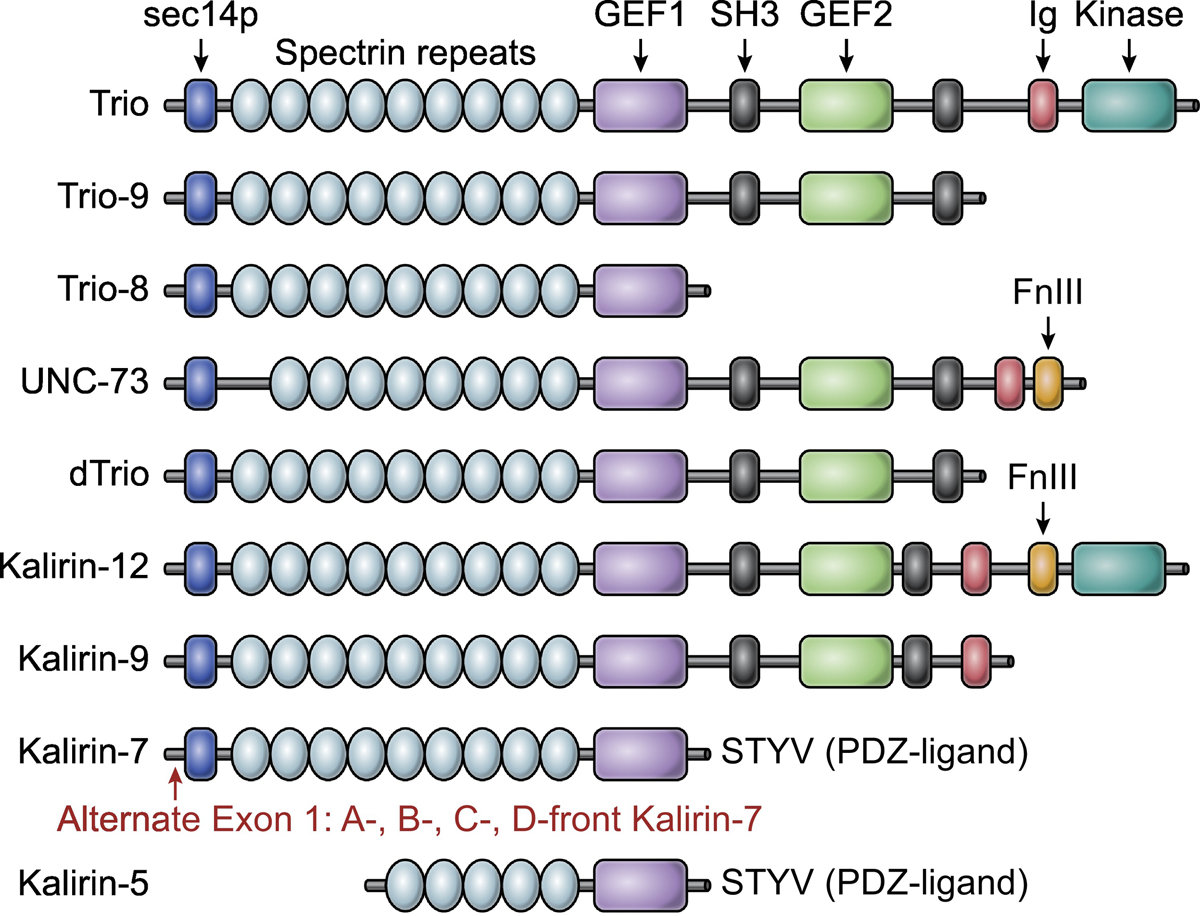
Alternative splicing of Kalirin and Trio genes gives rise to numerous isoforms. This alignment displays the dominant Trio and Kalirin species, along with the Drosophila (dTrio, row 5) and C. elegans orthologs (UNC-73, row 4). Kalirin and Trio contain two guanine nucleotide exchange factors (GEFs) that are composed of a catalytic Dbl homology domain, and a pleckstrin homology domain that functions allosterically. GEF1 activates Rac1 and RhoG, whereas GEF2 activates RhoA. Kalirin and Trio GEFs are well conserved, with the greatest sequence diversity observed in their non-GEF flanking sequences. Kalirin-7 and Trio-9 are the dominant species in the brain. Kalirin-7 is unique in that it harbors a C-terminal PDZ-ligand. Alternative start sites in exon 1 of Kalrn generate Kalirin-7 with various N-terminal sequences (so called a, b, c, and d-front Kalirin-7). For further structural highlights, see Box 1. Abbreviations: SH3, Src homology 3; FnIII, Fibronectin type III; Ig, Immunoglobulin; GEF, Guanine nucleotide exchange factor; PDZ, Postsynaptic density-95/discs large/zona occludens.
Box 1. Multiple Promoters and Alternative Splicing: Trio and Kalirin Proteomic Complexity.
The Kalrn and Trio genes each give rise to numerous isoforms, due to pronounced alternative splicing and promoter usage (Figure 1) [14, 20, 21, 114]. Both proteins, in their full-length form, are defined by two GEF domains (GEF1 and GEF2) and a C-terminal serine/threonine kinase domain, a unique feature of these Dbl family members. Kalirin and Trio each have an N-terminal Sec14p domain trailed by numerous repeating spectrin-like repeats, with two Src-homology 3 (SH3) domains following the GEF sequences, and a single immunoglobulin-like (Ig) domain. Kalirin is defined further by the addition of a fibronectin-like (FN3) domain leading the C-terminal kinase domain (Figure 1).
C-terminal sequence variations are the most common variants among brain-specific Trio isoforms. Alternative splicing of Trio exon 48 results in two distinct Trio isoforms, Trio-9S and Trio-9L, which are considered the dominant brain-specific species [20, 21]. Trio-9S and Trio-9L most resemble full-length UNC-73 and dTrio in that they do not contain the C-terminal kinase domain. Moreover, a cerebellum-specific isoform is also observed after postnatal day 30 (P30) (in mice), termed Trio-8, which is specific to Purkinje neurons [20]. Trio-8 is unique in that it contains only the first Rac1/RhoG GEF domain.
Like Trio, Kalirin is expressed ubiquitously and can be found in cardiac and skeletal muscle, as well as in the liver and in endocrine tissue [115]. Alterative 3’ splicing gives rise to the major Kalirin isoforms, Kalirin-12, Kalirin-9, and Kalirin-7. Kalirin-12 retains both GEF1 and GEF2 domains, as well as the kinase domain. Kalirin-9, however, lacks the kinase domain and is most similar to Trio-9 [14]. Kalirin-7 is the brain specific isoform of Kalirin, as well as the dominant species in the adult [116]. Kalirin-7 is most similar to Trio-8 in that it contains only the Rac1/RhoG GEF. It is, however, unique in that it contains a putative PDZ (postsynaptic density protein, Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein) interacting motif at its C-terminal end (Figure 1) [116]. Moreover, recent work has identified additional alternative promotor usage that gives rise to further Kalirin-7 species with differing N-terminal sequences; with bKalirin-7 and cKalirin-7 being most prevalent, differing in only 25 amino acids (Figure 1) [114]. These N-terminal differences are thought to mediate unique phosphoinositide-dependent interactions that control Kalirin-7 protein localization and function [114]. An additional Kalirin-7 variant, termed ΔKalirin-7 or −5, lacks the N-terminal Sec14 domain and first four spectrin repeats, but retains the PDZ-interacting motif [14].
