

Abstract
Increased proteotoxic stress (IPTS) resulting from the increased production or decreased removal of abnormally folded proteins is recognized as an important pathogenic factor for a large group of highly disabling and life-threatening human diseases, such as neurodegenerative disorders and many heart diseases. The proteasome is pivotal to the timely removal of abnormal proteins but its functional capacity often becomes inadequate in the disease conditions; consequently, proteasome functional insufficiency in return exacerbates IPTS. Recent research in proteasome biology reveals that the proteasome can be activated by endogenous protein kinases, making it possible to pharmacologically prime the proteasome for treating diseases with IPTS.
Keywords: proteasome, proteotoxicity, desmin-related cardiomyopathy, phosphodiesterases, cAMP-dependent protein kinase, cGMP-dependent protein kinase
Protein Quality Control and Proteotoxicity
To function properly, a polypeptide must attain and maintain its native 3-dimentional conformation through a process known as folding. Unfortunately, protein misfolding is inevitable. As the resultant misfolded proteins are toxic to a cell, the cell has developed multi-layered molecular mechanisms known as protein quality control (PQC) to minimize the level and toxicity of misfolded proteins. The best-delineated PQC pathway is the endoplasmic reticulum (ER) associated PQC, which prevents misfolded ER proteins from going to the secretary pathway but, instead, retrotranslocates them back to the cytosol for immediate degradation.[1] However, most cellular proteins do not pass through the ER; hence, ER-independent PQC is also important. In general, PQC is performed by the intricate collaboration between molecular chaperons and targeted protein degradation; the latter is carried out primarily by the ubiquitin-proteasome system (UPS) although the autophagy also play a complementary role in removal of misfolded proteins [2].
Proteotoxicity refers to all adverse effects of misfolded proteins in the cell [3]. Misfolded proteins, if failing to be repaired, become terminally misfolded proteins which have two possible fates: the first and the least harmful fate is immediate removal by the UPS; otherwise, the misfolded proteins undergo aberrant aggregation which forms sequentially soluble oligomers and insoluble aggregates [2]. The soluble oligomers in particular are highly toxic to the cell, including impairing the 26S proteasome (see Glossary) and causing cell dysfunction and ultimately cell death [4–11]. Thus, proteotoxicity poses a greater threat to organs with poor ability to regenerate their primary cells (e.g., brain and heart) and to the cells that must stay functional and alive for an exceptionally long time, such as mammalian cardiomyocytes and neurons.
Both neurodegeneration and heart failure (HF) are leading causes for mortality and morbidity in mankind but treatments currently available for these conditions are far from satisfactory. Proteotoxicity is a well-known pathogenic factor for neurodegenerative disease [12]; however, it has been underappreciated that increased proteotoxic stress (IPTS) is also strongly implicated in the genesis of a large subset of human HF [13–19]. Experimental studies have established that PQC inadequacy or IPTS plays an important pathogenic role in the progression from a large subset of common forms of heart disease to HF [13, 20–27]. IPTS via aberrant protein aggregation impairs proteasomal function [7, 8], which in turn leads to accumulation of misfolded proteins, such that proteasome functional insufficiency (PFI) and IPTS form a vicious circle. Thus, breaking the vicious circle by priming the proteasome or making the proteasome ready to work more efficiently is a conceivable strategy to treat diseases with IPTS. To this end, tremendous progress has been made in the pharmacological exploitation of proteasome phosphoregulation in the past several years, making it very exciting to predict that proteasome-priming based new therapeutic strategies for neurodegenerative diseases and HF are in the horizon.
Proteasome Functional Insufficiency Is a Major Pathogenic Factor for Neurodegeneration and Heart Failure
When escaping from or overwhelming chaperones and the UPS, misfolded proteins undergo aberrant protein aggregation, giving rise to sequentially soluble oligomers and insoluble aggregates in the cell [28]. The oligomers bind and inhibit the proteasome with low nanomolar affinity through allosteric impairment of the substrate gate in the 20S core, preventing the 19S regulatory particle from injecting substrates into the degradation chamber [7]. PFI is a proteasome function status in a cell where the proteasome fails to meet the demand for a timely degradation of the proteins designated for proteasomal degradation. Both proteasome impairment and increased demand for proteasome-mediated proteolysis can lead to PFI. PFI and aberrant protein aggregation have been seen in many neurodegenerative diseases, including Huntington’s disease (HD) [29, 30], Alzheimer’s disease (AD) [31], Parkinson’s disease (PD) [32], and amyotrophic lateral sclerosis (ALS) [33]. Moreover, cancer patients receiving pharmacological inhibitors of the proteasome (e.g., bortezomib) frequently develop peripheral neuropathy [34]. Similarly, multiple lines of evidence support that PFI is also a major pathogenic factor for HF. First, myocardial ubiquitinated proteins are increased in human HF resulting from ischemic heart disease or dilated cardiomyopathy [35], as well as in animal models of nearly all heart disease [27], suggesting insufficient proteasomal removal of ubiquitinated proteins in diseased hearts [19]. Second, mutations in UPS genes are linked to human cardiomyopathy and HF [36–38]. Third, cardiac dysfunction occurs in up to 7% of human patients receiving proteasome inhibitors (carfilzomib, bortezomib) as part of their chemotherapies [39]. Lastly, genetic proteasome enhancement protects against not only desmin-related cardiomyopathy (DRC, see Box 1) but also ischemia/reperfusion injury, pressure overloaded HF, and diabetic cardiomyopathy [40–43]. Therefore, proteasome enhancement should be explored as a potentially new therapeutic strategy for a large subset of heart disease [28], besides neurodegenerative disease [12].
Box 1. DRC Mouse Models to study Cardiac Proteotoxicity.
