

Abstract
Purpose:
We report an increased incidence of EBV-induced B-cell lymphoproliferative disease (LPD) in patients treated with siplizumab, an anti-CD2 antibody. The development of EBV-LPD has been associated with the use of immunosuppressive agents used in solid organ, bone marrow, and stem cell transplantation and in certain congenital immunodeficiencies.
Experimental Design:
We conducted a single-institution phase I dose-escalation trial of siplizumab, a humanized monoclonal antibody to CD2, in 29 patients withT-cell malignancies.
Results:
Although initial responses were encouraging, 4 (13.7%) patients developed EBV-LPD and the trial was stopped. Reductions in CD4+ and CD8+ cell count numbers in response to therapy were seen in all patients, but in those patients developing EBV-LPD a significantly greater reduction in natural killer (NK) cell number and CD2 expression onTcells was seen.These findings highlight the importance of NK-cell depletion and CD2 expression in addition toT-cell depletion in the etiology of EBV-LPD.
Conclusions:
The emergence of EBV-LPD may be associated with the ability of siplizumab to deplete bothTand NK cells without affecting B cells. Agents that depleteT-and NK-cellpopulations without affecting B cell number should be screened for this potentially serious adverse event.
The contribution of EBV to the pathogenesis of B-cell lymphoproliferative disorders in immunocompromised individuals is well established. The best described condition is post-transplant lymphoproliferative disorders, with the first series published in 1969 (1, 2). The WHO recognizes four broad clinical settings of immunodeficiency-associated lymphomas and lymphoproliferative disorders: primary immunodeficiency syndromes, infection with HIV, immunosuppression in patients who have received solid organ or bone marrow allograft, and iatrogenic immunosuppression associated with methotrexate therapy for autoimmune disease (3). This classification system omits other iatrogenic causes of immunodeficiency, and published data regarding the role of other immunosuppressive therapies in causing EBV lymphoproliferative disease (LPD) are limited (4–8). Defective immunosurveillance combined with chronic antigenic stimulation is believed to be responsible for the development of LPD in patients receiving immunosuppressive therapy. The highest rates of EBV-LPD are seen following lung transplantation and T-cell-depleted allogeneic bone marrow transplantation with up to 20% of patients developing this complication (9).
T-cell lymphomas constitute a diverse group of hematologic malignancies that account for ~10% of non-Hodgkin’s lymphomas (10). T-cell lymphomas are typically aggressive and infrequently cured by chemotherapy, and prospective randomized trials are rarely done (11–14). Our observation that siplizumab, a humanized monoclonal antibody (mAb) against CD2, is effective in an animal model of adult T-cell leukemia/lymphoma (ATLL) was the basis for considering a clinical trial using this agent in T-cell malignancies (15). Preliminary clinical trial results showed some similar objective responses as seen in preclinical studies. However, the trial was halted when four cases of EBV-LPD were identified following siplizumab therapy. We present the clinical cases identified and the data proposing potential pathogenic mechanisms.
Materials and Methods
Study design.
This was a single-institution phase I dose-escalation study of siplizumab, a humanized mAb directed against CD2, in patients with T-cell lymphoproliferative disorders. Whereas the primary endpoint was safety assessment, secondary endpoints included assessment of antitumor activity, pharmacokinetic studies, CD2 saturation kinetics, and T-cell and natural killer (NK)-cell elimination and recovery following therapy. The trial was approved by the National Cancer Institute Institutional Review Board and all patients provided written informed consent.
In the original trial design, cohorts of patients received escalating doses of intravenous siplizumab over 2 or 3 consecutive days per treatment week every 2 weeks. As the trial progressed, it became evident that the level of CD2 expression on the cell surface was dramatically reduced after the first infusion of siplizumab. It was proposed that maximal efficiency may be achieved by weekly drug administration; therefore, the study design was amended. In the revised design, patient cohorts received a single-day administration on days 0 and 14 and once weekly thereafter. The assigned doses and schedule per cohort are outlined in Table 1.
Table 1.
Schedule of siplizumab administration cohorts 1 to 10
| Siplizumab doses (mg/kg) | ||||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Patients enrolled | |
| Cohort 1 | 0.2 | 0.2 | — | 3 |
| Cohort 2 | 0.2 | 0.2 | 0.2 | 3 |
| Cohort 3 | 0.4 | 0.4 | — | 3 |
| Cohort 4 | 0.4 | 0.4 | 0.4 | 4 |
| Cohort 5 | 0.4 | 0.8 | 1.2 | 3 |
| Cohort 6 | 0.4 | 1.2 | 1.8 | 3 |
| Cohort 7 | 0.4 | 1.8 | 2.6 | 3 |
| Cohort 8 | 0.8 | — | — | 3 |
| Cohort 9 | 3.4 | — | — | 3 |
| Cohort 10 | 4.8 | — | — | 1 |
NOTE: Cohorts 1 to 7 received a cycle every 14 d, whereas cohorts 8 to 10 received a single-day dose days 1 and 14 and then weekly administration.
Molecular monitoring for cytomegalovirus (CMV) was undertaken on all patients at baseline, with every treatment course and monthly thereafter. Monitoring was done using quantitative real-time PCR. A positive PCR was defined as >250 CMV copies/mL blood.
Pathology review.
Diagnosis of EBV-LPD was established according to the WHO criteria for post-transplant lymphoproliferative disorders using immunohistochemistry on formalin-fixed, paraffin-embedded tissue sections. The following panel of antibodies was used for immunohistochemistry: CD20, CD3, CD4, CD8, CD2, CD25, and CD30 in all cases; in addition, CD5, CD7, CD15, Mum-1, and Pax-5 were used to rule out B-cell lymphoma in selected patients.
