

Summary
Although PD-L1 expression on tumor is related to the prognosis of immune checkpoint blockade (ICB) therapy, a recent study also demonstrated clinical benefits even in patients without PD-L1 expression. To understand the relationship between innate resistance and antitumor cytotoxic T lymphocyte (CTL) responses especially against neoantigens, the interaction between PD-L1+ or genetically PD-L1-deleted colorectal tumors and CTLs was assessed under an ICB therapy, finding the robust CTL activation in PD-L1-deleted tumor-bearing mice. Using antigen libraries based on immunogenomics, we identified three H2-Kb-restricted, somatic-mutated immunogenic neoantigens by utilizing enhanced CTLs responses due to PD-L1 deficiency. Furthermore, we identified three T cell receptor (TCR) repertoires relevant to the neoantigens, confirming the response of TCR-gene-transduced CTLs to parental tumor cells. Notably, neoantigen-pulsed dendritic cell (DC) therapy reversed the tumor tolerance. Thus, innate resistance of tumors determines their responsiveness to neoantigens and mixed neoantigen peptides may be useful in DC therapy against innate resistance type tumor.
Subject Areas: Biological Sciences, Immunology, Cancer
Graphical Abstract
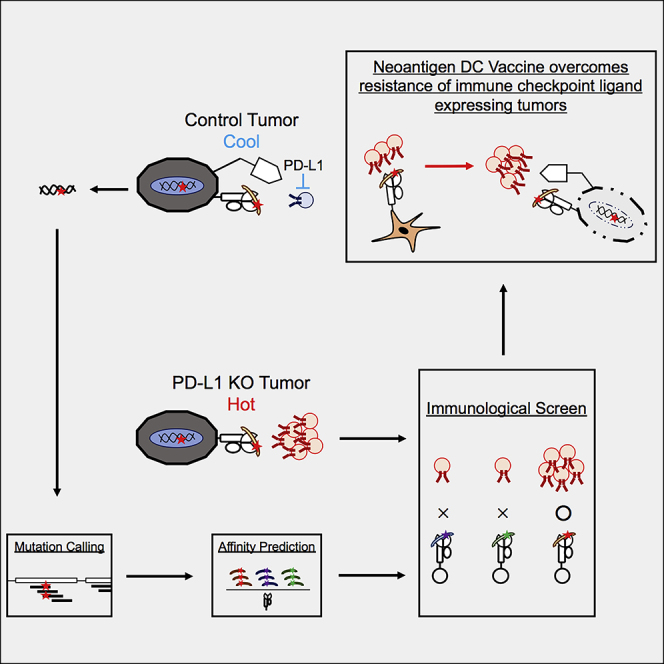
Highlights
-
•
Poor prognosis of PD-L1-expressing tumors in some cancers
-
•
Identification of TCRVα and Vβ repertoire responsive for H2-Kb-restricted neoantigens
-
•
Neoantigen-epitope-pulsed DC therapy demonstrates antitumor effect in vaccine
Biological Sciences; Immunology; Cancer
Introduction
Cancer immunotherapy has recently evolved into one of the most promising cancer treatment modalities. Although tumor cells hijack the immune system, in particular causing T cell exhaustion, several reports have demonstrated its reversal through immune checkpoint blockade (ICB), i.e., antibodies targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) (Hodi et al., 2016, Shimizu et al., 2018, Wei et al., 2018). Currently, these have been used as effective immunotherapeutic drugs. Specifically, ICB therapy has demonstrated improved overall survival (OS) and progression-free survival (PFS) in the treatment of many different tumor types, such as melanoma (Robert et al., 2015), non-small-cell lung cancer (NSCLC) (Brahmer et al., 2015), head and neck cancer (Ferris et al., 2016), and other cancers, due to the durable antitumor CD8+ cytotoxic T lymphocytes (CTLs). In addition, ICB-therapy-induced recruitment of T cells in the tumor increases in hot tumors, but not in cold tumors (Tumeh et al., 2014, Topalian et al., 2016). Despite its successful clinical activity, still limited patients received the clinical benefits, for which many studies have addressed the factors related to ICB sensitivity and resistance.
The interplay between PD-1 and PD-L1 regulates T cells in the tumor microenvironment (TME). PD-1 expression is upregulated in activated T cells and remains high in exhausted T cells from tumor-infiltrating lymphocytes (Ahmadzadeh et al., 2009). Constitutive PD-L1 expression caused by aberrant transactivation due to genetic or signaling alteration in an intrinsic manner defines the innate resistance. After exposure to IFN-γ released by effector T cells, PD-L1 and other inhibitory molecules were induced at the transcription level and triggers the adaptive resistance (Topalian et al., 2015). Clinically, PD-L1 expression on tumors or immune cells in the tumor microenvironment is associated with durable clinical responses to anti-PD-1/PD-L1 therapies in many tumor types (Topalian et al., 2012, Herbst et al., 2014). However, the significance of PD-L1 expression in each cell type is controversial. PD-L1 expression on tumors or immune cells can independently attenuate antitumor T cell immunity. PD-1+ T cells in the tumor are impaired due to interactions with PD-L1 expressed on either tumor cells (Juneja et al., 2017) or other tumor-infiltrating immune cells (e.g., myeloid cells or TAM) (Lin et al., 2018, Tang et al., 2018) in the TME. Furthermore, immune checkpoint knockout mice or wild-type (WT) mice treated with anti-PD-1 antibodies (Abs) suppressed tumor cells, indicating that the anti-PD-1 Ab has an antitumor effect on both PD-L1-expressing host tumor-associated macrophages (TAM) and tumor cells (Kleffel et al., 2015, Lin et al., 2018). Recently, the functional importance of immune cells relative to the tumor has been reported in the regulation of the antitumor T cell response in NSCLC patients (Kowanetz et al., 2018), but its importance on the tumor still has to be evaluated. Furthermore, despite low PD-L1 expression on tumor, recent several reports demonstrated that some patients respond well to PD-1 pathway blockade (Brahmer et al., 2015, Zou et al., 2016, Rittmeyer et al., 2017, Eggermont et al., 2018), indicating that the relation of PD-L1 expression and ICB sensitivity partly depends on pathological conditions, thus ascribing PD-L1 expression to innate or acquired resistance. To elucidate the underlying mechanisms of these phenomena, CTL responses in the presence or absence of ICB treatment, with focus on PD-L1 expression on tumor cells should be compared.
