

Summary
Feedback circuits are one of the major causes underlying tumor resistance. Thus, compounds that target one oncogenic pathway with simultaneously blocking its compensatory pathway will be of great value for cancer treatment. Here, we develop a new MEK inhibitor designated as KZ-02 that exhibits unexpectedly higher cytotoxicity than its starting compound AZD6244, a well-known MEK inhibitor, in colorectal cancer (CRC). Subsequent kinase selectivity study identified Pim-1 as an additional cellular target for KZ-02. Further studies showed that AZD6244 and Pim-1 1 (a Pim-1 inhibitor) have a synergistic effect on CRC suppression. Mechanistic study revealed that MEK inhibition by AZD6244 leads to increased Pim-1 expression, which could be a general mechanism behind the compromised cell-killing activity of MEK inhibitors. KZ-02, despite increasing Pim-1 mRNA expression, simultaneously promotes Pim-1 proteasomal degradation. Therefore, we uncover a new MEK inhibitor KZ-02 with significantly enhanced antitumor activity by co-targeting MEK and Pim-1.
Subject Areas: Biochemistry, Biological Sciences, Cancer, Chemistry
Graphical Abstract
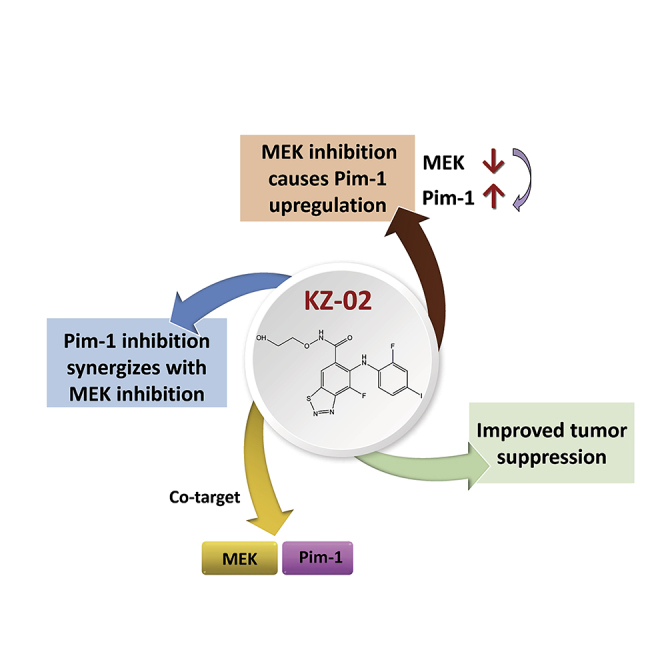
Highlights
-
•
Inhibition of MEK leads to Pim-1 upregulation
-
•
Pim-1 inhibition synergizes with MEK inhibition
-
•
A new compound is developed, which cotargets MEK and Pim-1
Biochemistry; Biological Sciences; Cancer; Chemistry
Introduction
The RAS/ERK pathway involved in the control of cell proliferation and apoptosis regulation is deregulated in more than 30% of human cancers (Zhang et al., 2018, Zhao and Adjei, 2014). The narrow substrate specificity and distinctive structural characteristics render MEK1/2 ideal targets for therapeutic development (Duncan et al., 2015, Fischmann et al., 2009). Since 2000, when the first phase I study of a MEK inhibitor (CI-1040) was carried out by Lorusso and his colleagues (Lorusso et al., 2005), more than a dozen of highly specific and potent MEK1/2 inhibitors have been developed and evaluated in clinical studies (Decaudin et al., 2018, Finn et al., 2018). Despite the increasing number of MEK inhibitors identified, only trametinib (GSK212), cobimetinib (GDC-0973), and binimetinib (MEK162) have gained U S Food and Drug Administration (FDA) approval for clinical use (Signorelli and Shah Gandhi, 2017, Wright and McCormack, 2013). Other agents exhibit limited efficacy when used as a single agent and fail to demonstrate substantial clinical activity in treating tumors. One leading cause for this lack of efficacy is due to cross talks between ERK pathway and other pathways, such as PI3K-AKT pathway (Brighton et al., 2018). These have resulted in the clinical study of MEK inhibitors combined with inhibitors of PI3K, AKT, or mTOR (Fukumoto et al., 2018, Rosenberg et al., 2018, Wainberg et al., 2017). Despite the progress obtained, these agents are only efficacious in a limited range of cancers. Therefore, there is a great need to develop new MEK inhibitors and find mechanisms that confer tumor cell resistance to MEK inhibition.
The proviral integration site for moloney murine leukemia virus (Pim-1) is a serine/threonine (Ser/Thr) kinase. The activity of Pim-1 is exclusively dependent on its protein levels, which are controlled by gene transcription and proteasome-mediated degradation (Qian et al., 2005, Shay et al., 2005). Pim-1 displays oncogenic potential with upregulated expression in human malignancies of hematopoietic and epithelial origin (Brasó-Maristany et al., 2016). Pim-1 overexpression predicts worse outcome for several human malignancies, but it also correlates with better prognosis in human prostate and pancreatic cancers (Narlik-Grassow et al., 2013, Reiser-Erkan et al., 2008). Pim-1 is recognized as a therapeutic target, although serious side effects have stopped a clinical trial of one Pim-1 inhibitor (Cortes et al., 2018, Fan et al., 2017). Pim-1 is a critical effector mediating cross talks among different signaling pathways, especially those involving AKT and ERK (Le et al., 2016). It has been reported that Pim-1 plays an important role in the regulation of ERK pathway (Lin et al., 2010). Inhibition of Pim-1 is associated with increased ERK phosphorylation, suggesting that activation of ERK-mediated survival pathways could result from Pim-1-targeted inhibition. However, whether the ERK/MAPK pathway may regulate Pim-1 activity remains unclear.
Herein, we developed a new MEK inhibitor designated as KZ-02 with AZD6244 as the starting point. KZ-02 showed ∼5-fold greater kinase inhibitory activity than AZD6244. However, KZ-02 exhibited unexpectedly high cytotoxicity of three orders of magnitude more than AZD6244. This leads to identification of Pim-1 as an additional target for KZ-02. Further studies reveal that MEK inhibition leads to enhanced Pim-1 expression, which dampens the cell-killing ability of MEK inhibitors. However, KZ-02, despite increasing Pim-1 expression due to its inhibition of MEK activation, also promotes proteasome-dependent degradation of Pim-1, ultimately inhibiting Pim-1 activation.