Desmin-related cardiomyopathy (DRC) is the cardiac manifestation of desmin-related myopathy (DRM) which belongs to myofibrillar myopathies (MFMs) [90]. MFMs are a group of rare genetic neuromuscular disorders characterized by aberrant protein aggregates in muscle cells and disruption of myofibrils. The pathological hallmark of DRM is the presence of desmin-positive aberrant aggregates in affected muscle cells. Mutations in the desmin gene (DES), αB-crystallin gene (CRYAB, e.g., R120G missense mutation), and other genes encoding DES-related proteins are linked to human DRM. Most mutations cause misfolding of the affected proteins. CRYAB is a heat shock protein that is both constitutively expressed and stress-inducible, and is highly enriched in striated muscle, especially cardiac muscle. The R120G-missense mutation and other mutations of CRYAB are associated with human inherited myopathies and, in some cases, cataracts and hearing loss. CryABR120G is a bona fide misfolded protein and dominantly suppresses CRYAB chaperone functions [91, 92]. The transgenic mouse model of cardiomyocyte (CM)-restricted transgenic overexpression of CryABR120G (R120G mice) was one of the first animal models of MFMs [20, 21]. R120G mice develop restrictive cardiomyopathies recapitulating most aspects of clinical DRC: aberrant protein aggregates in CMs, CM death, cardiac hypertrophy, cardiomyopathy, HF, and shortened lifespan [20]. R120G mice have played a remarkable role in advancing the research into cardiac proteotoxicity [3, 20, 93]. It was one of the mammalian models employed for the first in vivo demonstration of UPS impairment by misfolded proteins [9, 10], the mammalian model to demonstrate the pathogenic role of PFI in vivo [41], and the animal model of proteotoxicity used to show in vivo that priming the proteasome can be therapeutically beneficial [50]. Human patients harboring the CryABR120G are rare but these mice not only serve a useful model for studying the genetic disease per se but are used for in vivo investigations into cardiac protein quality control and proteotoxicity in general. The frequent presence of pre-amyloid oligomers (PAOs) within the cardiomyocytes of explanted human hearts with end-stage HF resulting from hypertrophic or dilated cardiomyopathies indicates the involvement of IPTS in the genesis of HF of common etiology [13]. R120G mouse hearts display PAOs [13]. In DRC, aberrant protein aggregation is essential to proteasome impairment by misfolded proteins [8]. A major mechanism is that the oligomers bind and inhibit the proteasome with low nanomolar affinity through allosteric impairment of the substrate gate in the 20S core, preventing the 19S regulatory particle from injecting substrates into the degradation chamber [7]; alternatively, the inefficiently cleaved disease-associated proteins may cause the 26S proteasome to become clogged, leading to its impairment [94].
Proteasome Functionality Is Highly Regulated
Recent advances in UPS biology have unveiled that the functionality of the 26S proteasome is vigorously regulated and dictates the fate and the degradation efficiency of ubiquitinated proteins [44–47]. Phosphorylation is by far the most prevalent and best studied post-translational modification of the proteasome. It seems to occur to virtually all proteasome subunits; and mass spectrometry identified >450 phosphosites on human proteasomes [44]. However, biochemical analyses reveal less than 8 proteasome subunits show significant phosphorylation, and only two have so far been shown to have physiological consequences: RPT3 during the S-M phase of the cell cycle and RPN6 in response to cAMP and the cAMP-dependent kinase (PKA) [44, 48]. This is perhaps because most cellular proteins, especially abundant ones, undergo phosphorylation on many amino acid residues but the phosphorylation at these residues in the proteasome proteome is rare and of no obvious physiological significance [44]. To date, several kinases have been found to phosphorylate the proteasome [44]. In vitro assays show that proteasome phosphorylation by most kinases, including PKA and the cGMP-dependent kinase (PKG) [49, 50], increases proteasome activity (Figure 1) and thereby regulates various cellular processes [44].
Figure 1. The composition of the 26S proteasome and an illustration of a 26S proteasome nanodomain.
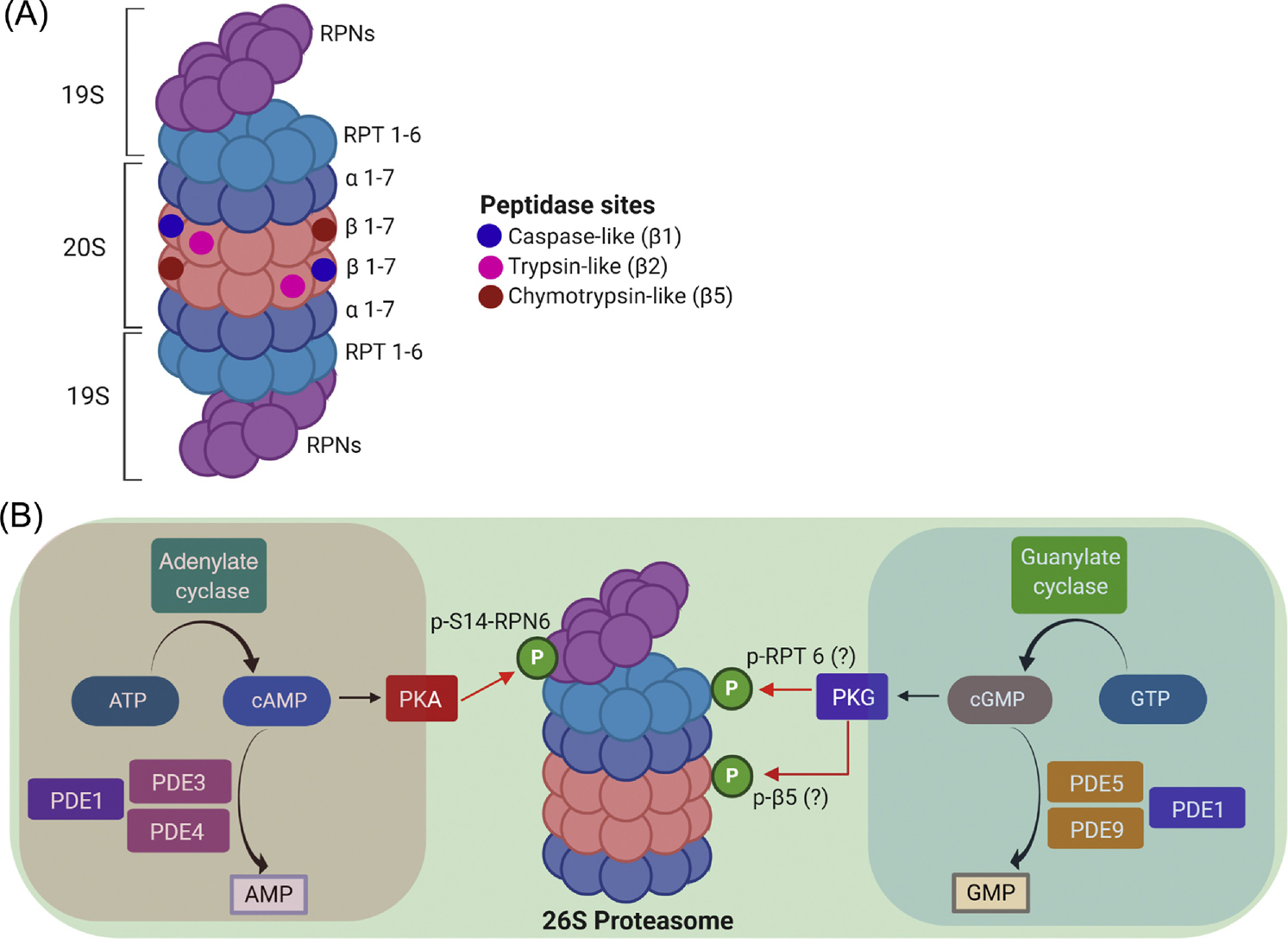
A, The 26S proteasome contains a cylinder-shaped 20S core particle capped by a 19S regulatory particle at one or both ends. The 20S is an axial stack of four rings: two outer α-rings and two inner β-rings. Each ring is formed by 7 protein subunits: α1 through α7 for the α ring and β1 through β7 for the β ring. The proteasomal peptidase activities reside in β1, β2, and β5 subunits while the α ring gates substrate access to the proteolytic chamber of the 20S. The 19S consists of a lid and a base subcomplex; the lid is formed primarily by the non-ATPase subunits RPNs and the base is composed of six ATPase subunits (RPT1 through RPT6). B, An illustration of a 26S proteasome nanodomain. Main regulators of the cAMP/PKA and the cGMP/PKG signaling modules are highlighted in the pink and the blue zone, respectively. Both cAMP/PKA and cGMP/PKG activate the 26S proteasome. PKA does so via the selective phosphorylation of the Ser14 of RPN6 (p-S14-RPN6), a 19S lid subunit strategically positioned to interact with both the base of the 19S and the α-ring of the 20S. PKG primes the proteasome perhaps through phosphorylating RPT6 and β5subunits.