The underlying genomic features of tumor cells contribute to ICB responses, and increased tumor mutation burden has been shown to be associated with survival benefits from both anti-CTLA-4 and anti-PD-1 therapy in multiple malignancies (Snyder et al., 2014, Le et al., 2015, Rizvi et al., 2015, Hugo et al., 2016). High mutation burden was frequently diagnosed in mutagen-associated cancers, such as melanoma and NSCLC as well as cancers associated with DNA-mismatch repair gene defects, such as microsatellite instability in colorectal cancers (Schumacher and Schreiber, 2015, Riaz et al., 2016). These high mutation burden tumors potentially generate immunogenic neoantigens. In fact, tumors with clonal neoantigens may significantly elicit effective immune responses (McGranahan et al., 2016), and peptides containing these mutations presented on MHC class I (MHC-I) can be recognized as “non-self” by T cells. For this purpose, neoantigens in high mutation burden tumors can be identified by whole-exome sequence and RNA sequence (RNA-Seq), and accompanying bioinformatics approaches can predict specific neoepitopes in individual cancers using MHC-I-binding algorithms and immunogenomics methods (Castle et al., 2012, Karasaki et al., 2017). Furthermore, ICB immunotherapy has sometimes been found to be effective even in cancer with a low number of mutations but containing some indels, such as renal cell carcinoma, indicating that the number of mutations present in tumors does not solely determine ICB responsiveness (Turajlic et al., 2017, Motzer et al., 2018, Yang et al., 2019). Based on these reports, there are many possible implications regarding ICB sensitive and resistant cases, particularly their PD-L1 expressions and potential neoantigen responses under ICB therapy. Nonetheless, comprehensive analyses are yet to be performed.
Development of a therapeutic strategy for ICB-resistant cancers is an important issue. Of which, dendritic cell (DC) vaccine was identified as a candidate, which plays a central role in linking innate and adaptive immune system responses (Fujii et al., 2004, Fujii et al., 2007, Steinman, 2012, Bottcher and Reis, 2018, Fujii and Shimizu, 2019). Ex vivo-expanded DCs pulsed with MHC-I-restricted cancer testis antigen peptides (Bezu et al., 2018) and DCs electroporated with transfected mRNA coding tumor antigen (Bol et al., 2015) and tumor lysates or irradiated tumor cells (Fujii et al., 1999, Rojas-Sepulveda et al., 2018, Tanyi et al., 2018) have been demonstrated in many basic and clinical studies (Steinman and Banchereau, 2007, Saxena et al., 2018). It is noteworthy that utilizing neoantigens, several reports applied DC vaccines in clinical study and showed that neoantigen-pulsed DC can induce tumor regression through neoantigen-specific T cell responses in refractory solid tumors such as melanoma (Carreno et al., 2015, Chen et al., 2019).
Regarding PD-L1 expression in ICB treatment, PD-L1-mediated immune suppression by IFN-γ-induced adaptive resistance in T cell infiltrating hot tumors has been described (Spranger et al., 2013, Tumeh et al., 2014, Wilky, 2019). In contrast, the mechanism of PD-L1-mediated immune suppression by the tumor in a cell intrinsic manner, i.e., innate resistance, and its reversal by ICB therapy remain to be fully resolved. Furthermore, little is known about whether PD-L1 expression on tumors controls T cell immunity against neoantigens, which can partly determine the ICB response. Therefore, in the current study, we used several models—PD-L1-expressing or PD-L1-deleted tumor cells, with or without ICB (anti-PD-1 Ab and anti-CTLA-4 Ab) therapy—to explore how PD-L1 expression on tumor cells leads to innate resistance to ICB therapy and also elucidated potential mechanism of PD-L1-deleted tumor responses in ICB therapy. Further, we demonstrated the availability of our identified neoantigens-based DC therapy to compensate for ICB resistance, resulting in the generation of neoantigens responding T cell immunity as well as enhanced pre-existing immunity. Therefore, DCs vaccination has potential application for resistant tumors.
Results
TCGA Analysis Indicates the Association between PD-L1 and Survival in Cancer Patients
PD-L1 expression in tumor sites is generally known to be associated with overall survival but differs in patients and their contexts. To clarify the variable association of PD-L1 expression and overall survival, we first analyzed all study cohorts in publicly available TCGA database (The Cancer Genome Atlas; Cancer Genome Atlas Research Network, 2014). PD-L1 expression in some cancers were related to better prognosis but varied among study (Figure 1A). Because the cytolytic score (CYT), that is, the level of granzyme A and perforin, was previously reported to be mainly associated with antitumor immunological effects (Rooney et al., 2015, Roufas et al., 2018), we sub-grouped based on PD-L1 expression, CYT score, and estimated survival. We found that PD-L1 low expression on hot tumors (CYT high) remarkably correlated with better prognosis, than the other three groups—PD-L1 low expression on the cold tumor (CYT low) or PD-L1 high expression—in colorectal adenocarcinoma (COAD) and uterine corpus endometrial carcinoma (UCEC) (Figure 1B). Some other cancers (breast invasive carcinoma [BRCA], cervical squamous cell carcinoma [CESC], diffuse large B cell lymphoma [DLBC], acute myeloid leukemia [LAML], liver hepatocellular carcinoma [LIHC], and sarcoma [SARC]), but not all, also show similar tendencies (Figure S1). In addition to previous observations of survival in patients with high microsatellite instability (MSI) (Gryfe et al., 2000, Le et al., 2015), a group of cancer patients with low PD-L1 expression and high MSI were also associated with better prognosis in these COAD and UCEC studies (Figure 1C). Thus, our results indicated that low PD-L1 expression with strong antitumor immunity or with potential mutation load is related to survival benefits in patients with certain tumors.
Figure 1.

The Association between PD-L1 Expression in Tumor Tissue and Survival
(A) Forest plot of hazard ratios and 95% confidence intervals associated with PD-L1 expression. PD-L1 low expression groups in each study were referenced. NA, not analyzed due to low hazard ratios.
(B and C) Analysis of the association between PD-L1 expression and patient survival across TCGA colon cancer (left) and uterine corpus endometrial carcinoma (right). (B) Kaplan-Meier survival curves (time is measured on the x axis) of patients with high versus low CYT score. Log rank statistics: COAD, p = 0.019; UCEC, p = 0.0054. (C) Kaplan-Meier survival curves of patients with high versus low MSI. Data were analyzed by log rank statistics: COAD, p = 0.14; UCEC, p = 0.22.