Results
Development of a New MEK Inhibitor KZ-02
The clinical importance of MEK signaling in human cancer and the limitations of existing MEK inhibitors promoted us to develop new MEK inhibitors. The starting point we used was the well-known MEK inhibitor AZD6244. AZD6244 has been reported to potently inhibit MEK1 with IC50 (50% inhibitory concentration) of 14 nmol/L (Yeh et al., 2007). It is currently under clinical evaluation as a single agent or in combination with other cytotoxic chemotherapy drugs or radiation therapy in various tumor types (Carvajal et al., 2018, Song et al., 2018, Tai et al., 2016). Based on the unsatisfactory potency of AZD6244, we proposed that a large substituent group on 3-nitrogen might result in a decreased antitumor activity. In addition, the de-methylation of 3-nitrogen methyl of AZD6244 could occur by in vivo enzyme metabolism (Dymond et al., 2016). Therefore, we replaced the 3-nitrogen methyl in the benzoheterocyclic ring of AZD6244 with S atom, generating a benzothiadiazole core. We also replaced the -Cl and -Br substituent on the aniline with -F and -I, respectively. The resulting new compound was designated as KZ-02 (Figure 1A). The step-by-step synthesis details for KZ-02 were described in Figure S1. The corresponding analysis spectra for key intermediates and target compound were presented in Data S1. A complex structure of MEK1/KZ-02 was built based on the MEK1/AZD6244 co-crystal structure (PDB code: 4U7Z) (Figure 1B). The replacement of the N-methyl by a smaller group of S atom may improve the affinity between KZ-02 and MEK1. Besides, compared with AZD6244 that has one H-bond accepting nitrogen for Ser212, KZ-02 has two on the thiadiazole ring (Figure 1B), which may facilitate the binding between KZ-02 and the Ser212 of MEK1. The iodine and fluorine atom on lipophilic ring of KZ-02 may also contribute to an improved affinity by modifying the interaction of KZ-02 with the Val127 and Lys97 of MEK1. As expected, KZ-02 indeed exhibited improved potency versus AZD6244 as reflected by in vitro kinase inhibition assay that was carried out as a commercial service by Cerep Drug Discovery Services Co. LTD (France). The IC50 for KZ-02 was 1.1 nmol/L, whereas the IC50 of AZD6244 was determined as 5.1 nmol/L (Figures 1C and 1D). Thus, we obtained a new compound, KZ-02, that is ∼5-fold more potent than AZD6244 in MEK inhibition.
Figure 1.

Development of a New MEK Inhibitor KZ-02
(A) Chemical structure of KZ-02 and AZD6244.
(B) KZ-02 structure (purple) overlapped with AZD6244 (yellow) with the co-crystal structure of MEK1/ATP/AZD6244 (PDB code: 4U7Z) as the model. KZ-02 shows similar binding with MEK1, and key residues of MEK1 that are important for interaction with KZ-02/AZD6244 are shown.
(C) In vitro kinase inhibition assay determines IC50 for AZD6244 to 5.09 nmol/L.
(D) In vitro kinase inhibition assay determines IC50 for KZ-02 to be 1.07 nmol/L. Data are represented as mean ± SD.
KZ-02 Exhibits Unexpectedly Higher Cytotoxicity over AZD6244
Next, we compared the inhibitory effect of KZ-02 and AZD6244 on colorectal cancer (CRC) cell growth. Two CRC cell lines, Colo205 and HT29, were used, and these two cell lines both harbor a constitutively activated RAS/ERK pathway (Janku, 2018). CRC cells were treated with increasing concentrations of compounds and cell proliferation was determined at 24, 48, and 72 h by using the MTT (methylthiazolyldiphenyl-tetrazolium bromide) method. KZ-02 significantly inhibited cell viability in a dose-dependent manner; moreover, KZ-02 exhibited higher activity than ADZ6244 in both CRC cell lines at nearly all time points (Figures 2A and 2B). The IC50 of KZ-02 against Colo205 and HT29 at 48 h were 0.015 and 4.1 nmol/L, respectively (Figure 2C). Compared with AZD6244, KZ-02 was three orders of magnitude more potent in inhibiting the growth of Colo205 cells (0.015 versus 15.5 nmol/L), whereas the IC50 of AZD6244 for inhibiting HT29 could not be determined at concentrations ranging from 0.01 to 1,000 nmol/L (Figure 2C). Considering the surprisingly increased superiority of KZ-02 to AZD6244 in MTT assay (∼1,000-fold) compared with that in MEK inhibition assay (∼5-fold), we suspected that KZ-02 might have cellular target(s) in addition to MEK.
Figure 2.

KZ-02 Exhibits Higher Activity Than AZD6244 in Inhibiting CRC Growth
(A and B) Examination of activity of KZ-02 versus AZD6244 in CRC cell growth inhibition. Colo205 (A) or HT29 (B) cells were treated with increasing concentrations of KZ-02 or AZD6244 and cell proliferation was determined at 24, 48, and 72 h by using the MTT method. Error bars are based on the standard deviations of triplicate samples.
(C) IC50 values for KZ-02 and AZD6244 calculated from data shown in (A) and (B), and the ratio of their IC50s was also calculated.
(D) Examination of antitumor effects of KZ-02 versus AZD6244 in xenograft models. BALB/c mice were subcutaneously injected with 1×107 Colo205 cells. Mice were then randomly assigned to three groups (n = 8), vehicle, KZ-02, and AZD6244, which were orally administered once daily for 20 days. Tumor size and body weight were measured every 3 days from day 23 to day 41.
(E) Body weight variations of mice used in (D).
Data are represented as mean ± SD. Comparisons of two groups are performed with Student's t test. ∗p < 0.05, ∗∗∗∗p < 0.0001.
To further evaluate the antitumor effect of KZ-02 in vivo, we generated Colo205 colon cancer xenograft to examine the antitumor effects of KZ-02. After 20 days of oral administration, KZ-02 inhibited Colo205 xenograft growth by 56.6% at a dosage of 1 mg/kg/day; by contrast, AZD6244 showed a similar inhibition when the dosage was used as high as 10 mg/kg/day (Figure 2D). Of note, even the highest dose of drugs tested in these experiments had no apparent effect on the body weight of tumor-bearing mice (Figure 2E). These results further confirmed the superior activity of KZ-02 over AZD6244.
