

Abstract
Objective
Investigations of autophagy in β-cells have usually focused on its homeostatic function. More dynamic roles in inhibiting glucose-stimulated insulin secretion (GSIS), potentially involving remodelling of cellular lipids, have been suggested from in vitro studies but not evaluated in vivo.
Methods
We employed temporally-regulated deletion of the essential autophagy gene, Atg7, in β-cells. Mice were fed chow or high-fat diets (HFD), in conjunction with deletion of Atg7 for the last 3 weeks (short-term model) or 9 weeks (long-term model). Standard in vivo metabolic phenotyping was undertaken, and 450 lipid species in islets quantified ex vivo using mass spectroscopy (MS). MIN6 cells were also employed for lipidomics and secretory interventions.
Results
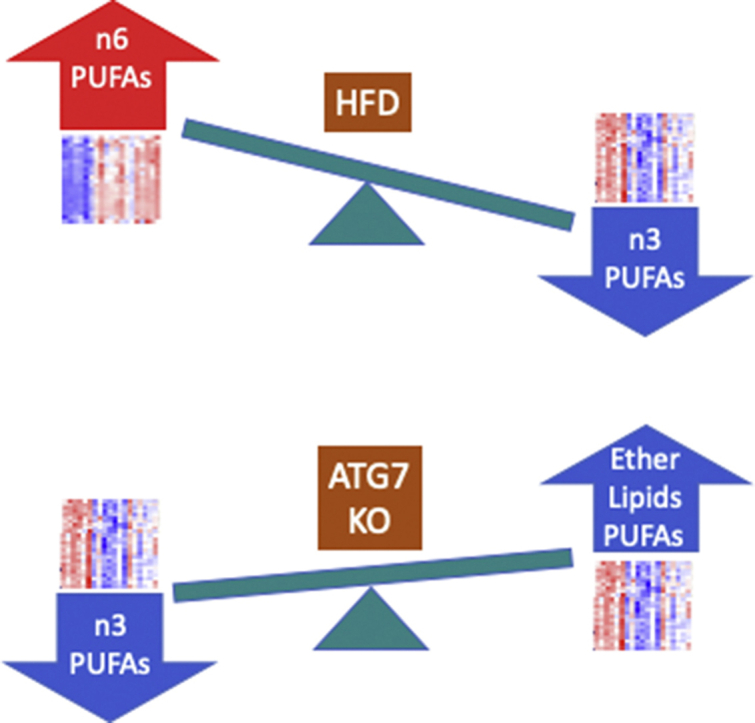
β-cell function was impaired by inhibiting autophagy in the longer-term, but conversely improved by 3-week deletion of Atg7, specifically under HFD conditions. This was accompanied by augmented GSIS ex vivo. Surprisingly, the HFD had minimal effect on sphingolipid and neutral lipid species, but modulated >100 phospholipids and ether lipids, and markedly shifted the profile of polyunsaturated fatty acid (PUFA) sidechains from n3 to n6 forms. These changes were partially countered by Atg7 deletion, consistent with an accompanying upregulation of the PUFA elongase enzyme, Elovl5. Loss of Atg7 separately augmented plasmalogens and alkyl lipids, in association with increased expression of Lonp2, a peroxisomal chaperone/protease that facilitates maturation of ether lipid synthetic enzymes. Depletion of PUFAs and ether lipids was also observed in MIN6 cells chronically exposed to oleate (more so than palmitate). GSIS was inhibited by knocking down Dhrs7b, which encodes an enzyme of peroxisomal ether lipid synthesis. Conversely, impaired GSIS due to oleate pre-treatment was selectively reverted by Dhrs7b overexpression.
Conclusions
A detrimental increase in n6:n3 PUFA ratios in ether lipids and phospholipids is revealed as a major response of β-cells to high-fat feeding. This is partially reversed by short-term inhibition of autophagy, which results in compensatory changes in peroxisomal lipid metabolism. The short-term phenotype is linked to improved GSIS, in contrast to the impairment seen with the longer-term inhibition of autophagy. The balance between these positive and negative inputs could help determine whether β-cells adapt or fail in response to obesity.
Keywords: Autophagy, High-fat diet, Insulin secretion and pancreatic islets
Graphical abstract
Highlights
-
•
High fat diets markedly increase the n6:n3 PUFA ratio in β-cells.
-
•
Three-week inhibition of autophagy in β-cells improves in vivo glucose metabolism.
-
•
This is accompanied by enhanced peroxisomal function, notably increases in ether lipids and n3 PUFAs.
-
•
Intervention in ether lipid synthesis similarly alters insulin secretion.
-
•
Impact of autophagy in β-cells depends on duration and dietary context.
Abbreviations
- Atg7
autophagy related 7
- CE
cholesteryl ester
- DG
diacylglycerol
- ER
endoplasmic reticulum
- GSIS
glucose-stimulated insulin secretion
- GTT
glucose tolerance test
- HFD
high-fat diet
- KO
knockout
- KRHB
Krebs Ringer Hepes Buffer
- LAL
lysosomal acid lipase
- LC3
microtubule-associated protein 1 light chain 3
- MS
mass spectrometry
- n-3
omega-3
- n-6
omega-6
- MHC
monohexosylceramide
- MUFA
monounsaturated FA
- (O)
alkyl lipid
- (P)
plasmalogen
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PUFA
polyunsaturated fatty acid
- SFA
saturated FA
- T2D
type 2 diabetes
- TG
triacylglycerol
- WT
wildtype.
1. Introduction
Autophagy is a cellular degradation mechanism that is important for cell growth and survival, and which serves to remove impaired organelles and protein aggregates [1,2]. It is particularly critical, under states of cellular stress, for the elimination of toxic substances and for the provision of catabolites that are salvaged as metabolic fuels. Autophagy is therefore tightly regulated to maintain optimal cellular homeostasis, and sustained or dysregulated autophagy is likely to trigger programmed cell death [1,2].
Defective autophagy is implicated in multiple human diseases, including Type 2 diabetes (T2D) [[3], [4], [5]]. The loss of glucose homeostasis defining this condition is ultimately caused by the failure of pancreatic β-cells to secrete sufficient insulin to overcome the loss of insulin sensitivity that occurs during obesity [6,7]. At a cellular level, there are multiple contributors to this β-cell failure: loss of β-cell mass due to apoptosis; reductions in insulin content; impaired processing of pro-insulin to mature insulin; and disruption of the metabolic and signalling pathways whereby release of insulin is acutely triggered in response to glucose stimulation [8]. T2D is also associated with changes in morphology and gene expression in human β-cells that are consistent with alterations of autophagy [9]. This has been complemented with evidence from mouse studies in which the essential autophagy gene, Atg7, was specifically deleted in β-cells [[10], [11], [12], [13], [14]]. The emerging view is thus that autophagy is essential for maintaining β-cell function, especially in the context of obesity or other stressors [[3], [4], [5]]. Indeed, activation of autophagy could constitute part of the normal protective mechanism whereby β-cells initially compensate for the extra demands of obesity. This is potentially explained by the capacity for autophagy to inhibit endoplasmic reticulum (ER) stress, which is one mechanism postulated to contribute to β-cell failure in T2D [[15], [16], [17]].
In contrast, some in vitro studies have suggested that autophagy is inhibitory for glucose-stimulated insulin secretion (GSIS) in the short term [[18], [19], [20]]. Underlying mechanisms potentially include an enhanced degradation of proinsulin [19,20]. We have also highlighted a negative role for lipophagy [18], a specialized form of autophagy, whereby neutral lipid stores (in the form of lipid droplets) are catabolized to generate free fatty acids (FAs) [21]. We proposed that this depletes the lipid stores in β-cells that are otherwise mobilized in response to glucose as part of the mechanism contributing to stimulus-secretion coupling [18]. However, a separate line of evidence suggests that the expansion of neutral lipid stores during obesity or high-fat feeding is beneficial to β-cells, sequestering FAs in a relatively inert form such that they are not incorporated in toxic intermediates such as the sphingolipid or ceramide [22]. Conversely, cellular damage also results when these storage mechanisms are overwhelmed [23,24]
As such, neutral lipid and sphingolipid metabolism have been intensively investigated in β-cells in conjunction with lipotoxicity models, whereby chronic cellular treatment with saturated FAs (SFAs) (such as palmitate) is employed to recapitulate many of the features of β-cell failure, including apoptosis and impaired insulin secretion [7,15,22]. In contrast, mono-unsaturated FAs (MUFAs), such as oleate, are protective against apoptosis [22,25], although they do disrupt GSIS [26]. Notably, we have recently shown that oleate is more potent than palmitate in promoting autophagy [27]. Polyunsaturated FAs (PUFAs) have been less well studied in β-cells. These are derived from C18 dietary precursors and undergo extensive elongation and desaturation in two parallel pathways to form omega-3 (n-3) and omega-6 (n-6) PUFAs respectively, depending on the positioning of the first double bond (see Figure 4C) [28,29]. PUFAs are particularly enriched in abundant phospholipids, such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE). In canonical phospholipids, the two FA side chains are linked to the glycerol backbone by esterification, but alternatively the FA in sn1 position can be attached by one of two forms of ether bond, to generate alkyl lipids and plasmalogens, respectively. These ether lipids are highly enriched in PUFAs at the sn2 position and display differences in both their biophysical properties and cellular roles compared to esterified lipids [30,31]. They have been detected in β-cells [[32], [33], [34]] and shown to be turned over in response to acute glucose stimulation [33], but their cellular function has not been addressed. Critical steps in both PUFA maturation and ether lipid synthesis occur in peroxisomes, in addition to the better-known role that organelle plays in oxidation of very long chain FAs [35,36].
