

Abstract
Recent advances in the knowledge of the EGFR pathway have revealed its contribution to distinct immune/inflammatory functions of the epidermis. The purpose of our study was to evaluate the role of EGFR in the regulation of keratinocyte GM-CSF expression. In cultured human keratinocytes, proinflammatory cytokines synergized with TGF-α to induce GM-CSF expression. Accordingly, high epidermal levels of EGFR activation are associated with enhanced expression of GM-CSF in lesional skin of patients with psoriasis or allergic contact dermatitis. In cultured keratinocytes, pharmacological inhibition of EGFR activity reduced GM-CSF promoter transactivation, whereas genetic inhibition of AP-1 reduced expression of GM-CSF. Furthermore, EGFR activation enhanced TNF-α-induced c-Jun phosphorylation and DNA binding, whereas c-Jun silencing reduced GM-CSF expression. Using two different mouse models, we showed that the lack of a functional EGFR pathway was associated with reduced cytokine-induced phosphorylation of ERK1/2, JNK1/2, c-Jun and reduced keratinocyte-derived GM-CSF expression both in vitro and in vivo. Finally, the analysis of GM-CSF expression in the skin of cancer patients treated with anti EGFR drugs showed an association between ERK activity, c-Jun phosphorylation, and epidermal GM-CSF expression. These data demonstrate that the EGFR pathway is critical for the upregulation of keratinocyte GM-CSF expression under conditions of cytokine stimulation.
INTRODUCTION
The epidermal growth factor receptor (EGFR) and its ligands participate in the response of the epithelium to injury in several target sites. In the skin, EGFR ligands are actively synthesized by keratinocytes during acute wound healing, and their sustained overexpression is a characteristic feature in chronic inflammatory disorders including psoriasis, atopic dermatitis and allergic contact dermatitis (Mascia et al., 2003). Apart from their well-established mitogenic role (Jost et al., 2000; Wetzker and Böhmer, 2003), these growth factors are involved in a number of immune protective mechanisms. Transforming growth factor (TGF)-α, the major endogenous EGFR ligand in human skin, promotes the expression of several anti-microbial peptides (Sørensen et al., 2003) and of Toll Like Receptor (TLR) -5 and -9, and synergizes with these receptors in the induction of leukocyte chemoattractants, which include anti-microbial peptides themselves and CXCL8/IL-8 (Miller et al., 2005). Through these mechanisms, the EGFR-ligand system creates a link between innate and adaptive immune responses. Moreover, TGF-α regulates tumor necrosis factor (TNF)-α and interferon (IFN)-γ induced levels of a cluster of chemokines implicated in the massive recruitment of T cells, monocytes and immature dendritic cells, including the CXCR3 ligand IP-10/CXCL10, MCP-1/CCL2 and RANTES/CCL5 (Mascia et al., 2003). While activation of the EGFR-ligand system opposes microbial colonization and promotes efficient wound closure, it prevents excessive leukocyte recruitment into the skin. Noteworthy, the absence of a fully functional EGFR pathway is associated with a mixed inflammatory infiltration in the skin of both EGFR null mice and patients undergoing chemotherapy with EGFR antagonists (Roberts et al., 2004).
EGFR is overexpressed and mutated in a variety of malignancies and a number of different approaches that aim to inhibit EGFR activation are currently in use or being developed. The most common adverse effect seen with EGFR inhibition therapy is a pronounced skin inflammatory response (papulo-pustular rash) that can affect different areas of the body with variable severity (Robert et al., 2005). Recent studies suggest that the skin rash may represent a valuable tool that helps in monitoring the efficacy of some EGFR targeted drugs (Pérez-Soler et al., 2005; Melosky et al., 2009). However, it is not known if the cutaneous rash is an independent effect of the presence of the drug target in the epidermis or if the inflammatory response is of more relevance to the therapeutic response within the tumor. Thus more investigative efforts are needed to better understand the role of EGFR in the modulation of inflammatory mediators expression in epithelial cells.
Granulocyte/macrophage-colony stimulating factor (GM-CSF) is recognized as a major immune regulator governing survival of granulocyte and macrophage lineage populations at all stages of maturation (Hamilton, 2008). GM-CSF is secreted by keratinocytes shortly after injury, and it is considered an early response factor in tissue regeneration (Florin et al., 2006). Transgenic mice overexpressing keratinocyte-derived GM-CSF exhibit an accelerated wound healing process with higher numbers of proliferating keratinocytes at wound edges, increased formation of granulation tissue and enhanced neovascularization (Mann et al., 2001). Besides its activity in promoting angiogenesis and wound healing in normal tissues (Valdembri et al., 2002), GM-CSF has an important role in promoting angiogenesis and malignant growth in squamous cell carcinomas of head and neck (Gutschalk et al., 2006).