Pim-1 Is an Additional Target for KZ-02
To uncover possible additional targets of KZ-02, we used the commercial service provided by the Cerep Drug Discovery Services Co. LTD (France) to perform the kinase selectivity experiment for KZ-02. The inhibitory activity values were expressed as percent of inhibitory activity of 5 mmol/L stausporin, a pan kinase inhibitor. Among the selected 77 kinases, Pim-1 ranked the second only next to MEK1, exhibiting significantly higher sensitivity to KZ-02 than the rest of the kinases (Table S1). In vitro kinase inhibition assay determined the IC50 of KZ-02 against Pim-1 to be 1.34 μmol/L (Figure 3A). To further confirm the inhibition of KZ-02 toward Pim-1 kinase activity, we examined the effect of KZ-02 on the phosphorylation of Cdc25C, a known substrate of Pim-1 (Yuan et al., 2014). Treatment of KZ-02, in addition to blocking phosphorylation of ERK (the substrate for MEK), also led to inhibition of Cdc25C phosphorylation in both Colo205 and HT29 cells (Figure 3B). As expected, inhibition of Pim-1 activity by either Pim-1 1 (a Pim-1 inhibitor) or siRNA resulted in decreased phosphorylation of Cdc25C (Figure 3C). Together, these results demonstrate that KZ-02 is also an inhibitor for Pim-1.
Figure 3.

KZ-02 Additionally Targets Pim-1
(A) Kinase selectivity screening identified Pim-1 as an additional target for KZ-02 with IC50 being 1.34 μmol/L.
(B) Examination of KZ-02 inhibition on Pim-1 activity. Colo205 and HT29 cells were treated with KZ-02 (10 nmol/L) or DMSO (control) for 2 h, followed by western blot analysis. Cdc25C is a known substrate of Pim-1.
(C) Either Pim-1 1, a Pim-1 inhibitor, or Pim-1 siRNA treatment decreased Cdc25C phosphorylation. Cells were treated with 100 nmol/L Pim-1 1 for 2 h, or 40 nmol/L Pim-1 siRNA for 48 h, and then were lysed and subjected to western blot analysis. Data are represented as mean ± SD.
Pim-1 Inhibition Has a Synergistic Effect with MEK Inhibition
If the additional Pim-1 inhibition accounts for the surprisingly high activity of KZ-02, it is reasonable to surmise that Pim-1 inhibition may increase the cell-killing efficiency of MEK inhibition. To test this possibility, we treated Colo205 and HT29 cells with AZD6244 and Pim-1 1, individually or in combination, for 24, 48, and 72 h. The inhibitory effects of these treatments on tumor cell growth were shown in Figures 4A and 4B. Of note, Pim-1 1 itself barely inhibited proliferation of both cell lines. We then calculated the coefficient of drug interaction (CDI), which is used to evaluate drug combination effect (Xu et al., 2007). We observed a synergistic effect in both CRC cell lines (CDI<1), with significant synergy (CDI<0.7) occurring for most of Colo205 cell points and a portion of HT29 cell points (Figures 4C and 4D). This result suggested that Pim-1 1 has a synergistic action with AZD6244 in suppressing tumor cell growth.
Figure 4.

Pim-1 Inhibition Synergizes with MEK Inhibition in Inhibiting Tumor Growth
(A and B) AZD6244 and Pim-1 1 show synergism in inhibiting tumor cell growth. Colo205 (A) or HT29 (B) cells were treated with AZD6244 and Pim-1 1, individually or in combination (1:1, nmol/L), for 24, 48, and 72 h. The inhibitory effects of these treatments on tumor cell growth were then measured by the MTT assay.
(C) CDIs were calculated for data from (A).
(D) CDIs were calculated for data from (B).
(E) Evaluation of AZD6244 and Pim-1 1 combination in xenograft models. HT29 bearing xenograft mice were orally administrated with AZD6244 (20 mg/kg), Pim-1 1 (20 mg/kg), or their combination (both 20 mg/kg), and tumor volumes were examined every 3 days for 15 consecutive days.
(F) CDIs were calculated for data in (E).
(G) Body weights of experimental mice used in (E).
Error bars are based on the standard deviations of triplicate samples for in vitro cell viability test and eight duplicate results for in vivo tumor inhibition. Data are represented as mean ± SD. Comparisons of two groups are performed with Student's t test. ∗p < 0.05.
To confirm the synergism between AZD6244 and Pim-1 1, we evaluated the in vivo antitumor efficacy of AZD6244 and Pim-1 1 in combination versus either agent alone by using the mouse xenograft model. AZD6244, Pim-1 1 or their combination was administered orally to HT29 cell-bearing mice. We found that the combination of AZD6244 and Pim-1 1 exhibited a significant improvement of tumor suppression compared with AZD6244 and Pim-1 1 used alone (Figure 4E). The minimum relative tumor volume (tumor volume on day 15/tumor volume on day 0) for AZD6244 plus Pim-1 1 was calculated to be 1.34 with CDI values all being less than one (Figure 4F). In comparison, the relative tumor volumes for AZD6244 and Pim-1 1 when used alone were 3.13 and 5.25, respectively, and the value for the vehicle control was 5.43. Negligible body weight fluctuations indicated few adverse effects of these therapeutic treatments on laboratory animals (Figure 4G). Taken together, these results showed that AZD6244 and Pim-1 1 have a synergistic effect on killing CRC tumor cells.
MEK Inhibition Resulted in Increased Pim-1 Expression
Next, we set out to investigate the mechanism behind such a synergistic action between MEK inhibition and Pim-1 inhibition. As mentioned before, inhibition of Pim-1 is previously reported to increase ERK phosphorylation (Lin et al., 2010). We asked whether ERK/MAPK pathway might affect Pim-1 activity in return. To test this possibility, we first examined the effect of AZD6244 treatment on Pim-1 expression in Colo205 and HT29 cells. As shown in Figure 5A, AZD6244 treatment (100 nmol/L) led to a decrease of ERK phosphorylation but an increase of Pim-1 protein expression and Cdc25C phosphorylation. Consistent with this, knockdown of ERK by siRNA produced similar results (Figure 5B). We then included two additional MEK inhibitors, trametinib and MEK162, which are both approved by FDA for cancer treatment, and tested their effects on Pim-1 expression in HT29 cells. Both MEK inhibitors inhibited ERK phosphorylation and meanwhile led to an increase of Pim-1 levels as did AZD6244 (Figure 5C). Together, these results suggest that MEK inhibition increases Pim-1 expression, which could be an inherent feature for MEK inhibitors.
Figure 5.

