

Abstract
Objectives
This study aims to discuss the clinical, laboratory and genetic findings, and treatment options for six patients who were diagnosed with Blau syndrome (BS)/early-onset sarcoidosis (EOS).
Patients and methods
The study included four patients (2 males,2 females; mean age 7 years; range 4 to 10 years) with EOS and two siblings (1 male, 1 female; mean age 10 years; range, 9 to 11 years) with BS. Age, age of initial symptoms, age of diagnosis; articular involvement, presence of uveitis, dermatitis, or fever, other organ involvement, laboratory findings, results of metabolic tests for mucopolysaccharidosis and mucolipidosis, results of genetic, pathologic, and immunologic tests, radiologic findings to evaluate skeletal dysplasia, and treatment options were collected.
Results
The median age at diagnosis of all patients was 6 years (range, 1 to 10 years). Five patients had camptodactyly and bilateral boggy synovitis in the wrists and ankles, one had granulomatous inflammatory changes in the liver and kidney biopsy, and one had attacks of fever and granulomatous dermatitis. None had uveitis. The detected mutations in nucleotide-binding oligomerization domain containing 2 (NOD2) were P268S (rs2066842), M513T (rs104895473), R702W (rs2066844), V955I (rs5743291), H343Y (rs199858111), and M491L (16:50745293). The treatments of patients included corticosteroids, non-steroid anti-inflammatory drugs, methotrexate, infliximab, adalimumab, anakinra, and canacinumab.
Conclusion
Camptodactyly and boggy synovitis are important signs of BS/EOS. Methotrexate and tumor necrosis factor blockers are more effective in patients with predominantly articular symptoms. In patients 5 and 6 and their mother, we determined a novel M491L mutation in the NOD2 gene. Currently, this work is in progress towards identifying the pathogenesis and treatment options for this disease.
Keywords: Blau syndrome, clinical findings, genetic, nucleotide-binding oligomerization domain containing 2
Introduction
Granulomatous autoinflammatory diseases are monogenic syndromes caused by mutations in the region encoding the nucleotide-binding domain of the nucleotide-binding oligomerization domain containing 2 (NOD2) gene, mapped to chromosome 16q12, as defined in 2001.[1] These syndromes are the familial and sporadic forms of the same disease (Blau syndrome [BS], online mendelian inheritance in man [OMIM] 186580 and early-onset sarcoidosis [EOS], OMIM 609464). In 1985, pediatrician Edward Blau described the BS for the first time as a dominantly inherited, chronic inflammatory syndrome characterized by the clinical triad of granulomatous dermatitis, symmetric arthritis, and recurrent uveitis with onset before four years of age.[2] Most of the studies on BS/EOS are case reports in the literature. Until 2014, 146 familial cases (BS) and 62 sporadic cases (EOS) with NOD2 mutations had been reported.[3] An international registry study group identified 31 cases of BS from 11 countries in 2015.[4] In 2018, 50 BS cases with uveitis were reported from 25 clinical centers.[5] In this study, we aimed to discuss the clinical, laboratory and genetic findings, and treatment options for six patients who were diagnosed with BS/EOS.
Patients and Methods
This study was performed at the Department of Department of Child Health and Diseases, Division of Pediatric Rheumatology at Erciyes University Faculty of Medicine between May 2013 and January 2017. The study included four patients (2 males, 2 females; mean age 7 years; range 4 to 10 years) with EOS and two siblings (1 male, 1 female; mean age 10 years; range, 9 to 11 years) with BS. Patients' characteristics were obtained from patient charts. Age, age of initial symptoms, age of diagnosis; articular involvement, presence of uveitis, dermatitis, or fever, other organ involvement, laboratory findings, results of metabolic tests for mucopolysaccharidosis and mucolipidosis, results of genetic, pathologic, and immunologic tests, radiologic findings to evaluate skeletal dysplasia, and treatment options were collected. The study protocol was approved by the Erciyes University Faculty of Medicine Ethics Committee (29/09/2017-442). A written informed consent was obtained from the legal guardians of each patient. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Peripheral blood samples for deoxyribonucleic acid (DNA) extraction were obtained. The genomic DNA isolation and purification were performed using DNeasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany). Quality control and quantitation of the isolated DNA sample were performed using a Nanodrop (Thermo Fisher Scientific, Wilmington, DE, USA). The structural integrity of the DNA sample was confirmed by gel electrophoresis.
We performed targeted gene sequencing for all NOD2 exonic and intronic regions in the 16q12.1 (39.497 bp) genomic coordinate in all patients. Sequencing and bioinformatic data analysis were performed on an Illumina NextSeq500® according to the Illumina Nextera XT® Kit’s (Illumina Inc., San Diego, CA, USA) standard protocol as described previously.[6]
The sequencing data were aligned based on the GRCh37 human reference genome. A minimum depth of 200 reads was obtained for all sequences. Missense, splice site and stop codon variants, and frame shift and non-frame shift insertions and deletions were filtered according to 1000 Genomes, Exome Aggregation Consortium, dbSNP138 and the ClinVar database based on a minor allele frequency below 1%. The sorting intolerant from tolerant and polymorphism phenotyping tools were used to assess the possible effects of the variants on the proteins.