PKA was the first kinase implicated in phosphorylating proteasomes [51]. PKA and PP2A can be co-purified with the proteasome from cardiomyocytes [52]. Phosphorylation at Ser120 of RPT6 was shown to be responsible for proteasome activation by PKA [53]; but this is convincingly refuted by a more recent study [49]. It turns out Ser14 of RPN6/PSMD11, rather than Ser120-RPT6, is selectively phosphorylated by PKA and Ser14-RPN6 phosphorylation (pS14-Rpn6) is fully responsible for the activation of 26S proteasomes by PKA in cells [49]. More recently, it was reported that pS14-Rpn6 is selectively increased when proteasomes are activated not only by pharmacologically raising cAMP in cells and mouse brain but also by hormones (in heart by epinephrine) and physiological states (skeletal muscles upon exercise and fasting) that raise cAMP [54]. In cultured cardiomyocytes, raising cAMP increases pS14-Rpn6 in a PKA-dependent manner and enhances UPS performance [55]. Several reports provided associative evidence that cardiac proteasome activation by PKA might play a role in cardiac (patho)physiology [52, 54, 56–58].
PKG
In cultured cardiomyocytes, both genetic and pharmacological stimulation of PKG increased proteasome peptidase activities, shortened the half-life of a surrogate UPS substrate (GFPu), facilitated the proteasomal degradation and reduced the cytotoxicity of a DRC-linked misfolded protein (CryABR120G), demonstrating for the first time that PKG activates the proteasome and thereby promotes proteasomal degradation of misfolded proteins in the cell [50]. Two-dimensional western blot analyses revealed that the isoelectric points of Rpt6 and Psmb5 in cultured cardiomyocytes were shifted to the acidic side by overexpression of a constitutive PKG, suggesting PKG may phosphorylate these proteasome subunits. This proposition remains to be tested. In a subsequent study, Ranek et al. further demonstrated that PKG mediates the proteasome activation by muscarinic receptor 2 in cardiomyocytes [59], unveiling a physiological relevance for proteasome activation by PKG.
The (patho)physiological significance of proteasome phosphoregulation by PKA, PKG or any other kinases in the heart or any organs remains undefined. However, this has not prevented researchers from exploiting proteasome phosphoregulation to target proteotoxicity.
Proteasome Enhancement Protects against Proteotoxicity
A pathogenic role of proteasome malfunction was proposed for neural degeneration much earlier than for cardiac proteotoxicity [10, 60]; however, the first definitive evidence that establishes the pathogenic role for PFI in any disease or organs came from the cardiac field [40, 41]. Li et al. reported that overexpression of PA28α stabilizes PA28β and thereby increases 11S-associated 20S proteasomes, increases proteasome proteolytic function in cultured cardiomyocytes [40]. Moreover, they have further demonstrated that mice with cardiomyocyte-restricted PA28α overexpression show enhanced myocardial UPS performance and resistance to cardiac proteotoxicity and ischemic/reperfusion injury [41]. This genetic approach to priming cardiac proteasomes was also adopted to establish the pathogenic role of PFI in HF induced by pressure overload and diabetic cardiomyopathy [42, 43]. More recently, this method has migrated into the neural science field. In a mouse retinal degenerative disease model, PA28α overexpression protected cells against retinal degeneration and delayed the disease onset by increasing proteasome activities [61]. This report also shows that increasing proteasome activity through overexpression of Psmd11 can achieve similar but less robust effects, compared to PA28a overexpression [61]. In an aging-related study using a fly model, overexpression of the proteasome β5 subunit enhanced proteasome function, slowed down age‐related declines in learning and memory, and increased longevity [70]. As most HF and neurodegeneration are age-dependent, these studies using genetic enhancement of the proteasome not only establish the pathogenic role of PFI but also provide compelling evidence that proteasome enhancement is benign and can effectively treat disease with IPTS.