Anti-tumor CTL Responses in PD-L1+ or PD-L1-KO Tumors
In accordance with clinical observation, several murine tumors have been characterized, including B16 melanoma showing low PD-L1 expression and low immunogenicity with low T cell infiltration (known as a cold tumor type) and a murine colorectal MC38 expressing high PD-L1 and high immunogenicity with high T cell infiltration (known as a hot tumor type). In this study, MC38, which is known to be highly immunogenic and relatively responds to ICB treatment, was used to compare PD-L1 expression and T cell responses in ICB therapy(Yadav et al., 2014, Tanegashima et al., 2019). To examine an effect of PD-L1 of tumor cells on the antitumor immunity, we first generated genetically PD-L1-deleted tumors (hereafter MC38-PD-L1-KO cells) by plasmid transfection-based transient CRISPR-Cas9 expression in the MC38 cell line. MC38-PD-L1-KO cells expressed MHC-I, but not PD-L1 (Figure S2A). We then examined the tumor proliferation activity in vitro and in vivo. There was no difference in the proliferation of tumor cells between MC38 and MC38-PD-L1-KO in an in vitro culture (Figure 2A). Subsequently, we compared tumor growth after a subcutaneous injection of MC38 and MC38-PD-L1-KO and found that tumor growth of MC38-PD-L1-KO was more slowly progressive than MC38 in WT mice, but not in Rag1−/− mice (Figure 2B), indicating T cell dependence. We also established the PD-L1-deficient murine breast cancer cell line, E0771, and confirmed similar phenomena (Figure S2B), but were not found with B16F10, probably due to the cold tumor type (Figure S2C). These imply that PD-L1 expression and tumor antigen presentation in tumor cells directly dampened the T-cell-dependent tumor suppression. Further, anti-PD-1 Ab treatment alone showed an antitumor effect by reactivating T cells in the tumor, although somewhat insufficient (Figure S2D). The combination of anti-PD-1 Ab plus anti-CTLA-4 Ab has been shown to be more promising for effective CTL generation (Hodi et al., 2016, Shimizu et al., 2018, Wei et al., 2018). To understand the relationship between CTL induction and PD-L1 expression on tumors, we evaluated the combined ICB treatment (anti-PD-1 Ab plus anti-CTLA-4 Ab) against MC38 or MC38-PD-L1-KO tumor cells in vivo. The tumor regression was more effective in ICB-treated MC38-PD-L1-KO tumor-cell-bearing mice than ICB-treated MC38 tumor-bearing mice or non-treated MC38-PD-L1-KO tumor-cell-bearing mice (Figure 2C).
Figure 2.

Enhanced Immune Responses of CTLs from PD-L1-KO Tumor
(A) Proliferating activity of MC38 or MC38-PD-L1-KO cells in vitro by WST-1 assay. Data are pooled from three independent experiments, and the values represent mean ± SD.
(B) C57BL/6J (left) or Rag1−/− (right) mice were injected with 5 × 105 MC38 or MC38-PD-L1-KO cells, subcutaneously (s.c.). Tumor growth was monitored at indicated time points by measuring three perpendicular diameters. MC38 or MC38-PD-L1-KO in C57BL/6J mice n = 20 and 21/group, respectively, in Rag1−/− mice, n = 6/group. Data are pooled from four and two independent experiments, and the values represent mean ± SEM.
(C) C57BL/6J mice were injected with 5 x 105 MC38 cells or MC38-PD-L1-KO cells s.c. and then treated with anti-PD-1 and anti-CTLA-4 on day 7, 10, and 13. Tumor growth was monitored at indicated time points by measuring three perpendicular diameters. (n = 17/group). Data are pooled from six independent experiments, and the values represent mean ± SEM.
(D) The frequency of immune cells in tumor sites was analyzed at day 15 after tumor inoculation by flow cytometric analysis. The values represent mean ± SD (n = 4/group). Closed and open bar indicated MC38 and MC38-PD-L1-KO tumor-bearing mice, respectively; ns indicates not significant.
(E) Tumor antigen-specific T cell response in ICB-treated MC38 or MC38-PD-L1-KO tumor-bearing mice. Frequency of MutAdpgk tetramer+ in CD8+ T cells was plotted. Data are pooled from four independent experiments; n = 10/group. Data were analyzed by unpaired Student's t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Due to the enhanced antitumor effect in MC38-PD-L1-KO mice, we hypothesized two possibilities: (1) the balance of immune responses was altered in MC38-PD-L1-KO and (2) T cells responded to neoantigens that were more expressed in MC38-PD-L1-KO. As such, we analyzed the immune responses two weeks later in MC38 or MC38-PD-L1-KO mice when no difference in tumor size was observed. As shown in Figure 2D, there was no statistical difference in the frequency of various types of immune cells including T and natural killer (NK) cells. Next, we assessed the CTL function. As shown in Figure 2C, growth of MC38-PD-L1-KO cells in ICB-treated mice progressed slowly and retarded two weeks after subcutaneous tumor inoculation. Therefore, we analyzed the T cell response for the previously identified H2-Db restricted antigen, mutant (Mut) Adpgk, and H2-Kb restricted antigen, p15E, on day 15. In ICB-treated MC38-PD-L1-KO bearing mice, we detected the apparent MutAdpgk-specific CD8+ T cell infiltration in the tumor (Figure 2E). We also assessed their functions using exhaustion/activation markers and found the differential presence of PD-1+Tim-3+ or PD-1+CD69+ responsive CD8+ T cells in MC38-KO tumor compared with MC38 WT tumor (Figure S3). Moreover, CD8+ T cells in the spleen and lymph nodes of these mice responded well to p15E and MutAdpgk antigens (Figure S4). Thus, the ICB-treated mice injected with MC38-PD-L1-KO enhanced the apparent antigen-specific T cell response.
Screening of H2-Kb-Restricted Neoantigens by Exome and Transcriptome Analyses
MutAdpgk was previously reported as a type of neoantigen, expressed on H2-Db of MC38 (Yadav et al., 2014). We speculated that neoepitopes on other MHCs could be identified, and thus a mixture of antigens, composed of different MHC-restricted neoantigens, would be more efficient as vaccines. Therefore, we conducted exome and transcriptome sequences of MC38 and MC38-PD-L1-KO tumor tissues to identify tumor-specific point mutations with amino acid substitutions, including stop gain, frameshift, and missense mutations (Figure 3A). Further, putative neoepitopes were found to be randomly distributed throughout the genome. First, to identify single-nucleotide variants (SNVs), data derived from the exome and RNA sequences obtained from MC38 or MC38-PD-L1-KO were compared with normal mouse blood (Figure S5A). As a result, 7,683 and 6,876 coding variants with almost similar mutation signature were identified by exome sequencing (Figure S5B), and subsequently 826 and 775 by RNA-seq, which overlapped with the exome-based variants, in MC38 and MC38-PD-L1-KO, respectively. Among these, 808 and 755 missense mutations were found in MC38 and MC38-KO, respectively. Common major mutated epitopes in MC38 and MC38-PD-L1-KO were observed to harbor 645 potential epitopes that might be stable or universal (Figure 3B). By contrast, mutated epitopes in MC38 alone or MC38-PD-L1-KO alone were 163 and 110, respectively (Figure 3B). Nevertheless, the rates of mutation detected in both transcriptome and exome analyses were similar: 826/7683 = 0.107 (WT) and 755/6876 = 0.113 (PD-L1-KO). Although some mutations disappeared and other novel mutations possibly appeared in PD-L1-KO tumor probably due to immunoediting or clonal evolution in a broad sense, majority of the missense mutations was shared. Therefore, we hypothesized that common CTL responses against parental tumors may be present in PD-L1-KO tumor-bearing mice, by which augmented CTL responses enable high sensitive screening of tumor-specific response as shown in Figures 2E and S4.