Figure 4.

Short-term inhibition of autophagy increases mass of n-3 PUFA species of PC and PE that are selectively depleted by HFD. Mice were dieted and treated with tamoxifen as indicated in the legend to Figure 1. Islets were isolated and total cellular lipids extracted and quantified by MS. Data are means ± SEM of five to seven independent values. Shown are mass (per mg of cellular protein) of PC and PE containing sidechains of: (A) 22:6 n-3; (B) 22:5 n-6; (D) 22:5 n-3; (E); 20:5 n-3 (F) 20:3 n-6; and 22:4 n-6 (E). Also shown is (H) a representative image and densitometric quantification of expression of Elov5 and (C) n-3 and n-6 PUFA pathways. LA, linoleic acid; ALA, α-linoleic acid; EPA, eicosapentaenoic acid; ARA, arachidonic acid; DHA, docosahexaenoic acid. Statistical analyses (A–G) were by two-way ANOVA with Tukey post hoc test, or (H) one-way ANOVA with Sidak post hoc test. ##P < 0.005, ###P < 0.0005, ####P < 0.0001 versus chow control, or ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.0005, ∗∗∗∗P < 0.0001 as indicated.
To date the in vivo functions of autophagy in β-cells have been mainly assessed using standard models for gene deletion where Atg7 (for example) is absent from birth [10,11,13,17]. This approach is not well suited to addressing alternative, potentially more subtle and dynamic roles. To circumvent these issues, we have used a conditional and tissue-specific knockout mouse model and compared the outcomes of short-versus long-term autophagy inhibition, with a particular emphasis on lipid metabolism. Our results demonstrate that β-cell function is impacted very differently depending on the context and duration of autophagy inhibition. Surprisingly, our study also highlights novel links between autophagy, the metabolism of PUFAs and ether lipids, and the dysregulation of insulin secretion during lipid oversupply.
2. Material and methods
2.1. Animals
All studies were approved by the Garvan/St. Vincent's Hospital Animal Ethics Committee. The β-cell-specific conditional Atg7 knockout mice were generated by breeding Pdx1-CreER+ mice [37] with Atg7flox (Atg7f/f) mice (RIKEN, Wako, Saitama, Japan). To delete Atg7 from the β-cells, Cre induction was performed by subcutaneous injection of tamoxifen (8 mg/day) for 2 days consecutively [38]. The knockout mice were denoted as Atg7f/f:Pdx1-CreER+ (KO) and their control littermates were Atg7f/f (WT), although in some instances Pdx1-CreER+ controls were used.
Mice were fed ad libitum with a standard chow (10.88 kJ/g; 8% fat, 21% protein and 71% carbohydrate) or a lard and sucrose supplemented HFD (19.67 kJ/g; 45% fat, 20% protein and 35% carbohydrate), based on diet D12451 (Research Diets, New Brunswick, NJ, USA). After 5 weeks, starting from 8-weeks old, the mice were all injected with tamoxifen and then studied after a further 3 or 9 weeks as short or long-term deletion models.
2.2. In vivo mouse studies
Oral glucose tolerance tests (GTT; 3 g glucose/kg of body weight) were performed after a 6-hour fast. Plasma insulin was measured with the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem, IL, USA). Total fat mass was determined by dual energy X-ray absorptiometry (DEXA) using a PIXImus small-animal densitometer and associated software (PIXImus II; GE Medical Systems, Madison, WI, USA).
2.3. Islet isolation, insulin secretion assays and proinsulin content
Mouse islets were isolated as described previously [38]. For the GSIS assay, batches of five islets were picked with at least four replicates per groups and were preincubated for 1 h in Krebs–Ringer HEPES buffer (KRHB) containing 0.1% (wt/vol.) BSA and 2 mmol/l glucose. Then they were incubated for 1 h at 37°C with KRHB containing either 2 or 20 mmol/l glucose. An aliquot of the buffer was taken, and secreted insulin determined by the insulin RIA kit (Linco/Millipore, Billerica, NJ, USA). The islets were lysed for the measurement of insulin and proinsulin content by the homogeneous time-resolved fluorescence assay using the Insulin Ultra Sensitive Assay Kit (Cisbio, Codolet, France) and the Proinsulin rat/mouse ELISA kit (Mercodia, Uppsala, Sweden), respectively.
2.4. Cell culture and treatments
MIN6 insulinoma cells (passages 29–37) were maintained at 37 °C and 5% CO2 in DMEM (25 mM glucose), supplemented with 10% FCS, 10 mM HEPES, 50 units/ml of penicillin and 50 μg/ml streptomycin [26,39]. Cells were seeded 4 × 105 cells per well in 12-well plates, or at 1 × 106 in 6-well plates. For lipid treatments, cells were exposed to either oleate or palmitate (both 0.4 mM final pre-coupled to 0.92 g BSA/100 ml), or BSA control in DMEM (5 mM glucose) for 48 h [26]. In some studies, GFP-control, or short and long variants of Myc-DDK-tagged mouse Dhrs7b/PexRAP (OriGene Technologies, Rockville, MD, USA) were overexpressed 24 h prior to lipid treatments using Nanojuice transfection reagent (Merck Millipore, Bayswater, Victoria, Australia) [39]. For knockdown studies, MIN6 cells were transfected with control non-targeting siRNA or Dhrs7b ONTARGETplus SMARTpool siRNA, using Dharmafect3 reagent (Dharmacon, Pittsburgh, PA, USA) in DMEM (25 mM glucose). Media was replaced after 24 h and cells maintained for a further 48 h [18]. GSIS was measured in KRBH in 2.8 or 20 mM glucose for 1 h, and insulin quantified using a FRET-based Insulin Ultra-Sensitive Assay (Cisbio, Codolet, France).
2.5. Lipidomics and lipid nomenclature
Analyses of MIN6 cells (2 x 6-well plates per treatment) were exactly as described previously [33]. Islets (200 islets per replicate) were pelleted in ice cold PBS after isolation. Samples (50 μg of protein) were extracted [40] and then analysed using an expanded methodology as recently described [41]. Briefly, cells were suspended in 10 μL PBS and extracted in a single-phase extraction using an addition of 20 times volume of chloroform:methanol 2:1 containing lipid internal standards, followed by sonication and centrifugation to pellet any protein precipitate. The supernatant was removed and dried under vacuum before the samples were reconstituted in equal volumes of water saturated butanol and 10 mM ammonium formate in methanol. Samples were run through an Agilent 1200 HPLC, with an Agilent 2.1 × 100 mm C18 column, coupled to an AB SCIEX Qtrap 4000 mass spectrometer and subjected lipidomic analysis as previously described [33,39]
The lipid naming convention used in this study follows the guidelines of the Lipid Maps Consortium with subsequent additions [42,43]. Glycerophospholipids typically contain two fatty acid chains and in the absence of detailed characterisation are expressed as the sum composition of carbon atoms and double bonds (i.e. PC(38:6)). However, where the acyl chains have been determined but the position is unknown, we have employed an underscore between the acyl chains (i.e. PC(38:6) becomes PC(16:0_22:6)). Where the position of the acyl chains is known, the acyl chains are separated by a “/” with the sn1 and sn2 acyl chains in order (i.e. PC(16:0_22:6) becomes PC(16:0/22:6)). This is also extended into other lipid classes and subclasses. In some cases, lipid species may be separated chromatographically but incompletely characterised. Here we have adopted an (a) and (b) nomenclature, for example PC(P-18:1/22:6) (a) and (b) where (a) and (b) represent the elution order [41]. In this instance we believe this represents the positional isomers of the n7 and n9 double bonds on the 18:1 alkenyl chain, although suitable standards to verify this are not currently available.
2.6. Western blotting
Protein extracts from isolated islets were resolved on a 12% SDS-PAGE gel and transferred onto a PVDF membrane. After blocking with 5% non-fat milk, the membrane was incubated with primary antibodies against: Atg7 (Cell Signalling Technology, Danvers, MA, USA), LC3, Elovl5 (Abcam, Cambridge, UK), or β-actin (Sigma) overnight at 4°C, and then probed with peroxidase-labelled secondary antibodies. Chemiluminescence intensity of bands from autoradiography film was quantified using Image J software (National Institute of Health, Bethesda, MD, USA).