In situ hybridization analysis for EBV RNA was done on 4-Am-thick formalin-fixed, paraffin-embedded tissue using the INFORM EBV-encoded nontranslated RNA probe (Ventana Medical Systems). The signal was visualized using the ISH iVIEW Blue Detection kit (Ventana Medical Systems) with nitroblue tetrazolium/BCIP and a Fast Red nuclear counterstain. All the procedures were done on a BenchMark XT autostainer (Ventana Medical Systems) according to the manufacturer’s instructions. Clonal rearrangement of the IgH gene was assessed using DNA extracted from formalin-fixed, paraffin-embedded tissue sections and PCR amplified using primers to framework region III and the joining region of the immunoglobulin heavy gene as described previously (16).
Flow cytometry.
Flow cytometry was used to assess the surface expression of selected T, B, and NK cell markers on peripheral blood mononuclear cells at baseline and before each cycle of siplizumab. We used a whole blood lysis-no wash method and analyzed with a FACSCalibur (Becton Dickinson) using Multiset software (Becton Dickinson). T-cell subsets were identified by directly conjugated mAbs; anti-CD3, anti-CD4, and anti-CD8; B cells by anti-CD19; and NK cells by a combination of anti-CD16 and anti-CD56 evaluated on CD3− lymphocytes. All mAbs were obtained from Becton Dickinson. To calculate absolute numbers of each lymphocyte subset, the percentage of cells staining positive was multiplied by the absolute peripheral blood lymphocyte count determined by a Celldyne 3500 (Abbott).
CD2 receptor saturation assays.
T lymphocytes from aliquots of whole blood were assessed for CD2 expression on normal and malignant T cells, before and after siplizumab therapy, using flow cytometric immunophenotyping, within 24 h of collection. Whole blood lysis was done using ammonium chloride before staining for 30 min at room temperature with cocktails containing four antibodies. The antibody panels were chosen based on the number of cells, previous histologic diagnosis, and available clinical history and designed to contain disease-specific combinations that detected the malignant cells (antibody concentration according to the manufacturer’s recommendations). Four-color flow cytometry was done using a Becton Dickinson FACSCalibur flow cytometer. The sensitivity of the fluorescent detectors was set and monitored using Calibrite beads (Becton Dickinson) according to the manufacturer’s recommendations. Data (collected in List mode) were analyzed with CellQuest software (Becton Dickinson). At least 5,000 lymphocytes were acquired per tube. For analysis, cell populations were analyzed by gating on forward scatter, side scatter, CD45, and CD3.
Receptor occupancy was determined using conjugated antibodies reactive with distinct CD2 epitopes. The siplizumab antibody was conjugated with phycoerythrin at a 1:1 ratio to allow determination of antibody-binding capacity using the BD Biosciences QuantiBRITE system for fluorescence quantitation. A second FITC-conjugated and a phycoerythrin-conjugated antibody (S5.2) conjugated at a 1:1 ratio that recognizes CD2 (Becton Dickinson) but does not compete with siplizumab binding was used to quantitate the total level of CD2 expression on normal and malignant T cells by measuring antibody-binding capacity at saturation using the BD Biosciences QuantiBRITE system.
EBV quantitation.
When possible, quantitative real-time PCR was done on peripheral blood mononuclear cell suspensions prepared from whole blood as described previously (17). The EBV viral load was expressed per 106 mononuclear cell genome equivalents. Calculated EBV genome equivalents up to 200 copies per 1 million mononuclear cells can be detected using this assay on peripheral blood mononuclear cell preparations from healthy EBV seropositive adults. Duration of PCR positivity was defined as the time from the identification of EBV reactivation to the first negative PCR.
Statistical analysis.
The longitudinal biomarkers CD4, CD8, B, and NK cell counts and CD2 expression were assessed over time in the 29 individuals on this study. The data was log (natural) transformed to aid analysis. Using a linear mixed model, the data were examined for changes in mean (log-scale) over time (ref. 18; how the mean transformed saturation levels changed across the baseline, post-cycle 1, and post-cycle 2 measurements). A conditional F test was used to test for changes across time. Furthermore, we compared the patterns over time by whether a patient developed EBV-LPD using linear mixed models with a likelihood ratio test (χ2 test with 3 df).
Results
Clinical findings.
Twenty-nine patients were enrolled over 2 years, the majority of whom had either ATLL (n = 15) or large granular lymphocyte leukemia (n = 7). The characteristics of the patients who developed EBV-LPD are outlined in Table 2. The first recognized case of EBV-LPD was diagnosed in a 72-year-old Asian male with T-cell large granular lymphocyte leukemia and red cell aplasia who had stable disease after 8 weeks (6.4 mg/kg total dose) of siplizumab. When dose-limiting neutropenia developed, siplizumab was stopped. Subsequent cyclosporine therapy resulted in a hematologic remission, but after 2 months the patient developed an EBV-related diffuse large B-cell lymphoma of the sigmoid colon (Fig. 1). At diagnosis, peripheral blood EBV PCR copies were only moderately elevated at 1,600 equivalents and became undetectable <8 weeks after treatment with combination chemotherapy (etoposide, prednisone, vincristine, cyclophos-phamide, and doxorubicin) and rituximab.
Table 2.