MEK Inhibition Increases Pim-1 Expression
(A) AZD6244 increases Pim-1 levels in CRC cells. Colo205 and HT29 cells were treated with AZD6244 (100 nmol/L) for 2 h, and expression of proteins was then detected by western blot analysis with indicated antibodies.
(B) Knockdown of ERK increases Pim-1 levels. CRC cells were incubated with 40 nmol/L of ERK1/2 siRNA or a scramble control siRNA for 48 h, and protein levels were detected by western blot analysis with indicated antibodies.
(C) Effects of two additional MEK inhibitors, trametinib and MEK162, on Pim-1 expression. HT29 cells were treated with trametinib (100 nmol/L) or MEK162 (100 nmol/L) for 2 h, and then cells were lysed and Pim-1 levels were detected by western blot analysis.
(D) The effect of MEK inhibition on Pim-1 expression in xenograft models. Four control or AZD6244-treated HT29 xenografted tumors were randomly selected to test the impact of AZD6244 on the expression of Pim-1 using western blot analysis.
(E) Correlation analysis of ERK phosphorylation levels and Pim-1 protein levels in 20 CRC patient tumor specimens. Clinical colon carcinoma specimens were analyzed by immunohistochemistry (IHC). The immunoreactive score (IRS) of Pim-1 versus p-ERK for each sample was calculated. Five IRSs that have overlapped ones were labeled.
(F) Representative image shows that reduced ERK phosphorylation corresponding to overexpression of Pim-1 in human colorectal tumors detected by IHC (solid red line indicated the enlarged view of the red box in each picture).
(G) Representative image shows that enhanced ERK phosphorylation corresponding to the reduction of Pim-1 expression in human colorectal tumors detected by IHC (solid red line indicated the enlarged view of the red box in each picture).
To further confirm the effect of MEK inhibition on Pim-1 expression, we carried out in vivo analysis in HT29 cell-bearing mice. AZD6244 was administered orally at a dosage of 20 mg/kg/day for 15 days and then Pim-1 levels in control and AZD6244-treated tumor issues were analyzed by western blot analysis. As shown in Figure 5D, AZD6244 treatment led to a decrease of ERK phosphorylation, which was accompanied by an increase of Pim-1 protein levels. Finally, we examined the relationship of ERK phosphorylation levels and Pim-1 protein levels in serial slides of twenty clinical colon carcinoma specimens by immunohistochemistry (IHC). As shown in Figure 5E, ERK phosphorylation levels showed a negative correlation with Pim-1 protein levels in these clinical specimens with the negative correlation slope being −0.741. Consistently, quintessential ERK inhibition was associated with high Pim-1 positivity and vice versa as reflected by IHC assay (Figures 5F and 5G). Combined together, these results demonstrated that Pim-1 is subjected to negative regulation of ERK activation.
MEK Regulates Pim-1 via ERK/AMPK/IL-23/STAT3/Pim-1 Pathway
Next, we sought to explore the possible signal transduction cascade bridging ERK and Pim-1. Since interleukin-induced STAT3 activation is the major mechanism behind Pim-1 gene transactivation (Block et al., 2012), we first tested the effect of AZD6244 on the expression of interleukins. We treated HT29 cells with AZD6244 for 2 h (100 nmol/L) and examined by real-time PCR the expression of several interleukins (ILs), including IL-8, IL-10, IL-16, and IL-23, which have been reported to be expressed in CRC cells (Gao et al., 2009, Geng et al., 2017, Hu et al., 2017, Shi et al., 2016, Ting et al., 2013). We found that only IL-23 was significantly upregulated by AZD6244 (Figure 6A). IL-23 is a proinflammatory cytokine that is involved in many autoimmune diseases (Shi et al., 2016); moreover, IL-23 has been implicated in the progression of CRCs (Hu et al., 2017). Next, we investigated whether IL-23 may affect STAT3 phosphorylation and Pim-1 levels. We treated HT29 cells with recombinant IL-23 (10 ng/mL) for 2 h. As shown in Figure 6B, IL-23 treatment increased STAT3 phosphorylation and Pim-1 expression, whereas knockdown of IL-23 by siRNA had a reverse effect. We then asked whether IL-23 is required for AZD6244-induced increase of Pim-1 expression. For this, we treated HT29 cells with AZD6244 combined with IL-23 siRNA. As shown in Figure 6C, AZD6244 treatment increased STAT3 phosphorylation and Pim-1 expression, whereas knockdown of IL-23 abrogated these effects of AZD6244 without affecting its inhibitory effect on ERK/AMPK pathway. Of note, AMPK activation lead to a decrease of IL-23 expression (Shi et al., 2016). Indeed, we observed that an AMPK-specific inhibitor (compound C) significantly increased the expression of IL-23, which is accompanied by an increase of STAT3 phosphorylation and Pim-1 levels; however, these effects could be reversed by knockdown of IL-23 (Figure 6D). Similarly, either MEK inhibition by AZD6244 or AMPK inhibition by compound C led to an increase of IL-23 expression (Figures 6E and 6F). Taken together, these results reveal a feedback regulation of Pim-1 by MEK through ERK/AMPK/IL-23/STAT3/Pim-1 pathway, which accounts for MEK inhibitor-induced Pim-1 upregulation and thus confers tumor cell resistance to MEK inhibition (Figure 6G).
Figure 6.

MEK Regulates Pim-1 via ERK/AMPK/IL-23/STAT3 Cascade
(A) IL-23 is specifically upregulated by AZD6244. HT29 cells were treated with AZD6244 (100 nmol/L) for 2 h, and mRNA levels of IL-8, IL-10, IL-16, and IL-23 were then detected by real-time PCR.
(B) HT29 cells were treated with recombinant IL-23 (10 ng/mL) for 2 h or IL-23 siRNA (50 nmol/L) for 48 h. STAT3 phosphorylation and Pim-1 expression were then detected by western blot analysis.
(C) HT29 cells were treated with AZD6244 (100 nmol/L) for 2 h with or without 48-h pretreatment with a scramble or IL-23 siRNA (50 nmol/L), and protein expression or phosphorylation levels were then detected by specific antibodies as indicated.
(D) Same experiment as (C) with AZD6244 replaced by an AMPK inhibitor, compound C.
(E) HT29 cells were treated either with AZD6244 (100 nmol/L) for 2 h, and then IL-23 expression was detected by real-time PCR.
(F) HT29 cells were treated either with compound C (40 μmol/L) for 2 h, and then IL-23 expression was detected by real-time PCR.
(G) Mechanistic model for MEK inhibition-induced Pim-1 upregulation. MEK inhibition increases IL-23 expression through ERK/AMPK signaling, which then activates STAT3, thereby promoting Pim-1 transactivation.