Results
Patient 1: A four-year-old male patient was referred for an evaluation for fever, lymphadenopathy, and non-pruritic skin eruption which was most prominent on the arms and legs. These symptoms began when the patient was four months old. The fever persisted for 7-10 days, accompanied by a maculopapular rash, and cervical and axillary lymphadenopathy. The longest of the disease convalescence periods was a month. He had no uveitis or articular involvement. There was no consanguinity or family history of any autoinflammatory disorders. A physical examination showed generalized, maculopapular eruption, particularly on the arms and legs, and an enlarged lymph node in the left cervical chain and axillary region (Figure 1). Laboratory tests conducted for etiology; complete blood count, blood smear, immunoglobulins, neutrophil function test by flow-cytometry, and a ratio of lymphocyst subgroups by flow-cytometry gave normal results. The serology of Epstein- Barr virus, cytomegalovirus, toxoplasma, brucella, tularemia, Bartonella henselae, and results of the quantiferron test for tuberculosis were negative. A skin biopsy taken from a papule showed subepidermal non-Langerhans histiocytes and a non-caseating granuloma (Figure 2). A lymph node biopsy showed granulomatous inflammation as well (Figure 3). There was no mediastinal lymph node involvement on chest computed tomography. The NOD2 mutations detected were P268S (rs2066842, exon 4) and V955I (rs5743291, exon 9). The patient could not achieve remission with steroid, methotrexate, anakinra, adalimumab, and infliximab. He is still on a low dose steroid and canakinumab treatment.
Figure 1. Maculopapular rash on patients’ legs.
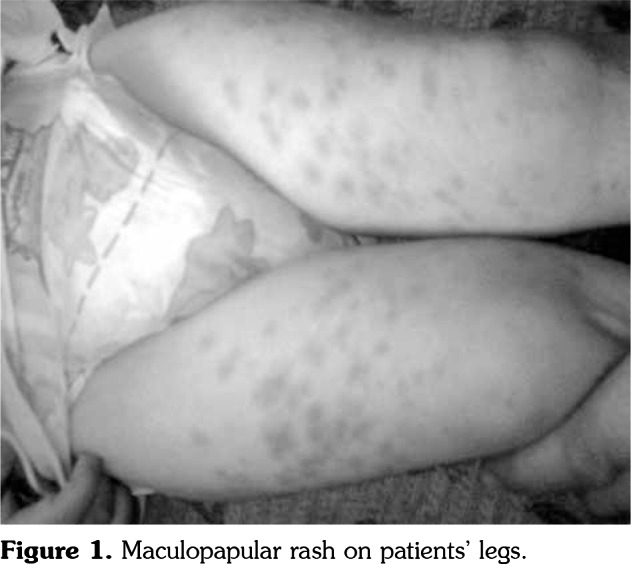
Figure 2. Skin biopsy showed granulomatous inflammation.

Figure 3. Lymph node biopsy showed granulomatous inflammation.

Patient 2: A 10-year-old female patient was diagnosed with juvenile idiopathic arthritis (JIA) at the age of two. While she was on methotrexate and etanercept therapy, she developed multiorgan failure, and was referred to our department at the age of seven. Hepatic and renal biopsies revealed granulomatous inflammation in both organs. She had no uveitis. There was consanguinity but no family history. We re-evaluated her diagnosis due to polyarticular involvement, boggy synovitis on the wrists and ankles, camptodactyly, and the renal and hepatic biopsy results. The NOD2 mutation detected was M513T (rs104895473, exon 4) heterozygote. The patient was put on methotrexate and infliximab, which led to remission.
Patient 3: A six-year-old female patient was admitted for polyarticular involvement, boggy synovitis on both wrists, camptodactyly, and hyperpigmented macular eruption on the anterior legs. Her complaints had started one year before. She was evaluated due to rapidly progressive deformities. Enzyme levels for mucopolysaccharidosis and mucolipidosis were in the normal ranges. Radiographic findings for skeletal dysplasia were normal. She had no uveitis. A skin biopsy could not be performed. There was no consanguinity or family history. The NOD2 mutation detected was R702W (rs2066844, exon 4) heterozygote. Remission was achieved with methotrexate.
Patient 4: An eight-year-old male patient was admitted for boggy synovitis on both wrists and legs, polyarthritis, and camptodactyly. His complaints started at nine months of age. He had no uveitis or dermatitis. There was consanguinity but no family history. He was evaluated due to mucopolysaccharidosis, mucolipidosis, and skeletal dysplasia. The NOD2 mutation detected was H343Y (rs199858111, exon 4). Remission could not be achieved with corticosteroids, methotrexate, etanercept, tocilizumab, adalimumab, infliximab, and canakinumab. He is on a low dose steroid and siklosporin treatment.