Patient characteristics
| Case | Sex | Age (y) | Diagnosis | Prior therapy | Cohort/dose schedule | Total |
|---|---|---|---|---|---|---|
| 1 | M | 72 | T-cell large granular lymphocyte leukemia | Three cycles: Cyclophosphamide Vindesine Prednisone IFN; Three cycles: Fludarabine Cyclophosphamide IFN |
Cohort 8: day 0, 0.8 mg/kg Weekly | 6.4 mg/kg over 8 wk |
| 2 | F | 65 | T-cell large granular lymphocyte leukemia | Cyclosporine Methotrexate |
Cohort 9 Day 0, 3.4 mg/kg Weekly | 64.6 mg/kg over 19 wk |
| 3 | F | 58 | ATLL Acute subtype |
Eight cycles: Cyclophosphamide Doxorubicin Vincristine Prednisone |
Cohort 9 Day 0, 3.4 mg/kg Weekly | 34 mg/kg over 10 wk |
| 4 | F | 56 | Cutaneous T-cell lymphoma Stage IV | Topical steroids Psoaralen and UV light therapy Local radiation |
Cohort 6 Day 0, 0.4 mg/kg Day 1, 1.2 mg/kg Day 3, 1.8 mg/kg Every other week |
6.8 mg/kg over 2 wk |
| 3 mo cyclosporine | 189 | Extranodal non-Hodgkin’s lymphoma of gastrointestinal tract Monoclonal process Peak EBV viral load 1,600 |
Complete remission post-chemotherapy | |||
| None | 147 | Mediastinal adenopathy Unable to determine clonality Peak EBV viral load 910 |
Complete remission after stopping siplizumab | |||
| One cycle: denileukin diftitox | 55 | Extranodal non-Hodgkin’s lymphoma Monoclonal process EBV viral load unavailable |
Death from ATLL progression | |||
| Six cycles: romidepsin Two cycles: gemcitabine |
309 | Extensive adenopathy Polyclonal process Peak EBV viral load 1.4 × 106 |
Complete remission with rituximab | |||
Fig. 1.
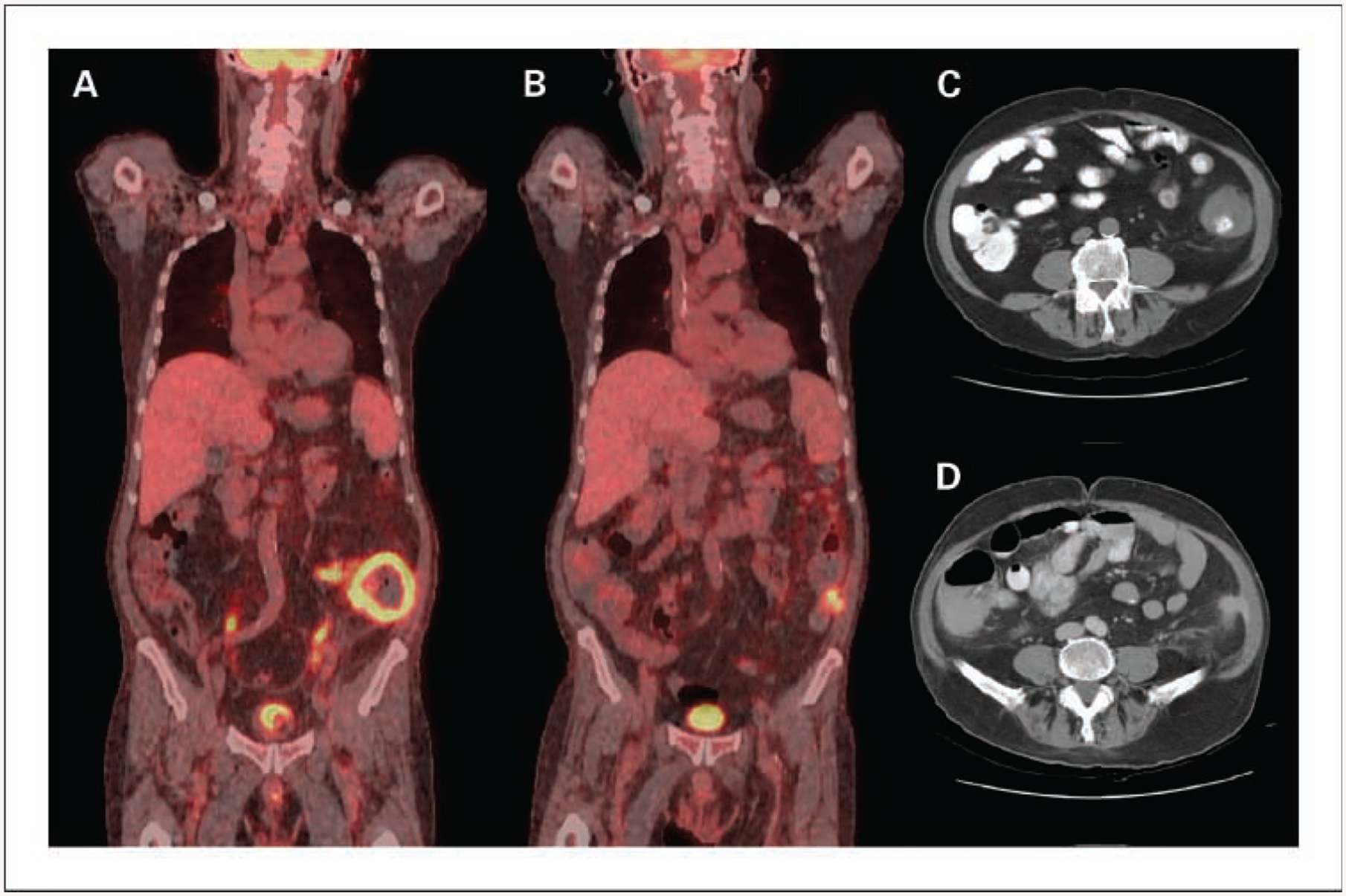
Radiographic imaging of the 72-year-old patient with extranodal non-Hodgkin’s lymphoma of gastrointestinal tract. A, sagittal 18-fluorodeoxyglucose-positron emission tomography image at diagnosis of EBV lymphoma showing circumferential metabolic activity within the wall of the descending colon. B, sagittal 18-fluorodeoxyglucose-positron emission tomography imaging after completion of chemotherapy showing small residual focus of metabolic activity. Computed tomographic images showing left lower quadrant mass involving descending colon at diagnosis (C) and imaging post-therapy shows minor residual abnormality (D).