2.7. Histology and immunohistochemistry
Mouse pancreata were harvested and fixed in 4% paraformaldehyde in PBS overnight, followed by 30% sucrose incubation at 4°C for 6 h. The tissues then were embedded in paraffin for sectioning. Sections were subjected to either hematoxylin and eosin (H&E) or immunofluorescent staining. Primary antibodies used in the immunofluorescent staining were mouse anti-insulin (Sigma), rabbit Pmp70, rabbit anti-p62 (Enzo Life Sciences, Farmingdale, NY, USA). Secondary antibodies used were Alexa 647-labeled anti-mouse or Alexa 488-labeled anti-rabbit (Molecular Probes, Eugene, OR, USA). The sections were mounted with ProLong® Gold antifade reagent containing DAPI (Molecular Probes). The fluorescence signal was detected by fluorescence microscopy (DM5500; Leica Microsystems, Sydney, NSW, Australia). For detection of apoptosis, sections were stained using mouse anti-insulin antibody (Sigma) and the Click-iT™ TUNEL Assay kit (Thermo Fisher Scientific) as per the manufacturer's protocol. Sections were counterstained with DAPI (Molecular Probes) and mounted using Fluoromount™ Mounting Medium (Sigma Aldrich). Images were captured on a Leica DM 6000 microscope using LASX software. Total cell numbers were quantified using Fiji software and TUNEL positive cell staining was quantified manually. Staining for Pmp70 was quantified using Image J. Briefly, the threshold was set for each image to define the area of the islet as a region of interest. The region of interest was then applied to the Pmp70 channel and the positive pixels were counted using integrated density normalised to islet area.
2.8. mRNA analyses
RNA was extracted using an RNEasy kit (Qiagen, Melbourne, Australia). RNA concentration was determined using the Nanodrop and reverse transcription was performed using the QuantiTect® Reverse Transcription kit (Qiagen). RT-PCR was performed using the LightCycler 480 instrument with UPL hydrolysis probes (Roche, North Ryde, Australia). Primer sequences are provided in Supplementary Table 1. The Ct values were determined using the absolute quantification/2nd derivative maximum function using the LightCycler480 software. Relative gene expression was then calculated using the 2−ΔΔCt method, with TBP as the housekeeping gene.
2.9. Statistical analysis
Heatmaps, cluster analysis (Pearson Correlation) and t-tests for Figure 4 were generated with MultiExperiment Viewer (MeV_4_8_1) [44]. Calculations of the integrated area under the curve, and all other statistics were performed using GraphPad Prism 8 software (GraphPad software La Jolla, CA, USA), as detailed in the figure legends.
3. Results
3.1. Mouse model for β-cell specific, inducible inhibition of autophagy
We made use of the inducible Pdx1CreER+ model, which allows for temporally controlled deletion of Atg7 in β-cells in adult mice [37]. Because the Atg7 protein has a long half-life (∼1 week) [45], initial studies focused on optimizing the shortest window for inhibiting autophagy without compromising the extent of deletion. Atg7 deletion in the islets was apparent within 2 weeks of tamoxifen injection (40% loss), and maximal loss (60%) occurred by 3 weeks (Supplementary Figure 1). The residual Atg7 expression was probably due to other (non-β) cell types in the islets. Because Atg7 is required for the conversion of microtubule-associated protein 1 light chain 3 (LC3) I into the autophagic marker LC3II, these downstream proteins were also assessed. At 3 weeks post injection accumulation of LC3II was decreased, together with a reciprocal increase in the LCI precursor (Supplementary Figure 1), thereby confirming a functional inhibition of autophagy.
3.2. Prolonged inhibition of autophagy in adult mice compromises β-cell function
To test the effects of Atg7 deletion in adult β-cells, we used mice pre-fed a chow or HFD for 5 weeks, then injected with tamoxifen and maintained on their respective diets for another 9 weeks. Staining for p62, which accumulates during the inhibition of autophagy, was not detected in WT islets (Supplementary Figure 2A,i,ii), but visible in the KO islets (Supplementary Figure 2A,iii,iv). Embryonic deletion of Atg7 has been shown to promote the appearance of cyst-like structures in islet cells [10,11]. These cysts were also detected in our inducible model, most notably in combination with high-fat feeding (Supplementary Figure 2Biv). To test the metabolic correlates of these morphological changes we next conducted GTTs. As expected, the HFD impaired glucose tolerance in WT mice, and this was exacerbated by the loss of Atg7 (Supplementary Figure 2C and D). The additional impairment was accompanied by a significant inhibition of insulin release during the GTT, when comparing the WT and KO animals on HFD (Supplementary Figure 2E). Broadly speaking, both these functional and morphological changes are consistent with earlier studies [10,12,13,46], suggesting firstly that our inducible system works as expected, and secondly that these consequences of Atg7 deletion are independent of potential roles in early β-cell development.
3.3. Short-term autophagy deficiency during prolonged high-fat feeding improves glucose tolerance and insulin secretion
We next examined in vivo glucose homeostasis under conditions where the duration of Atg7 deletion was shortened to 3 weeks. No differences between WT and KO mice were apparent immediately prior to tamoxifen injection, although the preceding 5 weeks of HFD did impair glucose-tolerance as expected (Supplemental Figs. 3A and C). Loss of Atg7 did not alter overall adiposity (Figure 1A) but improved both glucose tolerance (Figure 1B,C) and insulin excursions (Figure 1D), but only in the mice maintained on the HFD. This is the opposite of the results obtained in the longer-term deletion models (Supplementary Figure 2B) and was observed irrespective of comparing the KO responses to Pdx1-CreER+ controls (Supplemental Figs. 3B and D), or Atg7f/f controls (Figure 1). Consistent with the in vivo results, GSIS was augmented by Atg7 deletion in β-cells, but only in those islets isolated from mice fed the HFD (Figure 1E). In contrast, there was no significant alteration in the actual contents of either insulin (Figure 1F), or proinsulin (Figure 1G). β-cell apoptosis was likewise unaffected by the loss of ATG7 (Figure 1H, Supplementary Figure 4A). Collectively, these results suggest that, in contrast to the effects of longer-term deletion of ATG7, short-term inhibition of autophagy directly improves the secretory responsiveness of β-cells to glucose stimulation.
Figure 1.

Short-term (3 weeks) inhibition of autophagy in mouse β-cells improves glucose tolerance and augments insulin secretion after high-fat feeding. Atg7f/f:Pdx1-CreER+ (KO) or Atg7f/f:WT (Control) mice were fed with chow or HFD starting at 8-weeks and then injected with 8 mg/day tamoxifen consecutively for 2 days at 13-weeks. (A) DEXA scans for fat mass, (B,C) oGTT, and (D) insulin excursions were performed at 16 weeks. Mouse islets were isolated for (E) GSIS, (F) insulin content and (G) proinsulin content. (H) Pancreas was sectioned for analysis of TUNEL staining. Data shown are the mean values ± SEM from three cohorts with a total of seven to nine (A–F) or three to four mice (G). (B,D) Statistical analyses were by two-way ANOVA with Sidak post hoc test #P < 0.05 and ####P < 0.0001 for the overall effect of genotype in the HFD cohorts, and ∗P < 0.05 or ∗∗∗P < 0.0005 for differences between HFD WT and HFD KO at individual points as indicated. One-way ANOVA with Sidak post hoc test, ∗∗∗P < 0.005, ∗∗∗∗P < 0.0001 versus corresponding chow value (A), or ∗∗∗P < 0.0005, ∗∗∗∗P < 0.0001 as indicated (C,E).
3.4. Minimal effects of HFD or short-term autophagy deficiency on sphingolipids and neutral lipids in β-cells
In vitro studies have established that inhibition of lipophagy augments GSIS by expanding a reservoir of neutral lipids, which is subsequently mobilized to generate coupling factors during acute glucose stimulation [18]. We therefore undertook a comprehensive lipidomic analysis to determine the in vivo effects of both HFD and Atg7 deletion in islets. Summary data for the 474 individual lipid species that were detected are shown in Supplementary Table 2. Surprisingly, we found no evidence for changes in the total mass of major neutral lipids in response to diet, although diacylglycerol (DG) was augmented by genotype (Figure 2A). This is addressed further below. Likewise, there were no alterations in the total mass of various sphingolipids due to the HFD (Figure 2B). In contrast, the loss of Atg7 increased levels of both ceramide and its immediate glycosylated derivative, monohexosylceramide (MHC) (Figure 2B). This was most apparent in abundant FA species such as C16:0 (Figure 2C,D). Although our earlier in vitro studies had highlighted the benefits of enhancing MHC [39,47], the accumulation of ceramide in the Atg7 KO islets seems inconsistent with improved secretory function [48,49]. Atg7 deletion impacted only modestly on total phospholipid mass with enhancement of relatively minor forms such as phosphatidylglycerol (PG) and lyso-phosphatidylinositol (LPI) (Figure 2E). Most surprisingly, however, our results revealed a profound effect of genotype on ether lipids, notably an augmentation of both alkyl (O) and plasmalogen (P) forms of PC (Figure 2F).