Kalirin and Trio are both present throughout development and into adulthood, with differing isoform-specific expressions and subcellular localizations. A distinct expression difference is that Trio, but not Kalrn, is expressed in migrating neural crest cells in Xenopus laevis, suggesting Trio isoforms may be more important in organization of neural tissue relative to Kalirin [11, 16]. This is consistent with their respective knockout phenotypes in mice [17]. In the CNS of vertebrates, larger Kalirin isoforms are expressed abundantly early in development and localize to growth cones, whereas the brain-specific isoform Kalirin-7 localizes to the postsynaptic density (PSD), with expression paralleling that of synaptogenesis [18]. Kalirin-7 is expressed abundantly in the adult cortex and hippocampus, and persists as the dominant brain-specific isoform in the adult. However, there is an age-dependent increase in larger Kalirin isoform expression in orbitofrontal cortex [19]. Trio, likewise, displays dynamic expression patterns in embryonic and adult tissues, though the available isoform-specific data are less specific. Trio expression is apparent by embryonic day 10 in the neocortex and cerebellum, with the exception of Trio-8, which is limited to the cerebellar Purkinje neurons at P30 onward [20, 21]. While Trio is known to localize to growth cones, the presynaptic terminal, and the PSD, the exact temporal and isoform-specific expression patterns are not clearly delineated.
Neurite Outgrowth and Axon Guidance
Actin cytoskeleton regulation by Rho GTPases is central for cell migration and polarization, with Trio and Kalirin participating as key players in these processes. In particular, studies have established the importance of Trio in neurite growth and axon guidance. Trio has been shown to function in neurite formation downstream of the neurotrophin nerve growth factor (NGF) via RhoG activation in PC12 cells [22]. These data are challenging to interpret, as extensions in PC12 cells are not neurites precisely. However, the NGF downstream target Kidins220 has been shown to interact with Trio and activate it, resulting in Rac1 activation and neurite extension both in PC12 cells and in hippocampal neurons, supporting the role of NGF in Trio-mediated neurite extension [23]. dTrio is also known to interact with the Netrin receptor Frazzled as part of a signaling network regulating CNS midline axon guidance in Drosophila [24]. Indeed, Netrin-1 mediated Rac1 activation in the mouse is dependent on Trio for normal axon guidance, as revealed by the reduction in Netrin-1-induced Rac1 activation and axon guidance in the absence of Trio [25, 26]. This mechanism also involves the interaction with heat shock cognate protein 70 (Hsc70) and the phosphorylation of Trio at Y2622 by the Src kinase Fyn [27, 28].
The bulk of developmental studies on Trio have focused on Rac1/RhoG regulation; indeed, Trio’s GEF1 domain has been shown to be sufficient for neurite outgrowth [29]. Several studies have shown Trio’s GEF2 domain to be natively inhibited, and its activation results in the restriction of neurite extension and dendritic branching [29–32]. In this regard, Trio’s dual GEF domains provide dynamic regulation of neurite outgrowth and dendritic branching, with GEF1 mediating neurite extension and GEF2 destabilizing this process via RhoA activation. Recent studies of Trio’s activation of RhoA have suggested it is additionally involved in axon guidance. Slit homolog 2 protein (Slit2), a secreted glycoprotein involved in axon guidance and neuronal migration, induces RhoA activation through Trio, which is important for telencephalic wiring [33]. Slit2 is a ligand for the Roundabout (Robo/SAX-3) receptors, which are regulated by unc-73, possibly linking Slit2’s regulation of Trio RhoA GTPases via this receptor [34]. It is evident that both Trio GEF domains are necessary for regulated neurite outgrowth and guidance; however, the role of additional Trio domains in this process has remained largely unknown. Interestingly, it has been demonstrated that Golgi-resident Trio regulates membrane trafficking in developing cerebellar granule cells via interactions of its N-terminal spectrin repeats with RABIN8, resulting in RAB8 and RAB10 activation [35]. Expression of active RAB8 and RAB10 is able to restore neurite outgrowth deficits induced by loss of Trio. RAB8 and RAB10 activation is critical for vesicle trafficking to the growth cone, suggesting that Trio activation of these proteins through its N-terminal domains is an additional factor important for neurite outgrowth. These new data, along with previous work, together establish both Trio GEF domains as regulators of axon development, while highlighting emerging roles of additional Trio domains in this process [36].
Kalirin is also involved in neurite and dendritic outgrowth and in dendritic arborization. Cortical neurons in Kalirin KO mice display simplified dendritic arborization and smaller neurite length [37]. Like Trio, distinct roles in this process are mediated by different Kalirin isoforms and functional domains [36]. Knocking out Kalirin in sympathetic neuronal cultures stunts preexisting axonal extension, whereas overexpression of Kalirin results in the prolific sprouting of new axonal fibers [38]. Interestingly, expression of the GEF1 domain alone increases growth cone size, whereas overexpression of the GEF2 domain results in over elongation of axons and increased neurite formation [15]. It has also been shown that Kalirin’s kinase domain, which is unique to Kalirin-12, may mediate neurite formation and length [39]. Kalirin’s Rac1 GEF1 domain was also found to be important for dendrite elaboration [39]. These data demonstrate the salience and isoform specific contributions of Kalirin in dendritic maturation and development. Nevertheless, upstream signals mediating Kalirin GEF activation in neurite outgrowth and guidance are not as well established as Trio’s; however, work has shown that brain-derived neurotrophic factor (BDNF) activation of Rac1 in neurite outgrowth is mediated by Kalirin (Figure 2, Key Figure) [40].
Figure 2, Key Figure. Kalirin and Trio serve postsynaptic and presynaptic functions.
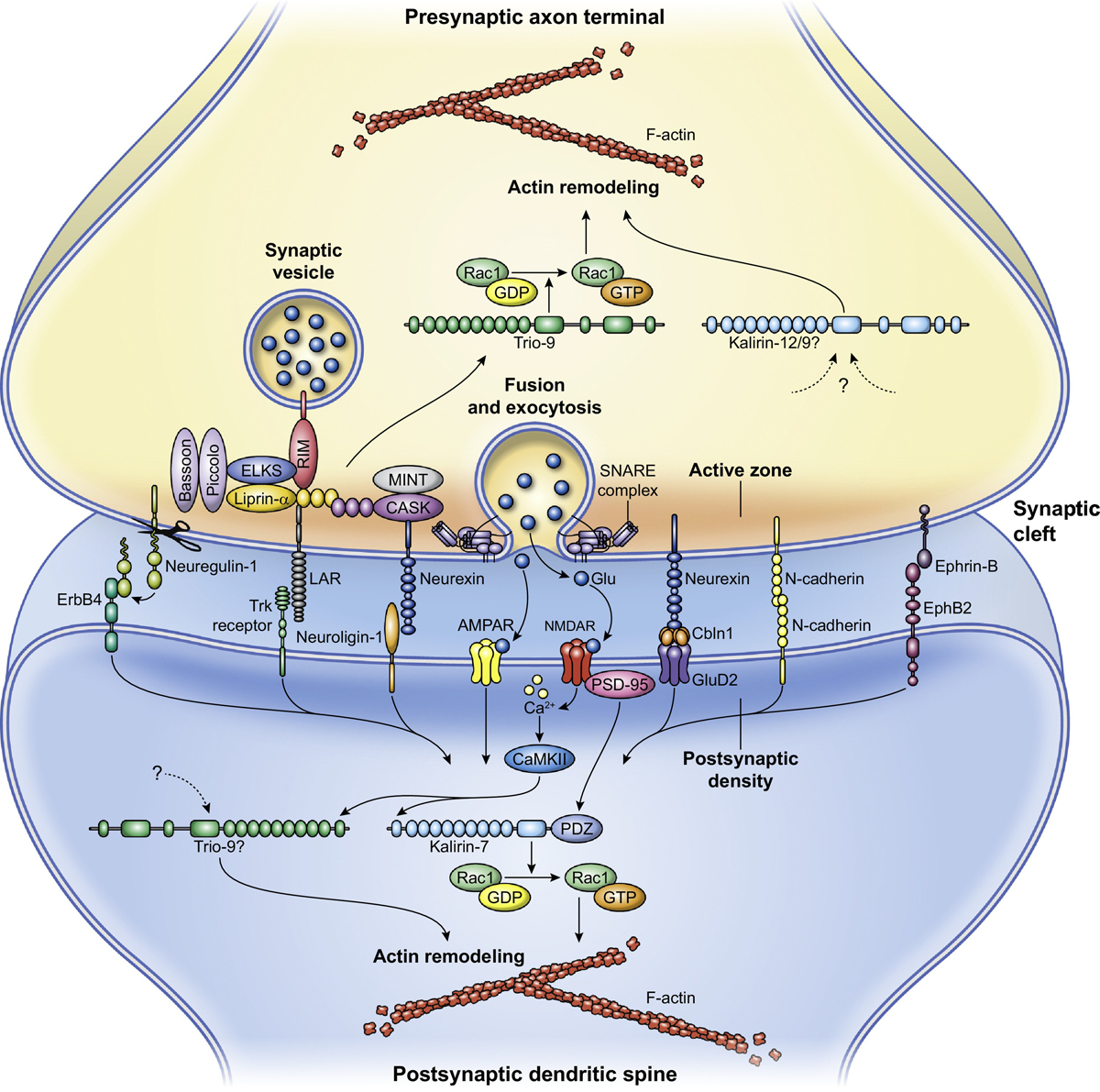
Kalirin and Trio interact with multiple proteins at the pre- and post-synaptic sites. These interactions integrate upstream signals into dynamic changes in the actin cytoskeleton that work to regulate spine formation, dendritic morphology, and presynaptic release. Kalirin-7 interacts with several neurotransmitter receptors and adhesion molecules, translating glutamate-mediated signals and synaptic adhesion into changes in the actin cytoskeleton. Presynaptically, Trio interacts with many proteins involved in vesicle release and trafficking, regulating the actin dynamics facilitating these processes. Upstream signaling regulating Trio at postsynaptic spines and Kalirin at the presynaptic site is less understood. Abbreviations: Glu, Glutamate; Cbln1, Cerebellin-1; NMDAR, N-methyl-D-aspartate receptor; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; LAR, LCA-related protein tyrosine phosphatase; PDZ, Postsynaptic density-95/discs large/zona occludens; CaMKII, Ca2+/calmodulin-dependent protein kinase II; EphB2, EPH receptor B2; CASK, calcium/calmodulin dependent serine protein kinase; RIM, Rab interacting molecule; ELKS, ERC/RAB6-interacting/CAST; MINT, Munc18 interacting protein; Trk, Tropomyosin receptor kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; GTP, Guanosine triphosphate; GDP, Guanosine diphosphate.
