

Abstract
Natural evolution has generated an impressively diverse protein universe via duplication and recombination from a set of protein fragments that served as building blocks. The application of these concepts to the design of new proteins using subdomain-sized fragments from different folds has proven to be experimentally successful. To better understand how evolution has shaped our protein universe, we performed an all-against-all comparison of protein domains representing all naturally existing folds and identified conserved homologous protein fragments. Overall, we found more than 1000 protein fragments of various lengths among different folds through similarity network analysis. These fragments are present in very different protein environments and represent versatile building blocks for protein design. These data are available in our web server called F(old P)uzzle (fuzzle.uni-bayreuth.de), which allows to individually filter the dataset and create customized networks for folds of interest. We believe that our results serve as an invaluable resource for structural and evolutionary biologists and as raw material for the design of custom-made proteins.
Abbreviations: HMM, hidden Markov model; HisF, imidazole glycerol phosphate synthase; TPR, tetratricopeptide repeat; MIT, microtubule interacting and trafficking
Keywords: protein design, evolution, protein recombination, protein fragments
Graphical abstract
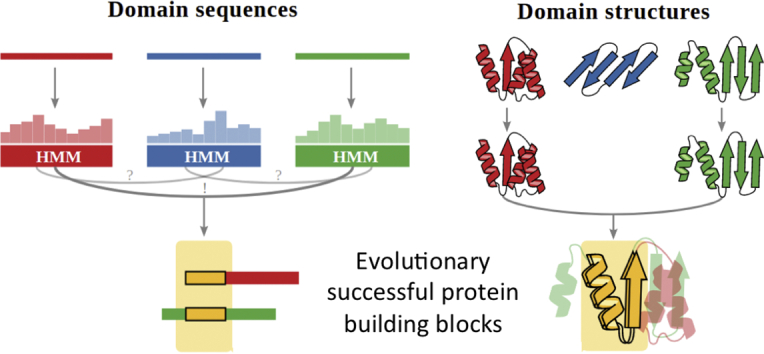
Highlights
-
•
Nature has created an impressive diversity of proteins via the recombination, replication and differentiation of a set of protein fragments that act as building blocks.
-
•
We have performed an all-against-all search comparison of proteins in structural databases via sensitive homology detection methods and identified fragments that appear across the protein universe in very different environments.
-
•
We have identified more than a 1000 sub-domain sized fragments that Nature has reused to create new proteins and that can be used as an innovative route for protein design.
-
•
The results are publicly available in our Fuzzle database at fuzzle.uni-bayreuth.de.
Introduction
Proteins are critical components in all cellular processes and display an impressive number of diverse functionalities. In order to be active, most proteins need to assemble into a three-dimensional structure that is encoded by its amino acid sequence. A basic protein unit is the domain, which is defined as an independently folding unit. Significant efforts have been placed on hierarchically classifying domains into categories, such as the CATH [1], SCOP [2], and ECOD [3] databases. To date, SCOP classifies over 290,000 domains according to their architecture and topology into more than 1200 folds that have been previously considered to be evolutionarily unrelated. Although domains have thus long been defined as the basic evolutionary unit [[4], [5], [6]], their origin itself has been investigated in recent years.
Although a de novo assembly of folded proteins or domains by random concatenation of amino acids is not entirely impossible [7], it is statistically implausible [8]. Several recent studies suggest that domains originate from smaller fragments that have been duplicated, recombined, and differentiated. Alva et al. clustered representative domains by their sequence similarity and represented them in a Cartesian space. The distance among points was inversely proportional to their sequence similarity. The authors found in many instances links between superfamilies of the same fold, and in some cases, between different folds [9]. Nepomnyachiy et al. represented the protein universe via networks [10], where the nodes represent domains and the links connected nodes that share a fragment, namely, a sub-domain sized protein segment of similar sequence and structure. By varying the thresholds that define a fragment, the authors tried to reproduce the SCOP definition of folds, i.e. separate island-like network components that encompass structurally similar domains. While they were able to find optimal parameters that successfully separate folds from all-β and α + β classes, this was not the case for the all-α and α/β classes: trying to isolate domains from the same folds inevitably connected domains from other folds. The results suggested that several fragments have been reused throughout evolution and thus appear today in different protein contexts.
We previously identified one of these α/β fragments. The (βα)8-barrel or TIM-barrel fold is one of the most successful scaffolds in evolution. In an extensive database search, structural similarity between a half-barrel and the flavodoxin-like fold was identified [11]. Based on these findings, it was possible to design a chimeric (βα)8-barrel through the combination and optimization of parts of imidazole glycerol phosphate synthase (HisF) and the chemotaxis response regulator CheY both from Thermotoga maritima [12]. In a follow-up study, the combination of HisF with another member of the flavodoxin-like fold, the nitrite response regulator NarL from Escherichia coli, led to a protein with even higher stability [13].
While the first comparison of the (βα)8-barrel and flavodoxin-like folds was based on structure similarity, the field of sequence analysis had advanced enormously in the past years with the introduction of profile hidden Markov model (HMM) comparisons. Profile HMMs are similar to simple sequence profiles (such as position site-specific scoring matrices) [14], but in addition to the amino acid frequencies in the columns of a multiple sequence alignment, they contain the position-specific probabilities for insertions and deletions along the alignment [15]. In short, profile HMM comparisons help to detect distantly related sequences and to generate high-quality alignments below the twilight zone [16] of sequence similarity for homologous proteins [17].
We thus revisited the (βα)8-barrel-flavodoxin-like relationship using profile HMM comparisons and found that their structural similarity is not due to convergent evolution, but in fact due to homology [18]. Further comparisons of members of these two folds and the vast natural sequence space allowed identification of an intermediate protein family, of which one of its members, N-TM0182, was crystallized. Notably, a detailed look at the structure-based sequence alignments revealed that the highest similarity of N-TM0182 to either the (βα)8-barrel or the flavodoxin-like fold resides in a 45-amino-acid (αβ)2 fragment. Following a similar methodology, Alva et al. pursued the hypothesis that today's domains have arisen from fusion and differentiation from a set of primordial peptides present in the RNA-world. The authors identified a set of 40 peptides of up to 38 amino acids whose similarity is representative of a common descent despite being present in different folds [19].
Based on this evidence, it is plausible to propose that current folds arose through the reuse of fragments in different structural contexts. The first experiments in recombining fragments from the same [[20], [21], [22], [23]], as well as from different folds [12,13,24], have been successful in building hybrid proteins. These findings open up great opportunities for protein design, as we can now identify which fragments have been particularly successful in the course of evolution and recombine them to create new chimeras [25,26]. Here, we investigate how many fragments can be found in today's protein universe. To that end, we took the SCOP database clustered at 95% sequence identity and performed a sequence- and structure-based comparison of all domains. We have made our results available as an online database named Fuzzle (Fold Puzzle) at fuzzle.uni-bayreuth.de. Our study aims to (i) understand evolutionary relationships between different folds that might not be evident at first sight and (ii) identify protein fragments that can be useful building blocks for the design of new proteins through recombination. We believe Fuzzle will be an invaluable resource for protein engineers and to elucidate the evolution of protein folds.