Figure 2.

Short-term inhibition of autophagy alters total mass of phospholipids and ether lipids in mouse β-cells more than sphingolipids or neutral lipids. Mice were dieted and treated with tamoxifen for 3 weeks as indicated in the legend to Figure 1. Islets were isolated and total cellular lipids extracted and quantified by MS. Data are means ± SEM of 5–7 independent values. Masses of total species of (A) NLs, (B) SLs, (E) PLs or (F) ether lipids were summed, and expressed relative to the corresponding chow control. Mass of major species of (C) ceramide or (D) MHC per mg of cellular protein are shown. Statistical analyses were by two-way ANOVA with Tukey's (A,B,E,F) or Dunnett's (C,D) post hoc tests. #P < 0.05, ##P < 0.005, ####P < 0.0001 versus chow control, or ∗P < 0.05, ∗∗P < 0.005, as indicated.
3.5. High-fat feeding and short-term autophagy deficiency impact predominately on PUFA-containing lipids in β-cells
A complementary analysis, focusing on the individual species that were significantly altered by diet, revealed a more subtle remodelling of phospholipids (Figure 3A) than was apparent from the summed mass data (Figure 2E). Indeed, of the 134 metabolites that were increased or decreased between the chow and HFD groups, phospholipids comprised >90%, mostly canonical PC or PE or their ether lipid equivalents (Figure 3A). Moreover, in terms of fatty acid side-chain, these were overwhelmingly enriched in PUFAs, especially the n-3 species, 22:6 (Figure 3B). Overall, these individual species clustered into three distinct groups (Figure 3C): i) a large set encompassing mainly 18:2-esterified phospholipids and 22:6 plasmalogens that were depleted by HFD in WT but partially reverted by loss of Atg7; ii) a cohort increased by diet independently of genotype, mostly n-6-containing (22:4, 22:5) plasmalogens; and iii) a more diverse set, albeit enriched for 20:5 and some 22:6 species, which were reduced by diet regardless of genotype.
Figure 3.

HFD predominately alters PUFA species of phospholipids and ether lipids, and these are variously impacted by short-term inhibition of autophagy. Mice were dieted and treated with tamoxifen for 3 weeks as indicated in the legend to Figure 1. Islets were isolated and total cellular lipids extracted and quantified by MS. Data are from five to seven independent values. Individual lipid species were tested for significant modulation by HFD (P < 0.05 unpaired t-test, corrected for multiple comparison with 0.05 false discovery rate. (A) Shown are significantly altered species categorized by (A) lipid class or (B) side-chain. (C) Cluster analysis (Pearson correlation) and heat map (Log2 and row adjusted) of significantly altered species: i) decreased by HFD partly reverted by ATG7 deletion ii) increased by HFD independently of ATG7 deletion, and iii) decreased by HFD independently of ATG7 deletion.
We next analysed the alterations in PUFA-containing species of canonical PC and PE more closely. In WT mice, the HFD exerted reciprocal effects on the end-products of the n-3 and n-6 pathways (see Figure 4C) with 22:6 sidechains diminished (Figure 4A) and 22:5 species (Figure 4B) augmented. In contrast to the effects of diet, the loss of ATG7 appeared to impact solely on the n-3 pathway, partially (30–40%) reverting the loss of 22:6 sidechains due to the HFD in WT mice (Figure 4A). Tracking backwards up this pathway (Figure 4C), we determined that the effect of ATG7 deletion was also apparent in n-3 22:5 species (Figure 4D), but not their immediate 20:5 precursors (Figure 4E). We also checked upstream metabolites in the n-6 pathway, which shares enzymes with that of the n-3 sequence (Figure 4C). Indeed, 20:3 (Figure 4F) and 22:4 intermediates (Figure 4G) were also augmented by the loss of ATG7, even though final n-6 22:5 products were unaltered (Figure 4B). These findings would implicate elongase enzymes, in particular ELOVL5, as a site potentially upregulated by ATG7 inhibition. Indeed, protein expression of this enzyme was augmented in islets of ATG7 KO mice, independently of diet (Figures 4H). mRNA levels corresponding to this enzyme, and other components in the pathway, were unaltered by the inhibition of autophagy (Supplementary Figure 5). Collectively these results show that high-fat feeding promotes an n-3 to n-6 switch in islet PUFA accumulation, which is partially countered via a selective enhancement of the n-3 pathway when autophagy is inhibited.
3.6. Short-term autophagy deficiency augments ether lipids, in ways additional to the repletion of PUFA sidechains
We next addressed the ether lipids, focusing on the n-3 PUFA metabolites since n-6 PUFA sidechains were only detected in very low abundance (not shown). We firstly established that the 22:6-containing ether lipids were also reduced by the HFD (Figure 5A–D), but to a lesser extent than the 60% decrease observed in their canonical PE and PC equivalents (Figure 3A). This decrement was strongly reverted by ATG7 deletion, especially in the alkyl lipids (60–70% protection) (Figure 5A,C). We reasoned that this might be explained not only by the sparing of PUFA sidechains (as in the canonical phospholipids) but also by separate and more specific effects of autophagy on the turnover of ether lipids, per se. We therefore focused on alterations in SFA or MUFA sidechains, to exclude contributions due to remodelling of PUFAs (Figure 5E–G). The canonical forms of PC and PE were only modestly reduced by the HFD, an effect mainly explained by the loss of 18:1-containing, rather than di-saturated species. This reduction was partially overcome by the loss of Atg7 (Figure 5E). SFA and MUFA species of ether lipids (Figure 5F,G) were unaltered by diet in WT mice but enhanced in the Atg7 KOs, an effect that was most pronounced in the alkyl lipid species (Figure 5F) but also present in plasmalogens (Figure 5G). This clearly suggests that autophagy normally restricts ether lipid synthesis, acting upstream of the generation of alkyl derivatives. These proximal steps also overlap with a salvage pathway (see Figure 6H) for forming phospholipids and potentially DG (by generation of lysophosphatidic acid), as would be consistent with an enhancement of total DG mass following Atg7 deletion (Figure 2A). This was now confirmed in terms of abundant SFA, and MUFA species (Figure 5H), revealing a similar pattern to that observed with the corresponding PC/PE(O) sidechains (Figure 5F).
Figure 5.

Short-term inhibition of autophagy increases mass of ether lipids independently of side-chain saturation. Mice were dieted and treated with tamoxifen as indicated in the legend to Figure 1. Islets were isolated and total cellular lipids extracted and quantified by MS. Data are means ± SEM of five to seven independent values. Shown are mass of major species (per mg of cellular protein) of PUFA-containing ether lipids (A–D); and SFA- and MUFA-containing canonical lipids (E), ether lipids (F,G) and DAG (H). Statistical analyses were by two-way ANOVA with Tukey post hoc test. #P < 0.05, ##P < 0.005, ####P < 0.0001 versus chow control, or ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.0005, ∗∗∗∗P < 0.0001 as indicated.
Figure 6.

Lonp2, a regulator of peroxisomal function, is enhanced by deletion of Atg7. Mice were dieted and treated with tamoxifen as indicated in the legend to Figure 1. (A) Double immunofluorescence staining for insulin (red) and Pmp70 (green) from representative images as indicated of pancreatic sections from chow control, HFD control, and HFD KO mice. Scale bar = 25 μm. (B) Quantification of Pmp70 IHC staining. (C–G) qPCR of islet gene expression as indicated. (H) Pathways of peroxisomal lipid metabolism. Abbreviations: LCF, long chain fatty; DHAP, dihydroxyacetone phosphate; G3P, glycerol 3-phosphate; LPA, lysophosphatidic acid; PL, phospholipid. (I–K) Lipidomic signatures of peroxisomal β-oxidation. Results are means and SEM of metabolite ratios calculated from 6 to 8 individual mice. Statistical analyses were by two-way ANOVA with Tukey post hoc test (D,E) or by one-way ANOVA with Sidak post hoc test (I–K) ∗P < 0.05, ∗∗∗∗P < 0.0001 as indicated.