Patient 5: An 11-year-old male patient was admitted with boggy synovitis on both wrists and ankles, polyarthritis, and camptodactyly. His complaints started during infancy. There were no skin lesions or uveitis. The NOD2 mutations detected were V955I (rs5743291, exon 9) and M491L (16:50745293, exon 4). There was no consanguinity. He was put on methotrexate and infliximab treatment, which led to remission.
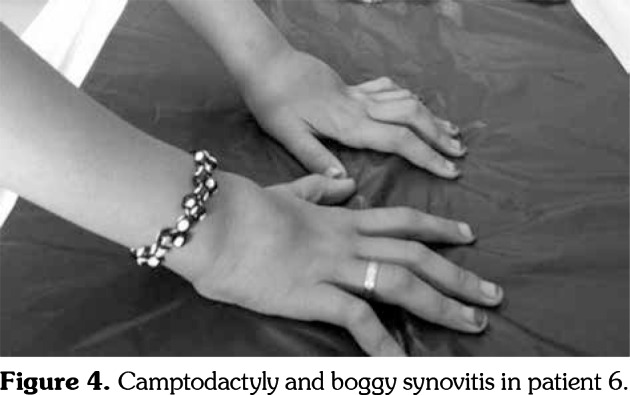
Patient 6: A nine-year-old female patient was admitted with boggy synovitis on both wrists and ankles, polyarthritis, and camptodactyly (Figure 4). Her complaints started during infancy. There were no skin lesions or uveitis. There was no consanguinity. She is the sibling of patient 5. We screened the NOD2 gene and found the M491L (16:50745293, exon 4) mutation. To our knowledge, this mutation has not been reported before. She was put on methotrexate and infliximab treatment, which led to remission.
Figure 4. Camptodactyly and boggy synovitis in patient 6.
The mother of patients 5 and 6 also had camptodactyly and articular complaints. The mother's NOD2 gene was found to have the M491L (16:50745293, exon 4) mutation, the same as in her daughter (patient 6). The father of patients 5 and 6 also had a V955I mutation in the NOD2 gene. The father did not have any complaints or clinical findings. The pedigree of patients 5 and 6 is shown as Figure 5.
Figure 5. Pedigree of patients 5 and 6. CA: Cancer.

All patients’ complete blood count, blood smears, immunoglobulin levels, urine calcium/ creatinine ratios, serum angiotensin-converting enzyme levels, and enzyme levels for mucopolysaccharidosis and mucolipidosis were in the normal ranges (except for patient 1). Radiographic findings of the chest, extremities, and vertebrae were evaluated to exclude skeletal dysplasia (except for patient 1). The ophthalmological examinations of all patients had normal results. Tuberculin skin tests were negative in all patients. The patients' clinical and laboratory findings are given in Table 1. The patients' genetic findings are provided in Table 2.
Table 1. Clinical features of patients.
| Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6/sibling of patient 5 |
| Age (year)/sex | 4/M | 10/F | 6/F | 8/M | 11/M | 9/F |
| Age of initial symptoms | 4 month | 2 year | 5 year | 9 month | Infancy | Infancy |
| Age of diagnosis (year) | 1 | 9 | 6 | 6 | 10 | 8 |
| Fever | + | - | - | - | - | - |
| Arthritis | - | + | + | + | + | + |
| Dermatitis | + | - | + | - | - | - |
| Uveitis | - | - | - | - | - | - |
| Tenosynovitis | - | + | + | + | + | + |
| Camptodactyly | - | + | + | + | + | + |
| Kidney | - | + | - | - | - | - |
| Liver | + | - | - | - | - | |
| LAP | + | - | - | - | - | - |
| Lung | - | - | - | - | - | - |
| High APR | + | + | - | - | - | + |
| High ACE | - | - | - | - | - | - |
| Mutations | P268S/V955I | M513T/- | R702W/SNP8 | H343Y/- | V955I/M491L | M491L |
| MEFV mutations | R761H/- | - | - | E148Q/- | E148Q/- | - |
| Previous treatment | Steroid, MTX, | Steroid, MTX, | NSAI, MTX | NSAI, MTX, | NSAI, MTX | NSAI, MTX |
| ANA, ADA, | ETA, TOS | ETA, TOS, | ||||
| IFN | ADA, INF, CAN | |||||
| Current treatment | LDS, CAN | MTX, INF | MTX | LDS, Siklosporin | MTX, INF | MTX, INF |
| LAP: Lymphadenopathy; APR: Acute Phase Reactans; ACE: Angiotensin converting enzyme; MEFV: Mediterranean fever; MTX: Methotrexate; ANA: Anakinra; ADA: Adalimumab; IFN: Infliximab; ETA: Etanercept; TOS: Tosilizumab; NSAI: Non-steroid anti-inflammatory drugs; CAN: Canakinumab; LDS: Low dose steroid. | ||||||
Table 2. Genetic findings of patients.