Kalirin and Trio in Synaptic Physiology
Kalirin is a central signaling hub in excitatory synaptic transmission and plasticity; however, the role of Trio in these processes is less understood [18]. Trio interacts with the presynaptic LAR/Liprin complex and clusters with the scaffolding proteins Piccolo and Bassoon, likely through regulating synaptic membrane exocytosis protein 1 (RIM1) (Figure 2) [41, 42]. Trio is targeted to presynaptic terminals through association with Piccolo and Bassoon via spectrin repeats 3 and 4 [43]. Trio may influence the regulated assembly of actin in presynaptic terminals, perhaps through interactions with Piccolo and Bassoon (Figure 2). Indeed, Trio knockout and haploinsufficiency have been shown to increase paired-pulse ratios, indicating deficits in presynaptic release probability [44]. These findings are consistent with prior data showing Rac1 mediated synaptic release at the neuromuscular junction is blocked with mutants in dTrio [45]. These data support a crucial role for Trio in presynaptic neurotransmitter release that is distinct from Kalirin. This idea is supported by data showing Trio to be a regulator of exocytosis in other cell types, such as pituitary endocrine cells [46]. Moreover, recent proteomic mapping of the Trio and Kalirin-7 interactomes has shown Trio, but not Kalirin-7, interacts with many presynaptic proteins known to regulate vesicle release and active zone dynamics [47].
Both Kalirin and Trio are found at the PSD and have roles in postsynaptic plasticity (Figure 2) [47–49]. Knocking down Kalirin and Trio expression in neurons has a dramatic effect on postsynaptic structure and function [50, 51]. For example, knocking down Kalirin or Trio individually in postsynaptic CA1 pyramidal neurons produce similar reductions in AMPAR-eEPSC amplitude following Schaffer collateral stimulation [50]. However, knocking down both Kalirin and Trio simultaneously in CA1 pyramidal neurons resulted in a near complete loss of glutamatergic synapses. Recombinant expression of either Kalirin or Trio alone was able to restore glutamatergic synapse function in these neurons [50]. These findings suggest a post-development redundancy in postsynaptic function between these two RhoGEFs in synaptic transmission. The influence Kalirin and Trio have on postsynaptic glutamatergic synapse function is also bidirectional, given that biolistic overexpression of Kalirin-7 or Trio-9 in CA1 pyramidal neurons in organotypic hippocampal slice cultures produces nearly identical increases in AMPAR-eEPSC, but not NMDAR-eEPSC amplitude [50].
Kalirin-7 has been shown to interact with many proteins of the PSD, including the NMDAR subunit GluN2B and PSD-95 (Figure 2) [16, 49]. Prior studies in the Kalirin and Kalirin-7 specific knockout mice demonstrated impaired, but not eliminated, hippocampal LTP induction [16, 50, 52]. Interestingly, molecular replacement of Kalirin-7 with an N-terminal point mutation (T95A) of a purported CaMKII phosphorylation site is sufficient to block LTP induction- and overexpression-mediated increases in AMPAR-eEPSCs [50, 53]. This LTP block is also observed when making the homologous mutation in Trio (T66A), suggesting that the N-terminal domain of these proteins, like other GEFs, is sensitive and critical for protein function.
Phosphorylation of Kalirin and Trio
Kalirin and Trio, as large and complex multidomain proteins, are subject to extensive posttranslational modifications that regulate their functions. Despite having C-terminal kinase domains, Trio and Kalirin have not been shown (to our knowledge) to autophosphorylate [54]. Several studies have investigated the role of phosphorylation, with CaMKII being reported to phosphorylate Kalirin-7 at several sites. One of these residues, T95, is important for Kalirin-7 function by regulating GEF activity, linking NMDAR activation to that of small GTPases through CaMKII [53]. However, recent phosphoproteomic analysis of Kalirin-7 has proposed T79 and S83 as alternative N-terminal CaMKII phosphorylation sites [55, 56]. Nevertheless, mutations of T95, and the analogous residue on Trio, have been shown to modulate plasticity and GEF activity, demonstrating this residue to be critical to the synaptic function of Kalirin [50, 55]. How this N-terminal residue modulates distal GEF1 actively is unclear, though it likely involves dynamic changes in protein folding that increase GEF1 activity through an allosteric process or perhaps facilitates greater Rac1 access to this catalytic domain.
Kalirin phosphorylation is additionally isoform- and brain region-specific [56]. For instance, tyrosine phosphorylation of Kalirin-7 modulates sensitivity to calpain, influencing its subsequent cleavage [56]. Kalirin-7 is also a substrate for CdK5, which targets Kalirin-7 at T1590. Cdk5 phosphorylation increases GEF activity and its regulation is involved in Kalirin-7 mediated spine formation and function [57, 58]. Phosphorylation of Trio in the context of synaptic function is less understood, though it has been shown that tyrosine phosphorylation via Fyn is an essential event in Trio activation of Rac1 in Netrin-1 mediated axon guidance [27].