Data are represented as mean ± SD. Comparisons of two groups are performed with Student's t test. ∗p < 0.05, ∗∗∗∗p < 0.0001.
KZ-02 Inhibits Pim-1 Activation by Promoting Its Proteasomal Degradation
According to the findings above, as an MEK inhibitor, KZ-02 should increase Pim-1 expression. To verify this, we first assessed the effect of KZ-02 on Pim-1 protein levels in Colo205 and HT29 cells. However, unlike AZD6244, KZ-02 treatment decreased, rather than increased Pim-1 protein levels (Figure 7A). Intracellular Pim-1 protein levels were controlled by both transcriptional regulation and proteasome-mediated turn over. Considering the opposing effects of AZD6244 and KZ-02 on Pim-1 protein levels, we next compared them on their impact on Pim-1 transcriptional regulation and proteasome-mediated turn over. We first performed real-time PCR to examine possible effects of KZ-02 or AZD6244 on mRNA levels of Pim-1. MEK inhibition, either by KZ-02 or by AZD6244, is accompanied with an increase of Pim-1 mRNA levels in Colo205 cells and HT29 cells (Figures 7B and S2A). This result is consistent with our findings above that MEK inhibition leads to an increased Pim-1 expression.
Figure 7.

KZ-02 Inhibits Pim-1 Function by Promoting Its Proteasomal Degradation
(A) KZ-02 treatment decreases Pim-1 protein levels. Colo205 and HT29 cells were treated with KZ-02 (10 nmol/L) for 2 h, and Pim-1 protein levels were then detected by western blot analysis.
(B) Both KZ-02 and AZD6244 increase Pim-1 mRNA levels. Tumor cells were treated with KZ-02 (10 nmol/L) or AZD6244 (100 nmol/L) for 2 h, and Pim-1 mRNA levels were then analyzed by real-time PCR. Similar experiment in HT29 cells was shown in Figure S2A.
(C) Tumor cells were treated with either KZ-02 (10 nmol/L) or AZD6244 (100 nmol/L) for 2 h with or without addition of the proteasome inhibitor MG132 (10 μg/mL) for 30 min, and Pim-1 expression was then detected by western blot analysis. Similar experiment in HT29 cells was shown in Figure S2B.
(D) KZ-02 treatment inhibits Pim-1 and its downstream effectors, whereas AZD6244 increases their activation in Colo205 cells. Similar experiment in HT29 cells was shown in Figure S2D.
(E) Tumor cells were treated with various concentrations of KZ-02, AZD6244+Pim-1 1 (1:1), or AZD6244+Pim-1 siRNA (8 nmol/L) as indicated for 24, 48, and 72 h. MTT assay was then used to characterize the inhibition of tumor cell proliferation. Similar experiment in HT29 cells was shown in Figure S2E.
(F) IC50 values were calculated for data in (D) and the ratios of AZD6244+Pim-1 1 to KZ-02 IC50 were shown. Pim-1 knockdown efficiency was confirmed in Figure S2C. Error bars are based on the standard deviations of triplicate samples.
Data are represented as mean ± SD. Comparisons of two groups are performed with Student's t test. ∗∗∗p < 0.001.
Next, we examined whether these two compounds could affect the proteasomal degradation of Pim-1. We treated tumor cells with either KZ-02 (10 nmol/L) or AZD6244 (100 nmol/L) for 2 h with or without addition of the proteasome inhibitor MG132. Pre-incubation of cells with 10 μg/mL MG132 for 30 min did not affect the increase of Pim-1 protein expression induced by AZD6244; however, MG132 abrogated the KZ-02-mediated decrease of Pim-1 protein levels in both tumor cells (Figures 7C and S2B). These data suggested that, although both compounds upregulate Pim-1 mRNA levels, KZ-02 simultaneously promotes proteasome-dependent degradation of Pim-1.
It has been reported that Pim-1 is involved in cell-cycle regulation through phosphorylating P21Cip1/Waf1/P27Kip1 (Morishita et al., 2008, Zhang et al., 2007). Moreover, Pim-1 could phosphorylate Bad to affect cell apoptosis (Aho et al., 2004). To further confirm different effects on Pim-1 activation by KZ-02 and AZD6244, we examined their effects on activation of P21Cip1/Waf1/P27Kip1 as well as Bad in Colo205 cells or HT29 cells. As shown in Figures 7D and S2D, although both compounds inhibited ERK phosphorylation, they exhibited opposite effects on activation of P21Cip1/Waf1/P27Kip1 and Bad. KZ-02 lead to decreased Pim-1 levels, accompanied by decreased phosphorylation of P21Cip1/Waf1/P27Kip1 and Bad; instead, AZD6244 increased phosphorylation of P21Cip1/Waf1/P27Kip1 and Bad. These results further confirmed our conclusion that KZ-02 inhibits Pim-1 activation, whereas AZD6244 increases it.
We then compared the antitumor activity of KZ-02 with that of AZD6244 and Pim-1 1 in combination via the MTT assay. The combination of AZD6244 and Pim-1 siRNA was also included for comparison. The effective Pim-1 knockdown by siRNA was confirmed (Figure S2C). KZ-02 exhibited higher cell-killing ability than either the combination of AZD6244 plus Pim-1 1 (red versus black lines) or the combination of AZD6244 and Pim-1 siRNA (red versus blue lines) in both CRC cell lines (Figures 7E and S2E). We then calculated IC50 values for KZ-02 and AZD6244 plus Pim-1 1 at 48 h of treatment, respectively. The ratio of IC50 value of AZD6244+Pim-1 1 versus KZ-02 was 6.8 and 72 for Colo205 and HT29 cells, respectively (Figure 7F). These results showed that KZ-02, as a dual-target inhibitor, is more effective than the combination of AZD6244 and Pim-1 1 in inhibiting CRC tumor cell growth.