| Gene NOD2 | SNP | Mutation type | Aminoacid changes | Nucleotide changes | Mutation | Effect | Result |
| Exon 4 | rs2066842 | snv | P/S | C/T | P268S | Pathogenic | BS/EOS |
| Exon 4 | rs104895473 | snv | M/T | T/C | M513T | Pathogenic | BS/EOS |
| Exon 4 | rs2066844 | snv | R/W | C/T | R702W | Pathogenic | BS/EOS |
| Exon 4 | rs199858111 | snv | H/Y | C/T | H343Y | Pathogenic | BS/EOS |
| Exon 9 | rs5743291 | snv | V/I | G/A | V955I | Pathogenic | BS/EOS |
| Exon 4 | 16:50745293 | snv | M/L | A/T | M491L | US | US |
| NOD2: Nucleotide-binding oligomerization domain containing 2; SNP: Single nucleotide polymorphism; snv: Single nucleotide variation; EOS: Early-onset sarcoidosis; US: Unknown significance. | |||||||
Discussion
Blau syndrom and EOS are rare autoinflammatory systemic granulomatus diseases that have been associated with NOD2 mutation. The NOD2 gene encodes a protein which is a member of the intracellular pattern- recognition receptors family. This NOD2 protein is involved in the inflammatory response through microbe-associated molecular pattern recognition.[7] This protein is mainly expressed in monocytes, macrophages, and dendritic cells, and plays a crucial role in innate immune defense against invading pathogens. The engagement of the muramyl dipeptide ligand leads to the unfolding of NOD2 from its autoinhibited state.[6] The consequent activation of the key downstream signaling molecule nuclear factor kappa B (NF-κB) ultimately results in the production of inflammatory cytokines, chemokines, and adhesion molecules.[7,8] The NOD2 mutation is a gain of function mutation leading to hyperactivation of the NF-kB response.[8]
Blau syndrome and EOS are familial and sporadic forms of the same disease, respectively. The occurrence of BS/EOS is primarily among Caucasians, although it has been reported in Asians as well as Afro-Americans.[9,10] There is no information about the distribution of the disease in the world. The classic BS/EOS symptom triad is skin rash, arthritis, and uveitis. Our sibling patients 5 and 6 and their mother were assessed as BS because of the familial form of the disease.
The disease begins typically before three- four years of age.[11] Onset occurs mostly with cutaneous and articular symptoms.[12-15] However, symptoms sometimes appear after the age of 10.[12] Our patients' initial symptoms started between the age of four months and five years.
Two main types of symptomatic eruptions are reported: rash and multiple subcutaneous nodules. The rash usually appears as an erythema with maculopapular configuration. Pinhead-sized, lichenoid, yellow to brown papules typically appear in clusters and might become confluent. The appearance is variable, often symmetric, and is located on the trunk and/or extremities.[16] Histology of the lesions consistently demonstrates non-caseating granulomas with multinucleated giant cells.[17] BS is diagnosed with confirmatory histology in more than 90% of patients.[13] A non-pruritic skin eruption was on the extremities of patients 1 and 3. Patient 1’s skin biopsy taken from a papule showed subepidermal non-Langerhans histiocytes and a non-caseating granuloma (Figure 2). This finding is characteristic of BS, and the skin biopsy gives a better diagnostic yield when compared to the synovium.[13]
Joint manifestations usually appear such as symmetric polyarthritis, involving the wrists, metacarpophalangeal, first metatarsophalangeal, and proximal interphalangeal (PIP) joints of the hands and feet, ankles, and occasionally the elbows. Contractures often develop in the PIP joints early in the course of the disease. This early deformity was originally described as camptodactyly.[8] Additionally, granulomatous inflammation in the periarticular structures leads to marked periarticular swelling and tenosynovial cysts particularly affecting the wrists and dorsa of the hands.[16] These appearances are characteristic phenotypic findings of the disease.[8] Rosé et al.[4] reported that the characteristics of arthritis in BS, comprising boggy synovitis and tenosynovitis, are distinct from those observed in both JIA and rheumatoid arthritis. Despite the chronicity of exuberant arthritis, destruction of the larger joints is not commonly reported.[8] Our patients’ most common manifestations were boggy synovitis and camptodactyly (Figure 4). These clinical findings allowed us to establish an early diagnosis for patients 3, 4, 5, and 6. Since camptodactyly with negative acute phase reactants should also be investigated for mucopolysaccharidosis, mucolipidosis, and skeletal dysplasia, our patients (except for patient 1) were investigated for these etiologies.
Eye symptoms generally appear later at around age 12.[8] Uveitis presents as an insidious granulomatous iridocyclitis with posterior uveitis in many patients, and a potential evolution can be seen into severe panuveitis with multifocal choroiditis. It is usually bilateral and associated with an increased risk of visual morbidity.[8] Most patients require continuing follow-up and treatment for eye symptoms throughout their lives. Chorioretinitis, cataracts, glaucoma, and retinal detachment can lead to significant visual impairment and blindness.[16] None of our patients had eye symptoms, and their regular ocular examinations were normal as a result of the early treatment. However, uveitis could be seen as a late finding of the disease in the future.