Results and Discussion
Database construction and content
The analysis is based on the hierarchically organized SCOP database [2]. To remove redundancy, we took the SCOP95 subset clustered at 95% sequence identity that contains 28,010 domains. Using HHsearch [15], we performed all-against-all pairwise profile HMM comparisons of this dataset. In order to remove structural constraints that theoretically can lead to analogous and hence false-positive hits, the predicted secondary structure of the HMM profiles was not scored when calculating the probabilities. In fact, no HHsearch hits were found for most of the analogous motifs described in Cheng et al. [27]. All pairwise sequence alignment hits were stored in the database. Subsequently, we measured the structural similarity for each hit in two ways: (i) by superposing and calculating the RMSD strictly using only residues aligned in the pairwise sequence alignment (RMSDSeq) and (ii) by using TM-align [28] to allow a structure-based superposition of the protein fragments identified by the sequence alignment (RMSDTM). We refer to these hits as fragments, i.e. significantly sized homologous protein segments of similar structure. This pipeline led to over 8 million pairwise hits that we stored in our Fuzzle database (Figure 1(a)). Each entry comprises the two SCOP domain identifiers (query and subject), their SCOP classification, the HHsearch statistics, the TM-score, and RMSD, among others (Figure 1(b)). The online database allows filtering individually for each of these parameters. For example, we observe more than 1.8 million pairwise hits (about 20% of the database) between domains that were previously assumed to be unrelated because they belong to different folds.
Figure 1.

Fuzzle database construction and content. (a) Database construction. Domain sequences and structures from SCOP95 were used as input. Sequence profile HMMs were generated for each domain and compared all-against-all. Matches between pairs of domains were structurally superimposed. The sequence and structural data collected for each pair constitutes a hit, which is stored in Fuzzle, summing up to more than 8 million pairwise hits. (b) Contents of a hit. Each hit in Fuzzle contains information about the two domains (query and subject), start and end of the fragment they share, HHsearch probability, and RMSD, among others. (c) Distribution of hits according to their probability and TM-score. The majority of hits present probabilities below 35% but there is also a non-negligible portion of hits with high probability and TM-score. Hits in this upper right quadrant (probabilities > 70% and TM-scores > 0.3) were plotted with their probabilities versus lengths (d).
Similarly to Alva et al. [19], we also observe a bimodal distribution for our dataset when plotting TM-score versus HHsearch probabilities (Figure 1(c)). Hits with high sequence and structural similarity reveal the presence of unrecognized homologous relationships between different folds (upper right quadrant in Figure 1(c)). While the work by Alva and colleagues focused on fragments ranging in length up to 38 residues, our search is not limited in length, as larger fragments could present interesting properties for protein design. We further explored the set of hits with probabilities over 70% and a TM-score higher than 0.3 (Figure 1(d)). When representing this set of hits according to their length and probability, we observe that most of the hits are relatively short (up to 40 amino acids). There are, however, a non-negligible proportion of hits of larger lengths (50–70 amino acids) and a few fragments over 100 amino acids.
These data are made available in an interactive web interface. The web server allows to individually filter the dataset and to create a customized analysis for domains of interest. The results can be visualized as tables, matrices, or graphs, and interesting domain pairs can be picked for structural superpositions to be shown (Figure 2). Documentation is provided to guide the user through the different views (fuzzle.uni-bayreuth.de/about/). Customized protein similarity networks can be downloaded as JSON files. The full database is also made available for download (fuzzle.uni-bayreuth.de/about/database). In this study, we use the SCOPe nomenclature [2]. For example, c.23.1.1 references the CheY-related family, where “c” represents the class, “23” represents the fold, “1” represents the superfamily, and the last “1” represents the family.
Figure 2.

Snapshot of the Fuzzle website. Relationships between domains of the superfamilies c.23.1 and c.1.3 are shown in the graph on the left side. Domains are represented as nodes and similarities as links. The parameters for the links can be changed above the graph and the network can be downloaded. Upon clicking on one link the superposition between the two connected nodes, representing domains, is shown on the right side. Other presentation modes such as tables and matrices can also be chosen instead.
The protein universe as a network
Several approaches have been used successfully in representing the protein universe. The hierarchical classifications as mentioned above [[1], [2], [3]], Cartesian representations [29,30] and networks [9,10,31,32] all provide complementary ways to visualize protein space and study the similarity of protein domains therein. The Fuzzle database can also be represented as a similarity network in which nodes represent protein domains and links represent connections with similarities above a given threshold. Since these links represent protein fragments, it allows us to analyze high-scoring fragments in a global context and to explore the evolutionary relations of domains. Fragments that have been reused in evolution can be identified as individual sub-graphs or so-called components. These components enable us to quickly identify interesting fragments for design, as each component constitutes a set of domains from different folds that have a fragment in common. Fragments that are found in different protein contexts are expected to have an intrinsic advantage, e.g. in stability or foldability. Due to these properties, such fragments can be used as building blocks in protein design [13,24].
Clustering of the database hits
Fuzzle comprises 28,010 domains with a total of 8,109,195 pairwise hits between them. Thus, many domains show similarities to more than one other domain. However, these similarities may occur in different regions of a domain, and hence, a domain may share distinct fragments with other domains. To unravel this complication, we first started to cluster these regions. The process is illustrated in Figure 3 with an example: domain d1jnfa2 [33] (transferrin), which belongs to the periplasmic-binding protein-like II domains (c.94.1.2; Figure 3(a)), appears in 12 hits with domains from other folds (Figure 3(b)). One can easily identify three areas that (i) are shared with domains of the flavodoxin-like fold (c.23), (ii) show a weak similarity to pre ATP-grasp domains (c.30), or (iii) are shared with domains of the knottins (g.3). Each alignment starts and ends at individual positions in the d1jnfa2 sequence. Using a density-based clustering method [34], these 12 regions can be grouped into three clusters (Figure 3(c)). We applied this procedure to the full dataset.
Figure 3.

Clustering of the database. (a) Domain d1jnfa2 [33], a periplasmic-binding protein-like II (c.94) domain, was selected for this example. (b) Schematic representation of the 12 alignments in which d1jnfa2 appears. Domain d16vpa_ is represented as a maroon box. Below appear the other domains ordered by their start position. SCOP families and probabilities are stated for each domain. Sequence positions where each alignment starts are depicted relative to d1jnfa2's sequence (20, 35, and 185). The coloring scheme is the same as in (c), where each alignment has been plotted according to start and end position. The length of each fragment is thus the distance to the diagonal. The 12 sequence regions were combined into three clusters, depicted within the d1jnfa2's structure.
Network construction and representation
Before constructing the network itself, Fuzzle hits are filtered using cutoffs for HHsearch probability, RMSDTM, TM-score, and alignment length. We pre-computed networks for several cutoffs of domains belonging to different folds (Figure S1). Networks with other parameters can also be computed dynamically on the Fuzzle website. In this article, we focus on a network filtered for a HHsearch probability of ≥ 70% (P value of 4.1 × 10−5), a length of 10 to 200 amino acids (based on the structural alignment), an RMSDTM ≤ 3 Å, and a TM-score ≥ 0.3. We enforce that the hits contain domains from different folds. Because TM-align finds the optimal superposition inside the sequence alignment, the TM-alignments can end up being considerably shorter than the original sequence alignments. Hence, we further restricted the sequence alignments length to be at most 25% larger than the structural superposition. This filtered dataset contained 208,944 hits in total.