Finally, we examined whether KZ-02 exclusively targets tumor cells. A potential drug candidate for cancer therapy should specifically target tumor cells with normal and benign cells spared. As shown above, KZ-02 exerted a minimal or tolerable effect on the body weight of the experimental animals (Figure 2E). To further confirm that KZ-02 specifically targets tumor cells while sparing normal or benign cells, we evaluated the potential toxicity of KZ-02, AZD6244, Pim-1 1, or AZD6244+Pim-1 1 on the immortalized normal colon cell line (CCD841CoN) over a range of dosages (0.0–10,000 nmol/L) by using the MTT assay. The corresponding tumor cell line Colo205 was used as a control (0.0–1,000 nmol/L). As shown in Figure S2F, more than 95% of cells in the benign cells remained viable after 48 h of treatment with any of the above compounds, or their combination, even at a concentration as high as 10,000 nmol/L. By contrast, in tumor cells, a combination of AZD6244 and Pim-1 1 exhibited a concentration-dependent cell-killing activity beginning at concentration as low as 0.01 nmol/L, which was more potent compared with AZD6244 alone, but weaker than KZ-02; Pim-1 1, consistent with our results above, and had no apparent cell-killing activity when used alone (Figure S2G). Combined together, these results showed that KZ-02 specifically inhibits tumor cell growth with better performance than the combination of AZD6244 and Pim-1 1.
Discussion
A recent large retrospective study detected mutations in the RAS/ERK pathway for nearly half of the patients with CRC (Lee et al., 2018), suggesting that MEK-targeted therapy could benefit a great number of patients with CRC. However, the development of acquired resistance, predominantly caused by reciprocal feedback of signal transduction pathways, will inevitably result in tumor recurrence in many cases. Understanding the feedback mechanisms underlying acquired resistance would thus facilitate the development of more effective drugs, as well as clinically relevant neoadjuvant regimens to improve the efficacy of anticancer treatment.
In this study, we initially aimed at developing a new MEK inhibitor based on the structure of the well-known MEK inhibitor AZD6244. However, the observation that KZ-02 shows remarkably more superiority to AZD6244 in the MTT assay than in the kinase assay urged us to explore the potential mechanism underlying the surprisingly high inhibitory activity of KZ-02. By using an in vitro kinase screening, we identified Pim-1 as an additional target for KZ-02. To our knowledge, this is the first MEK/Pim-1 dual-target inhibitor reported. Of note, KZ-02 shows less superiority over AZD6244 in xenograft mice model (∼10-fold) than in the MTT assay (∼1,000-fold). This is likely due to the inferior oral bioavailability of KZ-02 in mouse models. Further studies of pharmacokinetics and pharmacodynamics for KZ-02 would be required for evaluating and improving “drug-like” properties of KZ-02.
Pim-1 activity is dependent on its protein levels, which are controlled by transcription as well as by proteasomal degradation. Our data showed that both AZD6244 and KZ-02 increased Pim-1 mRNA levels, which could be a general mechanism underlying a compromised cell-killing ability of MEK inhibitors. Consistent with an elevation of Pim-1 activity following MEK inhibition, we find that AZD6244 induces cellular phosphorylation of Cdc25C, a substrate of Pim-1. This provides evidence that MEK pathway plays an important role in the feedback regulation of Pim-1. Further mechanistic studies revealed that inhibition of MEK could impede AMPK-mediated repression of IL-23 expression through decreasing phosphorylation of AMPK, which subsequently upregulates the transcription of Pim-1 by activating STAT3. It has been reported that Pim-1 inhibition could activate ERK (Lin et al., 2010). Thus, our study herein reveals a reciprocal feedback loop between ERK and Pim-1. In the current study, the Pim-1 inhibitor Pim-1 1 alone exhibits no apparent inhibition against CRC tumor cells. However, when combined with AZD6244, Pim-1 1 displays a synergistic effect with AZD6244 on killing tumor cells both in vitro and in vivo. Although further clinical studies are required to address the effectiveness of this combination in treating patients with CRC, our data here clearly demonstrated that Pim-1 is an important survival factor that affects MEK-targeted treatment of CRCs, thereby identifying Pim-1 as a druggable target for enhancing the sensitivity of CRCs to MEK inhibitors. Moreover, our findings that MEK inhibition leads to AMPK suppression and STAT3 activation also suggest a possible synergistic effect between MEK inhibitors and AMPK activators or STAT3 inhibitors in CRC treatment.
KZ-02, despite increasing Pim-1 mRNA levels owing to its inhibition of MEK, causes a decrease of Pim-1 protein levels. Further studies revealed that KZ-02 could promote proteasome-dependent Pim-1 degradation. This unique activity of KZ-02 as a dual-target inhibitor endows it with superior performance to AZD6244, and even to a combination of AZD6244 and Pim-1 1. Thus far, the mechanism by which KZ-02 mediates proteasomal degradation of Pim-1 remains unknown. Pim-1 protein stability is largely dependent on its associated molecular chaperones, Hsp90 or Hsp70. Hsp90-bound Pim-1 is more stable, whereas an association with Hsp70 will lead to ubiquitination and degradation of Pim-1 (Shay et al., 2005). Thus, one possibility is that KZ-02 may switch Pim-1 to a conformation that prefers an interaction with Hsp70. The mechanism behind how KZ-02 mediates Pim-1 degradation warrants further studies.
In conclusion, this study shows that Pim-1 activation is an important factor contributing to tumor cell desensitization to MEK inhibitors, rendering Pim-1 as a druggable target in MEK-targeted cancer therapy. Combination of Pim-1 and MEK inhibitors may serve as an effective anticancer strategy to improve CRC treatment. Moreover, a new MEK/Pim-1 dual-target inhibitor KZ-02 is developed, which exhibits a high tumor cell-killing activity by simultaneously inhibiting ERK phosphorylation and promoting proteasome-dependent Pim-1 degradation.
Limitations of the Study
In this study, we developed a new MEK inhibitor that exhibits significantly improved antitumor activity by simultaneously targeting Pim-1. The new MEK inhibitor, designated as KZ-02, inhibits Pim-1 kinase activity as reflected by the kinase selectivity assay and the in vitro kinase inhibition assay. Notably, in addition to inhibiting Pim-1 kinase activity, we found that KZ-02 also decreases Pim-1 protein levels by promoting proteasomal degradation of Pim-1. Considering a mild inhibitory activity of KZ-02 against Pim-1 in the in vitro kinase inhibition assay but a significantly improved tumor cell-killing activity compared with AZD6244 as reflected by the MTT assay and the mouse tumor growth assay, we tend to believe that KZ-02 inhibits Pim-1 activity mainly through decreasing Pim-1 protein levels rather than inhibiting its kinase activity directly. However, so far, the cellular target and molecular mechanism behind the KZ-02-mediated Pim-1 degradation remain unclear, which absolutely warrant further investigations.
Resource Availability
Lead Contact
Dr. Yukun Cui (yukuncui@yahoo.com).
Materials Availability
Materials are available from the corresponding author on request.