Pharmacologically, proteasome priming by cGMP/PKG and cAMP/PKA has shown great promise (Table 1). The synthesis of cAMP and cGMP is catalyzed by adenylate cyclase (AC) and guanylate cyclase (GC), respectively, whereas their breakdown is catalyzed by phosphodiesterases (PDEs) which are classified into 11 families (PDE1 thru PDE11) [62]. Thus, activating AC and GC will increase intracellular levels of cAMP and cGMP, respectively; and inhibition of a PDE will increase cAMP, cGMP, or both, depending on which PDE is targeted (Figure 1). Ranek et al. reported the first study rationally using a pharmacological compound to prime the proteasome to treat a disease with IPTS [50], which shows that increasing myocardial cGMP by PDE5 inhibition (sildenafil) attenuates myocardial aberrant protein aggregation and slows down disease progression in a mouse model of DRC. Later, Myeku et al. reported that PDE4 inhibition (rolipram) promoted PKA-mediated proteasome activation and was efficacious in the early treatment of a mouse tauopathy model [63]. Similarly, administration of a potent adenylate cyclase activator (forskolin), another way to elevate cAMP, induced neuroprotection in the APP/PS1 AD mouse model [64]. Inhibition of PDE10 with Papaverine or PF-02545920 attenuated both motor/cognitive dysfunction and neuropathological abnormalities, besides improving cortico-basal ganglia function in an HD mouse model when treated at pre-symptomatic ages [65, 66]. Cilostazol, a FDA-approved PDE3 inhibitor that primarily increases cAMP, was also shown to activate proteasome and attenuate tauopathy and cognitive decline in an mouse AD model [67]. In a PD cell model, treatment with dipyridamole, a non-selective PDE1 inhibitor, increased cGMP level and rescued cells from α-synuclein induced toxicity [68]. More recently, PDE1 inhibition (IC86430) has been demonstrated to activate cardiac proteasomes, facilitate proteasomal degradation of misfolded proteins in cardiomyocytes in a PKA- and PKG-dependent fashion, and show striking therapeutic benefits in a mouse model of advanced cardiac proteinopathy [55]. Hence, evidence is rapidly emerging for pharmacologically stimulating PKA and PKG to treat both neural and cardiac diseases with IPTS.
Table 1.
Studies harnessing proteasome phosphoregulation to treat proteotoxicity
| PDEs inhibited | Drugs | Disease models | Effects | References |
|---|---|---|---|---|
| PDE1 | IC86430 | Tg mice of DRC | Increase p-S14-Rpn6 and proteasome activities, improve cardiac diastolic function, and delay mouse premature death | [55] |
| PDE1 | Dipyridamole | Cell model of PD | Reduce a-synuclein induced toxicity | [68] |
| PDE3 | Cilostazol | AD patients | Reduce risk to develop dementia | [70] |
| PDE4 | Rolipram | Tg mice of Tauopathy | Improve cognition with early-stage disease | [63] |
| PDE4 | Piclamilast | Tg mice of DRC | Increase p-S14-Rpn6 and proteasome activities | [55] |
| PDE5 | Sildenafil | Tg mice of DRC | Slow down disease progression | [50] |
| PDE5 | Sildenafil | AD patients | Improve vascular and metabolic function | [69] |
| PDE10 | Papaverine; PF-02545920 | Tg mice of HD | Improve cognition and correct basal ganglion circuitry deficits | [65] [66] |
AD, Alzheimer’s disease; DRC, desmin-related cardiomyopathy; HD, Huntington’s disease; PD, Parkinson’s disease; PDE, phosphodiesterase; p-S14-Rpn6, Ser14-phosphorylated Rpn6; Tg, transgenic
Taken together, these experimental findings strongly indicate that enhancing proteasome functionality with either genetic or pharmacological means can protect against IPTS in different neural and cardiac diseases. It is reasonable to predict that studies aimed at translating these exciting experimental findings to the clinic are on the horizon. This is also because FDA-approved drugs for augmentation of cAMP or cGMP are readily available in the clinic for treating other conditions and, excitingly, there are reported clinical observations that corroborate this new strategy (see Clinician’s Corner). For example, sildenafil, a highly selective PDE5 inhibitor that augments cGMP/PKG signaling was shown to benefit AD patients [69]. Use of Cilostazol, a PDE3 inhibitor that predominantly increases cAMP, was associated with a reduced risk of developing dementia [70].
Clinician’s Corner.
Proteotoxicity participates in the development and progression of a broad spectrum of common and yet highly disabling and life-threatening diseases, such as neurodegenerative disease, a large subset of heart failure (HF) and diabetes. However, no current clinical therapies for these diseases are targeting increased proteotoxic stress (IPTS) yet.
Through aberrant protein aggregation, IPTS impairs the proteasome whereas proteasome impairment further aggravates IPTS, forming a vicious circle in pathogenesis. Hence, enhancing the proteasome holds a key to breaking the vicious circle. Phosphorylation of 26S proteasomes by PKA or PKG increases proteasome activities.
FDA-approved drugs to stimulate PKA or PKG are readily available: Augmenting cGMP/PKG signaling by PDE5 inhibition (sildenafil) protects against cardiac IPTS. Similarly, cAMP/PKA activation via PDE4 inhibition (rolipram) improves cognition in a mouse model of AD. Clinically, PDE5 inhibition (sildenafil) improves cerebral hemodynamic function and increases cerebral oxygen metabolism in AD patients; and a reduced risk of developing dementia was observed in humans taking Cilostazol, a PDE3 inhibitor that predominantly increases cAMP.
On the other hand, long-term use of a PDE3 inhibitor (milrinone) increased the mortality of patients with chronic HF; and clinical trials on HF treatment with PDE5 inhibition (sildenafil) failed to show a positive outcome. It is unknown whether the results of these clinical trials are applicable or not to the exploitation of PKA- and PKG-mediated proteasome activation for IPTS treatment. Regardless, duo-activation of PKA and PKG would be a better strategy than solo-activation because it takes advantage of both their shared proteasome-priming property and their opposing actions on other targets. This proposition remains to be formally tested but it is strongly supported by the striking therapeutic benefits from PDE1 inhibition (IC86430) on a well-established mouse model of advanced cardiac proteinopathy. The outcome of the clinical trials on a PDE1 inhibitor to treat Parkinson’s disease (NCT03257046)I and HF (NCT03387215)II will shine light on this as well although they have a different rationale.