A second case of EBV-LPD arose in a 65-year-old Caucasian female with transfusion-dependent red cell aplasia secondary to large granular lymphocyte leukemia, who achieved a complete hematologic and pathologic remission with siplizumab, receiving a total of 19 cycles (64.6 mg/kg total dose). She developed an enlarging mediastinal mass. Biopsy confirmed a polymorphic monotypic B-cell EBV-associated LPD. Blood EBV DNA was detectable by PCR at 910 copies per 1 million mononuclear cells. Withdrawal of siplizumab resulted in regression of the mediastinal mass by computed tomography imaging and normalization of 18-fluorodeoxyglucose uptake by positron emission tomography (Fig. 2).
Fig. 2.

Computed tomography and 18-fluorodeoxyglucose-positron emission tomography images showing anterior mediastinal mass with 18-fluorodeoxyglucose metabolic activity at baseline (A and C) and after withdrawal of siplizumab therapy (B and D).
Subsequently, a 58-year-old Japanese female with primary refractory acute ATLL developed EBV-LPD. Although an initial objective response to weekly siplizumab was observed after three cycles, therapy was stopped after 10 cycles (34 mg/kg total dose) due to biopsy-proven disease progression in the skin. At the time of progression, she was also noted to have developed new lesions in the liver, colon, and kidney that were assumed to be ATLL. After receiving one cycle of salvage therapy with denileukin diftitox, she was admitted to her local hospital with a bowel perforation and hypovolemic shock. Pathologic review of the resected bowel showed both ATLL and EBV-positive monomorphic lymphoma (Fig. 3).
Fig. 3.

Histologic examination of colonic biopsy in 58-year-old Asian female showing involvement with ATLL (A) and EBV-associated B-cell lymphoma (B). A, paraffin-embedded sections of colonic mucosa containing ATLL. H&E staining at ×20 (i) and ×40 (ii) shows involvement by ATLL, which is CD25+ (iii) and CD30+ (iv). B, colonic mucosa with an area of extensive atypical lymphoid infiltrate visualized with H&E staining at ×5 (i) and ×20 (ii).These lymphocytes are CD20+ (iii) and the majority are EBV+ (iv). Images were taken using an Olympus Bx41microscope, objective UPlanFI 20×/0.50 ∞/0.17 and 40×/0.75 ∞/0.17, with an adaptor U-TV0.5×C using a digital camera Q-imaging Micropublisher 5.0RTV.The images were captured using “Q-Capture version 3.1”and imported into Adobe Photoshop 7.0.
Considering that EBV-LPD was identified in three patients on study, we reviewed all patients enrolled and identified an additional previously unrecognized case of EBV-LPD in a 57-year-old female with cutaneous T-cell lymphoma. She was enrolled on protocol and received two cycles (6.8 mg/kg total dose) of siplizumab administered on the biweekly schedule; however, restaging at that point showed increasing lymphadenopathy suggestive of disease progression. Therapy was stopped after progression was confirmed by lymph node excision biopsy. Within 6 weeks of stopping siplizumab, lymphopenia resolved and lymphadenopathy improved marginally. Subsequently, she was enrolled on protocol using romidepsin and received 5 months of therapy, when she developed CMV pneumonia and lymph node biopsy-proven polymorphic B-cell lymphoma positive for EBV-encoded nontranslated RNA staining. The peak blood EBV levels were 1,400,000 EBV equivalents per 1 million human mononuclear cells. Treatment with rituximab resulted in EBV DNA clearance in peripheral blood by PCR and regression of adenopathy. The duration of EBV DNA PCR positivity was 18 weeks. EBV-LPD in this patient has been attributed to romidepsin therapy, an investigational histone deacetylase inhibitor, which has been associated with other cases of EBV-LPD.6 In retrospect, the lymph node biopsy done after two cycles of siplizumab showed clusters of EBV-positive B cells. The presence of EBV staining at the time of the initial lymph node biopsy and subsequent lymphoma diagnosis suggests that siplizumab may have had a role in the pathogenesis of the EBV-LPD, perhaps accelerated by the subsequent romidepsin therapy.
Peripheral blood mononuclear cell subpopulation analysis.
Siplizumab resulted in ≥90% reduction in the geometric mean of log-transformed CD4 (P < 0.001) and CD8 (P < 0.001) T-cell counts after two cycles of therapy. There were no statistically significant differences in CD4 or CD8 T-cell depletion between those developing EBV-LPD and those that did not. Similar levels of CD4 and CD8 T-cell depletion were seen regardless of siplizumab administration schedule (Supplementary Data). NK cell numbers also showed a significant reduction in response to siplizumab (P < 0.001), with an 86.1% reduction from pretreatment to post-cycle 1 measurements and a 76.5% reduction from pretreatment to post-cycle 2 measurements. In contrast with CD4 and CD8 T-cell counts, however, there was a statistically significant (P = 0.04) difference in NK-cell reductions over time between those who developed EBV-LPD and those who do not. NK cell numbers fell more significantly in those who developed EBV-LPD (Fig. 4). The statistically significant result should be interpreted cautiously due to the limited number of patients with longitudinal NK cell numbers and who developed EBV-LPD. There was, however, no difference in NK cell number reduction between patients on the weekly and biweekly schedule (P = 0.66). Siplizumab does not bind to B cells, and as expected, no statistically significant changes over time were seen in B-cell counts among the 29 patients (P = 0.73); in particular, there was no change in peripheral blood B cell numbers in those developing EBV-LPD.