Synaptic Adhesion Molecules
Kalirin-7 is a downstream effector for many postsynaptic adhesion molecules, such as N-cadherin and ephrinB-EphB (Figure 2) [59, 60]. These studies demonstrate Kalirin-7 function is required for N-cadherin and ephrin-induced spine development, morphology, and plasticity, providing mechanisms linking cellular adhesion to dynamic changes in the actin cytoskeleton. Trio is similarly thought to be involved in integrating cellular adhesion signals. Various cadherin-mediated processes have been shown to involve Trio as a downstream regulator of Rac1/RhoA actin dynamics in several model systems. For instance, Trio forms a complex with Cadherin-11 that is important for cranial neural crest cell migration and may control the mature organization of neuronal clusters in the hindbrain [61–63]. Additionally, Trio is an integral part of the N-cadherin adhesion complex in endothelial cells, though the function of this complex is unexplored in neurons [12]. Recently, Kalirin-7 was discovered to be an interactor of Neuroligin-1, and functions as a downstream regulator of Neuroligin-1-mediated synaptic enhancement in an interaction dependent manner (Figure 2) [47]. This is consistent with a recent study showing Neuroligin-1 promotes actin assembly associated with spine enlargement and plasticity [64]. Interestingly, it was shown that Neuroligin-1 interacts with the N-terminal domains of Kalirin-7 that are lacking in Kalirin-5 [47]. Analogous to other RhoGEFs, the non-enzymatic domains of Kalirin-7 regulate its subcellular localization and influence on synaptic structure and function. The GEF domains of Kalirin-7 and Kalirin-5 are equally active and both retain the PDZ-ligand that targets these proteins to the synapse, suggesting additional divergent functions of the N-terminal domains of Kalirin-7 in controlling dendritic morphology and synaptic plasticity [49]. These may be distinct from those of Trio at the PSD, as it was shown from proteomics that Trio does not interact with Neuroligin-1 as robustly, which highlights a particular divergence from Kalirin-7 [47]. The phenotypic overlap between Kalirin-7 and Neuroligin-1 suggests reciprocity in function, with Kalirin-7’s ability to control cell morphology being dependent on Neuroligin-1. This is an exciting area for further study.
Additional adhesion molecules have been suggested to interact with Kalirin-7. Proteomic analysis of pan-Kalirin interacting partners shows Kalirin clusters synaptic adhesion molecules [65]. Kalirin-7-specific proteomic analysis has identified additional adhesion molecules, including the ionotropic glutamate receptor delta 2 (GluD2) as a specific interactor [47]. GluD2 is unique in that it is predominantly expressed in Purkinje neurons of the cerebellum [66]. GluD2 functions similarly to neuroligins, acting as an adhesion molecule and synaptic organizer interacting trans-synaptically with neurexin via the intermediary secreted protein cerebellin [67]. Further functional analysis of this interaction is needed. Given the diversity of adhesion molecules functioning through Kalirin-7, we propose Kalirin-7 as a fundamental downstream regulator of upstream synaptic adhesion (Figure 2). More work is needed to delineate further the role of other adhesion proteins in this process and the role of Trio in this pathway.
Kalirin and Trio RhoGEFs in Disease
Aberrant activity of RhoGEFs is implicated across human disease, mediating changes in the density and morphology of spines observed in neuropsychiatric, neurodegenerative, and neurodevelopmental disorders [68]. Pathological signaling involving Kalirin and Trio is associated with various disorders, specifically autism spectrum disorder (ASD), intellectual disability (ID), Alzheimer’s disease (AD), Huntington’s disease (HD), and schizophrenia [51, 69–71]. Most if not all of these disorders are characterized by disruptions in synaptic structure and function, with many sharing similar genetic etiologies, highlighting conservation in the underlying pathobiology and aberrant signaling. Therefore, it is not surprising that Trio and Kalirin dysfunction is associated with overlapping and distinct pathologies, reflective of their discrete and overlapping functions, expression patterns, and protein-protein interactions [11, 47, 50].
Autism spectrum disorder and intellectual disability
Convergent evidence has accumulated that abnormalities in the development and function of glutamatergic synapses are involved in many cognitive disorders, including ASD and ID [72, 73]. It is thought most forms of severe ID, and many cases of ASD, are genetic in origin [74, 75]. Many ASD risk factors identified in genetic screens are indeed key regulators of synaptic plasticity, and included genes that code for scaffolding molecules, glutamate receptors, and cell adhesion molecules [76]. Furthermore, dysregulation of synaptic actin organization, including dysregulation of Rac1, has been implicated in ASD-related phenotypes [75, 77, 78]. The role of Trio in regulating actin dynamics in synapse development and plasticity demonstrate it is particularly sensitive to disruptions associated with ASD/ID. Indeed, many de novo mutations and rare variants in TRIO have been identified in patients (Figure 3). Recent whole exome sequencing studies in ASD and public database mining have increased the number of disease-related TRIO mutations identified, and have confirmed that de novo mutations in TRIO are a significant risk factor for ASD/ID [71, 79–81]. Recent findings from various groups reveal that a large number of these ASD and ID-related de novo mutations are strikingly clustered in the GEF1 domain of Trio, with an even stronger association with ASD/ID for the Rac1 interacting DH1 subdomain (Figure 3) [71, 82–85]. This cluster of ASD-related mutations in Trio’s GEF domain represents one of a very small number of ASD/ID mutation “hotspots” that have been identified to date. A majority of these disease-related TRIO mutations are hypomorphic, reducing Trio’s ability to activate Rac1, potentially reflecting a phenotype similar to Trio haploinsufficiency [44]. When expressed in neurons, Trio harboring these mutations were found to decrease postsynaptic AMPA receptor function [71]. However, other mutations have been identified that are hypermorphic [71]. One ASD/ID-related mutation identified in the GEF1 domain of Trio increases Rac1 activation, with neuronal expression resulting in dramatic increases in postsynaptic AMPA and NMDA receptor function [71]. This increase in glutamatergic neurotransmission was caused by this hypermorphic mutation, making Trio abnormally synaptogenic [71]. It is now apparent that mutations in either the GEF1 or GEF2 domains dysregulate GTPase activation, and the location of mutation and the resulting functional phenotype contribute to the ASD/ID diversity associated with Trio dysfunction. Indeed, recent work has suggested that opposite modulation of Rac1 via mutations in TRIO is associated with a distinct and domain-specific ASD/ID phenotype [85]. In particular, specific Trio-mediated Rac1 activation phenotypes correlate with head size and degree of ID, with the majority of mutations in GEF1 associated with microcephaly and a second cluster of mutations in the spectrin repeats associated with macrocephaly (Figure 3) [85]. Additionally, a recent report of a disease-related mutation associated with bipolar disorder, located in the GEF2 domain that was found to increase RhoA activation, demonstrating domain specific phenotypes across many disorders [84].
Figure 3: Disease-related mutations in Trio and Kalirin.