3.7. Short-term autophagy deficiency augments peroxisomal lipid metabolism in β-cells
We hypothesised that the increased accumulation of ether lipids, and potentially terminal PUFA metabolites, could be explained by an expansion of peroxisomes, the site of key steps in both these pathways [35,36]. Indeed, such an expansion might be expected following the loss of ATG7 because peroxisomes are normally degraded by a form of autophagy (pexophagy) [50]. However, there was no effect of either diet or genotype on peroxisomal area, as assessed by the marker, peroxisomal membrane protein 70 (PMP70) (Figure 6A,B). Expression of the corresponding gene (Abcd3) was also unaffected under these conditions (Figure 6C), although a loss of ATG7 in the KO samples was confirmed (Figure 6D). Independently of size, functional defects have been recently attributed to mutations in enzymes of peroxisomal β-oxidation, which are characterized not only by the expected accumulation of very long-chain FAs but also by the loss of ether lipids and n-3 22:5-containing PUFAs [51]. Specific lipidomic signatures were developed to dissociate changes restricted to loss of particular enzymes, versus those occurring in common, regardless of the targeted enzyme [51]. Unfortunately, only three of these ratios could be calculated from FA species present in our data set, but all were increased by the HFD in WT islets (Figure 6I–K). The shift in SM species, commonly observed in general instances of peroxisomal disruption [51], was completely reverted by the loss of ATG7 (Figure 6I). In contrast, the two PC signatures, diagnostic of more specific defects in Acox1 and/or Hsd17b4, were similar between the two genotypes (Figure 6J,K). Collectively, these results provide strong evidence that peroxisomal function is impaired by the HFD, and that some, but not all aspects of this impairment can be rescued in the absence of autophagy. We next analysed mRNA of key genes (Figure 6H) that might account for these features but found no significant changes due to genotype in components of either β-oxidation or ether lipid synthesis (Figure 6F,G). In contrast, the loss of ATG7 enhanced the expression of the peroxisomal chaperone/protease, Lonp2 (Figure 6E). This encodes a protein that is essential for the degradation of oxidized proteins, but also the maturation of many enzymes targeted to the peroxisomal matrix, including those for peroxisomal β-oxidation and synthesis of ether lipids [52]. Thus the upregulation of Lonp2 potentially contributes to the overall lipid remodelling phenotype we observe under conditions of inhibited autophagy.
3.8. Impaired synthesis of ether lipids is linked to defects in GSIS due to FA oversupply
MIN6 cells have been widely employed for studying the mechanisms whereby chronic exposure to FAs, generally palmitate, causes β-cell dysfunction [26,27,47], but ether lipids have not been addressed. We now found that total alkyl lipids and plasmalogens were consistently diminished by 48 h of exposure to moderately elevated levels of oleate, whereas palmitate only reduced PC(O) species (Figure 7A). Changes in canonical phospholipids were less apparent, although oleate did slightly suppress PE (Figure 7B). Further analysis of the ether lipid species that were significantly altered by these FAs revealed that oleate downregulated many more species than it increased (Figure 7C). In contrast, the effects of palmitate were both less obvious and more variable. (Figure 7C). Quantitative evaluation confirmed that most ether lipids were selectively depleted by oleate, regardless of whether they contained saturated, mono-unsaturated, or 22:6 sidechains (Figure 7D,E,F). An exception was the abundant alkyl lipid PC(O-34:1) that was reduced by both FAs (not shown). Although effects of mass action cannot be excluded completely in this model, the presence of species with mono-unsaturated sidechains amongst those depleted in response to oleate treatment, would suggest a specific disruption in ether lipid synthesis rather than a competition between FAs. The precise mechanism of action of oleate, however, remains to be determined as it was not associated with alterations in the expression of Lonp2, or of genes encoding enzymes of ether lipid synthesis or peroxisomal β-oxidation (not shown).
Figure 7.

Ether lipid accumulation in MIN6 cells is preferentially inhibited by pre-treatment with oleate compared to palmitate. MIN6 cells were cultured for 48 h with 0.4 mM oleate or palmitate complexed to 0.9% BSA. Total cellular lipids were extracted from cells and quantified by MS. Data are means ± SEM of four independent values. Shown are summed masses of total species of (A) ether lipids, or (B) canonical phospholipids, expressed relative to the corresponding BSA control. (C) Individual species of ether lipid significantly modulated by FA pre-treatment (P < 0.05 unpaired t-test, corrected for multiple comparison with 0.05 false discovery rate), are presented using cluster analysis (Pearson correlation) and heat map (Log2 and row adjusted). (D–F) Mass of major species of SFA- and MUFA-containing alkyl lipids (D), plasmalogens (E), or PUFA-containing plasmalogens (F) are shown per mg of cellular protein. Statistical analyses were by two-way ANOVA with Tukey post hoc test. #P < 0.05, ###P < 0.0005, ####P < 0.0001 versus chow control, or ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.0005 as indicated.
Regardless of the exact mechanism, the selectivity of oleate versus palmitate suggests that this model could be exploited to investigate the role of ether lipids in regulating insulin secretion. As the point of intervention we chose Dhrs7b, which encodes the final enzyme (PexRAP) in the peroxisomal portion of the pathway of ether lipid synthesis (Figure 6H) [53,54]. Knocking down this enzyme (Figure 8A) inhibited GSIS (Figure 8B) without any change in the total cellular content of insulin (Figure 8C). We next investigated whether overexpression of Dhrs7b could protect against secretory dysfunction due to chronic lipid exposure. We employed two constructs, a shorter form previously shown to augment ether lipid synthesis [53], as well as a longer version whose function remains uncharacterized (Figure 8D). Notably the active form (i) reverted defective GSIS due to pre-treatment of MIN6 cells with oleate, but not that due to palmitate (Figure 8E). The longer form of Dhrs7b (ii) was without effect, and neither construct altered insulin content (Figure 8F). These results suggest that the defects in GSIS due to chronic exposure to oleate are mediated, at least in part, by a depletion of ether lipids.
Figure 8.

Dhrs7b expression in MIN6 modulates GSIS. MIN6 cells were maintained under standard culture conditions (A–D) or for 48 h with 0.4 mM oleate or palmitate complexed to 0.9% BSA (E,F). Cells were treated with siRNA for Dhrs7b and extracts analysed for (A) mRNA, (B) GSIS or (C) total insulin content. Cells overexpressing the shorter (i) and longer (ii) Dhrs7b constructs were extracted for western blotting (D) and analysed for (E) GSIS or (F) total insulin content. Data are means ± SEM of the number of independent values as shown. Statistical analyses were by unpaired Students t-test (A), or one-way ANOVA with Sidak's post hoc test (B–F) ∗P < 0.05, ∗∗P < 0.005 as indicated.
4. Discussion
Although autophagy was the initial focus of our investigation, the first major finding relates more generally to high-fat feeding, per se. Thus an HFD promoted the accumulation of n-6 linked phospholipids in islets, together with a marked depletion of n-3 FA sidechains. This is important because n-3 FAs are generally thought to be beneficial, through anti-oxidant, anti-inflammatory and other properties [29]. Notably, a whole-body transgenic mouse in which n-3 PUFAs were enriched at the expense of n-6 varieties showed improvements in insulin secretion [55] and protection against islet ER stress due to an HFD [56]. Benefits of supplementing β-cells with docosahexaenoic acid (22:6; DHA) and other n-3 FAs have also been reported, both in vivo and in vitro, and potentially explained by receptor-dependent [57] and independent effects [58]. Moreover, a genetic screen identified Elovl2 as a critical regulator of GSIS [59] and a protective factor against glucolipotoxicity [58]. We also found that an HFD disrupts the n-3 synthetic pathway at or above the generation of eicosapentaenoic acid (EPA, 20:5) and conversely enhances n-6 metabolites prior to the generation of 22:4 sidechains. Identification of the precise mechanism underlying this PUFA remodelling will require further work, but because metabolites in the n-6 and n-3 pathways compete for the same enzymes, it is conceivable that flux through the two arms might be reciprocally regulated. For example, enhanced provision of n-6 substrates (such as arachidonic acid from phospholipid remodelling) would not only feed into the n-6 arm but also limit access of endogenous n-3 metabolites to the common repertoire of elongases and desaturases. Regardless, our findings now suggest that dysregulation of the balance between n-3 and n-6 PUFA pathways might contribute to the onset of β-cell failure during pre-diabetes and potentially help explain why supplementation with EPA improves insulin secretory function in models of obesity/T2D in vivo and counters SFA lipotoxicity in vitro [60,61].
To our knowledge, this profound shift in n-3 to n-6 metabolism in β-cells following a HFD is a novel finding, although other instances of PUFA remodelling have been previously demonstrated in response to nutrient overload in vitro [62]. Our failure to observe major alterations in sphingolipids and neutral lipids is also surprising in the light of an extensive literature detailing their accumulation and negative consequences during β-cell failure [[22], [23], [24],39,[47], [48], [49]]. These findings, however, generally pertain to in vitro models of lipotoxicity. Our in vivo results are consistent, however, with early lipidomic analyses of islets from genetically obese, ob/ob mice [32,63], which showed only modest alteration, or even slight decreases, in neutral lipids and sphingolipids during obesity. It is known that intracellular storage depots of cholesteryl ester (CE) and TG are tightly regulated and actively turned over in β-cells, at least in response to changes in glucose concentration [7,64], so perhaps homeostatic mechanisms defend better against chronic lipid loading in vivo compared to in vitro. Another potential caveat is that our whole-cell analyses do not exclude the potential contributions of localized changes in ceramide (or other lipids). Indeed, in our own relatively mild model of in vitro lipotoxicity, we observed increases in ER-localized ceramide in response to chronic treatment with palmitate [39], which were not apparent at the whole cell level [47].