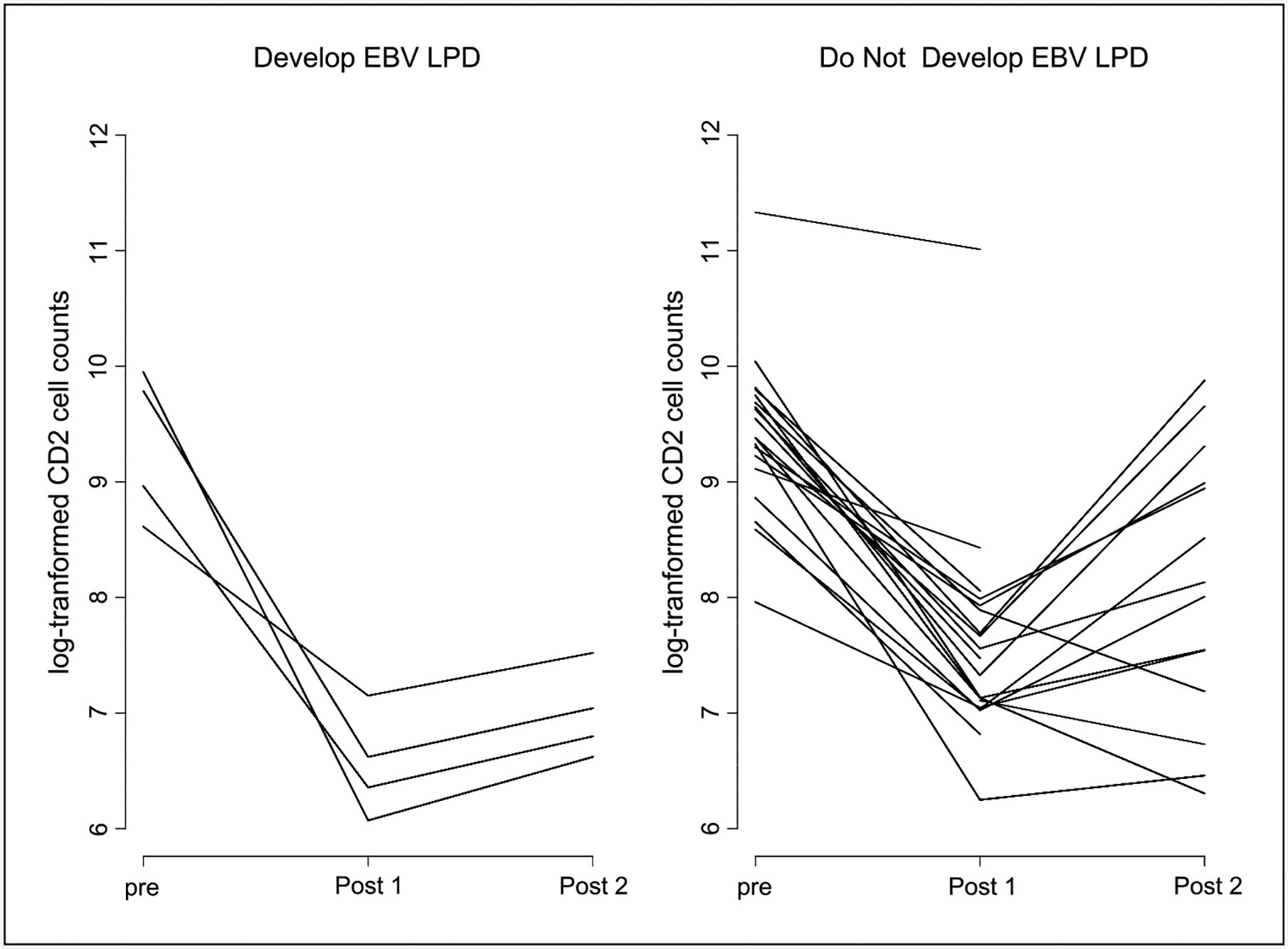
Fig. 4.

Comparison of NK cell number changes between those who develop EBV-LPD and those who do not, showing a greater reduction in NK cell number in those who develop EBV-LPD (P = 0.04).
CD2 expression.
An 86.1% reduction in expression of the CD2 geometric mean was seen after the first cycle compared with baseline and a 76% reduction from baseline after two cycles of siplizumab (P < 0.001). A statistically significant difference in the pattern of CD2 expression by EBV-LPD status was observed (P = 0.03), with more profound declines in CD2 expression from baseline after cycles 1 and 2 in those who developed EBV-LPD (Fig. 5). There was also a statistically significant difference in the pattern of CD2 expression when comparing patients treated with the weekly and biweekly schedules (Fig. 6). Lymphocytes from patients treated with the weekly schedule showed a more significant reduction in CD2 expression across time (P = 0.007) compared with patients on the biweekly schedule.
Fig. 5.
Comparison of trends in CD2 saturation between those who do and do not develop EBV-LPD (P = 0.03) with more sizable changes from baseline for the post-cycle 1and post-cycle 2 measurements for those who develop EBV-LPD.
Fig. 6.

Comparison of trends in CD2 saturation with different administration schedules of siplizumab with statistically significant differences between changes from baseline in weekly and biweekly schedules. Weekly showed more pronounced changes (P = 0.007).
Discussion
EBV is a member of the human γ-herpes virus family that has evolved to exist in equilibrium with its human host. Transmission to a naive host results in viral amplification during acute infection and then persistence for life as an asymptomatic latent infection in B cells. During primary EBV infection, healthy individuals mount a robust humoral and cellular immune response associated with infectious mononucleosis. Antibodies to the viral capsid proteins reduce infectivity; however, the cellular response consisting of CD4+ and CD8+ T cells is essential for controlling both primary and latent EBV infection. Most EBV-associated disease, except for infectious mononucleosis, occurs when the host cellular immune responses are impaired (19). EBV-LPD classically presents as polyclonal or oligoclonal expansions of B cells expressing the full spectrum of EBV latent viral proteins but can progress to aggressive non-Hodgkin’s lymphoma. A classification for virally driven lymphoproliferative processes such as EBV-LPD has not been outlined by the WHO; however, a working consensus has emerged based on morphology (20–22).