Given the relevance of EGFR and GM-CSF in skin and cancer immunobiology, we investigated the in vitro and in vivo control of the EGFR pathway on GM-CSF expression in human and mouse keratinocytes and skin.
RESULTS
GM-CSF is overexpressed in chronic inflammatory skin disorders
We previously reported that GM-CSF is strongly up regulated in chronic lesions of atopic dermatitis patients (Pastore et al., 1997). Here we report that high levels of this cytokine could also be detected in the skin of patients suffering from psoriasis (Figure 1b) or affected by allergic contact dermatitis (Figure 1c), whereas extremely low epidermal GM-CSF expression was observed in healthy controls (Figure 1a). The epidermis of the inflammatory conditions showed cytoplasmic, nuclear and perinuclear GM-CSF staining (see Figure 1 insets). Moreover a strong GM-CSF positivity was also associated with inflammatory cells and endothelial cells in the upper part of the dermis in the lesional skin (Figure 1b and c) and in dermal endothelium of healthy skin (Figure 1a).
Figure 1. GM-CSF is highly expressed by epidermal keratinocytes in chronically inflamed skin.
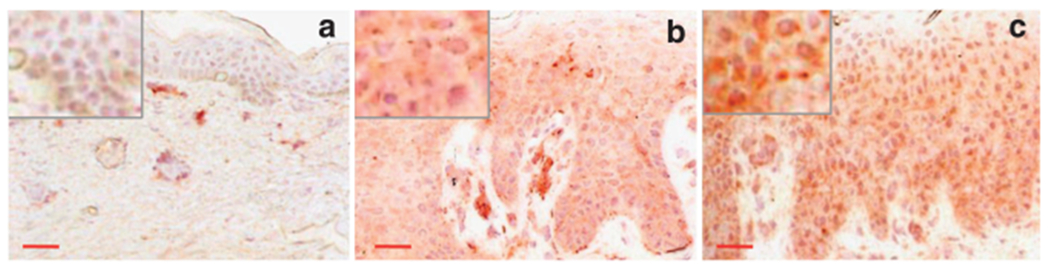
Expression of GM-CSF in the epidermis of a healthy donor (a), in lesional skin of plaque psoriasis (b) and chronic allergic contact dermatitis (c). Representative immunohistochemistry results from seven healthy donors, five patients with psoriasis, and three patients with allergic contact dermatitis. Normalization bar = 100 μm for panel a (× 200) and 50 μm for panels b and c (× 100).
TGF-α synergizes with TNF-α and IFN-γ for the induction of GM-CSF in cultured human keratinocytes
In T cell-mediated inflammatory skin disorders including atopic dermatitis, psoriasis and allergic contact dermatitis, the epidermal compartment displays strong up-regulation of TGF-α (Mascia et al., 2003). Although no data are presently available on the specific role of EGFR signaling during GM-CSF expression in keratinocytes in vivo, the concomitant high levels of both TGF-α and GM-CSF in lesional skin could be suggestive of a potentiation of GM-CSF expression by TGF-α.
We found that exogenous TGF-α alone was a poor stimulus for GM-CSF mRNA expression in cultured human keratinocytes. In these cells, TGF-α strongly synergized with the pro-inflammatory cytokine TNF-α (Figure 2a) and induced an earlier and stronger response to IFN-γ stimulation (Figure 2b). By contrast, keratinocyte pretreatment with the selective EGFR inhibitor PD168393 (PD16) prevented GM-CSF induction both by TNF-α (Figure 2a) and IFN-γ (Figure 2b), indicating that EGFR signaling is actively involved in keratinocyte response to T cell-derived cytokines. These effects were dependent on the concentration of TGF-α and PD16, as confirmed at the protein level by ELISA in 24 hours culture supernatants (Figure 2c and d).
Figure 2. TGF-α enhances, whereas EGFR signaling inhibition suppresses, the expression of GM-CSF by cytokine-stimulated human keratinocytes.