As described in “Clustering of the database hits,” these hits are clustered according to their regional position within a domain. This extra procedure is necessary since a protein domain might contain several distinct regions/fragments: each of these fragments might be shared with other domains that are not related to each other. The clustering ensures that all the hits in a domain can be categorized according to the regions they are involved in. For example, for a domain termed A, cluster A1 could span residues 3–50, while cluster A2 residues 110–190. Therefore, although the domain remains the same, fragments A1 and A2 belong to different clusters (Figure 3). Our analysis resulted in 24,999 unique clusters, each containing fragments that are shared across different folds. In the network, domains are depicted as nodes that are linked whenever they have a fragment in common. Please note that since a domain might contain several distinct fragments, a domain can occur multiple times in the network (c.f. Figure 3). However, the similarity network ensures that it is transitive: this means if domain A is linked with B and domain B is linked with domain C, then all three domains contain the same fragment.
The occurrence of common fragments in protein domains
The network is undirected (the links have no direction), unweighted, and each pair of nodes is at most connected by one link between them, thus being a so-called simple network (Figure 4). Overall, 8288 unique domains are represented as 24,999 nodes with 162,852 links. More than 50% of the domains occur at least two times in the graph due to the fact that they contain different distinct fragments (c.f. 2.1). Domains from all seven SCOP classes are contained in the network with varying proportions (16.3% all-α, 13.3% all-β, 45.2% α/β, 15.3% α + β). Since there is not always a path between nodes in the graph, the network is disconnected (Figure 4(a)). In fact, our intention was to build a disconnected network, each island-like subgraph represents a set of domains that share a common fragment. In network theory, these subgraphs are termed connected components or simply components. The component that contains the greatest number of nodes is termed major component. Throughout this work, we label these components as a numbered fragment (e.g. “fragment 0”), since all the nodes (domains) in a component contain the same protein fragment. The network contains 1337 components or fragments that cover 519 folds with lengths ranging from 11 to 198 amino acids. The mean number of links per component, excluding the major component, is 6.5 (median, 3.0).
Figure 4.

(a) Network representation of the dataset. Each node represents a domain. Each domain is colored according to its SCOP class. The links connect domains that have a fragment in common. Since a domain might contain several distinct fragments, it can appear several times in the network. Cutoffs to build this network were: probability > 70%, RMSDTM < 3 Å, TM-score > 0.3, and the length of the sequence alignment was at most 25% larger than the structural alignment. (b) A closer view to the major component (fragment 0) and the folds that populate it in its different regions. Labels indicate where the relationships between two folds occur, for example, c.1–c.23. Note that most connections occur with the Rossmann fold (c.2) in the blue region and with the DNA/RNA-binding 3-helical bundle (a.4) in the orange region. The largest distance between any two pair of nodes in this network is 21.
In network theory, the neighbors of a node are those nodes that are directly connected via links. The degree of a node is computed as the number of links of a node to others. Because our graph is simple, i.e. two nodes can be at most connected by one link, the degree of a particular node is equal to the number of neighbors it has. We computed the node degree for each node in the network and observed that most nodes present a low degree, with a median of 2 degrees/neighbors per node. However, a very small number of nodes show a very high degree. These nodes, which present a degree greatly above the average, are termed hubs. In this case, the top 2% of most connected nodes possess 80% of the graph links. Indeed, the degree distribution of the network is highly right-skewed (Figure S2). This power-law distribution behavior is observed in different fields, such as in social networks [35], in the network of the World Wide Web [36], and more specifically in the reuse of protein segments of different lengths [31,37,38]. The 10 hubs with the highest degree belong to the folds c.5, c.3, c.111, and c.4; all of which are contained in the major component (Table S1). The four most connected domains belong to fold “c.5” and correspond to the N-terminal domain of UDP-N-acetylmuramate-LL-alanine ligase MurC, a bacterial protein involved in cell wall formation [39] that has been the object of pharmaceutical research as a target for antibacterial compounds [40]. Although a high connectivity with a large number of nodes gives an impression of structure reusability, it could also be the result of many connections between two folds with very similar topology, such as c.2 (Rossmann-like domains) and c.5 (MurC N-terminal domains). We therefore looked for “promiscuous” nodes, i.e. those with the largest variety of folds among their neighbors (Table S2). Out of these 10 most “promiscuous” nodes, we found that the first 4 are also the four most connected ones, but that the other 6 nodes do not present such high degrees. For instance, the domain of the anti-repressor SinI (d1b0nb_ [41,42], fold a.34) is connected to 18 different folds (Table S2), although presenting a low degree (with 106 neighbors). The sinI anti-repressor protein consists of a helix-turn-helix motif (HTH), a recurrent structural motif capable of binding DNA that is present in many folds in the all-α SCOP class.
In addition to the presentation of the network in Figure 4, we added a simplified graph on the web server for better viewing (fuzzle.uni-bayreuth.de/fragments/network): while the nodes in this manuscript always represent domains, the nodes in the graph on the web server initially show the identified components. Upon clicking on a node, it expands to show the domains contained in one of them (Figure S6).
More than half of the protein universe is connected
The most connected and promiscuous nodes exclusively belong to the major component that we denote as “fragment 0.” With 14,856 nodes, it contains 59.4% of all the nodes of the network (Figure 4(b)). Most of the folds in this component belong to the all-α (19.0%) or α/β (73.6%) class. In general, a fragment shared among domains in a single component only slightly deviates in length and sequence. The largest length deviation occurs in “fragment 42,” which contains 598 domains with an average length of 25 ± 11.4 amino acids. In contrast, “fragment 0” has an average alignment length of 49.3 ± 25.5 amino acids as a consequence of small alignment differences accumulating through the network construction over long distances. Due to this behavior, we do not to show an average structure of the fragment in this large component. However, this is not an artifact due to an over-representation of domains in the all-α and α/β classes, since we also find larger components when randomly sampling 1000 domains from each class (Figure S3). It is neither an artifact of the choice of cutoffs; regardless of the chosen cutoffs, we observe always two large all-α and α/β components that are sometimes linked (Figure S1). We also changed the density-based clustering method to an overlapping function. Here, “fragment 0” only disintegrated to smaller entities when forcing the hits to overlap 100% (Text S1, Figure S4).
Our analysis shows a robust behavior of the protein network. It decomposes into two well-defined regions, a sparse and a dense, well-connected region. This has been also observed in previous publications [10,43,44]. The data further suggest that the all-α and α/β class fragments might have been more successfully reused in evolution and, thus, are interesting targets for protein design by recombination.
The network analysis indicates that the two large all-α and α/β regions are connected. In total, there are 461 links connecting domains of the all-α and α/β SCOP classes, which comprise 22 different fold pairs including 8 all-α and 15 α/β folds (Table S3). While many of these connections are spread over the major component of the network, three of them mainly facilitate the link between the two regions (Figure S5). The connection with the most links occurs between the P-loop containing nucleoside triphosphate hydrolases (c.37) and the DNA-binding SAM domain-like fold (a.60). Remarkably, these folds have been described as being some of the most ancestral [45]. The alignments between these two folds are short, with an average length of 22 amino acids and average RMSDTM of 2.1 Å. The fragment corresponds to an α-helix that connects with a loop and the beginning of a second α-helix (a.60) or a β-strand (c.37).