In this clinical trial, the administration of siplizumab was associated with a high incidence of EBV-LPD; although not statistically significant, it approached 50% (3 of 7 patients) in patients treated with a weekly schedule in comparison with the biweekly dosing where it occurred in 1 of 22 patients. This high incidence is associated with depletion of both CD4+ and CD8+ T cells and NK cells and down-regulation of T-cell CD2 expression. CD2 expression on NK cells may also be down-regulated, but this was not monitored. The weekly administered schedule of siplizumab decreased CD2 expression to a greater extent than the biweekly schedule. In addition to the four cases of EBV-LPD observed in our trial, one fatal case of EBV-LPD has also been observed in a similar phase I trial of siplizumab in T-cell malignancies in a patient with peripheral T-cell lymphoma.7
A complicating issue in the cases of EBV-LPD observed in this trial is the potential additive or synergistic role of prior or subsequent immunosuppressive therapy. Two patients developing EBV-LPD received additional T-cell-modulating therapy, cyclosporine and romidepsin, following siplizumab therapy and one received prior treatment with cyclosporine. Although there are scattered case reports documenting lymphoma in the setting of immunosuppression, the absolute risk associated with individual immunosuppressive agents is not known. One study concluded that no single immunosuppressive agent preferentially caused post-transplant lymphoproliferative disorders, but the risk was attributable to the whole transplantation process (23). However, preclinical data suggest that cyclosporine can specifically inhibit the cell-mediated response to EBV infection and transformation. In the laboratory, T-cell depletion and addition of cyclosporine to the culture medium are used to enhance the ability to produce EBV-positive lymphoblastoid cell lines (24).
Compared with post-transplant lymphoproliferative disorders, the median age at diagnosis of EBV-LPD is 45 years (25); in our patients, the median age was 61.5 years. Similar to other studies, extranodal presentation is not uncommon (25, 26). In our study population, there was a relatively brief interval between drug administration and onset of EBV-LPD similar to the onset of disease occurring in the setting of solid organ transplant populations (27, 28). CMV infection is associated with post-transplant lymphoproliferative disorder risk and may be an independent risk factor rather than simply evidence of significant level of immunosuppression (29). We detected CMV DNA by PCR in 3 of the patients developing EBV-LPD, whereas only one other non-ATLL patient treated with siplizumab showed CMV reactivation by PCR.
CD2 plays a key role in lymphocyte adhesion and cell signaling through binding to its receptor LFA3 (CD58; ref. 30). Anti-human CD2 mAbs inhibit T-cell responses to various stimuli (31). The importance of dose-timing and dose-dependent effects has been shown in preclinical studies. The inhibitory effect of anti-CD2-directed therapies is most potent during antigen presentation and higher doses result in similar levels of T-cell depletion but greater CD2 modulation (32). CD2 knockout mice produce T cells that have both reduced proliferative responses and reduced IFN-γ release in response to antigen stimulation compared with wild-type (33). However, CD2-deficient T cells maintain normal cytolytic activity. These results suggest that control of EBV infection may rely on the ability to increase T cell numbers in response to emergence of EBV-infected B cells and raises intriguing questions regarding the role of IFN-γ in the control of EBV infections (34).
NK cells are a key component of the early innate immune response to microbes and can inhibit the EBV immortalization of resting B cells (34, 35). Individuals with X-linked proliferative disease that is associated with fatal primary EBV infection are deficient in NK cells, which support the importance of NK cells (36). The critical role of NK cells in cytokine-based therapy for EBV-LPD has been shown in preclinical models, which may be in part related to both direct and indirect antitumor effects of IFN-γ production (27, 28). The exact role of NK cells in the development of EBV-LPD is not clear. For example, recipients of T-cell-depleted stem cell allografts are at greatest risk in the first 3 to 6 months, at which time NK cell numbers have recovered but the patients remain profoundly T-cell-deficient (37). In contrast, we found a significant reduction in NK cell numbers after siplizumab therapy in all patients and noted a statistically greater reduction noted in those patients developing EBV-LPD. In summary, following anti-CD2 therapy, the patients manifested the exceedingly rare immunologic condition of a combined deficiency of T-cell subsets and NK cells without B-cell depletion. Cytotoxic T cells normally provide effective therapy against MHC class I-expressing malignant cells, whereas NK cells may provide effective therapy against cells that have lost their class I MHC expression. Thus, a deficiency of both T and NK cells may increase the risk of the emergence of EBV-LPD.
A potential strategy for future studies with siplizumab is to eliminate B cells in conjunction with T-cell depletion in an attempt to prevent EBV-LPD emergence. Rituximab has shown activity in the treatment of EBV-LPD, with overall response rates of 44% to 75% (38, 39). In addition, rituximab has been tested in a preemptive fashion in patients undergoing T-cell-depleted stem cell transplantation (39, 40). In the Van Esser et al. study, patients who exhibited evidence of EBV reactivation were treated with one infusion of rituximab, which was effective in all but one patient. These observations suggest that EBV-LPD may be prevented by early administration of rituximab and that its use before T- and NK-cell depletion may improve this outcome. Visilizumab, a humanized, anti-CD3 mAb used to treat steroid refractory acute graft versus host disease, although effective, has been associated with an increased risk of EBV-LPD, which can be mitigated by monitoring plasma EBV DNA titers and preemptively administering rituximab (41). Preemptive therapy requires detection of a threshold EBV DNA viral loads or rising titers, and as shown in our cohort, some patients may have low viral loads in spite of histologically confirmed EBV-LPD. A recent prospective study showed that the combination of EBV DNA load and T-cell recovery was superior at identifying high-risk patients (42).