Figure 3.

Neoantigen Screening from PD-L1-KO Tumors
(A) Circos plot showing the detected mutations in the MC38 tumor. Tumor tissues were sampled at day 15 after tumor inoculation. Mutations of MC38 were identified by whole-exome sequence and RNA sequencing (RNA-Seq). FPKM value and MHC-I affinity values were also plotted.
(B) Venn diagram of identified missense mutations between MC38 WT and PD-L1-KO tumors. Reactive peptide sequences and their IC50 values as calculated by NetMHCpan ver3.0. H2-Kb-restricted peptides for MC38 neoantigen candidates were synthesized and prepared.
(C) Experimental scheme of neoantigen candidate screening. MC38-PD-L1-KO tumor-bearing mice were treated with anti-PD-1 and anti-CTLA-4 on day 7, 10, and 13. Fifteen days after tumor inoculation, CD8 T cells were purified from pooled splenocytes and the tumor draining lymph node (4 mice per experiment) and cocultured with 30 Gy-irradiated splenocytes in the presence or absence of 10 μg/mL neoantigen candidate peptides for 72 h.
(D) Upper, culture supernatants were measured for IFN-γ production by ELISA. Data are pooled from five independent experiments. Lower, character of neoantigen peptides.
High Immunogenic Neoantigens Identified by Immunome Analyses
SNVs were analyzed for their potential to generate MHC-I-restricted epitopes of the murine H2-Kb alleles using the NetMHCpan algorithm. We selected neoantigen candidates presented on H2-Kb (predicted IC50 < 200, rank <1) from common missense mutations in both WT and PD-L1-KO tumors using the NetMHCpan (ver 3.0) algorithm (Nielsen and Andreatta, 2016). Overall, 49 peptides derived from abundant transcripts were more likely to be presented on MHC-I of MC38 cells. Next, we evaluated the immunogenicity of mutated tumor antigens in vivo. By utilizing PD-L1-KO tumors, one week after tumor inoculation, the tumor-bearing mice were treated three times with ICB combination therapy. CD8+ T cells from the spleen and lymph nodes were harvested and stimulated with or without 49 types of epitope candidate peptides for 72 h. IFN-γ production as an indicator of CD8+ T cell responses was measured (Figure 3C). With this screening, significant IFN-γ production was shown by culturing in the presence of #1 (Zbtb40 R768P), #36 (Dpagt1 V213L), and #42 (Cry1 V416L) (Figure 3D).
To examine whether these three predicted peptides could elicit CD8+ T cell responses, we administered each peptide to WT mice together with anti-CD40 Ab plus poly(I:C). We demonstrated that CD8+ T cells specific for MutZbtb40, MutDpagt1, and MutCry1 antigens, respectively, produced IFN-γ, TNF-α, and IL-2 (Figure 4Ai) and expressed CD137+PD1+ CD8+ T cells in an antigen-specific manner (Figure 4Aii). We also demonstrated that antigen-specific CD8+ T cells responded to MutZbtb40 or MutDpagt1 neoantigen by multimer staining (Figure 4Bi, ii) and ELISA or ELISPOT assay (Figure 4C), but they did not respond to the non-mutated original antigen. Thus, through a computational approach and analysis of their immunogenic antigen-specific properties and biological analyses, these three predicted neoantigen peptides were determined to be immunogenic in the context of MC38 tumors.
Figure 4.

Bioactive Neoantigen Epitopes
Analysis of neoantigen-specific T cells in vaccinated mice. C57BL/6J mice were administered intravenously (i.v.) with neoantigen peptide (Zbtb40, Dpagt1, and Cry1) together with poly(I:C) and anti-CD40 Ab.
(A) One week later, neoantigen-specific T cell activation in these mice was assessed by intracellular staining for IFN-γ and TNF-α production (i) and upregulation of PD-1 and CD137 (ii) after culturing with or without each peptide for 6 h (i) or 24 h (ii).
(B) As shown in (A), neoantigen-specific T cell proliferating responses in these mice were assessed by multimer-APC, CD8a-FITC, and TCR-β-PE. Representative flow cytometry analysis (i) and each value was plotted (ii).
(C) Neoantigen-specific T cell response was quantified. Whole splenocytes (ELISA) or isolated CD8+ T cells (ELISPOT) from mice given each neoantigen were cultured with each neoantigen peptide or non-mutated relevant peptide for 24 h and assessed for IFN-γ production. Data are pooled from two independent experiments, and the values represent mean ± SD. Circles and dot lines link respective mice.
Vaccination with Neoantigen-Peptide-pulsed DCs Demonstrates Tumor Protection
Different from self-antigen-derived, conventional tumor antigens, neoantigens are specific and powerful. Because DCs are known as the most powerful antigen-presenting cells (APCs) for priming T cells in murine and humans, ex vivo-expanded DCs are of interest. We initially assessed the antitumor effect using neoantigen-pulsed DC therapy in prophylactic models. To determine if CD8+ T cells induced against neoepitopes could provide protective anti-tumor immunity, C57BL/6J mice were administered with the mutated peptide (Zbtb40, Dpagt1, and Cry1)-pulsed DCs and then subsequently challenged with 1 × 105 MC38 tumor cells (Figure 5A). Tumor growth was inhibited in most animals in each neoantigen-pulsed DC vaccine group (Figure 5A). This protection in mice immunized with MutCry1-peptide-pulsed DCs was absent in anti-CD8 Ab-treated mice, but not anti-CD4 Ab-treated mice, strongly supporting that CD8+ T cell responses specific to mutated peptides conferred protection (Figure 5B).
Figure 5.

Demonstration of Antitumor Response by Neoantigen-Pulsed DCs Therapy
(A) Immunogenicity of neoantigen in vivo using prophylactic tumor model. Bone-marrow-derived dendritic cells (BM-DCs) were generated as previously reported. Each peptide-pulsed BM-DC (1×106/mouse) was intravenously injected twice to WT mice at day 14 and 7. Immunized mice were challenged with 1 × 105 MC38 tumor cells s.c. Tumor growth was monitored at indicated time points (mean ± SEM, n = 8/group).
(B) MutCry1-peptide-pulsed DCs (1 × 106/mouse) were administered to WT mice. Vaccinated mice were challenged with 1 × 105 MC38 tumor cells. In some experiments, mice were treated with anti-CD4 Ab or anti-CD8 Ab 2 days before tumor injection and repeatedly during tumor monitoring. Tumor growth was monitored at indicated time points (mean ± SEM, n = 7/group). Data are pooled from two independent experiments and analyzed by one-way ANOVA followed by Turkey's multiple comparison test. ∗∗p < 0.001, ∗∗∗p < 0.001.