In addition to the above insights on high-fat feeding, our study provides novel findings relating to the regulation of β-cell function by autophagy. Firstly, we demonstrated that short-term inhibition of autophagy tends to counter the depletion of n3-PUFAs seen following an HFD. This was associated with enhanced expression of the elongase, Elovl5, in islets of Atg7 KO mice. This was apparent at the level of protein, but not mRNA, suggestive of a post-translational mechanism that is yet to be elucidated. As expected, however, there were accompanying increases in the concentrations of immediate elongase products comprising 20:3, 22:4, and 22:5 sidechains. A potential upregulation of the peroxisomal β-oxidation pathway might also contribute to the accumulation of n-3 22:6 PUFAs (see below). This unexpected link between autophagy and n-3 FA remodelling could potentially impact on multiple PUFA-regulated processes, and more specifically on ether lipid accumulation (see below). Unexpectedly, the loss of ATG7 also increased the sphingolipids, MHC and ceramide. These effects are difficult to interpret functionally as they would be expected to exert competing outcomes on β-cell function [48,49]. Contrary to our starting hypothesis, however, we did not observe any accumulation of TG and CE due to the inhibition of autophagy. We had previously defined a role for lipophagy as a negative regulator of GSIS, but this was based more specifically on manipulation of lysosomal acid lipase (LAL) [18]. Even so, knock down of Atg7 in vitro, albeit a more ambiguous intervention for disrupting lipophagy, was also associated with enhanced lipid droplet formation [18]. Perhaps, therefore, this effect is less apparent with in vivo interventions. As such, the ultimate test of the role of lipophagy in β-cells probably now requires targeted deletion of LAL rather than Atg7 as employed here. Another important route for future investigation would be to extend our lipidomic analyses to models of more overt β-cell failure, such as during long-term deletion of Atg7.
The current study has delineated a second role for autophagy in the regulation of ether lipid synthesis, which is additional to the remodelling of PUFA sidechains as highlighted above. This is exemplified by the changes in SFA- and MUFA-esterified species of alkyl lipids, which were nearly doubled following a loss of ATG7. Plasmalogens, however, were also significantly increased. The function of ether lipids in β-cells has never been formally addressed, but work in other fields suggests that they are likely to have diverse and important impacts on membrane biology in particular [30,31]. Compared to esterified lipids, they can be more closely packed and thus are concentrated particularly in signalling platforms (lipid rafts). The presence of plasmalogens, which are enriched in secretory vesicles, also helps impart negative curvature to membranes, which is a critical feature of membrane fusion during trafficking and exocytosis [30,31]. Indeed, we have previously shown that n-3 conjugated, PC plasmalogens are dynamically reduced during GSIS [33]. These are much less abundant than the corresponding PE plasmalogens, perhaps pointing to highly compartmentalized, and functionally-specialized roles. Ether lipids are also generally implicated in oxidant defence [30], and although this has not been addressed in the β-cell, it is a potentially important consideration. Regardless of their precise role, our study now strongly links ether lipids to β-cell function by showing that loss of the key biosynthetic enzyme, Dhrs7b [53,54,65], inhibited GSIS. Moreover, overexpression of Dhrs7b selectively reverted the inhibition of insulin secretion due to pre-treatment with oleate but not palmitate. This specificity is important because oleate also caused a more pronounced inhibition of ether lipid accumulation (Figure 7) and, as we showed previously [27], stimulates autophagy more potently than palmitate in our model. Collectively, these data strongly suggest that activation of autophagy, and depletion PUFA-linked ether lipids, contribute to the mechanism whereby MUFAs inhibit GSIS during high-fat feeding. In vivo, these mechanisms would potentially synergize with other (better characterized) pathways mediating lipotoxicity in response to SFAs [15,[22], [23], [24], [25],66].
Our third new insight into autophagy is its reciprocal relationship with peroxisomal biology. Prior work had highlighted the role of pexophagy in regulating peroxisome mass [50], and there are indications that autophagy is triggered in β-cells by defective peroxisomes [67]. But the converse upregulation of peroxisomal function by loss of autophagy is, we believe, a novel observation in mammalian cells. Lonp2 potentially plays a critical role in this context. This protein acts as both a chaperone and a protease, removing oxidized or misfolded proteins [52], which is critical in a compartment that is a major source of reactive oxygen species [35,36]. A more general role arises from the fact that proteins are directed to the peroxisomal matrix by targeting sequences, which then need to be removed for functional maturation [68]. Lonp2, working co-operatively with another protease, Tysnd1, thus promotes the processing and activation of a host of enzymes, including those catalysing β-oxidation and ether lipid formation [[69], [70], [71]]. Both pathways are augmented by ATG7 deletion in our model, consistent with the enhanced expression of Lonp2. Notably, an enhancement of peroxisomal β-oxidation could contribute to the repletion in n3 22:5 species we also observe under these conditions because some of these same enzymes also mediate the terminal step of PUFA formation [72,73]. Although more work is required to establish a direct link between these diverse observations, an earlier study has shown that ablation of Pex5, a receptor for one of the peroxisomal targeting sequences, both diminishes total plasmalogen levels in islets and impairs insulin secretion [67]. More specifically, a reciprocal relationship between autophagy and Lonp2 has only been elaborated in lower organisms [74]. But it would make sense that an enzyme critical for the optimization of peroxisomal function would also be upregulated in mammalian cells, precisely under those conditions in which clearance of damaged proteins and organelles would be impaired, such as with deletion of ATG7.
Another major finding is that short-term inhibition of autophagy is actually beneficial to β-cell function. This conclusion, underpinned by our strategy of temporally controlling deletion, is in marked contrast to the interpretation of models employing embryonic deletion of Atg7 [[10], [11], [12], [13], [14]]. In vitro studies, however, had suggested that GSIS could be inhibited by autophagy, especially in the form of lipophagy [18]. Although in broader terms this is not excluded (see above), the current findings would better implicate plasmalogen/PUFA turnover in the specific context of our short-term model of Atg7 deletion. We do not exclude the possibility that other mechanisms, independent of lipid remodelling, also contribute to this phenotype. Indeed, earlier studies had highlighted a role for autophagy in promoting the degradation of insulin (and proinsulin) such that less hormone was available for release by exocytosis [20,75]. In our model, however, no changes in the contents of insulin or proinsulin were observed in islets isolated from mice in any of the four experimental groups. This phenotype might require deletion of both Atg7 and Atg5 as employed in vitro [20], or a shorter period of inhibition than the 3 weeks we used here. On the other hand, insulin can be degraded via many alternative processes [19,76,77], which would be interesting to assess in the future to determine whether they might compensate for ATG7 deletion. It is also possible that the upregulation of Lonp2 that we observed in the ATG7 KO islets might mediate other protective effects independently of lipid metabolism. Indeed, the peroxisome has been postulated as a major source of oxidative stress in β-cells, at least in vitro in response to lipotoxicity [78]. Enhanced expression of Lonp2 would tend to counter this, both by removing oxidized proteins and by promoting the peroxisomal localization of anti-oxidant proteins [52]. Notably, however, the loss of Pex5 did not exacerbate oxidative damage in islets of mice maintained on an HFD [67].
The overall conclusion of our study is that autophagy plays multiple roles in β-cells, the ultimate consequences of which will depend on the extent, duration and context of activation. Earlier work, employing embryonic deletion of Atg7, had highlighted longer-term homeostatic roles of autophagy, including the restriction of β-cell apoptosis, maintenance of insulin secretory granules, and preservation of the mitochondrial function [10,11]. Another expected long-term benefit of autophagy would be counter-regulation of ER stress, and indeed constitutive deletion of Atg7 does result in distension of the ER [11,17]. Not surprisingly, the overall consequence of prolonged inhibition of autophagy in β-cells is impaired glucose tolerance, loss of insulin content and the development of islet-cell cysts [10,11]. That we were able to recapitulate these features using our the longer-term (9-week) protocol for gene ablation in adult mice argues against an obligatory role of Atg7 in islet development or maturation, but an ongoing requirement for autophagy in maintaining functional β-cell mass during adulthood. Clearly then, a critical contributor to the differing phenotypes of our 3- and 9-week models is the duration of deletion. But another consideration is that the consequences of knocking out Atg7 are likely to be more pronounced under conditions where autophagy is enhanced, such as during high-fat feeding, as opposed to chow feeding, where the rate of autophagy in islets is already very low [16,46]. Whereas we previously interpreted this activation of autophagy as beneficial, by protecting β-cells from ER stress [16], our current results suggest that this occurs at the expense of a diminished capacity for GSIS. The balance between these competing outputs might help determine the extent to which β-cells compensate or fail in the progression to T2D, but this also highlights the difficulty in targeting such a fundamental process such as autophagy as a disease therapy.
Acknowledgements
This work was supported by Project Grants and Research Fellowships from the National Health and Medical Research Council of Australia to T.J.B. and P.J.M
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.101023.
Contributor Information
Peter J. Meikle, Email: peter.meikle@baker.edu.au.