Dual Activation of PKA and PKG to Protect against Cardiac Proteotoxicity
Most human diseases with IPTS are chronic; thus, their pharmacological treatment will likely be long-term. Hence, we must consider minimizing the undesired effects of chronic kinase activation when exploiting kinase-based proteasome priming as a treatment strategy. Clinically, augmenting cAMP/PKA signaling by PDE3 inhibition (milrinone) effectively treats acute HF but its long-term use is hindered by causing fatal arrhythmia [71, 72]. Clinical trials on augmenting cGMP/PKG with PDE5 inhibition (sildenafil) showed only a neutral outcome in treating HF with preserved ejection fraction (HFpEF) [73, 74]. Understandably, these unfavorable outcomes of prior clinical trials cast doubt on the use of cAMP/PKA or cGMP/PKG augmentation in solo to treat heart disease with IPTS. However, both PKG and PKA prime the proteasome and they do so through apparently different mechanisms [49, 50, 75]; hence, duo-activation of PKA and PKG is expected to be additive, if not synergistic, in priming the proteasome. On the other hand, cAMP/PKA and cGMP/PKG have shown diverse actions non-proteasome targets, some of which oppose each other, serving to minimize each other’s undesired effects. For example, stimulating cAMP/PKA leads to cardiac hypertrophy and increases heart rate, which are undesirable because both elevated resting heart rate and cardiac hypertrophy are associated with poorer prognosis in patients with heart diseases [76, 77]. On the contrary, augmenting cGMP/PKG decreases heart rate and suppresses cardiac hypertrophy [78–81]. Hence, we propose that duo-activation of PKA and PKG should be more effective than solo-activation of either in treating HF with IPTS (Figure 2). In this rational strategy, PKA and PKG complementarily improve PQC while mutually alleviating some of their unfavorable effects on the heart. While it remains to be formerly tested, this proposition has garnered support from existing evidence. PDE1 hydrolyzes both cAMP and cGMP and confers the main PDE activities in human hearts [82]. Multiple studies have shown both genetic and pharmacological inhibition of PDE1 protect the heart [83–86]. Most excitingly, PDE1 inhibition enhances the proteasome in cardiomyocytes in a PKA- and PKG-dependent manner and, when administered at an overt disease stage, strikingly improves cardiac diastolic function and mouse survival in a mouse model of DRC [55].
Figure 2. A schema for proposed duo-activation of PKA and PKG to treat cardiac disease with IPTS.
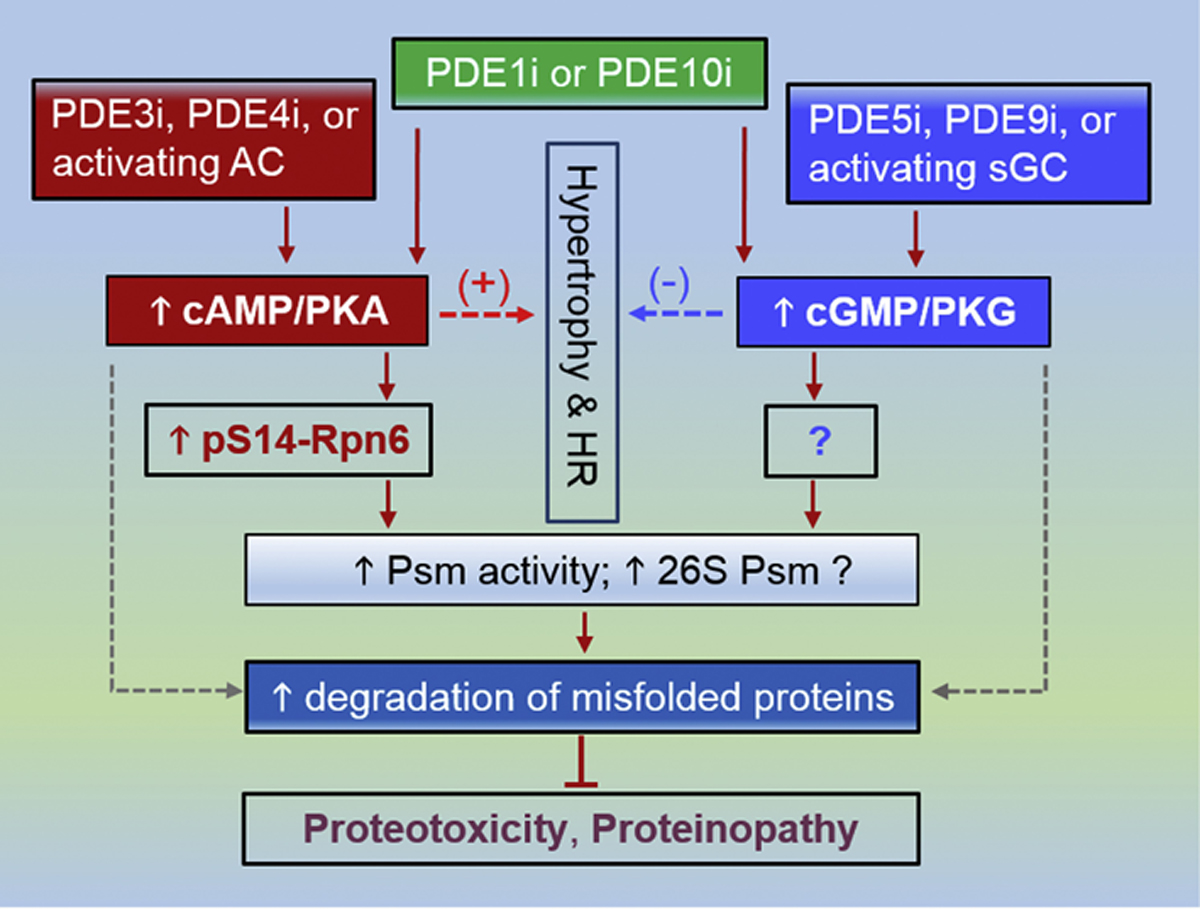
PDE3 inhibition (PDE3i), PDE4 inhibition (PDE4i) as well as activation of the adenylate cyclase (AC) (dark red) are expected to augment cAMP/PKA signaling, which leads to increased Ser14-phosphorylated RPN6/PSMD11 (p-S14-RPN6) and thereby increases proteasome (Psm) activities and perhaps 26S Psm assembly, which in turn facilitates the degradation of misfold proteins and protects proteotoxicity. In parallel, PDE5i, PDE10i or using an activator or stimulator of soluble guanylate cyclase (sGC) (blue) will augment cGMP/PKG signaling, which increases proteasome activities by phosphorylating Psm subunit(s) that remains to be identified and promotes the degradation of misfolded proteins. Stimulating cAMP/PKA promotes cardiac growth (hypertrophy) and increases heart rate (HR), which raises cardiac oxygen consumption and represents undesirable effects. Augmentation of cGMP/PKG signaling, on the other hand, suppresses hypertrophy and decreases HR, which in some cases may not be beneficial either. Hence, duo-activation of PKA and PKG is expected to cancel out some of their undesirable non-Psm effects while complementarily prime the Psm. The duo-activation may be achievable by PDE1 inhibition (PDE1i) or PDE10 inhibition (PDE10i) but PDE1 and PDE10i do not necessarily equally raise cAMP and cGMP in a given cell type; consequently, a proper combination of a method from the PKA activation side and one from the PKG activation side would likely better serve the purpose. The dot lines denote conceivable alternative pathways that currently have no clear support.