Next, we evaluated the therapeutic potential of each single peptide by comparing mixed neoantigens in the antitumor-specific T cell response. For this study, the treatment with a single neoantigen-pulsed DCs was followed by peptide with anti-CD40 Ab and Poly(I:C), which induces robust T cell responses (Nimanong et al., 2017). We monitored the number of tumor-bearing mice that did not exceed 400 mm3 of tumor size until day 25 and found that 40% of mice (i.e., 5/13, 6/13, and 7/13) treated with each of the neoantigen-pulsed DCs had smaller tumors on day 25 (Figure 6A). In addition, vaccinated mice with mixed peptide (Z + D + C)-pulsed DCs showed a greater antitumor effect, similar to MutAdpgk (Figure 6A). Thus, vaccination with neoantigens showed remarkable and sustainable inhibition of tumor growth. In addition, we found that the frequency of the three mixed-peptide-reactive CD8+ T cells that were capable of producing IFN-γ (left), IFN-γ/TNF-α (middle), or IL-2/IFN-γ/TNF-α (right) were increased at the tumor site (Figures 6B, 6C, S6A, and S6B). On the other hand, we detected the slight levels of MutAdpgk-specific IFN-γ-/TNF-α-producing T cells (Figures 6B and 6C). Thus, we observed an enhancement of the multifunctional CD8+ T cell response to three peptides in mice vaccinated with three neoantigen-peptide DCs. In addition, the CD8+ T cell response against MutAdpgk was generated in mice vaccinated with MutAdpgk-peptide DCs to a similar extent as with three peptide-pulsed DCs. These suggest that neoantigen-responding T cells by vaccination with peptide-pulsed DCs are multifunctional in an antigen-specific manner and imply that even when with neoantigens of low antigenicity, the mixed-peptide-pulsed DC therapy would be useful.
Figure 6.

Comparison of Therapeutic Effect by Single Epitope or Multiple Epitopes of Neoantigen
(A) Tumor therapeutic model. C57BL/6J mice were s.c. injected with MC38 tumor cells and treated with neoantigen single-peptide- or multipeptide-pulsed DCs at a week later and again with the relevant peptide together with poly(I:C) and anti-CD40 Ab. Tumor growth was monitored at indicated time points. Tumor growth curve in each group of mice was plotted. As shown by the red line, tumor volume <400 mm3 at day 25 was consider as a partial response. Data are pooled from five independent experiments (n = 11–13).
(B and C) Analysis of TIL in mice treated with multipeptides of neoantigens. TILs were stimulated by peptides in the presence of anti-CD28 and brefeldin. (B) The percentages of IFN-γ single-producing T cells (left), IFN-γ+TNF-α+-producing T cells (middle), and IFN-γ+TNF-α+IL-2+-producing T cells (right) were plotted. Data are pooled from three independent experiments (n = 6). (C) Representative flow cytometry analysis of CD8+ TILs shows IFN-γ and TNF-α.
(D) Therapeutic effect by neoantigen-pulsed DCs. MC38 cells were administered s.c. One week later, these mice were treated with neoantigen-pulsed DCs twice at 1-week intervals. Anti-PD-1 Ab was injected intraperitoneally (i.p.) at day 7, 10, and 13 (n = 10 and 11/group). Data are pooled from four independent experiments, and the values represent mean ± SEM. Data were analyzed by one-way ANOVA followed by Turkey's multiple comparison test. ∗p < 0.05.
Apparently, these are immunodominant neoepitopes. Further, these results show that CD8+ T cell responses are generated by vaccination with peptide-pulsed DCs against neoepitopes in MC38 tumors. To evaluate the immunotherapy using neoantigen-pulsed DC, we examined the antitumor therapeutic effect using mixed neoantigen peptides, including one Adpgk H2-Db- and three H2-Kb-restricted-peptide-pulsed (Z + D + C) DCs in MC38 tumor-bearing mice. As shown in Figure 6D, there was an extensive therapeutic effect. When we examined whether neoantigen-pulsed DCs and ICB therapy could synergistically inhibit tumor growth, we found that antitumor immunity induced by neoantigen-pulsed DCs was at a sufficient level, such that an additional effect by the anti-PD-1 Ab could not augment the response further (Figure 6D). In its therapeutic effect, single-agent anti-PD-1 Ab caused around 43% tumor reduction at day 25 (Figure S2D). In contrast, DC/Pep mix yielded 58% tumor reduction compared with WT tumors. Therefore, instead of ICB treatment, vaccination with DCs pulsed with neoepitope peptides generated sufficient T cell immunity to inhibit established tumors.
Neoantigen-Reactive TCR Recognizes Parental MC38 Tumor Cells
Because we showed that mutation-specific peptide selected by biological assay induced tumor-reactive T cells, we next attempted to identify the T cell receptor (TCR) repertoire of antigen-specific T cell clones that could be elicited by each neoantigen. A detailed analysis of the phenotypes of these T cell clonotypes and the biophysical properties of the TCR may shed light on the extent, depth, and requirements for efficient T cell responses against tumors. We isolated neoantigen-reactive T cells by FACS sorting of the CD8+multimer+ CTL reactive for MutDpagt1 or CD8+PD-1+CD137+ T cells reactive for MutCry1 (Figure S7A, left). We cloned each TCRα and TCRβ sequence into a retroviral vector (pMXs), followed by transfection into the TG40 cell line (Figure S7A, right). The top five sequences of repertoire analysis were selected, and orange- and green-colored ones were subcloned into the expression vector (Figure S7B). To verify that the sequences of the α and β chain were indeed candidates for neoantigen-reactive TCRα and β, we cocultured the TCR-gene-modified TG40 cells with peptide-pulsed EL4. As shown in Figures S7C and S7D, we found one combination of TCRα and TCRβ in MutDpagt1, whereas two combinations of TCRα and TCRβ were found in MutCry1 for CD69 upregulation, suggesting that they are unique and clonal TCRs. Because TCRs shared the conserved CDR3 sequences for recognizing the same MHC-I epitopes, clustering TCRs based on sequence motif enables epitope-specific recognition and diversity. We parsed randomly extracted repertoire sequences by TCRdist (Dash et al., 2017) for TCRB clustering. We observed the homology-based hierarchy and found that neoantigen reactive TCRs occurred in groups (red triangle) and these cluster-relevant newly established sequences in CDR3 during VDJ recombination (framed red; Figure 7A). These new motifs were shown to frequently interact with MHC-I epitopes by crystal structure analysis. Therefore, to confirm that these neoantigens could induce specific responses, we synthesized mutated di-amino acids sequences, AlaAla, into the CDR3 region. As expected, AlaAla mutation-harboring TCRs drastically lost their reactivity to neoantigens. Moreover, we performed mutant and WT peptide titration assays (Figure S8) and confirmed strong specificity of the neoantigen mutation for TCR8 and TCR10, which was weak for TCR36 (Figures 7B, 7C, and S8). We observed a slight, but apparent, specificity of TCR36 (for Dpagt1) and TCR8 and 10 (for Cry1) by a mutant and WT peptide titration. These suggested that several clonotypes could be generated from the same mutated specific peptide (Cry1 peptide).