The presence of fever is determined in expanded manifestations.[18] The main manifestation of the first patient was episodic fever, which was persistent for 7-10 days, accompanied by a rash. Aróstegui al.[14] and Okafuji et al.[15] also reported that 50% of their cohort had recurrent or persistent fever. These findings suggest that fever is one of the important symptoms of BS/EOS and is the reason why BS/EOS is misdiagnosed as systemic-onset JIA.[15]
Although interstitial pneumonitis and bronchial granulomas have been reported in patients with BS/EOS, the typical pulmonary involvement seen in patients with adult onset sarcoidosis has not been described.[19,20] Granulomatous lymphadenopathy has been defined.[20] Our patients did not have mediastinal lymph node or pulmonary involvement. There was cervical and axillary lymph node involvement accompanied by fever in patient 1. The lymph node biopsy showed non-caseating granulomatous inflammation in patient 1 (Figure 3).
Other expanded manifestations of BS/EOS are hepatosplenomegaly, sialadenitis, erythema nodosum, leukocytoclastic vasculitis, transient neuropathies, granulomatous glomerular and interstitial nephritis, arterial hypertension, pericarditis, pulmonary embolism, hepatic granulomas, splenic involvement, and chronic renal failure.[8] Hepatic and renal failure developed in patient 2 due to the JIA treatment. Although both of her involvements were initially thought to be related to the etanercept treatment, camptodactyly and the biopsy findings helped us establish the correct diagnosis.
The most commonly detected NOD2 mutations are R334W and R334Q.[18] The patients' mutations were determined as follows: P268S (rs2066842), V955I (rs5743291), M513T (rs104895473), R702W (rs2066844), H343Y (rs199858111), and M491L (16:50745293). To our knowledge, the last mutation that was determined in patient 5, 6 and their mother has not been reported before. These family members were evaluated as having BS inherited as autosomal dominant. Although the penetrance of BS/EOS associated with NOD2 mutations is very high, asymptomatic family members with NOD2 mutations have been defined in the literature.[21-23] The father of the patient 5, 6 who had a V955I mutation did not have any symptoms. It is unclear whether the type and/or location of the NOD2 mutation has an effect on the clinical features.[8] Rosé et al.[18] concluded that the presence of overlapping clinical features supports the hypothesis that additional genetic factors might contribute to the phenotype in pediatric granulomatous inflammatory disease. Therefore, we assessed Mediterranean fever (MEFV) mutations carried in up to 20% of healthy people in Turkey. However, we did not determine any significant MEFV mutations.
To the best of our knowledge, there has been no study on the optimal treatment for patients with BS/EOS, due to its rarity and the heterogeneity in its severity and the evolution of its expression. The main objective is to prevent ocular manifestations, which can be particularly severe.[17] Low-dose glucocorticoids are generally satisfactory at the quiescent stage. In some acute stages, high doses of glucocorticoids are necessary. Methotrexate, corticosteroids, and tumor necrosis factor (TNF) inhibitors continue to be the main treatments.[24] The patients treated with biological therapy after 2000 have been summarized in Table 3.[25-41] Although our patients with predominantly articular symptoms responded to methotrexate and TNF inhibitor therapies, we could not achieve remission with TNF, interleukin (IL)-6, or IL-1 inhibitor therapies in patient 4. In addition, remission was achieved with a low dose steroid and canakinumab treatment for patient 1 without articular symptoms. There is no consensus on which IL pathway predominates in the literature. Galozzi et al.[21] claimed that an exaggerated NOD2 response leading to pro-inflammatory release is not a direct mechanism explaining the pathophysiology of BS.
Table 3. Clinical features of patients receiving biologic therapy in the literature.