Examples of prominent fragments
“Fragment 0,” the major network component, represents 142,040 links, which connect 490 different fold pairs, belonging to 162 different folds. The 20 most frequent fold pairs accumulate 72% and 86% of the total component nodes and links, respectively (Table S4). Three examples of these fold pairs are highlighted in Figure 5.
Figure 5.

Examples for highly connected folds in the major component. (a) Highlighted regions show frequently connected nodes in the major component. In region ① and ②, the highly connected folds c.23 and c.1 are shown. Region ③’s links belong to pairs from the folds c.65 and c.2. In region ④, the links of the folds a.35 and a.4 are shown. (b) Examples for the four regions defined in (a). Each case shows a query (blue, left) and subject (green, right) and an overlay of the fragments they have in common (middle). ID 1 shows a special case where secondary structural elements in the fragment show sequence similarities but do not superimpose in the current folds (discrepancies shown in red).
For instance, we find the previously studied relationships between (βα)8-barrel and flavodoxin-like fold proteins [11,12,18]. Interestingly, domain pairs belonging to these folds populate two distinct regions in the network (Figure 5(a), regions 1 and 2, light green). For example, domain d3eula_ [46], a CheY-like protein from Mycobacterium tuberculosis (c.23.1), and d3jr2a [47], a ribulose-phosphate binding barrel from Vibrio cholerae, have two fragments in common that correspond to the two halves of the (βα)8-barrel (Figure 5(b), ID 1 and 2). This agrees with previously published work showing that the (βα)8-barrel fold arose through the duplication and fusion of a half-barrel like fragment [12,23,48]. We have investigated the relationship of (βα)8-barrel and flavodoxin-like protein fragments in detail [11,12,18]. Sometimes secondary structural elements are present in the sequence alignment that are not in the same spatial arrangement due to different structural constraints (Figure 5(b), red helices in ID 1).
Additionally, we highlight hits between Rossmann (c.2) and formyltransferase-like folds (c.65), which appear in two discrete areas in the center of the network (Figure 5, region 3). The hits in these areas are of different lengths, with means of 33.4 and 80.5 residues. However, both start in the N-terminal region of the domains. This may indicate either a duplication/differentiation of an ancestral, smaller sub-fragment, or at least two independent evolutionary events that formed the two folds from each other. Note that the smaller fragments in particular show hits with identities well above the significance value for this alignment length (> 30% [49]). In Figure 5(b) (ID 3), we show a 32-amino-acid-long βαβ-motif with 46% identity that occurs in human 10-formyltetrahydrofolate 2 dehydrogenase (d2bw0a2, c.65.1 [50] and the ketopantoate reductase PanE domain (d1yjqa2, c.2.1 [51]).
Another example is the most frequently observed connection among all-α domains between the DNA/RNA-binding 3-helical bundles (a.4) and the lambda repressor-like DNA-binding domains (a.35) (Figure 5(a), region 4), which corresponds to the “fragment 1” discussed in the work by Alva and colleagues [19]. In Figure 5(b) (ID 4), we show a 23-amino-acid-long HTH-motif that is shared between a lambda repressor-like DNA bonding domain (glucose-resistance amylase regulator CcpA d2jcga1, a.35.1 [52] and a C-terminal effector domain of the bipartite response regulators (d3ulqb_, a.4.6 [53]). The fragment corresponds to the DNA-binding HH motif, which has been widely studied in the past [[54], [55], [56], [57], [58]].
Some of the other fold-pairs in Table S4 have also been studied previously. For example, the two most frequent fold-pair interactions correspond to those between c.2 (NAD(P)-binding Rossmann-fold domains) and c.3 (FAD/NAD(P)-binding domain), and between c.2 and c.66 (S-adenosyl-l-methionine-dependent methyltransferases). Direct connections between c.3 and c.66 also occur but are less frequent (360 links). Recently, Laurino et al. [59] reported that these three folds present a common ribose binding site at the tip of the second Rossmann fold β-strand. Our analysis further indicates that today's Rossman-fold enzymes, which are able to bind SAM, NAD, and FAD cofactors, have a common origin.
In another example, Russell et al. [60] reported similarities between the histidine phosphocarrier protein kinase (c.91) and the phosphoenolpyruvate carboxykinase (c.37). They showed that these two proteins differ from other P-loop containing proteins no more than they differ from each other. The structure-based sequence alignment spanned over 89 residues with 19% identity. With 5804 links between nodes of these two folds, this connection is the fifth most common in the major component (Table S4), supporting thereby Russell's work.
Last but not least, we find over 1000 connections between the ribokinase (c.72) and the NAD(P)-binding Rossmann-folds. These similarities between proteins of these folds have been previously described by Manoj et al. [61] when they solved the structure of phosphopantothenoylcysteine synthetase, a member of the c.72 fold.
The network illustrates several evolutionary processes
We compared our fragment dataset to the one of Alva et al., to which we will refer as peptides. We expected our fragments to include many of the peptides in this work, as the main differences in methodology are only the input database (SCOP30 2.03) and no restriction to fragment lengths. Indeed, we find 37 out of the 40 peptides from Alva et al. [19] The missing three peptides did not pass our probability cutoff. In many cases, we find that these peptides are present not only in one of our fragments but in a collection of fragments of differing length. To exemplify this, we chose peptide 32 [19], a βαβ-motif present in the c.95 (theolase-like) and c.55 (ribonuclease H-like motif) folds (Figure 6). We found other fragments between these two folds that consist of the central βαβ-motif but extended on both termini. Some of these fragments might have been extended via duplication or decorations of smaller sub-fragments (Figure 6). This observation matches the notion that protein fragments are reused at different lengths, which has recently been studied by Nepomnyachiy et al. in detail.
Figure 6.

Common fragments in superfamilies c.95.1 and c.55.1. The first fragment in the series (1) has been previously reported [19] with a length of < 30 amino acids. We detected other fragments (numbers 2–6) shared by these superfamilies that have the same conserved core motif ①. The core motif is shown in white in examples 2–6, while the respective extensions are highlighted in blue and pink, which may have been added via duplication or differentiation and decoration.
Besides duplication and differentiation, we also found several cases of fragment recombination. When Alva et al. [19] studied this phenomenon their dataset did not initially indicate any domain that simultaneously contained two or more of the peptides in the set. Although the reasons remain unclear, it is plausible that (i) many fragments embody dominant cores that guide folding and (ii) that the combination of two compatible fragments puts them under evolutionary pressure, forcing one to adapt quickly to the new environment. When the authors relaxed the similarity criteria for one of the fragments following the second hypothesis, they found several instances of recombination.
In our dataset, we do find several domains that contain at least two of the 1337 identified fragments. Specifically, we looked for domains that contained two different fragments with lengths over 40 amino acids in non-overlapping regions. A total of 1155 domains fulfilled these criteria. Two examples are shown in Figure 7. The first example is the chemotaxis response regulator (CheY-3) like domain d3hzha_ from the flavodoxin-like fold (c.23; Figure 7(a)). The two fragments cover the five β-sheets dividing the flavodoxin into two halves. The N-terminal fragment is shared with a methionyl-tRNA formyltransferase (d1xg9a3) from the formyltransferase fold (c.65), a fragment that is contained in the major component of the network (Figure 5). The fragment is composed of a αβα-sandwich with three parallel strands. The C-terminal fragment on the other hand is found in a ribulose–phosphate binding barrel domain (d5a5wa_) from the (βα)8-barrel fold (c.1) [62]. This fragment is a (βα)2-motif corresponding to the fourth quarter barrel, a relationship that has been described in the past [12,13,18].