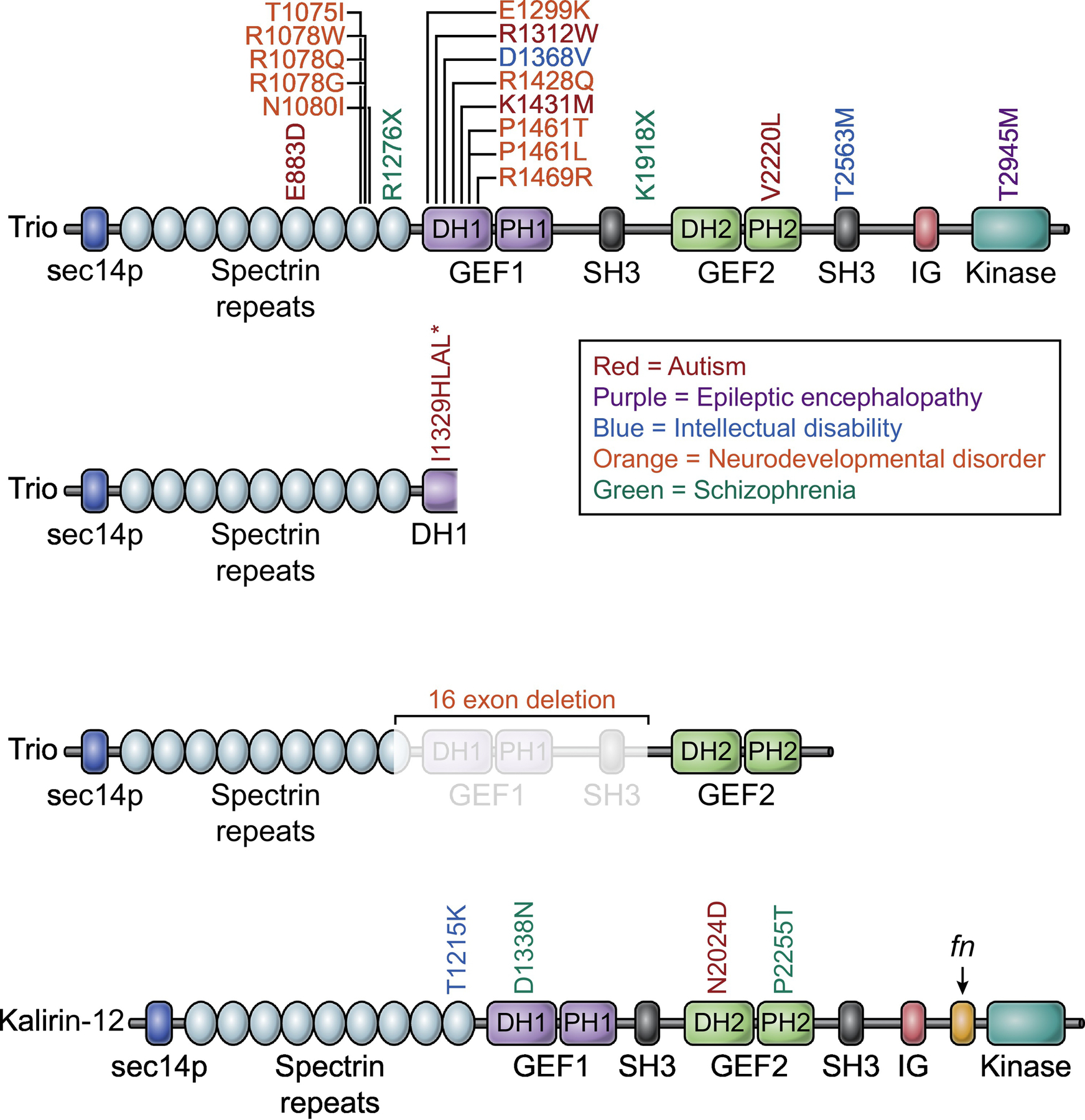
Mutations in Trio are associated with several neurological disorders. A majority of ASD/ID mutations occur in the first GEF domain, with an additional hotspot located in the second to last spectrin repeat. The top Trio row depicts missense mutations, the second and third rows depict nonsense mutations and copy number variants. The Asterix indicates a premature stop codon. Fewer disease-associated mutations have been identified in Kalirin. Abbreviations: DH, Dbl homology; PH, Pleckstrin homology; SH3, Src homology 3; FnIII, Fibronectin type III; IG, Immunoglobulin; GEF, Guanine nucleotide exchange factor; PDZ, Postsynaptic density-95/discs large/zona occludens.
Given Trio’s role in axon guidance and glutamatergic synapse regulation, we propose a model in which dysregulated Trio function results in a combination of disturbed synapse formation, developmental miswiring, and post-development aberrant glutamatergic transmission. Despite a number of previous large-scale human exome sequencing studies showing a genome-wide association of TRIO with ASD/ID, TRIO was not among the top 102 ASD-associated genes identified in a recent study [76]. In this study, of the ~17,500 genes analyzed, TRIO ranked 116th, putting it just below the gene identification threshold. It is important to note that TRIO’s association with ASD/ID is likely underestimated by human exome sequencing studies given that significance calculations are limited to entire genes rather than specific gene regions that encode functional protein domains. As discussed, ASD-related de novo mutations are largely clustered in Trio’s GEF1/DH1 domain (Figure 3) [71]. The exomic DNA encoding Trio’s DH1 domain exhibits a much higher degree of constraint relative to other coding regions of TRIO, and thus a higher level of genome-wide significance is observed when this region of the TRIO gene is considered independently [71].
Kalirin is not associated with ASD or ID as strongly as Trio, which is perhaps surprising given their shared genetic ancestry and parallel functions. Only a few mutations identified in Kalirin have been thought to cause ID. One such mutation, found in the GEF1 domain of Kalirin, was predicted to be pathogenic in a pair of siblings with ID and musculoskeletal abnormalities [86]. Moreover, Kalirin has come out as an ASD-associated gene in a study from individuals from the Faroe Islands [87]. Interestingly, the male carrying the de novo Kalirin mutation (N2024D) in the RhoA GEF2 domain did not have ID (Figure 3). It might be that Kalirin ASD mutations are underrepresented due to a milder phenotype, resulting in patients not seeking clinical assistance inclusive of genetic testing. It may also be that the absence of Kalirin expression in neural crest cells does not render it as sensitive to disruptions relative to Trio. Another intriguing hypothesis, however, is that Trio, but not Kalirin, may be part of a larger ASD/ID interaction network. Indeed, a protein interaction network analysis has identified converging molecular pathways among heterogenous ASD/ID [88]. Proteomic mapping of Trio and Kalirin-7 interactomes has revealed Trio, but not Kalirin-7, to be an interacting partner of many ASD/ID associated genes, such as DPYSL2 [47, 76].
Schizophrenia
The genetic architecture of schizophrenia is complex, involving both common and rare copy variants and single nucleotide mutations. Kalirin transcript levels and protein levels have been shown to be reduced in the dorsolateral prefrontal cortex of schizophrenia patients, with a loss of Kalirin protein correlating with spine loss [89, 90]. Interestingly, other brain regions in schizophrenia patients appear to have a dysregulation in Kalirin-9, which was found to be upregulated, but not in Kalirin-7 [91]. A genome-wide association study in a Japanese cohort found an association signal in the region of KALRN that did not reach genome-wide significance [92]. An additional resequencing and association study found that rare missense mutations in KALRN may be genetic risk factors for schizophrenia, adding further evidence that Kalirin may be involved in the etiology of schizophrenia [93]. In addition, a recent study found SZ patients in the South African Xhosa were more likely to have mutations in proteins involved in neurotransmission, including mutations in KALRN [94]. How schizophrenia-related variants directly relate to disease phenotypes is not entirely understood. However, characterization of several KALRN sequence variants have shown that single amino acid changes in KALRN result in impaired transcript level, protein function, neuronal morphology and cortical thickness, recapitulating many of the observations in Kalirin-mediated SZ [70, 95].
Interestingly, the largest whole exome sequencing study of schizophrenia parent-proband trios to date has identified TRIO as one of the strongest genes associated with schizophrenia, with a number of de novo loss of function mutations observed [96]. While no single gene reached genome wide significance in this study, TRIO is one of a small number of genes that has now reached genome wide significance following a recent meta-analysis of exome sequencing data taken from 24,248 schizophrenia cases, 97,322 controls, and 3,444 parent-proband trios (www.schema.broadinstitute.org). Moreover, recent assessments of transcriptomic organization have identified differential splicing changes in TRIO and KALRN in schizophrenia disease brains [97]. Kalirin-7 and Trio are known to interact with several proteins that have been implicated in SZ, including DISC1 (disrupted in schizophrenia) [98]. DISC1 is thought to function as a scaffold for Kalirin-7, thereby directly regulating activation of Rac1 [99]. Trio also has been shown to interact with DISC1 via its spectrin repeats and is also thought to facilitate GEF1 activation of Rac1 [100]. However, advances in sequencing technology and larger data sets have questioned the genetic association of DISC1 with SZ [101, 102]. DISC1 fails to reach significance in larger association and de novo mutation studies [96, 103, 104]. Nevertheless, this does not preclude its involvement in schizophrenia through very rare and penetrant variants [101, 105]. Other known interactors or effectors of Kalirin and Trio are also thought to be schizophrenia risk genes, including CDC42 and PAK [106].