Concluding Remarks
Recent research advances in proteasome phosphoregulation and pharmacology have paved a new avenue to the search for effective measures to treat disease with IPTS, a large category of disorders that afflict millions of people but currently have essentially no evidence-based effective therapy. In particular, the discoveries of proteasome activation by PKA and PKG have prompted fruitful pharmacological studies on augmenting cAMP and cGMP to treat cardiac and neural diseases with IPTS although long-term proteasome activation by stimulating a kinase to treat chronic diseases with IPTS conceivably requires working out strategies to minimize the undesirable non-proteasome effects (see Outstanding Questions). It is important to recognize that all evidence from reported animal studies, which suggests a contribution of proteasome priming to the cardiac or neural protection, is not definitive but rather associative by nature. To fill this critical gap requires establishing animal genetic models in which proteasome activation by a specific kinase is selectively interrogated without directly altering its effects on other targets. To enable this, it is crucial to first identify the proteasome phosphosite that is responsible for the regulation by the kinase. This is particularly relevant to PKG as its proteasome activation has shown great therapeutic promise but its proteasome phosphosite(s) remains to be defined.
Outstanding Questions.
How does PKG activate proteasomes? Can any other kinases phosphorylate and regulate the proteasome? Can proteasomes be more effectively primed while undesired effects on non-proteasome targets are minimized by the combined activation of two or more kinases (e.g., duo-activation of PKA and PKG)?
Are the proteasome phosphosites identified so far for a kinase (e.g., Ser14-RPN6/PSMD11 for PKA, Ser120-RPT6 for CaMKII) fully or primarily responsible for proteasome activation by the kinase in vivo? How much of the protection against a neural or cardiac disease with IPTS by augmentation of cAMP/PKA or cGMP/PKG signaling comes from proteasome phosphorylation/activation?
What is the physiological and pathophysiological significance of proteasome phosphorylation by individual kinases? How is the proteome altered by the proteasome phosphoregulation by a specific kinase? What roles does the altered degradation of specific proteins induced by activation of a kinase play in the regulation of specific cellular processes or (patho)physiology by the kinase?
How is the kinase activity of PKA, PKG, or any other kinases regulated specifically at the microdomains or even the nanodomains of the 26S proteasome in a specific cell type (e.g., cardiomyocytes, neurons, adipocytes, etc.)? Can kinase activities be manipulated specifically at proteasome nanodomains but not other undesirable domains of a cell?
Are all proteasomes in different subcellular domains created equal? Are the activities of the proteasome at different subcellular domains regulated in the same way or by different mechanisms?
Can augmentation of cGMP/PKG signaling effectively protect against neural proteotoxicity? Can soluble guanylate cyclase activators or stimulators protect against cardiac or neural proteotoxicity? Is PDE1 inhibition a valid strategy to treat neurodegenerative diseases?
Can priming the proteasome with a pharmacological method be effective in treating neural and cardiac disease with IPTS in humans?
Another reason for optimism is that cAMP/PKA and cGMP/PKG signaling are highly compartmentalized in the cell [87–89]. Hence, an exciting future direction is to delineate the mechanism controlling the activity of PKA, PKG and perhaps other kinases within the proteasome nanodomains. This holds the potential to enable precise manipulation of a kinase specifically at the proteasome nanodomains without inducing the undesirable effects on other aspects of cellular function or processes that would otherwise be induced by a broad intervention of the kinase throughout the cell. For instance, selective inhibition of a PDE isoform enriched in the proteasome nanodomain will allow augmentation of cAMP/PKA and/or cGMP/PKG signaling specifically to the proteasome without affecting the signaling to other subcellular domains such as ion channels, sarcoendoplasmic reticulum calcium ATPase, and myofilaments. Besides phosphorylation, many other posttranslational modifications also occur on proteasomes [45]; further elucidation of their regulation on proteasomes may unveil additional ways to prime the proteasome.
Highlights.
The prior assumption that 26S proteasomes act automatically at its maximal capacity in UPS-mediated proteolysis proves to be false. Functioning of 26S proteasomes is precisely regulated and dictates the fate and degradation efficiency of ubiquitinated proteins.
Stimulation of PKA or PKG primes the proteasome and facilitates proteasomal degradation, thereby protecting against increased proteotoxic stress (IPTS).
IPTS occurs in common yet highly disabling and life-threatening diseases. Proteasome impairment is both a result and cause of IPTS. Genetic enhancement of the proteasome protects against IPTS.
Augmentation of cAMP and/or cGMP, likely through stimulating PKA and/or PKG, confers therapeutic benefits in animal models of neural and cardiac IPTS.
PKA and PKG duo-activation, via canceling some of the undesired effects of solo-activation, may be a better strategy to treat cardiac IPTS.
Acknowledgements
Work in Dr. X. Wang’s laboratory was supported in part by NIH grants HL072166, HL085629, and HL131667 and the work in Dr. H. Wang’s laboratory was supported by NIH NS088084.