Figure 7.

Domains indicating recombination of two fragments. Domains in the center combine two different conserved fragments (blue and green). (a) The flavodoxin CheY-like domain consists of fragments also found in fold c.65 (formyltransferases, in blue) and c.1 (TIM-barrels, green). (b) The TPR-like domain contains two fragments also found in fold a.4 (spectrin repeat-like, in blue) and e.61 (ImpE-like, green).
The other example corresponds to a tetratricopeptide repeat (TPR)-like domain (d1ihga1, a.118.8), which contains seven helices of variable lengths comprising three TPR-motifs [63]. The N-terminal fragment comprises a longer helix (calmodulin binder) and one TPR-motif. We also find this fragment in domains of the microtubule interacting and trafficking (MIT)-domain superfamily (d4u7ya_, a.7.14), a trihelical structural fold that binds MIT-interacting motifs [64]. A study already described the structural similarities between MIT-domains and the first three helices of the TPR domain, suggesting that the MIT-interacting motif completes the missing fourth TPR helix [65]. The C-terminal fragment also occurs in the ImpE-like fold (e.61). Taking together, TPR-domains have been the subject of studies in the past, as they present a model of protein evolution via peptide amplification. Recently, a stable TPR-domain was built via amplification of an intrinsically disordered peptide from an ancient ribosomal protein hairpin [66], illustrating that a TPR-domain can be built through the amplification of TPR-motifs [67]. With the example in Figure 7(b), one could argue that these two fragments are remote homologs since they both contain TPR-motifs. Certainly, we found that many of the domains have smaller-sized fragments in common, such as the TPR-motif. These findings reiterate again the notion that the reuse of protein sequences is a phenomenon occurring at all lengths [68] (Figure 6).
The examples above illustrate how the network analysis allows us to view protein space from an evolutionary perspective. We can observe how different classes and folds are connected to each other via homologous fragments. What we observe is that few fragments seem to have been widely reused.
Implications for protein design
The identification of different fragments in the same domain indicates that proteins can be built through recombination of such pieces. This observation provides great potential for protein design. Fragments that appear in thousands of domains give plenty of opportunities as each of the domains appears with slightly different sequences and functions. In fact, one of the motivations for creating the Fuzzle database was to detect cases such as those in Figure 7 and to identify useful building blocks for protein design. This is the reason why we also include less stringent criteria for fragment definition in our study. The design of stable and functional proteins has proven to be still very challenging to date [69]. There are by now a couple of successful examples for de novo designed protein structures [[70], [71], [72], [73]]. However, the design of proteins via fragment duplication and recombination, i.e. mimicking the natural mechanisms of protein evolution, has met a broader range of success. The design of symmetric protein structures through duplication of the same fragment has succeeded in the design of TIM-barrel [23,[74], [75], [76]], β-trefoil [21,77,78], β-propeller [79,80], TPR-proteins [66], Armadillo-repeat proteins [[81], [82], [83]], and outer-membrane β-barrel folds [84]. The design via recombination has also been effective. In a set of two pioneering studies, Riechmann and Winter obtained folded proteins through recombination of polypeptide segments [85,86]. The design of TIM-barrels through the combination of TIM-barrel and flavodoxin-like fold fragments has also proven successful [12,24]. While the design via recombination is fairly new, it provides an alternative to classical design approaches. The added benefit of this approach is that individual fragments can contribute their unique functional properties to the chimeric protein. Larger fragments, often bearing a hydrophobic core and presenting functionality, presumably offer a better starting point as building blocks. This offers tremendous opportunities for the design of custom-made proteins and provides an interesting, alternate, and generalizable route for design.
Conclusion
It has been shown that contemporary proteins have arisen via the replication, differentiation, and recombination of smaller subdomain-sized fragments. We previously identified one of these fragments and used it in the design of novel chimeric proteins via heterologous recombination [12,13,18]. Motivated by these results, we extended our search to the full protein universe to identify many more of such fragments. A high-throughput profile HMM-HMM comparison led to a set of more than 8 million hits among more than 28,000 domains. These raw data were stored in the Fuzzle database that is accessible via a dynamic web interface (fuzzle.uni-bayreuth.de). By analyzing Fuzzle via similarity networks, we obtain a global picture of protein evolution and discern as yet unrecognized relationships between folds. These data are made available to the general user and can be filtered to their needs.
Overall, we identified 1337 fragments of varying lengths, ranging from 11 to 229 amino acids, with an average of 64. They populate many folds: from the 1221 folds available in the SCOP/Fuzzle database, the identified fragments cover 519. Interestingly, our fragments include most of the folds that are considered to be ancient. In particular, from the 24 folds proposed to be the most ancient by Caetano et al. [45], we find our fragments in all but two. This enrichment toward ancient folds supports the idea that the identified fragments arose early in the origin of folded proteins and, due to their reuse in the course of evolution, are today spread across the protein universe.
Since the same fragments are frequently found in the most ancestral folds as well as more recent protein scaffolds, they might have been reused successfully during evolution due to their intrinsic properties, such as foldability, stability, and functionality. This provides opportunities for the design of proteins, because recombination of stable, independent fragments that contribute unique functional properties can provide a tremendous advantage. For example, in the CheYHisF chimera, the properties inherited from the parents were easily evolvable, and thus, the recombination of fragments provides a good starting point for the design of more complex functionalities [87]. Therefore, we believe that the Fuzzle database and webserver offer invaluable tools for evolutionary studies as well as for the design of custom-made proteins.
Materials and Methods
Fuzzle database
The database contains all structures from the SCOP95 2.06 database clustered for 95% sequence identity. Protein sequences were retrieved from the corresponding ASTRAL dataset [88]. For each domain, we constructed a multiple sequence alignment using PSI-BLAST [14] taking the nr20 database as representative dataset of protein sequence space [89]. We then created a profile HMM for each entry, which was compared pairwise against all the other entries using HHsearch [17]. Default parameters were used. However, to avoid bias introduced by secondary structure content, structure scoring was switched off. Pairs of domains constituting an alignment were superimposed with two different procedures: (i) by calculating the RMSD strictly using only residues aligned in the pairwise sequence alignment (RMSDSeq) and (ii) by using TM-align [28] to allow a structure-based superposition of the protein fragments identified by the sequence alignment (RMSDTM). Altogether the Fuzzle database contains 8,109,195 hits involving 28,010 unique domains.
Construction of the network
First, we clustered each domain in the Fuzzle database according to the start and end of the sequence alignment. We used the density-based clustering method DBSCAN, the epsilon radius was set to 3 and the minimum cluster size to 1 [34]. After clustering, each Fuzzle hit was expanded to include the cluster id to which the two domains belong.
We then constructed a matrix of hits that passed certain cutoffs. Namely, we filtered hits with pairs belonging to different folds, a probability of > 70% (which equals a P-value of 4.1 × 10−5), a length between 10 and 200 residues, and RMSDTM < 3 Å, and a TM-score ≥ 0.3. We further allowed the sequence alignments to be at a maximum 25% larger than the structural alignments. This final subset contained 208,944 pairwise hits, with 8288 unique domains belonging to 24,999 different clusters.