Trevor J. Biden, Email: t.biden@garvan.org.au.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Galluzzi L., Pietrocola F., Levine B., Kroemer G. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosn E.E., Cassado A.A., Govoni G.R., Fukuhara T., Yang Y., Monack D.M. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proceedings of the National Academy of Sciences of the U S A. 2010;107(6):2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y.H., Kim J., Park K., Lee M.S. β-cell autophagy: mechanism and role in β-cell dysfunction. Mol Metab. 2019;27S:S92–S103. doi: 10.1016/j.molmet.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stienstra R., Haim Y., Riahi Y., Netea M., Rudich A., Leibowitz G. Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia. 2014;57(8):1505–1516. doi: 10.1007/s00125-014-3255-3. [DOI] [PubMed] [Google Scholar]
- 5.Watada H., Fujitani Y. Minireview: autophagy in pancreatic β-cells and its implication in diabetes. Molecular Endocrinology. 2015;29(3):338–348. doi: 10.1210/me.2014-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahn S.E., Hull R.L., Utzschneider K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 7.Prentki M., Nolan C.J. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchetti P., Dotta F., Lauro D., Purrello F. An overview of pancreatic beta-cell defects in human type 2 diabetes: implications for treatment. Regulatory Peptides. 2008;146(1–3):4–11. doi: 10.1016/j.regpep.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Masini M., Bugliani M., Lupi R., del Guerra S., Boggi U., Filipponi F. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009;52(6):1083–1086. doi: 10.1007/s00125-009-1347-2. [DOI] [PubMed] [Google Scholar]
- 10.Ebato C., Uchida T., Arakawa M., Komatsu M., Ueno T., Komiya K. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metabolism. 2008;8(4):325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Jung H.S., Chung K.W., Won Kim J., Kim J., Komatsu M., Tanaka K. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metabolism. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Kim J., Cheon H., Jeong Y.T., Quan W., Kim K.H., Cho J.M. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. Journal of Clinical Investigation. 2014;124(8):3311–3324. doi: 10.1172/JCI69625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shigihara N., Fukunaka A., Hara A., Komiya K., Honda A., Uchida T. Human IAPP-induced pancreatic beta cell toxicity and its regulation by autophagy. Journal of Clinical Investigation. 2014;124(8):3634–3644. doi: 10.1172/JCI69866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J.J., Quijano C., Chen E., Liu H., Cao L., Fergusson M.M. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1(4):425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biden T.J., Boslem E., Chu K.Y., Sue N. Lipotoxic endoplasmic reticulum stress, β-cell failure and Type 2 diabetes. Trends in Endocrinology and Metabolism. 2014;25(8):389–398. doi: 10.1016/j.tem.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Chu K.Y., O'Reilly L., Ramm G., Biden T.J. High fat diet increases autophagic flux in pancreatic beta-cells in vivo and ex vivo. Diabetologia. 2015;58:2074–2078. doi: 10.1007/s00125-015-3665-x. [DOI] [PubMed] [Google Scholar]
- 17.Quan W., Hur K.Y., Lim Y., Oh S.H., Lee J.C., Kim K.H. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia. 2012;55(2):392–403. doi: 10.1007/s00125-011-2350-y. [DOI] [PubMed] [Google Scholar]
- 18.Pearson G.L., Mellet N., Chu K.Y., Cantley J., Davenport A., Bourbon P. Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells. Diabetologia. 2014;57(1):129–139. doi: 10.1007/s00125-013-3083-x. [DOI] [PubMed] [Google Scholar]
- 19.Marsh B.J., Soden C., Alarcon C., Wicksteed B.L., Yaekura K., Costin A.J. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Molecular Endocrinology. 2007;21(9):2255–2269. doi: 10.1210/me.2007-0077. [DOI] [PubMed] [Google Scholar]
- 20.Riahi Y., Wikstrom J.D., Bachar-Wikstrom E., Polin N., Zucker H., Lee M.S. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016;59:1480–1491. doi: 10.1007/s00125-016-3868-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cnop M. Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochemical Society Transactions. 2008;36(Pt 3):348–352. doi: 10.1042/BST0360348. [DOI] [PubMed] [Google Scholar]
- 23.Briaud I., Harmon J.S., Kelpe C.L., Segu V.B., Poitout V. Lipotoxicity of the pancreatic β-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50(2):315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moffitt J.H., Fielding B.A., Evershed R., Berstan R., Currie J.M., Clark A. Adverse physicochemical properties of tripalmitin in beta cells lead to morphological changes and lipotoxicity in vitro. Diabetologia. 2005;48(9):1819–1829. doi: 10.1007/s00125-005-1861-9. [DOI] [PubMed] [Google Scholar]
- 25.Morgan N.G., Dhayal S. Unsaturated fatty acids as cytoprotective agents in the pancreatic β-cell. Prostaglandins Leukotrienes and Essential Fatty Acids. 2010;82(4–6):231–236. doi: 10.1016/j.plefa.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 26.Busch A.K., Cordery D., Denyer G.S., Biden T.J. Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic β-cell function. Diabetes. 2002;51(4):977–987. doi: 10.2337/diabetes.51.4.977. [DOI] [PubMed] [Google Scholar]
- 27.Chu K.Y., O'Reilly L., Mellet N., Meikle P.J., Bartley C., Biden T.J. Oleate disrupts cAMP signaling, contributing to potent stimulation of pancreatic β-cell autophagy. Journal of Biological Chemistry. 2019;294(4):1218–1229. doi: 10.1074/jbc.RA118.004833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guillou H., Zadravec D., Martin P.G., Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Progress in Lipid Research. 2010;49(2):186–199. doi: 10.1016/j.plipres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 29.Russo G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochemical Pharmacology. 2009;77(6):937–946. doi: 10.1016/j.bcp.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Dean J.M., Lodhi I.J. Structural and functional roles of ether lipids. Protein Cell. 2018;9(2):196–206. doi: 10.1007/s13238-017-0423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul S., Lancaster G.I., Meikle P.J. Plasmalogens: a potential therapeutic target for neurodegenerative and cardiometabolic disease. Progress in Lipid Research. 2019;74:186–195. doi: 10.1016/j.plipres.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Medina-Gomez G., Yetukuri L., Velagapudi V., Campbell M., Blount M., Jimenez-Linan M. Adaptation and failure of pancreatic β cells in murine models with different degrees of metabolic syndrome. Disease Models & Mechanisms. 2009;2(11–12):582–592. doi: 10.1242/dmm.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearson G.L., Mellet N., Chu K.Y., Boslem E., Meikle P.J., Biden T.J. A comprehensive lipidomic screen of pancreatic β-cells using mass spectroscopy defines novel features of glucose-stimulated turnover of neutral lipids, sphingolipids and plasmalogens. Molecular Metabolism. 2016;5(6):404–414. doi: 10.1016/j.molmet.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanadham S., Zhang S., Ma Z., Wohltmann M., Bohrer A., Hsu F.F. D6-, Stearoyl CoA-, and D5-desaturase enzymes are expressed in beta-cells and are altered by increases in exogenous PUFA concentrations. Biochimica et Biophysica Acta. 2002;1580(1):40–56. doi: 10.1016/s1388-1981(01)00189-5. [DOI] [PubMed] [Google Scholar]
- 35.Lodhi I.J., Semenkovich C.F. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metabolism. 2014;19(3):380–392. doi: 10.1016/j.cmet.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wanders R.J. Metabolic functions of peroxisomes in health and disease. Biochimie. 2014;98:36–44. doi: 10.1016/j.biochi.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 37.Gu G., Dubauskaite J., Melton D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 38.Cantley J., Davenport A., Vetterli L., Nemes N.J., Whitworth P.T., Boslem E. Disruption of beta cell acetyl-CoA carboxylase-1 in mice impairs insulin secretion and beta cell mass. Diabetologia. 2019;62(1):99–111. doi: 10.1007/s00125-018-4743-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boslem E., Weir J.M., Macintosh G., Sue N., Cantley J., Meikle P.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. Journal of Biological Chemistry. 2013;288:26569–26582. doi: 10.1074/jbc.M113.489310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weir J.M., Wong G., Barlow C.K., Greeve M.A., Kowalczyk A., Almasy L. Plasma lipid profiling in a large population-based cohort. The Journal of Lipid Research. 2013;54(10):2898–2908. doi: 10.1194/jlr.P035808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huynh K., Barlow C.K., Jayawardana K.S., Weir J.M., Mellett N.A., Cinel M. High-throughput plasma lipidomics: detailed mapping of the associations with cardiometabolic risk factors. Cell Chemical Biology. 2018;26:71–84. doi: 10.1016/j.chembiol.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Fahy E., Subramaniam S., Murphy R.C., Nishijima M., Raetz C.R., Shimizu T. Update of the LIPID MAPS comprehensive classification system for lipids. The Journal of Lipid Research. 2009;50(Suppl):S9–S14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebisch G., Vizcaino J.A., Kofeler H., Trotzmuller M., Griffiths W.J., Schmitz G. Shorthand notation for lipid structures derived from mass spectrometry. The Journal of Lipid Research. 2013;54(6):1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saeed A.I., Sharov V., White J., Li J., Liang W., Bhagabati N. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 45.Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of Cell Biology. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheng Q., Xiao X., Prasadan K., Chen C., Ming Y., Fusco J. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Scientific Reports. 2017;7(1):16348. doi: 10.1038/s41598-017-16485-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boslem E., MacIntosh G., Preston A.M., Bartley C., Busch A.K., Fuller M. A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochemical Journal. 2011;435(1):267–276. doi: 10.1042/BJ20101867. [DOI] [PubMed] [Google Scholar]