Glossary
- 26S proteasome
a large multi-protein complex formed by a cylinder-shaped 20S core particle (CP) capped with a 19S regulatory particle (RP) at one or both ends; its peptidase activities resides in the interior chamber of the 20S while the 19S recruits and processes polyubiquitinated proteins for channeling them into the 20S for proteolysis
- Alzheimer’s disease
a neurodegenerative disorder pathologically characterized by accumulation of amyloid plaques in the brain containing amyloid-β peptides and by formation of neurofibrillary tangles composed of hyperphosphorylated tau, a microtubule-associated protein
- Amyotrophic lateral sclerosis (ALS)
a disease that selectively causes the loss of neurons controlling voluntary muscles, leading to progressive muscular atrophy, loss of strength, paralysis, and death. Proteasome impairment in neurons has been associated with ALS
- Autophagy
the process by which lysosomes in a cell remove a portion of the cell; based on the pathway for the cargo to be delivered into the lysosome, it is classified into 3 formsmacroautophagy, microautophagy, and chaperone-mediated autophagy
- Cyclic nucleotide phosphodiesterases (PDEs)
the enzymes that hydrolyze cAMP and cGMP; PDEs are classified into 11 families (PDE1 through PDE11). PDE4, 7, and 8 families selectively hydrolyze cAMP while PDE5, 6, and 9 are specific for cGMP. The remaining 5 families are so-called duo-PDEs and hydrolyze both cAMP and cGMP
- Huntington’s disease (HD)
an inherited progressive neurodegenerative disease caused by abnormal CAG trinucleotide repeat expansion in exon 1 of the huntingtin gene. The mutant huntingtin proteins can cause proteasome malfunction
- Parkinson’s disease (PD)
a progressive neurodegenerative disorder that is clinically characterized by tremor and stiffness or slowing of movement. Pathologically it is characterized by selective loss of dopaminergic neurons in the midbrain (substantia nigra) and by the presence of Lewy bodies. Lewy bodies or the process forming them impairs proteasome function
- Proteasome activator 28 (PA28 or REG) or 11S proteasome
a type of proteasome regulatory complexes that, like the 19S RP, can also associate with the 20S CP at one or both ends. A 20S with a 19S cap at one end and an 11S at the other (19S-20S-11S) is termed a hybrid proteasome. There are three isoforms of PA28 subunitsPA28α, β, and γ (also known as REGα, β, γ). The 11S proteasome associated with the 20S is either a hetero-heptamer (PAα4β3) or a homo-haptamer of PA28γ
- Proteasome nanodomain
an intracellular territory taken by a 26S proteasome along with its associated partners and regulators (e.g., cAMP/PKA and cGMP/PKG) in the immediate vicinity. A double-capped 26S proteasome is a cylindrically shaped nanoparticle with a dimeter of approximately 12 nm and a height of 40~45 nm
- Tauopathy
a family of neurodegenerative diseases pathologically characterized by the deposition of abnormal tau proteins in the brain, which include Pick disease, progressive supranuclear palsy, corticobasal degeneration, and argyrophilic grain disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
PDE1 inhibitor to treat Parkinson’s disease https://clinicaltrials.gov/ct2/show/NCT03257046
PDE1 inhibitor to treat Heart Failure https://clinicaltrials.gov/ct2/show/NCT03387215
References
- 1.Glembotski CC et al. (2019) Proteostasis and Beyond: ATF6 in Ischemic Disease. Trends Mol Med 25 (6), 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X et al. (2013) Posttranslational modification and quality control. Circ Res 112 (2), 367–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandri M and Robbins J (2014) Proteotoxicity: an underappreciated pathology in cardiac disease. J Mol Cell Cardiol 71, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su H et al. (2011) Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res 108 (1), 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su H et al. (2011) COP9 signalosome regulates autophagosome maturation. Circulation 124 (19), 2117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pattison JS et al. (2008) Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 117 (21), 2743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thibaudeau TA et al. (2018) A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun 9 (1), 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J et al. (2006) Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol 40 (4), 451–4. [DOI] [PubMed] [Google Scholar]
- 9.Liu J et al. (2006) Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J 20 (2), 362–4. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q et al. (2005) Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res 97 (10), 1018–26. [DOI] [PubMed] [Google Scholar]
- 11.Su H et al. (2013) The COP9 Signalosome Is Required for Autophagy, Proteasome-Mediated Proteolysis, and Cardiomyocyte Survival in Adult Mice. Circ Heart Fail 6 (5), 1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myeku N and Duff KE (2018) Targeting the 26S Proteasome To Protect Against Proteotoxic Diseases. Trends Mol Med 24 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanbe A et al. (2004) Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A 101 (27), 10132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed MI et al. (2016) Disruption of desmin-mitochondrial architecture in patients with regurgitant mitral valves and preserved ventricular function. J Thorac Cardiovasc Surg 152 (4), 1059–1070 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer LM et al. (2018) Amyloid precursor protein-fragments-containing inclusions in cardiomyocytes with basophilic degeneration and its association with cerebral amyloid angiopathy and myocardial fibrosis. Sci Rep 8 (1), 16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rainer PP et al. (2018) Desmin Phosphorylation Triggers Preamyloid Oligomers Formation and Myocyte Dysfunction in Acquired Heart Failure. Circ Res 122 (10), e75–e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Troncone L et al. (2016) Abeta Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. J Am Coll Cardiol 68 (22), 2395–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tublin JM et al. (2019) Getting to the Heart of Alzheimer Disease. Circ Res 124 (1), 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day SM (2013) The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am J Physiol Heart Circ Physiol 304 (10), H1283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X et al. (2001) Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res 89 (1), 84–91. [DOI] [PubMed] [Google Scholar]
- 21.Wang X et al. (2001) Mouse model of desmin-related cardiomyopathy. Circulation 103 (19), 2402–7. [DOI] [PubMed] [Google Scholar]
- 22.Tannous P et al. (2008) Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 117 (24), 3070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meijering RA et al. (2015) Reviving the protein quality control system: therapeutic target for cardiac disease in the elderly. Trends Cardiovasc Med 25 (3), 243–7. [DOI] [PubMed] [Google Scholar]
- 24.Bhuiyan MS et al. (2013) Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123 (12), 5284–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao T et al. (2017) The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl Neurodegener 6, 3.28184302 [Google Scholar]
- 26.Wang X and Cui T (2017) Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy. Am J Physiol Heart Circ Physiol 313 (2), H304–H319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X et al. (2008) Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol 45 (1), 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X and Robbins J (2014) Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 71, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J et al. (2008) Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol 180 (6), 1177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dasgupta S et al. (2015) Reduced Levels of Proteasome Products in a Mouse Striatal Cell Model of Huntington’s Disease. PLoS One 10 (12), e0145333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y et al. (2014) The proteasome function reporter GFPu accumulates in young brains of the APPswe/PS1dE9 Alzheimer’s disease mouse model. Cell Mol Neurobiol 34 (3), 315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emmanouilidou E et al. (2010) Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 31 (6), 953–68. [DOI] [PubMed] [Google Scholar]
- 33.Kabashi E et al. (2012) Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13 (4), 367–71. [DOI] [PubMed] [Google Scholar]
- 34.Argyriou AA et al. (2008) Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 112 (5), 1593–9. [DOI] [PubMed] [Google Scholar]
- 35.Weekes J et al. (2003) Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3 (2), 208–16. [DOI] [PubMed] [Google Scholar]