Data and Code Availability
All data are available in the main text or in Supplemental Information.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Stanley Lin for his critical editing. This work was supported by Strategic priority research program of CAS (XDB19000000) to L.L., National Natural Science Foundation of China (81903662) to Y.L., National Natural Science Foundation of China (31571456) to X.S., National Natural Science Foundation of China (81272931, 81572588) the running open grant of Guangdong Provincial Key Laboratory for Breast Cancer Diagnosis & Treatment (2017B030314116), Guangdong Provincial Special Fund of Sci-tech Innovation Strategy, Key Disciplinary Project of Clinical Medicine under the Guangdong High-level University Development Program, and an open grant from the State Key Laboratory of Molecular Biology to Yukun Cui.
Author Contributions
Y.L. performed most of the in vitro experiments and participated in manuscript preparation. Ying Cheng synthesized KZ-02 and contributed to manuscript preparation. M.Z. conducted part of mechanistic studies, assisted with clinical sample collection, and participated in xenograft model-related experiments. X.H. assisted with experimental design. L.K. and K.Z. contributed to validation of the selectivity of KZ-02. Y.Z. contributed to data interpretation. L.L. contributed to the design of the study. H.T. designed KZ-02 and assisted with project supervision. X.S. participated in project supervision and manuscript preparation. Yukun Cui conceived this study, organized the collaboration, obtained financial support, and participated in manuscript preparation.
Declaration of Interests
H.T. has filed patents regarding KZ-02. No potential conflicts of interest were disclosed by the other authors.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101254.
Contributor Information
Hongqi Tian, Email: tianhongqi@irm-cams.ac.cn.
Xiaomin Song, Email: xmsong01@sibcb.ac.cn.
Yukun Cui, Email: yukuncui@yahoo.com.
Supplemental Information
References
- Aho T.L., Sandholm J., Peltola K.J., Mankonen H.P., Lilly M., Koskinen P.J. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004;571:43–49. doi: 10.1016/j.febslet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Block K.M., Hanke N.T., Maine E.A., Baker A.F. IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines. Pancreas. 2012;41:773–781. doi: 10.1097/MPA.0b013e31823cdd10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasó-Maristany F., Filosto S., Catchpole S., Marlow R., Quist J., Francesch-Domenech E., Plumb D.A., Zakka L., Gazinska P., Liccardi G. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016;22:1303–1313. doi: 10.1038/nm.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighton H.E., Angus S.P., Bo T., Roques J., Tagliatela A.C., Darr D.B., Karagoz K., Sciaky N., Gatza M.L., Sharpless N.E. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res. 2018;78:542–557. doi: 10.1158/0008-5472.CAN-17-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal R.D., Piperno-Neumann S., Kapiteijn E., Chapman P.B., Frank S., Joshua A.M., Piulats J.M., Wolter P., Cocquyt V., Chmielowski B. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: a phase III, multicenter, randomized trial (SUMIT) J. Clin. Oncol. 2018;36:1232–1239. doi: 10.1200/JCO.2017.74.1090. [DOI] [PubMed] [Google Scholar]
- Cortes J., Tamura K., DeAngelo D.J., de Bono J., Lorente D., Minden M., Uy G.L., Kantarjian H., Chen L.S., Gandhi V. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br. J. Cancer. 2018;118:1425–1433. doi: 10.1038/s41416-018-0082-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaudin D., El Botty R., Diallo B., Massonnet G., Fleury J., Naguez A., Raymondie C., Davies E., Smith A., Wilson J. Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget. 2018;9:21674–21686. doi: 10.18632/oncotarget.24670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan K.E., Chang L.Y., Patronas M. MEK inhibitors: a new class of chemotherapeutic agents with ocular toxicity. Eye (Lond) 2015;29:1003–1012. doi: 10.1038/eye.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymond A.W., Howes C., Pattison C., So K., Mariani G., Savage M., Mair S., Ford G., Martin P. Metabolism, excretion, and pharmacokinetics of selumetinib, an MEK1/2 inhibitor, in healthy adult male subjects. Clin. Ther. 2016;38:2447–2458. doi: 10.1016/j.clinthera.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Fan R.F., Lu Y., Fang Z.G., Guo X.Y., Chen Y.X., Xu Y.C., Lei Y.M., Liu K.F., Lin D.J., Liu L.L. PIM-1 kinase inhibitor SMI-4a exerts antitumor effects in chronic myeloid leukemia cells by enhancing the activity of glycogen synthase kinase 3beta. Mol. Med. Rep. 2017;16:4603–4612. doi: 10.3892/mmr.2017.7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.S., Ahn D.H., Javle M.M., Tan B.R., Jr., Weekes C.D., Bendell J.C., Patnaik A., Khan G.N., Laheru D., Chavira R. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Invest. New Drugs. 2018;36:1037–1043. doi: 10.1007/s10637-018-0600-2. [DOI] [PubMed] [Google Scholar]
- Fischmann T.O., Smith C.K., Mayhood T.W., Myers J.E., Reichert P., Mannarino A., Carr D., Zhu H., Wong J., Yang R.S. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–2674. doi: 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- Fukumoto S., Kanbara K., Neo M. Synergistic anti-proliferative effects of mTOR and MEK inhibitors in high-grade chondrosarcoma cell line OUMS-27. Acta Histochem. 2018;120:142–150. doi: 10.1016/j.acthis.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Gao L.B., Rao L., Wang Y.Y., Liang W.B., Li C., Xue H., Zhou B., Sun H., Li Y., Lv M.L. The association of interleukin-16 polymorphisms with IL-16 serum levels and risk of colorectal and gastric cancer. Carcinogenesis. 2009;30:295–299. doi: 10.1093/carcin/bgn281. [DOI] [PubMed] [Google Scholar]
- Geng R., Tan X., Wu J., Pan Z., Yi M., Shi W., Liu R., Yao C., Wang G., Lin J. RNF183 promotes proliferation and metastasis of colorectal cancer cells via activation of NF-κB-IL-8 axis. Cell Death Dis. 2017;8:2994–3004. doi: 10.1038/cddis.2017.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.H., Chen H.H., Yen S.L., Huang H.Y., Hsiao C.C., Chuang J.H. Increased expression of interleukin-23 associated with progression of colorectal cancer. J. Surg. Oncol. 2017;115:208–212. doi: 10.1002/jso.24505. [DOI] [PubMed] [Google Scholar]