Neurodegeneration
It has been speculated that functional decrease or loss of Kalirin could precipitate spine collapse, leading to synapse loss and cognitive dysfunction in AD [106]. Indeed, postmortem analyses of AD patient brains have found reduced levels of Kalirin transcripts and protein, as well as decreased levels and activation of Kalirin targets, such as PAK, in the hippocampus and prefrontal cortex [107–109]. These findings are supported by studies demonstrating Kalirin-7 overexpression prevents dendritic spine loss induced by amyloid oligomers and work showing that the restorative effects of the transcription factor XBP1 on memory and plasticity in AD models is mediated through control of Kalirin-7 [110, 111]. Amyloid-β (Aβ) may directly lead to Kalirin-7 dysregulation or loss through interaction with EphB2, an important upstream regulator of Kalirin-7 (Figure 2) [59]. EphB2 levels are decreased in AD patient brains and Aβ has been shown to directly interact with EphB2 [112, 113]. Depletion of EphB2 is critical in Aβ-mediated neuronal dysfunction, and rescuing EphB2 levels improves cognition in AD models, perhaps through spine stabilization via restoration of Kalirin-7 function and PAK activation [113]. Spine loss in AD may also involve dysregulation of other GTPase-regulatory proteins, such as Trio. Recent proteomic analysis of the Tau-P301S mouse identified depletion of GTPase-regulatory proteins as a salient feature of actin dysregulation and spine collapse observed in tauopathies [51]. Both Kalirin and Trio were prominently dysregulated in this study, pointing to a conserved role of these dual RhoGEFs in the pathophysiology of tauopathies. It is not known, however, what upstream factors mediate Trio dysregulation, and whether they are distinct from those of Kalirin-7.
Concluding Remarks
Trio and Kalirin are critical synaptic RhoGEFs underlying many essential processes of synaptic plasticity and development. Renewed interest in their biology has arisen from their strong association with neurodevelopmental, neuropsychiatric, and neurodegenerative disorders. In particular, Trio has emerged as an important gene in SZ and ASD/ID, whereas both Kalirin and Trio have now both been associated with neurodegenerative disorders. It is becoming more evident that disruptions in the regulation of GTPase activity is a pathogenic mechanism in many neurological disorders. Indeed, mutations in GAPs, specifically SynGAP, are also strongly associated with ASD and ID [76]. The diversity of Kalirin and Trio disease associations is reflective of their varied roles in axon guidance, synapse structure, and synaptic plasticity, which are mediated through precise isoform-specific expression and function. Deeper characterization of their distinct and overlapping biology will assist in better understandings of their pathobiology. Many fundamental questions still remain unanswered, including what the isoform-specific functions of Trio are and how they relate to Kalirin’s (see Outstanding Questions). Additionally, most studies on the synaptic functions of Trio and Kalirin were performed in young animals (or in vitro models likely to be relevant to early developmental stages), and accordingly, the presynaptic and postsynaptic function of Trio in the adult remains relatively unexplored. This developing field will prove important in the evolving understanding of synapse biology, but also for the development of better therapeutic targets and interventions.
Outstanding Questions.
Though much work has been done exploring Kalirin as a PSD signaling hub, the role of Trio in this process remains largely unknown. What is the postsynaptic role of Trio in plasticity and excitatory neurotransmission?
How do Kalirin and Trio regulate presynaptic development, and do they function in presynaptic release?
How do mutations in various domains of Trio and Kalirin contribute to the pathophysiology of various neurodevelopmental disorders?
What is the mechanism by which Kalirin and Trio contribute to spine loss in neurodegenerative disorders?
How can Kalirin and Trio be targeted for drug development?
What are the functions of various Trio isoforms, such as Trio-8?
Highlights.
The synaptic RhoGEFs Kalirin and Trio are paralog proteins that have shared and distinct functions in neuronal development and in synaptic plasticity. Recent phylogenetic analyses have provided resolution surrounding the evolution of these related genes. The retention of both Trio and Kalrn in an organism is concomitant with an increase in Kalirin and Trio isoforms. This proteomic diversity and differential expression is an essential feature of their sub-functionalization, which mediates their distinct and conserved functions.
Kalirin and Trio are present at the pre- and post-synaptic sites, and exert their effects on glutamatergic neurotransmission through their ability to regulate the actin cytoskeleton at these synapses.
Kalirin-7 is a downstream effector of many postsynaptic adhesion molecules. Such findings implicate Kalirin and possibly Trio as major signaling hubs that regulate glutamatergic synapse development and function.
Human genetics studies have identified TRIO as a risk gene in both Autism Spectrum Disorder (ASD) and Schizophrenia. In animal models, ASD-related mutations in Trio have been found to produce marked alterations in glutamatergic synapse function.
Acknowledgements
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (J.D.P and K.W.R.), and the National Institute of Mental Health grant number MH103398 (B.E.H.), as well as the Simons and McKnight Foundations (B.E.H.).
Glossary
- Paralog
Separate gene created by a duplication event within the same genome.
- Phylogenetic analysis
Means of analyzing the evolutionary relationships between genes across species.
- Urbilateria
A hypothetical last common ancestor of all organisms with bilateral symmetry (bilaterian clade).
- Prebilateria
Non-bilaterally symmetric clade (i.e. sponges and placozoans)
- Subfunctionalization
Process in which each paralog retains a subset of its original ancestral function, resulting in the preservation of duplicated genes in the same genome.
- Postsynaptic density (PSD)
An electron-dense structure, 30–60 nm thick, situated subjacent to the postsynaptic membrane. The PSD is composed of neurotransmitter receptors, scaffolding proteins, signaling elements, and trans-synaptic adhesion molecules. Thousands of different proteins are known to be contained in the PSD, all of which coordinate together in the means of synaptic transmission, and the dynamic modulation of these PSD components is an essential feature of synaptic plasticity.
- Haploinsufficiency
Model in which the functional copy of a gene (wild-type allele) in combination with the variant allele is unable to produce a normal or standard phenotype.
- eEPSC
Evoked excitatory postsynaptic currents are postsynaptic currents induced through stimulation of presynaptic neurons.
- GluD2
Glutamate receptor, ionotropic, delta 2. This protein is an ionotropic glutamate receptor expressed primarily in the Purkinje neurons of the cerebellum. Unlike other glutamate receptors, GluD2 does not bind glutamate, and functions largely as an adhesion molecule. Mutations or deletions in the GluD2 result in cerebellar ataxia.
- De novo mutations
A genetic mutation that is not inherited and is present for the first time in a certain progeny. These mutations are present in the offspring, but not in the parents.
- Rare variants
A genetic variant which occurs at very low (<1%) frequency in the population. While infrequent in the normal populations, these variants can be involved in the etiology of many disorders.
- Hypomorphic
A mutation that results in a loss of gene function.
- Hypermorphic
A mutation that results in a gain of gene function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer Statement
The authors declare no competing interests.