- 36.Al-Hassnan ZN et al. (2016) A substitution mutation in cardiac ubiquitin ligase, FBXO32, is associated with an autosomal recessive form of dilated cardiomyopathy. BMC Med Genet 17, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen SN et al. (2012) Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 111 (7), 907–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hedberg-Oldfors C et al. (2019) Cardiomyopathy with lethal arrhythmias associated with inactivation of KLHL24. Hum Mol Genet 28 (11), 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang HM et al. (2017) Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J Am Coll Cardiol 70 (20), 2536–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J et al. (2011) Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J 25 (3), 883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J et al. (2011) Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121 (9), 3689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J et al. (2017) Cardiac proteasome functional insufficiency plays a pathogenic role in diabetic cardiomyopathy. J Mol Cell Cardiol 102, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajagopalan V et al. (2013) Altered Ubiquitin-Proteasome Signaling in Right Ventricular Hypertrophy and Failure. Am J Physiol Heart Circ Physiol 305 (4), H551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins GA and Goldberg AL (2017) The Logic of the 26S Proteasome. Cell 169 (5), 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui Z et al. (2014) Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol 71, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehrbach NJ et al. (2019) Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 177 (3), 737–750 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rousseau A and Bertolotti A (2018) Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol 19 (11), 697–712. [DOI] [PubMed] [Google Scholar]
- 48.Guo X et al. (2016) Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat Cell Biol 18 (2), 202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lokireddy S et al. (2015) cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A 112 (52), E7176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ranek MJ et al. (2013) Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128 (4), 365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereira ME and Wilk S (1990) Phosphorylation of the multicatalytic proteinase complex from bovine pituitaries by a copurifying cAMP-dependent protein kinase. Arch Biochem Biophys 283 (1), 68–74. [DOI] [PubMed] [Google Scholar]
- 52.Zong C et al. (2006) Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res 99 (4), 372–80. [DOI] [PubMed] [Google Scholar]
- 53.Zhang F et al. (2007) Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem 282 (31), 22460–71. [DOI] [PubMed] [Google Scholar]
- 54.VerPlank JJS et al. (2019) 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H et al. (2019) PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv 5 (5), eaaw5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu H et al. (2008) Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol Cell Proteomics 7 (11), 2073–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asai M et al. (2009) PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol 46 (4), 452–62. [DOI] [PubMed] [Google Scholar]
- 58.Drews O et al. (2010) Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res 107 (9), 1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ranek MJ et al. (2014) Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G-dependent manner. J Mol Cell Cardiol 69C, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X and Robbins J (2006) Heart failure and protein quality control. Circ Res 99 (12), 1315–28. [DOI] [PubMed] [Google Scholar]
- 61.Lobanova ES et al. (2018) Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat Commun 9 (1), 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bobin P et al. (2016) Cyclic nucleotide phosphodiesterases in heart and vessels: A therapeutic perspective. Arch Cardiovasc Dis 109 (6–7), 431–43. [DOI] [PubMed] [Google Scholar]
- 63.Myeku N et al. (2016) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 22 (1), 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owona BA et al. (2016) Protective Effects of Forskolin on Behavioral Deficits and Neuropathological Changes in a Mouse Model of Cerebral Amyloidosis. J Neuropathol Exp Neurol 75 (7), 618–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giralt A et al. (2013) PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus 23 (8), 684–95. [DOI] [PubMed] [Google Scholar]
- 66.Beaumont V et al. (2016) Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington’s Disease Models. Neuron 92 (6), 1220–1237. [DOI] [PubMed] [Google Scholar]
- 67.Schaler AW and Myeku N (2018) Cilostazol, a phosphodiesterase 3 inhibitor, activates proteasome-mediated proteolysis and attenuates tauopathy and cognitive decline. Transl Res 193, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hollerhage M et al. (2017) Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci Rep 7 (1), 11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sheng M et al. (2017) Sildenafil Improves Vascular and Metabolic Function in Patients with Alzheimer’s Disease. J Alzheimers Dis 60 (4), 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tai SY et al. (2017) Cilostazol as an add-on therapy for patients with Alzheimer’s disease in Taiwan: a case control study. BMC Neurol 17 (1), 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amsallem E et al. (2005) Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev (1), CD002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Movsesian M (2016) Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol Ther 163, 74–81. [DOI] [PubMed] [Google Scholar]
- 73.Redfield MM et al. (2013) Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309 (12), 1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim GE and Kass DA (2017) Cardiac Phosphodiesterases and Their Modulation for Treating Heart Disease. Handb Exp Pharmacol 243, 249–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.VerPlank JJS and Goldberg AL (2018) Exploring the Regulation of Proteasome Function by Subunit Phosphorylation. Methods Mol Biol 1844, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kannel WB et al. (1987) Heart rate and cardiovascular mortality: the Framingham Study. Am Heart J 113 (6), 1489–94. [DOI] [PubMed] [Google Scholar]
- 77.Levy D et al. (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322 (22), 1561–6. [DOI] [PubMed] [Google Scholar]
- 78.Ranek MJ et al. (2019) PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 566 (7743), 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakamura T et al. (2018) Prevention of PKG-1alpha Oxidation Suppresses Antihypertrophic/Antifibrotic Effects From PDE5 Inhibition but not sGC Stimulation. Circ Heart Fail 11 (3), e004740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsai EJ and Kass DA (2009) Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther 122 (3), 216–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takimoto E et al. (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11 (2), 214–22. [DOI] [PubMed] [Google Scholar]
- 82.Vandeput F et al. (2007) Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J Biol Chem 282 (45), 32749–57. [DOI] [PubMed] [Google Scholar]
- 83.Chen S et al. (2018) Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. J Cardiovasc Dev Dis 5 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knight WE et al. (2016) PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller CL et al. (2009) Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105 (10), 956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hashimoto T et al. (2018) Acute Enhancement of Cardiac Function by Phosphodiesterase Type 1 Inhibition. Circulation 138 (18), 1974–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steinberg SF and Brunton LL (2001) Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol 41, 751–73. [DOI] [PubMed] [Google Scholar]
- 88.Conti M et al. (2014) Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J Gen Physiol 143 (1), 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kokkonen K and Kass DA (2017) Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Annu Rev Pharmacol Toxicol 57, 455–479. [DOI] [PubMed] [Google Scholar]
- 90.Goldfarb LG and Dalakas MC (2009) Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 119 (7), 1806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perng MD et al. (1999) The cardiomyopathy and lens cataract mutation in alphaB-crystallin alters its protein structure, chaperone activity, and interaction with intermediate filaments in vitro. J Biol Chem 274 (47), 33235–43. [DOI] [PubMed] [Google Scholar]
- 92.Bova MP et al. (1999) Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A 96 (11), 6137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McLendon PM and Robbins J (2011) Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol 301 (4), H1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Venkatraman P et al. (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14 (1), 95–104. [DOI] [PubMed] [Google Scholar]