- Janku F. Advances on the BRAF front in colorectal cancer. Cancer Discov. 2018;8:389–391. doi: 10.1158/2159-8290.CD-18-0125. [DOI] [PubMed] [Google Scholar]
- Le X., Antony R., Razavi P., Treacy D.J., Luo F., Ghandi M., Castel P., Scaltriti M., Baselga J., Garraway L.A. Systematic functional characterization of resistance to PI3K inhibition in breast cancer. Cancer Discov. 2016;6:1134–1147. doi: 10.1158/2159-8290.CD-16-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.K., Hwang J.H., Choi K.Y. Interaction of the Wnt/β-catenin and RAS-ERK pathways involving co-stabilization of both β-catenin and RAS plays important roles in the colorectal tumorigenesis. Adv. Biol. Regul. 2018;68:46–54. doi: 10.1016/j.jbior.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Lin Y.W., Beharry Z.M., Hill E.G., Song J.H., Wang W., Xia Z., Zhang Z., Aplan P.D., Aster J.C., Smith C.D. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010;115:824–833. doi: 10.1182/blood-2009-07-233445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso P.M., Adjei A.A., Varterasian M., Gadgeel S., Reid J., Mitchell D.Y., Hanson L., DeLuca P., Bruzek L., Piens J. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 2005;23:5281–5293. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- Morishita D., Katayama R., Sekimizu K., Tsuruo T., Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008;68:5076–5085. doi: 10.1158/0008-5472.CAN-08-0634. [DOI] [PubMed] [Google Scholar]
- Narlik-Grassow M., Blanco-Aparicio C., Cecilia Y., Perez M., Munoz-Galvan S., Canamero M., Renner O., Carnero A. Conditional transgenic expression of PIM1 kinase in prostate induces inflammation-dependent neoplasia. PLoS One. 2013;8:60277–60289. doi: 10.1371/journal.pone.0060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian K.C., Wang L., Hickey E.R., Studts J., Barringer K., Peng C., Kronkaitis A., Li J., White A., Mische S. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005;280:6130–6137. doi: 10.1074/jbc.M409123200. [DOI] [PubMed] [Google Scholar]
- Reiser-Erkan C., Erkan M., Pan Z., Bekasi S., Giese N.A., Streit S., Michalski C.W., Friess H., Kleeff J. Hypoxia-inducible proto-oncogene Pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2008;7:1352–1359. doi: 10.4161/cbt.7.9.6418. [DOI] [PubMed] [Google Scholar]
- Rosenberg L., Yoon C.H., Sharma G., Bertagnolli M.M., Cho N.L. Sorafenib inhibits proliferation and invasion in desmoid-derived cells by targeting Ras/MEK/ERK and PI3K/Akt/mTOR pathways. Carcinogenesis. 2018;39:681–688. doi: 10.1093/carcin/bgy038. [DOI] [PubMed] [Google Scholar]
- Shay K.P., Wang Z., Xing P.X., McKenzie I.F., Magnuson N.S. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol. Cancer Res. 2005;3:170–181. doi: 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
- Shi Q., Yin Z., Liu P., Zhao B., Zhang Z., Mao S., Wei T., Rao M., Zhang L., Wang S. Cilostazol suppresses IL-23 production in human dendritic cells via an AMPK-dependent pathway. Cell Physiol. Biochem. 2016;40:499–508. doi: 10.1159/000452564. [DOI] [PubMed] [Google Scholar]
- Signorelli J., Shah Gandhi A. Cobimetinib. Ann. Pharmacother. 2017;51:146–153. doi: 10.1177/1060028016672037. [DOI] [PubMed] [Google Scholar]
- Song H., Zhang J., Ning L., Zhang H., Chen D., Jiao X., Zhang K. The MEK1/2 inhibitor AZD6244 sensitizes BRAF-mutant thyroid cancer to vemurafenib. Med. Sci. Monit. 2018;24:3002–3010. doi: 10.12659/MSM.910084. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tai W.M., Yong W.P., Lim C., Low L.S., Tham C.K., Koh T.S., Ng Q.S., Wang W.W., Wang L.Z., Hartono S. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC) Ann. Oncol. 2016;27:2210–2215. doi: 10.1093/annonc/mdw415. [DOI] [PubMed] [Google Scholar]
- Ting W.C., Chen L.M., Huang L.C., Hour M.J., Lan Y.H., Lee H.Z., You B.J., Chang T.Y., Bao B.Y. Impact of interleukin-10 gene polymorphisms on survival in patients with colorectal cancer. J. Korean Med. Sci. 2013;28:1302–1306. doi: 10.3346/jkms.2013.28.9.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg Z.A., Alsina M., Soares H.P., Brana I., Britten C.D., Del Conte G., Ezeh P., Houk B., Kern K.A., Leong S. A multi-arm phase I study of the PI3K/mTOR inhibitors PF-04691502 and gedatolisib (PF-05212384) plus irinotecan or the MEK inhibitor PD-0325901 in advanced cancer. Target Oncol. 2017;12:775–785. doi: 10.1007/s11523-017-0530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.J., McCormack P.L. Trametinib: first global approval. Drugs. 2013;73:1245–1254. doi: 10.1007/s40265-013-0096-1. [DOI] [PubMed] [Google Scholar]
- Xu S.P., Sun G.P., Shen Y.X., Peng W.R., Wang H., Wei W. Synergistic effect of combining paeonol and cisplatin on apoptotic induction of human hepatoma cell lines. Acta Pharmacol. Sin. 2007;28:869–878. doi: 10.1111/j.1745-7254.2007.00564.x. [DOI] [PubMed] [Google Scholar]
- Yeh T.C., Marsh V., Bernat B.A., Ballard J., Colwell H., Evans R.J., Parry J., Smith D., Brandhuber B.J., Gross S. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- Yuan L.L., Green A.S., Bertoli S., Grimal F., Mansat-De Mas V., Dozier C., Tamburini J., Recher C., Didier C., Manenti S. Pim kinases phosphorylate Chk1 and regulate its functions in acute myeloid leukemia. Leukemia. 2014;28:293–301. doi: 10.1038/leu.2013.168. [DOI] [PubMed] [Google Scholar]
- Zhang X., Liu G., Ding L., Jiang T., Shao S., Gao Y., Lu Y. HOXA3 promotes tumor growth of human colon cancer through activating EGFR/Ras/Raf/MEK/ERK signaling pathway. J. Cell. Biochem. 2018;119:2864–2874. doi: 10.1002/jcb.26461. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Wang Z., Magnuson N.S. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol. Cancer Res. 2007;5:909–922. doi: 10.1158/1541-7786.MCR-06-0388. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Adjei A.A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or in Supplemental Information.