In summary, profound T-cell depletion is associated with the development of EBV-associated neoplasms, which may complicate disorders requiring the use of T-cell-directed therapies. Further, the use of multiple T-cell-directed therapies, either together or sequentially, may further impair immunosurveillance, putting patients at increased risk of these neoplasms. Because patients are frequently exposed to multiple therapies that impair T- and NK-cell function, current preclinical models that may fail to predict this outcome are limited; thus, toxicities associated with potent T-cell-directed therapies may not become evident until clinical studies are conducted as evidenced in this report. The emergence of EBV-LPD as a consequence of the ability of siplizumab to deplete both T and NK cell numbers, without B-cell depletion, strongly suggests that clinical studies employing agents that deplete T cells, especially those that also deplete NK cells without affecting B cell numbers, should be designed to screen for this potentially serious toxicity.
Supplementary Material
Translational Relevance.
This original report details a concerning outcome from a phase I dose-escalation study of the T-cell-specific monoclonal antibody siplizumab. We present data examining the possible pathogenesis of EBV lymphoproliferative disorders in this setting. Although it is known that profound T-cell depletion is associated, in several clinical settings, with the development of EBV-associated neoplasms, our findings highlight the importance of natural killer cell depletion and loss of CD2 expression in addition to T-cell depletion in the etiology of EBV lymphoproliferative disease. This report is important as it highlights that clinical studies employing agents that deplete T cells, especially those that also deplete natural killer cells without affecting B cell numbers, should be designed to screen for this potentially serious toxicity. This report also highlights the difficulty in screening for the complication. Although published literature suggests that patients may be followed for evidence of EBV reactivation by monitoring plasma EBV DNA levels, the level of elevation in our study suggests that even minimal increases in EBV DNA viral load should be evaluated aggressively with other imaging modalities. This also suggests that, in trials of T-cell-depleting agents, there occasionally may be poor correlation between EBV lymphoproliferative disease development and viral loads.
Acknowledgments
We thank Drs. GiovannaTosato and Jeffrey Cohen for critical review of the article.
Grant support: Intramural Research Program, NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Disclosure of Potential Conflicts of Interest
D. Reitsma, K. Kaucic, and L. Hammershaimb are employed by MedImmune, Inc. J.E. Janik and T.A. Waldmann have a Cooperative Research and Development Agreement with MedImmune Inc., and T.A. Waldmann has a patent on siplizumab for the treatment of T cell lymphoma.
S.E. Bates, personal communication.
K. Kaucic and L. Hammershaimb, personal communication.
References
- 1.Penn I, Hammond W, Brettschneider L, Starzl TE. Malignant lymphomas in transplantation patients. Transplant Proc 1969;1:106–12. [PMC free article] [PubMed] [Google Scholar]
- 2.Paya CV, Fung JJ, Nalesnik MA, et al. Epstein-Barr virus-induced posttransplant lymphoproliferative disorders. ASTS/ASTP EBV-PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting. Transplantation 1999;68:1517–25. [DOI] [PubMed] [Google Scholar]
- 3.Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. [Google Scholar]
- 4.Abruzzo LV, Rosales CM, Medeiros LJ, et al. Epstein-Barr virus-positive B-cell lymphoproliferative disorders arising in immunodeficient patients previously treated with fludarabine for low-grade B-cell neoplasms. Am J Surg Pathol 2002;26: 630–6. [DOI] [PubMed] [Google Scholar]
- 5.Bhargava R, Barbashina V, Filippa DA, Teruya-Feldstein J. Epstein-Barr virus positive large B-cell lymphoma arising in a patient previously treated with cladribine for hairy cell leukemia. Leuk Lymphoma 2004;45:1043–8. [DOI] [PubMed] [Google Scholar]
- 6.Lenz G, Golf A, Rudiger T, Hiddemann W, Haferlach T. Epstein-Barr virus-associated B-cell non-Hodgkin lymphoma following treatment of hairy cell leukemia with cladribine. Blood 2003;102:3457–8. [DOI] [PubMed] [Google Scholar]
- 7.Shields DJ, Byrd JC, Abbondanzo SL, Lichy JH, Diehl LF, Aguilera NI. Detection of Epstein-Barr virus in transformations of low-grade B-cell lymphomas after fludarabine treatment. Mod Pathol 1997;10:1151–9. [PubMed] [Google Scholar]
- 8.Orlandi E, Paulli M,Viglio A, et al. Epstein-Barr virus-positive aggressive lymphoma as a consequence of immunosuppression after multiple salvage treatments for follicular lymphoma. Br J Haematol 2001;112: 373–6. [DOI] [PubMed] [Google Scholar]
- 9.Cohen JI. Benign and malignant Epstein-Barr virus-associated B-cell lymphoproliferative diseases. Semin Hematol 2003;40:116–23. [DOI] [PubMed] [Google Scholar]
- 10.The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997;89:3909–18. [PubMed] [Google Scholar]
- 11.Savage KJ, Chhanabhai M, Gascoyne RD, Connors JM. Characterization of peripheralT-cell lymphomas in a single North American institution by the WHO classification. Ann Oncol 2004;15:1467–75. [DOI] [PubMed] [Google Scholar]
- 12.Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood 2004;104:634–41. [DOI] [PubMed] [Google Scholar]
- 13.Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good-prognosis (normal LDH) aggressive lymphomas: results of the NHL-B1 trial of the DSHNHL. Blood 2004;104:626–33. [DOI] [PubMed] [Google Scholar]