To build the network, we iterated over these pairwise hits and placed a node for each of the 24,999 cluster domains. We linked two nodes whenever the cluster domains they represent appear in the same pairwise hit. The network thus contained 24,999 nodes and 162,852 links. The network is simple, undirected, and disconnected, and therefore presents several subgraphs or connected components. By definition, each component represents a fragment that is shared among domains that belong to different folds. These parameters led to 1337 independent components or fragments.
Web server
The Fuzzle webserver is based on the Django framework (version 1.11). The website interface is designed with Javascript and the style relies on the bootstrap framework (version 4.0). The data are stored in PostgreSQL relational databases. Other software technologies used include JQuery (jquery.com), graph_tool (raph-tool.skewed.de, Datatables (datatables.net), D3js (d3js.org) [90], and the NGL viewer [91]. The webserver allows to individually filter the dataset and to create a customized network analysis for folds of interest. A documentation is provided to guide the user through the different views (fuzzle.uni-bayreuth.de/about/). Customized protein similarity networks can be downloaded as JSON files. The full database is available for download (fuzzle.uni-bayreuth.de/about/database).
Acknowledgments
Acknowledgments
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG grant HO 4022/1-2), the VolkswagenStiftung (grant 94747), and the European Research Council (ERC Consolidator grant 647548 “Protein Lego”). We thank Florian Michel for his careful feedback on the webserver.
Declaration of competing interests
None declared.
Edited by Amy Keating
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.04.013.
Contributor Information
Steffen Schmidt, Email: steffen.schmidt@uni-bayreuth.de.
Birte Höcker, Email: birte.hoecker@uni-bayreuth.de.
Appendix A. Supplementary data
Supplementary Information
References
- 1.Dawson N.L., Lewis T.E., Das S., Lees J.G., Lee D., Ashford P., Orengo C.A., Sillitoe I. CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017;45:D289–D295. doi: 10.1093/nar/gkw1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox N.K., Brenner S.E., Chandonia J.-M. SCOPe: structural classification of proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014;42:D304–D309. doi: 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H., Schaeffer R.D., Liao Y., Kinch L.N., Pei J., Shi S., Kim B.-H., Grishin N.V. ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apic G., Russell R.B. Domain recombination: a workhorse for evolutionary innovation. Sci. Signal. 2010;3:pe30. doi: 10.1126/scisignal.3139pe30. [DOI] [PubMed] [Google Scholar]
- 5.Ponting C.P., Russell R.R. The natural history of protein domains. Annu. Rev. Biophys. Biomol. Struct. 2002;31:45–71. doi: 10.1146/annurev.biophys.31.082901.134314. [DOI] [PubMed] [Google Scholar]
- 6.Baron M., Norman D.G., Campbell I.D. Protein modules. Trends Biochem. Sci. 1991;16:13–17. doi: 10.1016/0968-0004(91)90009-K. [DOI] [PubMed] [Google Scholar]
- 7.Baalsrud H.T., Tørresen O.K., Solbakken M.H., Salzburger W., Hanel R., Jakobsen K.S., Jentoft S. De novo gene evolution of antifreeze glycoproteins in codfishes revealed by whole genome sequence data. Mol. Biol. Evol. 2018;35:593–606. doi: 10.1093/molbev/msx311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang P.-S., Boyken S.E., Baker D. The coming of age of de novo protein design. Nature. 2016;537:320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 9.Alva V., Remmert M., Biegert A., Lupas A.N., Söding J. A galaxy of folds. Protein Sci. 2010;19:124–130. doi: 10.1002/pro.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nepomnyachiy S., Ben-Tal N., Kolodny R. Global view of the protein universe. Proc. Natl. Acad. Sci. U. S. A. 2014;111:11691–11696. doi: 10.1073/pnas.1403395111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Höcker B., Schmidt S., Sterner R. A common evolutionary origin of two elementary enzyme folds. FEBS Lett. 2002;510:133–135. doi: 10.1016/S0014-5793(01)03232-X. [DOI] [PubMed] [Google Scholar]
- 12.Bharat T.A.M., Eisenbeis S., Zeth K., Höcker B. A beta alpha-barrel built by the combination of fragments from different folds. Proc. Natl. Acad. Sci. U. S. A. 2008;105:9942–9947. doi: 10.1073/pnas.0802202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shanmugaratnam S., Eisenbeis S., Hocker B. A highly stable protein chimera built from fragments of different folds. Protein Eng. Des. Sel. 2012;25:699–703. doi: 10.1093/protein/gzs074. [DOI] [PubMed] [Google Scholar]
- 14.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Söding J., Biegert A., Lupas A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doolittle R.F. Similar amino acid sequences: chance or common ancestry? Science. 1981;214(80):149–159. doi: 10.1126/science.7280687. [DOI] [PubMed] [Google Scholar]
- 17.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 18.Farías-Rico J.A., Schmidt S., Höcker B. Evolutionary relationship of two ancient protein superfolds. Nat. Chem. Biol. 2014;10:710–715. doi: 10.1038/nchembio.1579. [DOI] [PubMed] [Google Scholar]
- 19.Alva V., Söding J., Lupas A.N. A vocabulary of ancient peptides at the origin of folded proteins. Elife. 2015;4 doi: 10.7554/eLife.09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopec K.O., Lupas A.N. β-Propeller blades as ancestral peptides in protein evolution. PLoS One. 2013;8:e77074. doi: 10.1371/journal.pone.0077074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broom A., Doxey A.C., Lobsanov Y.D., Berthin L.G., Rose D.R., Howell P.L., McConkey B.J., Meiering E.M. Modular evolution and the origins of symmetry: reconstruction of a three-fold symmetric globular protein. Structure. 2012;20:161–171. doi: 10.1016/j.str.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Franklin M.W., Nepomnyachyi S., Feehan R., Ben-Tal N., Kolodny R., Slusky J.S. Evolutionary pathways of repeat protein topology in bacterial outer membrane proteins. Elife. 2018;7 doi: 10.7554/eLife.40308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Höcker B., Claren J., Sterner R. Mimicking enzyme evolution by generating new (betaalpha)8-barrels from (betaalpha)4-half-barrels. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16448–16453. doi: 10.1073/pnas.0405832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisenbeis S., Proffitt W., Coles M., Truffault V., Shanmugaratnam S., Meiler J., Höcker B. Potential of fragment recombination for rational design of proteins. J. Am. Chem. Soc. 2012;134:4019–4022. doi: 10.1021/ja211657k. [DOI] [PubMed] [Google Scholar]