Figure 7.

Primary T Cell Expressing Neoantigen-Specific TCR Recognizes Tumor
(A) TCRdist analysis of TCRB repertoire sequences showing the tree diagram of TCRB CDR3 homology. Clonality was not reflected. Red arrows indicate neoantigen peptide reactive CDR3 sequence. As a negative control, rearranged CDR3-derived di-amino acids (red rectangle) were mutated into AlaAla.
(B and C) TCRαβ derived from CTL responding to Dpagt1pep included TCR36, and TCRαβ derived from CTL responding to Cry1pep is termed as TCR8 and TCR10. Reactive TCRα and β chains were selected, and they were tandemly linked with Furin-SGSG-P2A sequence and cloned into pMXs-IRES-GFP. Selected combination of TCRα and TCRβ were transfected to TG40 CD8A/B (TCR36, B; TCR8, TCR10, C). These TG40 cells were cocultured with each peptide-pulsed EL4 for 24 h. The percentage of CD69 upregulation by TCR36 or TCR8 and TCR10 and their AA mutation transduction in GFP+ cells for MutDapgt1 peptide or MutCry1 peptide is shown. Data were pooled from six or three independent experiments, and the values represent mean ± SD. Each black circle shows independent values.
(D) TCR36-transduced or TCR8- and TCR10-transduced GFP+CD8+ T cells were cocultured with MC38 in the presence or absence of neoantigens peptide for 48 h. Subsequently, supernatants were measured for IFN-γ by ELISA. Data were pooled from seven to ten independent experiments, and the values represent mean ± SD. Data were analyzed by unpaired Student's t test, ∗p < 0.05, ∗∗p < 0.01.
Finally, we examined whether antigen-specific TCRα and β chain-bearing T cells could respond to each peptide antigen and parental MC38 tumor cells. In primary T cells transduced with TCRα and TCRβ gene, even without neoantigens peptides, TCRs reacted with MC38 and produced IFN-γ (Figure 7D). Regarding the tumor response, not only TCR36 but also TCR8 and TCR10 could respond to MC38. Thus, tumor neoantigen-specific T cell clones exhibited antigen-specific responses against MC38.
Discussion
The TME is associated with response to ICB therapy. Particularly, PD-L1 expression, the frequency of CD8+ T cell infiltration, and the mutation burden in tumors correlate well with the likelihood of response during or after ICB therapy. However, the biological relevance of each of these factors has to be clarified for effective ICB usages. In this study, we showed that the difference between resistance and sensitivity of ICB tumors depended on the responsiveness of T cells to neoantigens as a key T cell response in tumor sites. Particularly, we demonstrated the relationship between PD-L1 expression on tumors and the sensitivity to ICB therapy by comparing ICB-treated MC38-PD-L1-KO (non-innate resistant type) and parental MC38 (innate resistant type). In fact, when tumors express PD-L1, even if they have the potential to generate an antitumor T cell response by ICB therapy, these T cells are weak, resulting in faster tumor growth. In contrast, when tumors lack PD-L1, the antitumor T cell immunity against neoantigens can be significantly elicited by ICB, resulting in slowed growth of the tumor.
A tumor-intrinsic role of PD-L1 is to promote cancer initiation toward CSC metastasis, progression, and resistance to therapy (Dong et al., 2018, Fabrizio et al., 2018). In in vitro studies, disruption of intrinsic PD-L1 by CRISPR/Cas9 technique led to suppression of progressive cancer cells, inhibition of spheroid formation of osteosarcoma, and increased anticancer drug sensitivities to doxorubicin and paclitaxel (Liao et al., 2017). PD-L1 transactivation pathway is likely abnormal in many cancers. In intrinsic mechanisms underlying aberrant PD-L1 activation, genomic alterations (i.e., copy number amplification of CD274 residues (Cancer Genome Atlas Research Network, 2014, Straub et al., 2016, George et al., 2017) and 3′-UTR disruption (Schoenberg and Maquat, 2012, Kataoka et al., 2016)), constitutive oncogenic signaling activation (via PI3K/AKT or RAS/MAPK (Crane et al., 2009, Coelho et al., 2017)), and epigenetic changes (e.g., expression of microRNA 197 during PD-L1 inhibition in lung cancer and aberrant DNA methylation (Fujita et al., 2015, Dong et al., 2016)) have been described. Whereas, as extrinsic factors, IFN-γ (Garcia-Diaz et al., 2017) and other inflammatory cytokines (e.g., IL-17 and TNF-α) (Wang et al., 2017) and HIF-1α (Noman et al., 2014) can activate PD-L1. Hence, what determines the low PD-L1 expression in highly immunogenic TME, which presumably drives best clinical benefits from ICB therapy, needs to be investigated in the future studies.
Effective antitumor vaccines using neoantigens are often the most immunogenic. In the current study, we identified novel three neoantigens on MC38 in an H2-Kb-restricted manner and showed the potential utility of a neoantigen-mixed peptides-pulsed DC vaccine beyond the PD-L1-restricted mechanism. DCs are essential in immunity owing to their role in activating T cells, thereby promoting antitumor responses. Compared with the clinical success of ex vivo DC therapy based on conventional tumor antigens, neoantigen-pulsed DCs have been anticipated for showing potent efficacy (Carreno et al., 2015, Chen et al., 2019). In fact, in this study, we exhibited the efficacy of four types of neoantigen-responding T cells infiltrating in the tumor in mice treated with four neoantigen-peptide-pulsed DCs. Moreover, we demonstrated that neoantigen-specific T cells in TME by DC vaccination possess multifunctionality and an antitumor effect similar to ICB therapy. It is known that anti-CTLA-4 Ab acts through APCs, particularly on DCs, leading to CTL generation, whereas anti-PD-1 Ab therapy can reactivate an impaired CTL. Effectiveness of ICB therapy must depend on DC function. Further, neoantigen-pulsed DC vaccines improved T cell responses in tumors, at levels similar to those by ICB therapy. These support ICB sensitivity dependences on CD8+ T cell responses specific to “multiple” neoantigens, conferring protection against tumors. Combination therapy using ICB may have clinical benefits, but a major limitation remains its characteristic antigen loss. Particularly, it is known that ICB efficacy can be impaired by deleting neoantigens on tumors, which results in tumor progression (Tran et al., 2016). In this study, using different MHC-restricted tumor-associated neoantigens simultaneously with mature DCs, we suggested that starting therapy using multiple neoantigen-peptide-pulsed DCs at early phase generates clinically relevant neoantigen-specific T cells before possible deletion and immune evasion.