| Number of patients | Clinical findings | Mutation of NOD2 | Treatments | Biologic treatments | Remission | |
| Cuesta et al.[25] | 3 | Dermatitis, arthritis, uveitis | ETA | No | ||
| Milman et al.[26] | 2 | Dermatitis, arthritis, uveitis | R334W Heterozygous | Hydroxychloroquine Cyclosporine Prednisolone Methotrexate |
ETA INF |
No Yes |
| Arostegui et al.[14] | 1 | Fever, arthritis, papilledema | R587C | ANA | Yes | |
| Becker et al.[20] | 1 | Arthritis, uveitis, Lymphadenitis Interstitial pneumonitis | R344Q Heterozygous | Prednisolone Methotrexate |
INF | Yes |
| Martin et al.[27] | 2 | 1. Uveitis, arthritis | Prednisolone Methotrexate |
ETA, INF, ANA | No | |
| 2. Uveitis | Prednisolone Methotrexate Cyclosporine |
INF, ANA, ADA | No Yes |
|||
| Yasui et al.[28] | 2 | 1. Fever, dermatitis, arthritis, uveitis | N670K Heterozygous | Prednisolone Methotrexate Tacrolimus |
INF Thalidomide |
No Yes |
| 2. Fever, dermatitis, uveitis | Prednisolone Methotrexate Cyclosporine |
ETA Thalidomide |
No Yes |
|||
| Caracseghi et al.[29] | 1 | Dermatitis, arthritis | Autosomal dominant | Prednisolone Methotrexate |
ETA | No |
| Sharma et al.[30] | 1 | Fever, arthritis, Dermatitis, uveitis | H520Y Heterozygous | Prednisolone Methotrexate Intavenous immunoglobulin |
ANA, ETA, ADA INF |
No Yes |
| Raiji et al.[31] | 1 | Arthritis, Dermatitis, uveitis | D382E | Prednisolone Methotrexate Cyclosporine |
ETA, INF ADA |
No Yes |
| Bravo-Ljubetic et al.[32] | 1 | Uveitis | - | Methotrexate | ETA, INF, ANA | No |
| Simonini et al.[33] | 1 | Arthritis, Dermatitis, uveitis | R334W Heterozygous | Prednisolone Methotrexate Mycofenolat mofetil |
INF ADA ABA CANA |
No No No Yes |
| Inoue et al.[34] | 1 | Fever, dermatitis, Takayasu arteritis Uveitis | D382E Heterozygous | Prednisolone Methotrexate |
INF | Yes |
| Chauhan and Michet[35] | 1 | Arthritis, granulomatous hepatitis, bronchiolitis obliterans, uveitis | Glu498Gly Heterozygous | Prednisolone Methotrexate Hydroxychloroquine |
ADA | Yes |
| La Torre et al.[24] | 1 | Arthritis, Dermatitis, uveitis | E383G Heterozygous | Prednisolone Methotrexate |
INF | Yes |
| Otsubo et al.[36] | 1 | Arthritis, Dermatitis, uveitis | E383G Heterozygous | Prednisolone Methotrexate Mizoribine |
INF ADA |
No Yes |
| Rose et al.[4] | 19 | Arthritis, Dermatitis, uveitis, expanded disease manifestations |
R344Q, R334W, A755V, G481D, E383K | Prednisolone Methotrexate Topical steroids |
13 ADA (3 articular), 1 Ocular, 7 expanded) 4 INF (2 articular), 1 ocular, 2 expanded), 1 CANA (1 articular), 1 Thalidomide |
Yes Yes Yes No |
| Achille et al.[37] | 1 | Granulomatous uveitis | P268S Heterozygous | Prednisolone Methotrexate |
ADA | Yes |
| Nagakura et al.[38] | 6 | Arthritis, Dermatitis (except 3 patients), Uveitis (except 2 patients) | R334Q (4 patients), R587C,R471C |
Prednisolone Methotrexate |
INF (4 patients) ETA (2 patients) TCZ (2 patients) |
Yes No No |
| Arvesen et al.[39] | Arthritis, Dermatitis, uveitis | R334Q | Prednisolone Methotrexate |
INF ADA |
No Yes |
|
| Toral-Lopez et al.[40] | 1 | Arthritis, dermatitis | c.1808A>G | Prednisolone Methotrexate |
INF | Yes |
| Lu et al.[41] | 1 | Arthritis, Dermatitis, uveitis | R311W | Prednisolone Methotrexate, Sulfasalazine |
INF TOS |
No Yes |
| ETA: Etanercept; TOS: Tosilizumab; ADA: Adalimumab; INF: Infliximab; CAN: Canakinumab; ANA: Anakinra. | ||||||
This study has some limitations. We could not carry out genetic tests on all family members except for the sibling patients and a functional analysis could not be performed for the new mutation.
In conclusion, camptodactyly and boggy synovitis are important signs of BS/EOS. Methotrexate and TNF blockers are more effective in patients with predominantly articular symptoms. Moreover, we determined a novel M491L mutation in the NOD2 gene. On the other hand, there is still no consensus on either the pathogenesis or treatment of BS/EOS in the literature, necessitating further studies. Currently, this work is in progress towards identifying the pathogenesis and treatment options.
Footnotes
Conflict of Interest: The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Financial Disclosure: The authors received no financial support for the research and/or authorship of this article.