- 14.Escalon MP, Liu NS, Yang Y, et al. Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: the M. D. Anderson Cancer Center experience. Cancer 2005;103:2091–8. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Zhang M, Ravetch JV, Goldman C, WaldmannTA. Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD2 monoclonal antibody, MEDI-507. Blood 2003;102: 284–8. [DOI] [PubMed] [Google Scholar]
- 16.Ramasamy I, Brisco M, Morley A. Improved PCR method for detecting monoclonal immunoglobulin heavy chain rearrangement in B cell neoplasms. J Clin Pathol 1992;45:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheinberg P, Fischer SH, Li L, et al. Distinct EBV and CMV reactivation patterns following antibody-based immunosuppressive regimens in patients with severe aplastic anemia. Blood 2007;109:3219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinheiro JC, Bates DM. Mixed-effects models in S and S-Plus. NewYork: Springer; 2000. [Google Scholar]
- 19.Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu Rev Med 2005;56:29–44. [DOI] [PubMed] [Google Scholar]
- 20.Nalesnik MA, Jaffe R, Starzl TE, et al. The pathology of posttransplant lymphoproliferative disorders occurring in the setting of cyclosporine A-prednisone immunosuppression. Am J Pathol 1988;133:173–92. [PMC free article] [PubMed] [Google Scholar]
- 21.Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting-Airlie House, Virginia, November 1997. J Clin Oncol 1999;17:3835–49. [DOI] [PubMed] [Google Scholar]
- 22.Knowles DM, Cesarman E, Chadburn A, et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 1995; 85:552–65. [PubMed] [Google Scholar]
- 23.Birkeland SA, Hamilton-Dutoit S. Is posttransplant lymphoproliferative disorder (PTLD) caused by any specific immunosuppressive drug or by the transplantation per se? Transplantation 2003;76:984–8. [DOI] [PubMed] [Google Scholar]
- 24.Bird AG, McLachlan SM. Cyclosporin A and Epstein-Barr virus. Lancet 1980;2:418. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Gomez MA, Pena M, Cabello M, et al. Posttransplant lymphoproliferative disease: a series of 23 cases. Transplant Proc 2006;38:2448–50. [DOI] [PubMed] [Google Scholar]
- 26.Hoshida Y, Li T, Dong Z, et al. Lymphoproliferative disorders in renal transplant patients in Japan. Int J Cancer 2001;91:869–75. [DOI] [PubMed] [Google Scholar]
- 27.Nalesnik MA. Posttransplantation lymphoproliferative disorders (PTLD): current perspectives. Semin Thorac Cardiovasc Surg 1996;8:139–48. [PubMed] [Google Scholar]
- 28.Tsai DE, Hardy CL, Tomaszewski JE, et al. Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: analysis of prognostic variables and long-term follow-up of 42 adult patients. Transplantation 2001;71:1076–88. [DOI] [PubMed] [Google Scholar]
- 29.Manez R, Breinig MC, Linden P, et al. Posttransplant lymphoproliferative disease in primary Epstein-Barr virus infection after liver transplantation: the role of cytomegalovirus disease. J Infect Dis 1997;176:1462–7. [DOI] [PubMed] [Google Scholar]
- 30.Selvaraj P, Plunkett ML, Dustin M, Sanders ME, Shaw S, Springer TA. The T lymphocyte glycoprotein CD2 binds the cell surface ligand LFA-3. Nature 1987;326:400–3. [DOI] [PubMed] [Google Scholar]
- 31.Kozarsky KF, Tsai C, Bott CM, Allada G, Li LL, Fox DA. An anti-CD2 monoclonal antibody that both inhibits and stimulates T cell activation recognizes a subregion of CD2 distinct from known ligand-binding sites. Cell Immunol 1993;150:235–46. [DOI] [PubMed] [Google Scholar]
- 32.Ding Y, Qin L,Yang Q, et al. A novel murine model for the assessment of human CD2-related reagents in vivo. J Immunol 1996;157:1863–9. [PubMed] [Google Scholar]
- 33.Teh SJ, Killeen N, Tarakhovsky A, Littman DR, Teh HS. CD2 regulates the positive selection and function of antigen-specific CD4−CD8+ Tcells. Blood 1997;89: 1308–18. [PubMed] [Google Scholar]
- 34.Lotz M,Tsoukas CD, Fong S, Carson DA,Vaughan JH. Regulation of Epstein-Barr virus infection by re-combinant interferons. Selected sensitivity to interferon-γ. Eur J Immunol 1985;15:520–5. [DOI] [PubMed] [Google Scholar]
- 35.Williams H, McAulay K, Macsween KF, et al. The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol 2005;129:266–74. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima H, Cella M, Bouchon A, et al. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. EurJ Immunol 2000;30:3309–18. [DOI] [PubMed] [Google Scholar]
- 37.O’Reilly RJ, Small TN, Papadopoulos E, Lucas K, Lacerda J, Koulova L. Biology and adoptive cell therapy of Epstein-Barr virus-associated lymphoproliferative disorders in recipients of marrow allografts. Immunol Rev 1997;157:195–216. [DOI] [PubMed] [Google Scholar]
- 38.Milpied N, Vasseur B, Parquet N, et al. Humanized anti-CD20 monoclonal antibody (rituximab) in post transplant B-lymphoproliferative disorder: a retrospective analysis on 32 patients. Ann Oncol 2000;11 Suppl 1:113–6. [PubMed] [Google Scholar]
- 39.Choquet S, Leblond V, Herbrecht R, et al. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood 2006;107:3053–7. [DOI] [PubMed] [Google Scholar]
- 40.van Esser JW, Niesters HG, van der Holt B, et al. Prevention of Epstein-Barr virus-lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood 2002;99:4364–9. [DOI] [PubMed] [Google Scholar]
- 41.Carpenter PA, Lowder J, Johnston L, et al. A phase II multicenter study of visilizumab, humanized anti-CD3 antibody, to treat steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transplant 2005;11:465–71. [DOI] [PubMed] [Google Scholar]
- 42.Annels NE, Kalpoe JS, Bredius RG, et al. Management of Epstein-Barr virus (EBV) reactivation after allogeneic stem cell transplantation by simultaneous analysis of EBV DNA load and EBV-specific T cell re-constitution. Clin Infect Dis 2006;42:1743–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.