Taken together, our findings provide tumor immunological evidence that the level of T cell responses to neoantigens and PD-L1 expression on tumor determine the positive and negative cancer immunity cycle, and therefore, may shape immunoediting during tumor occurrence, which must be optimally targeted for clinical responses. In addition, our results suggested that PD-L1-KO tumor cells may be useful in isolating neoantigen-specific T cells and that identified neoantigen vaccination could block immune escape, highlighting the recently refocused development in DC cancer vaccines against immune checkpoint ligand-expressing tumors (Figure S9).
Limitation of the Study
In this study, we clarified the innate resistance roles of PD-L1-expressing tumor, which dampens tumor-specific CTLs responses including neoantigens. The fact that PD-L1-deficient tumors treated with anti-PD-1Ab plus anti-CTLA-4 Ab facilitated the neoantigens screen with higher sensitivity may indicate the usefulness of applicable DC-based vaccines. This approach may extend to the clinical application. However, in the current study, the identification of neoantigens were derived from in vivo studies in murine. There are still several problems to identify the neoantigens in human. We need to make new in vitro protocol or new method to use humanized mice for identification of neoantigens.
Resource Availability
Lead Contact
Further information is available from the Lead Contact, Shin-ichiro Fujii (shin-ichiro.fujii@riken.jp).
Materials Availability
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact.
Data and Code Availability
The accession number for the Exome and RNA sequence datasets reported in this paper is DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA):DRA010264. The request for additional data or codes not infringing on ethics restrictions is available to Lead Contact.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number JP19K07653.
Author Contributions
S.F., M.O., and K.S. conceived and designed the experiments. Y.M., T.W., and O.O performed bioinformatics analysis for potential neoepitopes. T.W. conducted and analyzed RNA sequencing data, and M.O. detected the TCR repertoire. J.S. established the cassette of TCRA and TCRB genes, and M.O. generated the final TCRA and TCRB. M.O. mainly, K.S., S.U., and T.I. performed the biological studies, and S.F., M.O., K.S., and T.I conducted data analysis. S.F. supervised the overall project. S.F. and M.O. wrote the manuscript with input from all the other authors.
Declaration of Interests
The authors have no financial conflicts of interest related to this manuscript.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101238.
Supplemental Information
References
- Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezu L., Kepp O., Cerrato G., Pol J., Fucikova J., Spisek R., Zitvogel L., Kroemer G., Galluzzi L. Trial watch: peptide-based vaccines in anticancer therapy. Oncoimmunology. 2018;7:e1511506. doi: 10.1080/2162402X.2018.1511506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol K.F., Figdor C.G., Aarntzen E.H., Welzen M.E., van Rossum M.M., Blokx W.A., van de Rakt M.W., Scharenborg N.M., de Boer A.J., Pots J.M. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. Oncoimmunology. 2015;4:e1019197. doi: 10.1080/2162402X.2015.1019197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher J.P., Reis E.S.C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. 2018;4:784–792. doi: 10.1016/j.trecan.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J., Reckamp K.L., Baas P., Crino L., Eberhardt W.E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E.E., Holgado E. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno B.M., Magrini V., Becker-Hapak M., Kaabinejadian S., Hundal J., Petti A.A., Ly A., Lie W.R., Hildebrand W.H., Mardis E.R. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle J.C., Kreiter S., Diekmann J., Lower M., van de Roemer N., de Graaf J., Selmi A., Diken M., Boegel S., Paret C. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- Chen F., Zou Z., Du J., Su S., Shao J., Meng F., Yang J., Xu Q., Ding N., Yang Y. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Invest. 2019;129:2056–2070. doi: 10.1172/JCI99538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho M.A., de Carne Trecesson S., Rana S., Zecchin D., Moore C., Molina-Arcas M., East P., Spencer-Dene B., Nye E., Barnouin K. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–1099.e6. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane C.A., Panner A., Murray J.C., Wilson S.P., Xu H., Chen L., Simko J.P., Waldman F.M., Pieper R.O., Parsa A.T. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene. 2009;28:306–312. doi: 10.1038/onc.2008.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong P., Xiong Y., Watari H., Hanley S.J., Konno Y., Ihira K., Suzuki F., Yamada T., Kudo M., Yue J. Suppression of iASPP-dependent aggressiveness in cervical cancer through reversal of methylation silencing of microRNA-124. Sci. Rep. 2016;6:35480. doi: 10.1038/srep35480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong P., Xiong Y., Yue J., Hanley S.J.B., Watari H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front. Oncol. 2018;8:386. doi: 10.3389/fonc.2018.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont A.M.M., Blank C.U., Mandala M., Long G.V., Atkinson V., Dalle S., Haydon A., Lichinitser M., Khattak A., Carlino M.S. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018;378:1789–1801. doi: 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- Fabrizio F.P., Trombetta D., Rossi A., Sparaneo A., Castellana S., Muscarella L.A. Gene code CD274/PD-L1: from molecular basis toward cancer immunotherapy. Ther. Adv. Med. Oncol. 2018;10 doi: 10.1177/1758835918815598. 1758835918815598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris R.L., Blumenschein G., Jr., Fayette J., Guigay J., Colevas A.D., Licitra L., Harrington K., Kasper S., Vokes E.E., Even C. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S., Fujimoto K., Shimizu K., Ezaki T., Kawano F., Takatsuki K., Kawakita M., Matsuno K. Presentation of tumor antigens by phagocytic dendritic cell clusters generated from human CD34+ hematopoietic progenitor cells: induction of autologous cytotoxic T lymphocytes against leukemic cells in acute myelogenous leukemia patients. Cancer Res. 1999;59:2150–2158. [PubMed] [Google Scholar]
- Fujii S., Liu K., Smith C., Bonito A.J., Steinman R.M. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 2004;199:1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S., Shimizu K., Hemmi H., Steinman R.M. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol. Rev. 2007;220:183–198. doi: 10.1111/j.1600-065X.2007.00561.x. [DOI] [PubMed] [Google Scholar]
- Fujii S.I., Shimizu K. Immune networks and therapeutic targeting of iNKT cells in cancer. Trends Immunol. 2019;40:984–997. doi: 10.1016/j.it.2019.09.008. [DOI] [PubMed] [Google Scholar]