- 25.Feldmeier K., Höcker B. Computational protein design of ligand binding and catalysis. Curr. Opin. Chem. Biol. 2013;17:929–933. doi: 10.1016/j.cbpa.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Lechner H., Ferruz N., Höcker B. Strategies for designing non-natural enzymes and binders. Curr. Opin. Chem. Biol. 2018;47:67–76. doi: 10.1016/J.CBPA.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Cheng H., Kim B.-H., Grishin N.V. MALISAM: a database of structurally analogous motifs in proteins. Nucleic Acids Res. 2008;36:D211–D217. doi: 10.1093/nar/gkm698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi I.-G., Kim S.-H. Evolution of protein structural classes and protein sequence families. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14056–14061. doi: 10.1073/pnas.0606239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osadchy M., Kolodny R. Maps of protein structure space reveal a fundamental relationship between protein structure and function. Proc. Natl. Acad. Sci. U. S. A. 2011;108:12301–12306. doi: 10.1073/pnas.1102727108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wuchty S. Scale-free behavior in protein domain networks. Mol. Biol. Evol. 2001;18:1694–1702. doi: 10.1093/oxfordjournals.molbev.a003957. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Tal N., Kolodny R. Representation of the protein universe using classifications, maps, and networks. Isr. J. Chem. 2014;54:1286–1292. doi: 10.1002/ijch.201400001. [DOI] [Google Scholar]
- 33.Hall D.R., Hadden J.M., Leonard G.A., Bailey S., Neu M., Winn M., Lindley P.F. The crystal and molecular structures of diferric porcine and rabbit serum transferrins at resolutions of 2.15 and 2.60 Å, respectively. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002;58:70–80. doi: 10.1107/S0907444901017309. [DOI] [PubMed] [Google Scholar]
- 34.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. http://www.jmlr.org/papers/v12/pedregosa11a.html [Google Scholar]
- 35.Moreira A.A., Paula D.R., Costa Filho R.N., Andrade J.S. Competitive cluster growth in complex networks. Phys. Rev. E. 2006;73 doi: 10.1103/PhysRevE.73.065101. [DOI] [PubMed] [Google Scholar]
- 36.Barabasi A.-L., Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/SCIENCE.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 37.Deeds E.J., Ashenberg O., Shakhnovich E.I. From the cover: a simple physical model for scaling in protein–protein interaction networks. Proc. Natl. Acad. Sci. 2006;103:311–316. doi: 10.1073/pnas.0509715102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levitt M. Nature of the protein universe. Proc. Natl. Acad. Sci. 2009;106:11079–11084. doi: 10.1073/pnas.0905029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spraggon G., Schwarzenbacher R., Kreusch A., Lee C.C., Abdubek P., Ambing E., Biorac T., Brinen L.S. Crystal structure of an Udp-n-acetylmuramate-alanine ligase MurC (TM0231) from Thermotoga maritima at 2.3 Å resolution. Proteins Struct. Funct. Bioinforma. 2004;55:1078–1081. doi: 10.1002/prot.20034. [DOI] [PubMed] [Google Scholar]
- 40.Zidar N., Tomašić T., Šink R., Rupnik V., Kovač A., Turk S., Patin D., Blanot D. Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,4-dione inhibitors of MurD ligase. J. Med. Chem. 2010;53:6584–6594. doi: 10.1021/jm100285g. [DOI] [PubMed] [Google Scholar]
- 41.Lewis R.J., Brannigan J.A., Offen W.A., Smith I., Wilkinson A.J. An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J. Mol. Biol. 1998;283:907–912. doi: 10.1006/jmbi.1998.2163. [DOI] [PubMed] [Google Scholar]
- 42.Wolberger C., Dong Y., Ptashne M., Harrison S.C. Structure of a phage 434 Cro/DNA complex. Nature. 1988;335:789–795. doi: 10.1038/335789a0. [DOI] [PubMed] [Google Scholar]
- 43.Valavanis I., Spyrou G., Nikita K. A similarity network approach for the analysis and comparison of protein sequence/structure sets. J. Biomed. Inform. 2010;43:257–267. doi: 10.1016/J.JBI.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 44.Greene L.H. Protein structure networks. Brief. Funct. Genomics. 2012;11:469–478. doi: 10.1093/bfgp/els039. [DOI] [PubMed] [Google Scholar]
- 45.Caetano-Anollés G., Kim H.S., Mittenthal J.E. The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture. Proc. Natl. Acad. Sci. U. S. A. 2007;104:9358–9363. doi: 10.1073/pnas.0701214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnell R., Agren D., Schneider G. 1.9 Å structure of the signal receiver domain of the putative response regulator NarL from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2008;64:1096–1100. doi: 10.1107/S1744309108035203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.B. Nocek, N. Maltseva, J. Stam, W. Anderson, A. Joachimiak, CSGID, Crystal structure of the Mg-bound 3-keto-L-gulonate-6-phosphate decarboxylase from Vibrio cholerae O1 biovar El Tor str. N16961. doi:10.2210/PDB3JR2/PDB
- 48.Höcker B., Beismann-Driemeyer S., Hettwer S., Lustig A., Sterner R. Dissection of a (βα)8-barrel enzyme into two folded halves. Nat. Struct. Biol. 2001;8:32–36. doi: 10.1038/83021. [DOI] [PubMed] [Google Scholar]
- 49.Sander C., Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins Struct. Funct. Genet. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- 50.Kursula P., Schüler H., Flodin S., Nilsson-Ehle P., Ogg D.J., Savitsky P., Nordlund P., Stenmark P. Structures of the hydrolase domain of human 10-formyltetrahydrofolate dehydrogenase and its complex with a substrate analogue. Acta Crystallogr. D. Biol. Crystallogr. 2006;62:1294–1299. doi: 10.1107/S0907444906026849. [DOI] [PubMed] [Google Scholar]
- 51.Lobley C.M., Ciulli A., Whitney H.M., Williams G., Smith A.G., Abell C., Blundell T.L. The crystal structure of Escherichia coli ketopantoate reductase with NADP + bound. Biochemistry. 2005;44:8930–8939. doi: 10.2210/PDB1YJQ/PDB. [DOI] [PubMed] [Google Scholar]
- 52.Singh R.K., Palm G.J., Panjikar S., Hinrichs W. Structure of the Apo form of the catabolite control protein a (Ccpa) from Bacillus megaterium with a DNA-binding domain. Acta Crystallogr.,Sect.F. 2007;63:253. doi: 10.2210/PDB2JCG/PDB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker M.D., Neiditch M.B. Structural basis of response regulator inhibition by a bacterial anti-activator protein. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sauer R.T., Yocum R.R., Doolittle R.F., Lewis M., Pabo C.O. Homology among DNA-binding proteins suggests use of a conserved super-secondary structure. Nature. 1982;298:447–451. doi: 10.1038/298447a0. [DOI] [PubMed] [Google Scholar]
- 55.Pabo C.O., Sauer R.T. Protein–DNA recognition. Annu. Rev. Biochem. 1984;53:293–321. doi: 10.1146/annurev.bi.53.070184.001453. [DOI] [PubMed] [Google Scholar]
- 56.Brennan R.G., Matthews B.W. The helix-turn-helix DNA binding motif. J. Biol. Chem. 1989;264:1903–1906. [PubMed] [Google Scholar]
- 57.Aravind L., Anantharaman V., Balaji S., Babu M.M., Iyer L.M. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. doi: 10.1016/j.fmrre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki M., Brenner S.E. Classification of multi-helical DNA-binding domains and application to predict the DBD structures of σ factor, LysR, OmpR/PhoB, CENP-B, Rap1, and XylS/Ada/AraC. FEBS Lett. 1995;372:215–221. doi: 10.1016/0014-5793(95)00988-L. [DOI] [PubMed] [Google Scholar]
- 59.Laurino P., Tóth-Petróczy Á., Meana-Pañeda R., Lin W., Truhlar D.G., Tawfik D.S. An ancient fingerprint indicates the common ancestry of Rossmann-fold enzymes utilizing different ribose-based cofactors. PLoS Biol. 2016;14:1002396. doi: 10.1371/journal.pbio.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russell R.B., Márquez J.A., Hengstenberg W., Scheffzek K. Evolutionary relationship between the bacterial HPr kinase and the ubiquitous PEP-carboxykinase: expanding the P-loop nucleotidyl transferase superfamily. FEBS Lett. 2002;517:1–6. doi: 10.1016/S0014-5793(02)02518-8. [DOI] [PubMed] [Google Scholar]