- 48.Boslem E., Meikle P.J., Biden T.J. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets. 2012;4:177–187. doi: 10.4161/isl.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veret J., Bellini L., Giussani P., Ng C., Magnan C., Le Stunff H. Roles of sphingolipid metabolism in pancreatic β cell dysfunction induced by lipotoxicity. Journal of Clinical Medicine. 2014;3(2):646–662. doi: 10.3390/jcm3020646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwata J., Ezaki J., Komatsu M., Yokota S., Ueno T., Tanida I. Excess peroxisomes are degraded by autophagic machinery in mammals. Journal of Biological Chemistry. 2006;281(7):4035–4041. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- 51.Herzog K., Pras-Raves M.L., Ferdinandusse S., Vervaart M.A.T., Luyf A.C.M., van Kampen A.H.C. Functional characterisation of peroxisomal β-oxidation disorders in fibroblasts using lipidomics. Journal of Inherited Metabolic Disease. 2018;41(3):479–487. doi: 10.1007/s10545-017-0076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pomatto L.C., Raynes R., Davies K.J. The peroxisomal Lon protease LonP2 in aging and disease: functions and comparisons with mitochondrial Lon protease LonP1. Biological Reviews of the Cambridge Philosophical Society. 2017;92(2):739–753. doi: 10.1111/brv.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lodhi I.J., Link D.C., Semenkovich C.F. Acute ether lipid deficiency affects neutrophil biology in mice. Cell Metabolism. 2015;21(5):652–653. doi: 10.1016/j.cmet.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 54.Lodhi I.J., Yin L., Jensen-Urstad A.P., Funai K., Coleman T., Baird J.H. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metabolism. 2012;16(2):189–201. doi: 10.1016/j.cmet.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei D., Li J., Shen M., Jia W., Chen N., Chen T. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic β-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes. 2010;59(2):471–478. doi: 10.2337/db09-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J., Song M.Y., Bae U.J., Lim J.M., Kwon K.S., Park B.H. n-3 Polyunsaturated fatty acids protect against pancreatic β-cell damage due to ER stress and prevent diabetes development. Molecular Nutrition & Food Research. 2015;59(9):1791–1802. doi: 10.1002/mnfr.201500299. [DOI] [PubMed] [Google Scholar]
- 57.Wang X., Chan C.B. n-3 polyunsaturated fatty acids and insulin secretion. Journal of Endocrinology. 2015;224(3):R97–R106. doi: 10.1530/JOE-14-0581. [DOI] [PubMed] [Google Scholar]
- 58.Bellini L., Campana M., Rouch C., Chacinska M., Bugliani M., Meneyrol K. Protective role of the ELOVL2/docosahexaenoic acid axis in glucolipotoxicity-induced apoptosis in rodent beta cells and human islets. Diabetologia. 2018;61:1780–1793. doi: 10.1007/s00125-018-4629-8. [DOI] [PubMed] [Google Scholar]
- 59.Cruciani-Guglielmacci C., Bellini L., Denom J., Oshima M., Fernandez N., Normandie-Levi P. Molecular phenotyping of multiple mouse strains under metabolic challenge uncovers a role for Elovl2 in glucose-induced insulin secretion. Mol Metab. 2017;6(4):340–351. doi: 10.1016/j.molmet.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato T., Shimano H., Yamamoto T., Ishikawa M., Kumadaki S., Matsuzaka T. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes. 2008;57(9):2382–2392. doi: 10.2337/db06-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neuman J.C., Schaid M.D., Brill A.L., Fenske R.J., Kibbe C.R., Fontaine D.A. Enriching islet phospholipids with eicosapentaenoic acid reduces prostaglandin E2 signaling and enhances diabetic beta-cell function. Diabetes. 2017;66(6):1572–1585. doi: 10.2337/db16-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cohen G., Shamni O., Avrahami Y., Cohen O., Broner E.C., Filippov-Levy N. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia. 2015;58(6):1333–1343. doi: 10.1007/s00125-015-3566-z. [DOI] [PubMed] [Google Scholar]
- 63.Medina-Gomez G., Gray S.L., Yetukuri L., Shimomura K., Virtue S., Campbell M. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genetics. 2007;3(4) doi: 10.1371/journal.pgen.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cantley J., Biden T.J. Targeting triacylglycerol/fatty acid cycling in beta cells as a therapy for augmenting glucose-stimulated insulin secretion. Islets. 2010;2:127–129. doi: 10.4161/isl.2.2.11240. [DOI] [PubMed] [Google Scholar]
- 65.Lodhi I.J., Wei X., Yin L., Feng C., Adak S., Abou-Ezzi G. Peroxisomal lipid synthesis regulates inflammation by sustaining neutrophil membrane phospholipid composition and viability. Cell Metabolism. 2015;21(1):51–64. doi: 10.1016/j.cmet.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Busch A.K., Gurisik E., Sudlow M., Cordery D.V., Denyer G.S., Laybutt D.R. Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic β-cells from lipoapoptosis. Diabetes. 2005;54:2917–2924. doi: 10.2337/diabetes.54.10.2917. [DOI] [PubMed] [Google Scholar]
- 67.Baboota R.K., Shinde A.B., Lemaire K., Fransen M., Vinckier S., Van Veldhoven P.P. Functional peroxisomes are required for β-cell integrity in mice. Mol Metab. 2019;22:71–83. doi: 10.1016/j.molmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gould S.J., Valle D. Peroxisome biogenesis disorders: genetics and cell biology. Trends in Genetics. 2000;16(8):340–345. doi: 10.1016/s0168-9525(00)02056-4. [DOI] [PubMed] [Google Scholar]
- 69.Okumoto K., Kametani Y., Fujiki Y. Two proteases, trypsin domain-containing 1 (Tysnd1) and peroxisomal lon protease (PsLon), cooperatively regulate fatty acid beta-oxidation in peroxisomal matrix. Journal of Biological Chemistry. 2011;286(52):44367–44379. doi: 10.1074/jbc.M111.285197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Omi S., Nakata R., Okamura-Ikeda K., Konishi H., Taniguchi H. Contribution of peroxisome-specific isoform of Lon protease in sorting PTS1 proteins to peroxisomes. Journal of Biochemistry. 2008;143(5):649–660. doi: 10.1093/jb/mvn020. [DOI] [PubMed] [Google Scholar]
- 71.Mizuno Y., Ninomiya Y., Nakachi Y., Iseki M., Iwasa H., Akita M. Tysnd1 deficiency in mice interferes with the peroxisomal localization of PTS2 enzymes, causing lipid metabolic abnormalities and male infertility. PLoS Genetics. 2013;9(2) doi: 10.1371/journal.pgen.1003286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferdinandusse S., Denis S., Mooijer P.A., Zhang Z., Reddy J.K., Spector A.A. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. The Journal of Lipid Research. 2001;42(12):1987–1995. [PubMed] [Google Scholar]
- 73.Su H.M., Moser A.B., Moser H.W., Watkins P.A. Peroxisomal straight-chain Acyl-CoA oxidase and D-bifunctional protein are essential for the retroconversion step in docosahexaenoic acid synthesis. Journal of Biological Chemistry. 2001;276(41):38115–38120. doi: 10.1074/jbc.M106326200. [DOI] [PubMed] [Google Scholar]
- 74.Aksam E.B., Koek A., Kiel J.A., Jourdan S., Veenhuis M., van der Klei I.J. A peroxisomal lon protease and peroxisome degradation by autophagy play key roles in vitality of Hansenula polymorpha cells. Autophagy. 2007;3(2):96–105. doi: 10.4161/auto.3534. [DOI] [PubMed] [Google Scholar]
- 75.Yamamoto S., Kuramoto K., Wang N., Situ X., Priyadarshini M., Zhang W. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Reports. 2018;23(11):3286–3299. doi: 10.1016/j.celrep.2018.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim Y.M., Lim H., Hur K.Y., Quan W., Lee H.Y., Cheon H. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nature Communications. 2014;5:4934. doi: 10.1038/ncomms5934. [DOI] [PubMed] [Google Scholar]
- 77.Pasquier A., Vivot K., Erbs E., Spiegelhalter C., Zhang Z., Aubert V. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nature Communications. 2019;10(1):3312. doi: 10.1038/s41467-019-11170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elsner M., Gehrmann W., Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes. 2011;60(1):200–208. doi: 10.2337/db09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.