- Fujita Y., Yagishita S., Hagiwara K., Yoshioka Y., Kosaka N., Takeshita F., Fujiwara T., Tsuta K., Nokihara H., Tamura T. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol. Ther. 2015;23:717–727. doi: 10.1038/mt.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A., Shin D.S., Moreno B.H., Saco J., Escuin-Ordinas H., Rodriguez G.A., Zaretsky J.M., Sun L., Hugo W., Wang X. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J., Saito M., Tsuta K., Iwakawa R., Shiraishi K., Scheel A.H., Uchida S., Watanabe S.I., Nishikawa R., Noguchi M. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin. Cancer Res. 2017;23:1220–1226. doi: 10.1158/1078-0432.CCR-16-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryfe R., Kim H., Hsieh E.T., Aronson M.D., Holowaty E.J., Bull S.B., Redston M., Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi F.S., Chesney J., Pavlick A.C., Robert C., Grossmann K.F., McDermott D.F., Linette G.P., Meyer N., Giguere J.K., Agarwala S.S. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558–1568. doi: 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja V.R., McGuire K.A., Manguso R.T., LaFleur M.W., Collins N., Haining W.N., Freeman G.J., Sharpe A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017;214:895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasaki T., Nagayama K., Kuwano H., Nitadori J.I., Sato M., Anraku M., Hosoi A., Matsushita H., Takazawa M., Ohara O. Prediction and prioritization of neoantigens: integration of RNA sequencing data with whole-exome sequencing. Cancer Sci. 2017;108:170–177. doi: 10.1111/cas.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K., Shiraishi Y., Takeda Y., Sakata S., Matsumoto M., Nagano S., Maeda T., Nagata Y., Kitanaka A., Mizuno S. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- Kleffel S., Posch C., Barthel S.R., Mueller H., Schlapbach C., Guenova E., Elco C.P., Lee N., Juneja V.R., Zhan Q. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowanetz M., Zou W., Gettinger S.N., Koeppen H., Kockx M., Schmid P., Kadel E.E., 3rd, Wistuba I., Chaft J., Rizvi N.A. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1) Proc. Natl. Acad. Sci. U S A. 2018;115:E10119–E10126. doi: 10.1073/pnas.1802166115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Chen L., Feng Y., Shen J., Gao Y., Cote G., Choy E., Harmon D., Mankin H., Hornicek F. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget. 2017;8:30276–30287. doi: 10.18632/oncotarget.16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Wei S., Hurt E.M., Green M.D., Zhao L., Vatan L., Szeliga W., Herbst R., Harms P.W., Fecher L.A. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 2018;128:805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N., Furness A.J., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R.J., Tannir N.M., McDermott D.F., Aren Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthelemy P., Porta C., George S. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018;378:1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimanong S., Ostroumov D., Wingerath J., Knocke S., Woller N., Gurlevik E., Falk C.S., Manns M.P., Kuhnel F., Wirth T.C. CD40 signaling drives potent cellular immune responses in heterologous cancer vaccinations. Cancer Res. 2017;77:1918–1926. doi: 10.1158/0008-5472.CAN-16-2089. [DOI] [PubMed] [Google Scholar]
- Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P., Bronte V., Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N., Morris L., Havel J.J., Makarov V., Desrichard A., Chan T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016;28:411–419. doi: 10.1093/intimm/dxw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittmeyer A., Barlesi F., Waterkamp D., Park K., Ciardiello F., von Pawel J., Gadgeel S.M., Hida T., Kowalski D.M., Dols M.C. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C., Schachter J., Long G.V., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C., Lotem M. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- Rojas-Sepulveda D., Tittarelli A., Gleisner M.A., Avalos I., Pereda C., Gallegos I., Gonzalez F.E., Lopez M.N., Butte J.M., Roa J.C. Tumor lysate-based vaccines: on the road to immunotherapy for gallbladder cancer. Cancer Immunol. Immunother. 2018;67:1897–1910. doi: 10.1007/s00262-018-2157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roufas C., Chasiotis D., Makris A., Efstathiades C., Dimopoulos C., Zaravinos A. The expression and prognostic impact of immune cytolytic activity-related markers in human malignancies: a comprehensive meta-analysis. Front. Oncol. 2018;8:27. doi: 10.3389/fonc.2018.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M., Balan S., Roudko V., Bhardwaj N. Towards superior dendritic-cell vaccines for cancer therapy. Nat. Biomed. Eng. 2018;2:341–346. doi: 10.1038/s41551-018-0250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg D.R., Maquat L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Shimizu K., Iyoda T., Okada M., Yamasaki S., Fujii S.I. Immune suppression and reversal of the suppressive tumor microenvironment. Int. Immunol. 2018;30:445–454. doi: 10.1093/intimm/dxy042. [DOI] [PubMed] [Google Scholar]
- Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S., Spaapen R.M., Zha Y., Williams J., Meng Y., Ha T.T., Gajewski T.F. Up-regulation of PD-L1, Ido, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R.M. Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- Steinman R.M., Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Straub M., Drecoll E., Pfarr N., Weichert W., Langer R., Hapfelmeier A., Gotz C., Wolff K.D., Kolk A., Specht K. CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget. 2016;7:12024–12034. doi: 10.18632/oncotarget.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanegashima T., Togashi Y., Azuma K., Kawahara A., Ideguchi K., Sugiyama D., Kinoshita F., Akiba J., Kashiwagi E., Takeuchi A. Immune suppression by PD-L2 against spontaneous and treatment-related antitumor immunity. Clin. Cancer Res. 2019;25:4808–4819. doi: 10.1158/1078-0432.CCR-18-3991. [DOI] [PubMed] [Google Scholar]
- Tang H., Liang Y., Anders R.A., Taube J.M., Qiu X., Mulgaonkar A., Liu X., Harrington S.M., Guo J., Xin Y. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest. 2018;128:580–588. doi: 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi J.L., Bobisse S., Ophir E., Tuyaerts S., Roberti A., Genolet R., Baumgartner P., Stevenson B.J., Iseli C., Dangaj D. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018;10:eaao5931. doi: 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- Topalian S.L., Drake C.G., Pardoll D.M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Carvajal R.D., Sosman J.A., Atkins M.B. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S.L., Taube J.M., Anders R.A., Pardoll D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E., Robbins P.F., Lu Y.C., Prickett T.D., Gartner J.J., Jia L., Pasetto A., Zheng Z., Ray S., Groh E.M. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., Litchfield K., Xu H., Rosenthal R., McGranahan N., Reading J.L., Wong Y.N.S., Rowan A., Kanu N., Al Bakir M. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- Wang X., Yang L., Huang F., Zhang Q., Liu S., Ma L., You Z. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017;184:7–14. doi: 10.1016/j.imlet.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- Wilky B.A. Immune checkpoint inhibitors: the linchpins of modern immunotherapy. Immunol. Rev. 2019;290:6–23. doi: 10.1111/imr.12766. [DOI] [PubMed] [Google Scholar]
- Yadav M., Jhunjhunwala S., Phung Q.T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T.K., Fritsche J., Weinschenk T. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- Yang W., Lee K.W., Srivastava R.M., Kuo F., Krishna C., Chowell D., Makarov V., Hoen D., Dalin M.G., Wexler L. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the Exome and RNA sequence datasets reported in this paper is DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA):DRA010264. The request for additional data or codes not infringing on ethics restrictions is available to Lead Contact.