- 61.Manoj N., Strauss E., Begley T.P., Ealick S.E. Structure of human phosphopantothenoylcysteine synthetase at 2.3 Å resolution. Structure. 2003;11:927–936. doi: 10.1016/S0969-2126(03)00146-1. [DOI] [PubMed] [Google Scholar]
- 62.Söderholm A., Guo X., Newton M.S., Evans G.B., Näsvall J., Patrick W.M., Selmer M. Two-step ligand binding in a (βα)8 barrel enzyme: substrate-bound structures shed new light on the catalytic cycle of HisA. J. Biol. Chem. 2015;290:24657–24668. doi: 10.1074/jbc.M115.678086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor P., Dornan J., Carrello A., Minchin R.F., Ratajczak T., Walkinshaw M.D. Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure. 2001;9:431–438. doi: 10.1016/S0969-2126(01)00603-7. [DOI] [PubMed] [Google Scholar]
- 64.Guo E.Z., Xu Z. Distinct mechanisms of recognizing endosomal sorting complex required for transport III (ESCRT-III) protein IST1 by different microtubule interacting and trafficking (MIT) domains. J. Biol. Chem. 2015;290:8396–8408. doi: 10.1074/jbc.M114.607903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scott A., Gaspar J., Stuchell-Brereton M.D., Alam S.L., Skalicky J.J., Sundquist W.I. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13813–13818. doi: 10.1073/pnas.0502165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu H., Sepulveda E., Hartmann M.D., Kogenaru M., Ursinus A., Sulz E., Albrecht R., Coles M. 2016. Origin of a folded repeat protein from an intrinsically disordered ancestor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lupas A.N., Alva V. Ribosomal proteins as documents of the transition from unstructured (poly)peptides to folded proteins. J. Struct. Biol. 2017;198:74–81. doi: 10.1016/j.jsb.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Nepomnyachiy S., Ben-Tal N., Kolodny R. Complex evolutionary footprints revealed in an analysis of reused protein segments of diverse lengths. Proc. Natl. Acad. Sci. 2017;114:11703–11708. doi: 10.1073/pnas.1707642114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schreier B., Stumpp C., Wiesner S., Höcker B. Computational design of ligand binding is not a solved problem. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18491–18496. doi: 10.1073/pnas.0907950106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 71.Huang P.-S., Oberdorfer G., Xu C., Pei X.Y., Nannenga B.L., Rogers J.M., DiMaio F., Gonen T. High thermodynamic stability of parametrically designed helical bundles. Science. 2014;346:481–485. doi: 10.1126/science.1257481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomson A.R., Wood C.W., Burton A.J., Bartlett G.J., Sessions R.B., Brady R.L., Woolfson D.N. Computational design of water-soluble α-helical barrels. Science. 2014;346:485–488. doi: 10.1126/science.1257452. [DOI] [PubMed] [Google Scholar]
- 73.Huang P.S., Feldmeier K., Parmeggiani F., Velasco D.F., Höcker B., Baker D. De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat. Chem. Biol. 2016;12:29–34. doi: 10.1038/nchembio.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Höcker B., Lochner A., Seitz T., Claren J., Sterner R. High-resolution crystal structure of an artificial (βα)8 -barrel protein designed from identical half-barrels. Biochemistry. 2009;48:1145–1147. doi: 10.1021/bi802125b. [DOI] [PubMed] [Google Scholar]
- 75.Claren J., Malisi C., Höcker B., Sterner R. Establishing wild-type levels of catalytic activity on natural and artificial (βα)8-barrel protein scaffolds. Proc. Natl. Acad. Sci. 2009;106:3704–3709. doi: 10.1073/pnas.0810342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fortenberry C., Bowman E.A., Proffitt W., Dorr B., Combs S., Harp J., Mizoue L., Meiler J. Exploring symmetry as an avenue to the computational design of large protein domains. J. Am. Chem. Soc. 2011;133:18026–18029. doi: 10.1021/ja2051217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee J., Blaber M. Experimental support for the evolution of symmetric protein architecture from a simple peptide motif. Proc. Natl. Acad. Sci. 2011;108:126–130. doi: 10.1073/pnas.1015032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee J., Blaber S.I., Dubey V.K., Blaber M. A polypeptide “building block” for the β-trefoil fold identified by “top-down symmetric deconstruction”. J. Mol. Biol. 2011;407:744–763. doi: 10.1016/j.jmb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 79.Yadid I., Tawfik D.S. Functional β-propeller lectins by tandem duplications of repetitive units. Protein Eng. Des. Sel. 2011;24:185–195. doi: 10.1093/protein/gzq053. [DOI] [PubMed] [Google Scholar]
- 80.Voet A.R.D., Noguchi H., Addy C., Simoncini D., Terada D., Unzai S., Park S.-Y., Zhang K.Y.J. Computational design of a self-assembling symmetrical β-propeller protein. Proc. Natl. Acad. Sci. 2014;111:15102–15107. doi: 10.1073/pnas.1412768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parmeggiani F., Huang P.-S., Vorobiev S., Xiao R., Park K., Caprari S., Su M., Seetharaman J. A general computational approach for repeat protein design. J. Mol. Biol. 2015;427:563–575. doi: 10.1016/j.jmb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Madhurantakam C., Varadamsetty G., Grütter M.G., Plückthun A., Mittl P.R.E. Structure-based optimization of designed Armadillo-repeat proteins. Protein Sci. 2012;21:1015–1028. doi: 10.1002/pro.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parmeggiani F., Pellarin R., Larsen A.P., Varadamsetty G., Stumpp M.T., Zerbe O., Caflisch A., Plückthun A. Designed Armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J. Mol. Biol. 2008;376:1282–1304. doi: 10.1016/J.JMB.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 84.Arnold T., Poynor M., Nussberger S., Lupas A.N., Linke D. Gene duplication of the eight-stranded β-barrel OmpX produces a functional pore: a scenario for the evolution of transmembrane β-barrels. J. Mol. Biol. 2007;366:1174–1184. doi: 10.1016/j.jmb.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 85.Riechmann L., Winter G. Early protein evolution: building domains from ligand-binding polypeptide segments. J. Mol. Biol. 2006;363:460–468. doi: 10.1016/J.JMB.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 86.Riechmann L., Winter G. Novel folded protein domains generated by combinatorial shuffling of polypeptide segments. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10068–10073. doi: 10.1073/pnas.170145497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farías-Rico J.A., Höcker B. Design of chimeric proteins by combination of subdomain-sized fragments. in: Methods Enzymol. 2013:389–405. doi: 10.1016/B978-0-12-394292-0.00018-7. [DOI] [PubMed] [Google Scholar]
- 88.Brenner S.E., Koehl P., Levitt M. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501. doi: 10.1093/nar/gki025. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.D3.js - Data-driven documents, (n.d.). https://d3js.org/ (accessed May 15, 2019).
- 91.Rose A.S., Hildebrand P.W. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015;43:W576–W579. doi: 10.1093/nar/gkv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information