References
- 1.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 2.Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107:689–693. doi: 10.1016/s0022-3476(85)80394-2. [DOI] [PubMed] [Google Scholar]
- 3.Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, et al. Caveats and truths in genetic, clinical, autoimmune and autoinflammatory issues in Blau syndrome and early onset sarcoidosis. Autoimmun Rev. 2014;13:1220–1229. doi: 10.1016/j.autrev.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Rosé CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford) 2015;54:1008–1016. doi: 10.1093/rheumatology/keu437. [DOI] [PubMed] [Google Scholar]
- 5.Sarens IL, Casteels I, Anton J, Bader-Meunier B, Brissaud P, Chédeville G, et al. Blau Syndrome- Associated Uveitis: Preliminary Results From an International Prospective Interventional Case Series. Am J Ophthalmol. 2018;187:158–166. doi: 10.1016/j.ajo.2017.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016;48:67–73. doi: 10.1038/ng.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosé CD, Doyle TM, McIlvain-Simpson G, Coffman JE, Rosenbaum JT, Davey MP, et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J Rheumatol. 2005;32:373–375. [PubMed] [Google Scholar]
- 8.Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014;12:33–33. doi: 10.1186/1546-0096-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early- onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–1197. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 10.Rybicki BA, Maliarik MJ, Bock CH, Elston RC, Baughman RP, Kimani AP, et al. The Blau syndrome gene is not a major risk factor for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16:203–208. [PubMed] [Google Scholar]
- 11.Becker ML, Rose CD. Blau syndrome and related genetic disorders causing childhood arthritis. Curr Rheumatol Rep. 2005;7:427–433. doi: 10.1007/s11926-005-0046-3. [DOI] [PubMed] [Google Scholar]
- 12.Milman N, Ursin K, Rødevand E, Nielsen FC, Hansen TV. A novel mutation in the NOD2 gene associated with Blau syndrome: a Norwegian family with four affected members. Scand J Rheumatol. 2009;38:190–197. doi: 10.1080/03009740802464194. [DOI] [PubMed] [Google Scholar]
- 13.Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54:3337–3344. doi: 10.1002/art.22122. [DOI] [PubMed] [Google Scholar]
- 14.Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56:3805–3813. doi: 10.1002/art.22966. [DOI] [PubMed] [Google Scholar]
- 15.Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S, et al. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009;60:242–250. doi: 10.1002/art.24134. [DOI] [PubMed] [Google Scholar]
- 16.Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012;12:44–51. doi: 10.1016/j.autrev.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 17.Alonso D, Elgart GW, Schachner LA. Blau syndrome: a new kindred. J Am Acad Dermatol. 2003;49:299–302. doi: 10.1067/s0190-9622(02)61772-4. [DOI] [PubMed] [Google Scholar]
- 18.Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009;60:1797–1803. doi: 10.1002/art.24533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borzutzky A, Fried A, Chou J, Bonilla FA, Kim S, Dedeoglu F. NOD2-associated diseases: Bridging innate immunity and autoinflammation. Clin Immunol. 2010;134:251–261. doi: 10.1016/j.clim.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Becker ML, Martin TM, Doyle TM, Rosé CD. Interstitial pneumonitis in Blau syndrome with documented mutation in CARD15. Arthritis Rheum. 2007;56:1292–1294. doi: 10.1002/art.22509. [DOI] [PubMed] [Google Scholar]
- 21.Galozzi P, Negm O, Greco E, Alkhattabi N, Gava A, Sfriso P, et al. Ex vivo and in vitro production of pro-inflammatory cytokines in Blau syndrome. Reumatismo. 2015;66:277–284. doi: 10.4081/reumatismo.2014.772. [DOI] [PubMed] [Google Scholar]
- 22.Ebrahimiadib N, Samra KA, Domina AM, Stiles ER, Ewer R, Bocian CP, et al. A Novel NOD2-associated Mutation and Variant Blau Syndrome: Phenotype and Molecular Analysis. Ocul Immunol Inflamm. 2018;26:57–64. doi: 10.1080/09273948.2016.1185529. [DOI] [PubMed] [Google Scholar]
- 23.Saulsbury FT, Wouters CH, Martin TM, Austin CR, Doyle TM, Goodwin KA, et al. Incomplete penetrance of the NOD2 E383K substitution among members of a pediatric granulomatous arthritis pedigree. Arthritis Rheum. 2009;60:1804–1806. doi: 10.1002/art.24532. [DOI] [PubMed] [Google Scholar]
- 24.La Torre F, Lapadula G, Cantarini L, Lucherini OM, Iannone F. Early-onset sarcoidosis caused by a rare CARD15/NOD2 de novo mutation and responsive to infliximab: a case report with long-term follow-up and review of the literature. Clin Rheumatol. 2015;34:391–395. doi: 10.1007/s10067-014-2493-6. [DOI] [PubMed] [Google Scholar]