References
- 1.Schmidt A and Hall A, Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev, 2002. 16(13): p. 1587–609. [DOI] [PubMed] [Google Scholar]
- 2.Wennerberg K, Rossman KL, and Der CJ, The Ras superfamily at a glance. J Cell Sci, 2005. 118(Pt 5): p. 843–6. [DOI] [PubMed] [Google Scholar]
- 3.Etienne-Manneville S and Hall A, Rho GTPases in cell biology. Nature, 2002. 420(6916): p. 629–35. [DOI] [PubMed] [Google Scholar]
- 4.Nowak JM, et al. , [The Rho protein family and its role in the cellular cytoskeleton]. Postepy Hig Med Dosw (Online), 2008. 62: p. 110–7. [PubMed] [Google Scholar]
- 5.Ridley AJ, Rho family proteins: coordinating cell responses. Trends Cell Biol, 2001. 11(12): p. 471–7. [DOI] [PubMed] [Google Scholar]
- 6.Ba W and Nadif Kasri N, RhoGTPases at the synapse: An embarrassment of choice. Small GTPases, 2017. 8(2): p. 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossman KL, Der CJ, and Sondek J, GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol, 2005. 6(2): p. 167–80. [DOI] [PubMed] [Google Scholar]
- 8.Eva A and Aaronson SA, Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature, 1985. 316(6025): p. 273–5. [DOI] [PubMed] [Google Scholar]
- 9.Cook DR, Rossman KL, and Der CJ, Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene, 2014. 33(31): p. 4021–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt S and Debant A, Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases, 2014. 5: p. e29769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kratzer MC, et al. , Evolution of the Rho guanine nucleotide exchange factors Kalirin and Trio and their gene expression in Xenopus development. Gene Expr Patterns, 2019. 32: p. 18–27. [DOI] [PubMed] [Google Scholar]
- 12.Kruse K, et al. , N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J Cell Biol, 2019. 218(1): p. 299–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward R and Durrett R, Subfunctionalization: How often does it occur? How long does it take? Theor Popul Biol, 2004. 66(2): p. 93–100. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RC, et al. , Isoforms of kalirin, a neuronal Dbl family member, generated through use of different 5’- and 3’-ends along with an internal translational initiation site. J Biol Chem, 2000. 275(25): p. 19324–33. [DOI] [PubMed] [Google Scholar]
- 15.Penzes P, et al. , Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. J Neurosci, 2001. 21(21): p. 8426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma XM, et al. , Kalirin-7 is required for synaptic structure and function. J Neurosci, 2008. 28(47): p. 12368–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Brien SP, et al. , Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc Natl Acad Sci U S A, 2000. 97(22): p. 12074–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Penzes P and Jones KA, Dendritic spine dynamics--a key role for kalirin-7. Trends Neurosci, 2008. 31(8): p. 419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grubisha MJ, et al. , Age-dependent increase in Kalirin-9 and Kalirin-12 transcripts in human orbitofrontal cortex. Eur J Neurosci, 2016. 44(7): p. 2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McPherson CE, Eipper BA, and Mains RE, Multiple novel isoforms of Trio are expressed in the developing rat brain. Gene, 2005. 347(1): p. 125–35. [DOI] [PubMed] [Google Scholar]
- 21.Portales-Casamar E, et al. , Identification of novel neuronal isoforms of the Rho-GEF Trio. Biol Cell, 2006. 98(3): p. 183–93. [DOI] [PubMed] [Google Scholar]
- 22.Estrach S, et al. , The Human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr Biol, 2002. 12(4): p. 307–12. [DOI] [PubMed] [Google Scholar]
- 23.Neubrand VE, et al. , Kidins220/ARMS regulates Rac1-dependent neurite outgrowth by direct interaction with the RhoGEF Trio. J Cell Sci, 2010. 123(Pt 12): p. 2111–23. [DOI] [PubMed] [Google Scholar]
- 24.Forsthoefel DJ, et al. , The Abelson tyrosine kinase, the Trio GEF and Enabled interact with the Netrin receptor Frazzled in Drosophila. Development, 2005. 132(8): p. 1983–94. [DOI] [PubMed] [Google Scholar]
- 25.Briancon-Marjollet A, et al. , Trio mediates netrin-1-induced Rac1 activation in axon outgrowth and guidance. Mol Cell Biol, 2008. 28(7): p. 2314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng YJ, et al. , Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J Biol Chem, 2010. 285(32): p. 24834–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeGeer J, et al. , Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol Cell Biol, 2013. 33(4): p. 739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeGeer J, et al. , Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J Cell Biol, 2015. 210(5): p. 817–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellanger JM, et al. , Different regulation of the Trio Dbl-Homology domains by their associated PH domains. Biol Cell, 2003. 95(9): p. 625–34. [DOI] [PubMed] [Google Scholar]
- 30.Bandekar SJ, et al. , Structure of the C-terminal guanine nucleotide exchange factor module of Trio in an autoinhibited conformation reveals its oncogenic potential. Sci Signal, 2019. 12(569). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iyer SC, et al. , The RhoGEF trio functions in sculpting class specific dendrite morphogenesis in Drosophila sensory neurons. PLoS One, 2012. 7(3): p. e33634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awasaki T, et al. , The Drosophila trio plays an essential role in patterning of axons by regulating their directional extension. Neuron, 2000. 26(1): p. 119–31. [DOI] [PubMed] [Google Scholar]
- 33.Backer S, et al. , Trio GEF mediates RhoA activation downstream of Slit2 and coordinates telencephalic wiring. Development, 2018. 145(19). [DOI] [PubMed] [Google Scholar]
- 34.Watari-Goshima N, et al. , C. elegans VAB-8 and UNC-73 regulate the SAX-3 receptor to direct cell and growth-cone migrations. Nat Neurosci, 2007. 10(2): p. 169–76. [DOI] [PubMed] [Google Scholar]
- 35.Tao T, et al. , Golgi-resident TRIO regulates membrane trafficking during neurite outgrowth. J Biol Chem, 2019. 294(28): p. 10954–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellanger JM, et al. , The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene, 1998. 16(2): p. 147–52. [DOI] [PubMed] [Google Scholar]
- 37.Xie Z, Cahill ME, and Penzes P, Kalirin loss results in cortical morphological alterations. Mol Cell Neurosci, 2010. 43(1): p. 81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.May V, et al. , Kalirin Dbl-homology guanine nucleotide exchange factor 1 domain initiates new axon outgrowths via RhoG-mediated mechanisms. J Neurosci, 2002. 22(16): p. 6980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan Y, Eipper BA, and Mains RE, Kalirin-9 and Kalirin-12 Play Essential Roles in Dendritic Outgrowth and Branching. Cereb Cortex, 2015. 25(10): p. 3487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan Y, Eipper BA, and Mains RE, Kalirin is required for BDNF-TrkB stimulated neurite outgrowth and branching. Neuropharmacology, 2016. 107: p. 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stryker E and Johnson KG, LAR, liprin alpha and the regulation of active zone morphogenesis. J Cell Sci, 2007. 120(Pt 21): p. 3723–8. [DOI] [PubMed] [Google Scholar]
- 42.Astigarraga S, et al. , Three Drosophila liprins interact to control synapse formation. J Neurosci, 2010. 30(46): p. 15358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terry-Lorenzo RT, et al. , Trio, a Rho Family GEF, Interacts with the Presynaptic Active Zone Proteins Piccolo and Bassoon. PLoS One, 2016. 11(12): p. e0167535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katrancha SM, et al. , Trio Haploinsufficiency Causes Neurodevelopmental Disease-Associated Deficits. Cell Rep, 2019. 26(10): p. 2805–2817 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ball RW, et al. , Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron, 2010. 66(4): p. 536–49. [DOI] [PubMed] [Google Scholar]
- 46.Xin X, et al. , Cdk5 and Trio modulate endocrine cell exocytosis. J Cell Sci, 2004. 117(Pt 20): p. 4739–48. [DOI] [PubMed] [Google Scholar]
- 47.Paskus JD, et al. , Synaptic Kalirin-7 and Trio Interactomes Reveal a GEF Protein-Dependent Neuroligin-1 Mechanism of Action. Cell Rep, 2019. 29(10): p. 2944–2952 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bemben MA, et al. , CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat Neurosci, 2014. 17(1): p. 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penzes P, et al. , The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron, 2001. 29(1): p. 229–42. [DOI] [PubMed] [Google Scholar]
- 50.Herring BE and Nicoll RA, Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc Natl Acad Sci U S A, 2016. 113(8): p. 2264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dejanovic B, et al. , Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron, 2018. 100(6): p. 1322–1336 e7. [DOI] [PubMed] [Google Scholar]
- 52.Xie Z, et al. , Hippocampal phenotypes in kalirin-deficient mice. Mol Cell Neurosci, 2011. 46(1): p. 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie Z, et al. , Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron, 2007. 56(4): p. 640–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Debant A, et al. , The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci U S A, 1996. 93(11): p. 5466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiraly DD, et al. , Identification of kalirin-7 as a potential post-synaptic density signaling hub. J Proteome Res, 2011. 10(6): p. 2828–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller MB, et al. , Brain Region and Isoform-Specific Phosphorylation Alters Kalirin SH2 Domain Interaction Sites and Calpain Sensitivity. ACS Chem Neurosci, 2017. 8(7): p. 1554–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li MX, et al. , Role of Cdk5 in Kalirin7-Mediated Formation of Dendritic Spines. Neurochem Res, 2019. 44(5): p. 1243–1251. [DOI] [PubMed] [Google Scholar]
- 58.Xin X, et al. , Regulation of Kalirin by Cdk5. J Cell Sci, 2008. 121(Pt 15): p. 2601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Penzes P, et al. , Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron, 2003. 37(2): p. 263–74. [DOI] [PubMed] [Google Scholar]
- 60.Xie Z, et al. , Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. J Neurosci, 2008. 28(24): p. 6079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Charrasse S, et al. , M-cadherin activates Rac1 GTPase through the Rho-GEF trio during myoblast fusion. Mol Biol Cell, 2007. 18(5): p. 1734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Backer S, et al. , Trio controls the mature organization of neuronal clusters in the hindbrain. J Neurosci, 2007. 27(39): p. 10323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kashef J, et al. , Cadherin-11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of Trio and the small GTPases. Genes Dev, 2009. 23(12): p. 1393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu A, et al. , Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J Cell Biol, 2016. 212(4): p. 449–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkinson B, Li J, and Coba MP, Synaptic GAP and GEF Complexes Cluster Proteins Essential for GTP Signaling. Sci Rep, 2017. 7(1): p. 5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuzaki M and Aricescu AR, A GluD Coming-Of-Age Story. Trends Neurosci, 2017. 40(3): p. 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elegheert J, et al. , Structural basis for integration of GluD receptors within synaptic organizer complexes. Science, 2016. 353(6296): p. 295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forrest MP, Parnell E, and Penzes P, Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci, 2018. 19(4): p. 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Penzes P and Remmers C, Kalirin signaling: implications for synaptic pathology. Mol Neurobiol, 2012. 45(1): p. 109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Russell TA, et al. , A sequence variant in human KALRN impairs protein function and coincides with reduced cortical thickness. Nat Commun, 2014. 5: p. 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sadybekov A, et al. , An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat Commun, 2017. 8(1): p. 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Volk L, et al. , Glutamate synapses in human cognitive disorders. Annu Rev Neurosci, 2015. 38: p. 127–49. [DOI] [PubMed] [Google Scholar]
- 73.Bourgeron T, From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci, 2015. 16(9): p. 551–63. [DOI] [PubMed] [Google Scholar]
- 74.de Ligt J, et al. , Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med, 2012. 367(20): p. 1921–9. [DOI] [PubMed] [Google Scholar]
- 75.Vissers LE, et al. , A de novo paradigm for mental retardation. Nat Genet, 2010. 42(12): p. 1109–12. [DOI] [PubMed] [Google Scholar]
- 76.Satterstrom FK, et al. , Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell, 2020. 180(3): p. 568–584 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeidan-Chulia F, et al. , Exploring the multifactorial nature of autism through computational systems biology: calcium and the Rho GTPase RAC1 under the spotlight. Neuromolecular Med, 2013. 15(2): p. 364–83. [DOI] [PubMed] [Google Scholar]
- 78.Tian C, et al. , An Intellectual Disability-Related Missense Mutation in Rac1 Prevents LTP Induction. Front Mol Neurosci, 2018. 11: p. 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanders SJ, et al. , Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron, 2015. 87(6): p. 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takata A, et al. , Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep, 2018. 22(3): p. 734–747. [DOI] [PubMed] [Google Scholar]
- 81.Stessman HA, et al. , Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet, 2017. 49(4): p. 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pengelly RJ, et al. , Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet, 2016. 53(11): p. 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ba W, et al. , TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum Mol Genet, 2016. 25(5): p. 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katrancha SM, et al. , Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum Mol Genet, 2017. 26(23): p. 4728–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barbosa S, et al. , Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. Am J Hum Genet, 2020. 106(3): p. 338–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Makrythanasis P, et al. , Exome sequencing discloses KALRN homozygous variant as likely cause of intellectual disability and short stature in a consanguineous pedigree. Hum Genomics, 2016. 10(1): p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leblond CS, et al. , Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med, 2019. 4: p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sakai Y, et al. , Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med, 2011. 3(86): p. 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hill JJ, Hashimoto T, and Lewis DA, Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry, 2006. 11(6): p. 557–66. [DOI] [PubMed] [Google Scholar]
- 90.Rubio MD, Haroutunian V, and Meador-Woodruff JH, Abnormalities of the Duo/Ras-related C3 botulinum toxin substrate 1/p21-activated kinase 1 pathway drive myosin light chain phosphorylation in frontal cortex in schizophrenia. Biol Psychiatry, 2012. 71(10): p. 906–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deo AJ, et al. , Increased expression of Kalirin-9 in the auditory cortex of schizophrenia subjects: its role in dendritic pathology. Neurobiol Dis, 2012. 45(2): p. 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ikeda M, et al. , Genome-wide association study of schizophrenia in a Japanese population. Biol Psychiatry, 2011. 69(5): p. 472–8. [DOI] [PubMed] [Google Scholar]
- 93.Kushima I, et al. , Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr Bull, 2012. 38(3): p. 552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gulsuner S, et al. , Genetics of schizophrenia in the South African Xhosa. Science, 2020. 367(6477): p. 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Russell TA, et al. , A Schizophrenia-Linked KALRN Coding Variant Alters Neuron Morphology, Protein Function, and Transcript Stability. Biol Psychiatry, 2018. 83(6): p. 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Howrigan DP, et al. , Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat Neurosci, 2020. 23(2): p. 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gandal MJ, et al. , Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science, 2018. 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Millar JK, Christie S, and Porteous DJ, Yeast two-hybrid screens implicate DISC1 in brain development and function. Biochem Biophys Res Commun, 2003. 311(4): p. 1019–25. [DOI] [PubMed] [Google Scholar]
- 99.Hayashi-Takagi A, et al. , Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci, 2010. 13(3): p. 327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen SY, Huang PH, and Cheng HJ, Disrupted-in-Schizophrenia 1-mediated axon guidance involves TRIO-RAC-PAK small GTPase pathway signaling. Proc Natl Acad Sci U S A, 2011. 108(14): p. 5861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Avramopoulos D, Recent Advances in the Genetics of Schizophrenia. Mol Neuropsychiatry, 2018. 4(1): p. 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sullivan PF, Questions about DISC1 as a genetic risk factor for schizophrenia. Mol Psychiatry, 2013. 18(10): p. 1050–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schizophrenia Working Group of the Psychiatric Genomics, C., Biological insights from 108 schizophrenia-associated genetic loci. Nature, 2014. 511(7510): p. 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rees E, et al. , De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nat Neurosci, 2020. 23(2): p. 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sachs NA, et al. , A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol Psychiatry, 2005. 10(8): p. 758–64. [DOI] [PubMed] [Google Scholar]
- 106.Remmers C, Sweet RA, and Penzes P, Abnormal kalirin signaling in neuropsychiatric disorders. Brain Res Bull, 2014. 103: p. 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Youn H, et al. , Kalirin is under-expressed in Alzheimer’s disease hippocampus. J Alzheimers Dis, 2007. 11(3): p. 385–97. [DOI] [PubMed] [Google Scholar]
- 108.Murray PS, et al. , beta-Amyloid 42/40 ratio and kalirin expression in Alzheimer disease with psychosis. Neurobiol Aging, 2012. 33(12): p. 2807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao L, et al. , Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci, 2006. 9(2): p. 234–42. [DOI] [PubMed] [Google Scholar]
- 110.Xie Z, et al. , Kalirin-7 prevents dendritic spine dysgenesis induced by amyloid beta-derived oligomers. Eur J Neurosci, 2019. 49(9): p. 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cisse M, et al. , The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol Psychiatry, 2017. 22(11): p. 1562–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Simon AM, et al. , Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J Alzheimers Dis, 2009. 17(4): p. 773–86. [DOI] [PubMed] [Google Scholar]
- 113.Cisse M, et al. , Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature, 2011. 469(7328): p. 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miller MB, et al. , Alternate promoter usage generates two subpopulations of the neuronal RhoGEF Kalirin-7. J Neurochem, 2017. 140(6): p. 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miller MB, et al. , Neuronal Rho GEFs in synaptic physiology and behavior. Neuroscientist, 2013. 19(3): p. 255–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Penzes P, et al. , An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J Biol Chem, 2000. 275(9): p. 6395–403. [DOI] [PubMed] [Google Scholar]