- 25.Cuesta IA, Moore EC, Rabah R, Bawle EV. Blau syndrome (familial granulomatous arthritis, iritis, and rash) in an african-american family. J Clin Rheumatol. 2000;6:30–34. doi: 10.1097/00124743-200002000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Milman N, Andersen CB, Hansen A, van Overeem Hansen T, Nielsen FC, Fledelius H, et al. Favourable effect of TNF-alpha inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS. 2006;114:912–919. doi: 10.1111/j.1600-0463.2006.apm_522.x. [DOI] [PubMed] [Google Scholar]
- 27.Martin TM, Zhang Z, Kurz P, Rosé CD, Chen H, Lu H, et al. The NOD2 defect in Blau syndrome does not result in excess interleukin-1 activity. Arthritis Rheum. 2009;60:611–618. doi: 10.1002/art.24222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasui K, Yashiro M, Tsuge M, Manki A, Takemoto K, Yamamoto M, et al. Thalidomide dramatically improves the symptoms of early-onset sarcoidosis/ Blau syndrome: its possible action and mechanism. Arthritis Rheum. 2010;62:250–257. doi: 10.1002/art.25035. [DOI] [PubMed] [Google Scholar]
- 29.Caracseghi F, Izquierdo-Blasco J, Sanchez-Montanez A, Melendo-Perez S, Roig-Quilis M, Modesto C. Etanercept-induced myelopathy in a pediatric case of blau syndrome. Case Rep Rheumatol. 2011;2011:134106–134106. doi: 10.1155/2011/134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma SM, Martin TM, Rosé CD, Dick AD, Ramanan AV. Distinguishing between the innate immune response due to ocular inflammation and infection in a child with juvenile systemic granulomatous disease treated with anti-TNFa monoclonal antibodies. Rheumatology (Oxford) 2011;50:990–992. doi: 10.1093/rheumatology/keq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/ CARD15 gene. J AAPOS. 2011;15:205–207. doi: 10.1016/j.jaapos.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Bravo-Ljubetic L, Peralta-Calvo J, Noval S, Pastora-Salvador N, Abelairas-Gómez J, Merino R. Adalimumab therapy for refractory childhood uveitis. J AAPOS. 2013;17:456–459. doi: 10.1016/j.jaapos.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Simonini G, Xu Z, Caputo R, De Libero C, Pagnini I, Pascual V, et al. Clinical and transcriptional response to the long-acting interleukin-1 blocker canakinumab in Blau syndrome-related uveitis. Arthritis Rheum. 2013;65:513–518. doi: 10.1002/art.37776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue Y, Kawaguchi Y, Shimojo N, Yamaguchi K, Morita Y, Nakano T, et al. A case of infantile Takayasu arteritis with a p. D382E NOD2 mutation: an unusual phenotype of Blau syndrome/early-onset sarcoidosis. Mod Rheumatol. 2013;23:837–839. doi: 10.1007/s10165-012-0720-z. [DOI] [PubMed] [Google Scholar]
- 35.Chauhan K, Michet C. A case of blau syndrome. Case Rep Rheumatol. 2014;2014:216056–216056. doi: 10.1155/2014/216056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otsubo Y, Okafuji I, Shimizu T, Nonaka F, Ikeda K, Eguchi K. A long-term follow-up of Japanese mother and her daughter with Blau syndrome: Effective treatment of anti-TNF inhibitors and useful diagnostic tool of joint ultrasound examination. Mod Rheumatol. 2017;27:169–173. doi: 10.3109/14397595.2014.964388. [DOI] [PubMed] [Google Scholar]
- 37.Achille M, Ilaria P, Teresa G, Roberto C, Ilir A, Piergiorgio N, et al. Successful treatment with adalimumab for severe multifocal choroiditis and panuveitis in presumed (early-onset) ocular sarcoidosis. Int Ophthalmol. 2016;36:129–135. doi: 10.1007/s10792-015-0135-x. [DOI] [PubMed] [Google Scholar]
- 38.Nagakura T, Wakiguchi H, Kubota T, Yamatou T, Yamasaki Y, Nonaka Y, et al. Tumor Necrosis Factor Inhibitors Provide Longterm Clinical Benefits in Pediatric and Young Adult Patients with Blau Syndrome. J Rheumatol. 2017;44:536–538. doi: 10.3899/jrheum.160672. [DOI] [PubMed] [Google Scholar]
- 39.Arvesen KB, Herlin T, Larsen DA, Koppelhus U, Ramsing M, Skytte AB, et al. Diagnosis and Treatment of Blau Syndrome/Early-onset Sarcoidosis, an Autoinflammatory Granulomatous Disease, in an Infant. Acta Derm Venereol. 2017;97:126–127. doi: 10.2340/00015555-2485. [DOI] [PubMed] [Google Scholar]
- 40.Toral-López J, González-Huerta LM, Martín-Del Campo M, Messina-Baas O, Cuevas-Covarrubias SA. Familial Blau syndrome without uveitis caused by a novel mutation in the nucleotide-binding oligomerization domain-containing protein 2 gene with good response to infliximab. Pediatr Dermatol. 2018;35:180–183. doi: 10.1111/pde.13475. [DOI] [PubMed] [Google Scholar]
- 41.LuL , ShenM , JiangD , LiY , ZhengX , LiY , etal Blau syndrome with good Reponses to Tocilizumab: A case report and focused literature review. Semin Arthritis Rheum. 2018;47:727–731. doi: 10.1016/j.semarthrit.2017.09.010. [DOI] [PubMed] [Google Scholar]