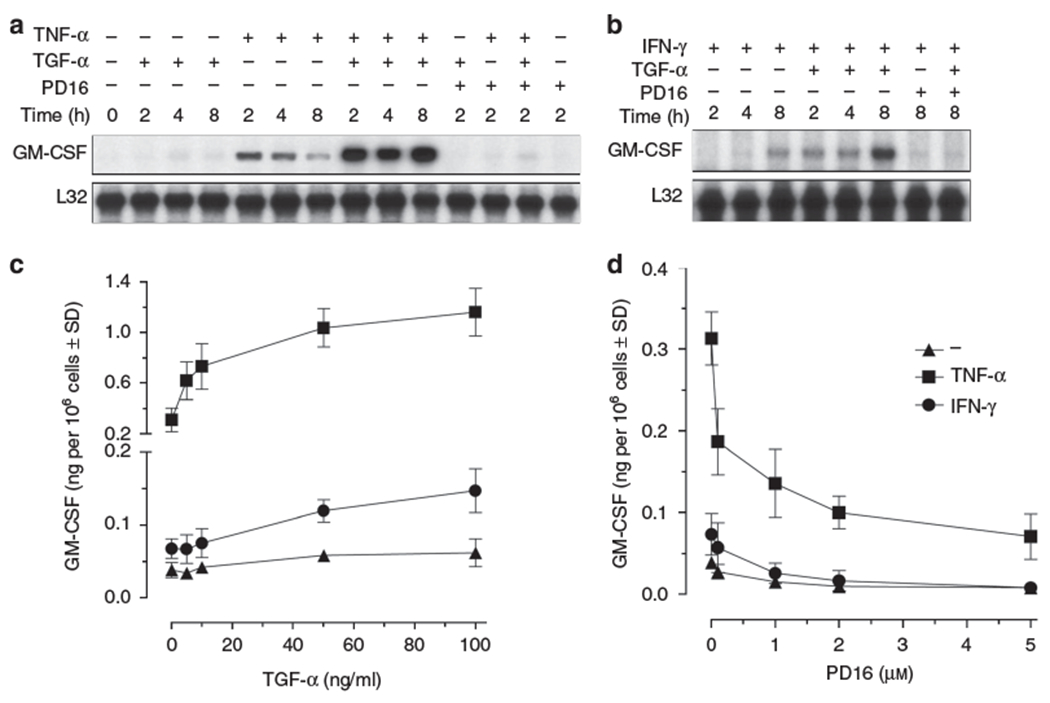
Cytokine gene expression was measured by RNase protection assay in total RNA (10 μg) extracted from human keratinocytes stimulated with TNF-α (50 ng/ml) (a) or IFN-γ (50 U/ml) (b) and/or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). The autoradiographies are representative of four independent experiments. ELISA detection of GM-CSF levels after 24 hours stimulation of human keratinocytes with escalating doses of TGF-α (c) or PD168393 (PD16) (d). Results are expressed as the mean of three independent experiments ± SD.
We verified if the synergistic response observed between TGF-α and TNF-α was restricted to this specific EGFR ligand or other members of the EGF family could evoke a similar response. To this aim we stimulated cultured keratinocytes with equimolar concentration (10 nM) of EGF, HB-EGF, Amphiregulin (AR) and TGF-α in presence or absence of TNF-α. ELISA tests showed that TNF-α induced GM-CSF expression was enhanced with a similar potency by TGF-α, EGF and HB-EGF while AR was less effective in synergizing with TNF-α (Supplementary Figure 1a).
EGFR activation is characterized by a homo/heterodimerization with EGFR itself or other members of the HER family of receptors. The preferred coreceptor of EGFR is represented by HER2. We investigated the role of HER2 in the regulation of TNF-α induced GM-CSF expression. In contrast to the reduction of GM-CSF expression observed in keratinocytes pretreated with PD16 (Figure 2a and Figure 2d), pretreatment with a specific HER-2 inhibitor, Herceptin, did not affect the expression of GM-CSF at the mRNA and protein level (Supplementary Figure 1c and Figure 1d). At the selected doses Herceptin was able to induce HER2 degradation while not affecting EGFR levels (Supplementary Figure 1b) (Hudis, 2007).
TGF-α upregulates TNF-α induced GM-CSF transcription via increased c-Jun phosphorylation and transactivation
We have previously reported that effective transactivation of the minimal GM-CSF promoter, namely p91CAT, requires cooperation between activator protein (AP)-1 and nuclear factor κB (NFκB) binding sites in keratinocytes (Pastore et al., 2000). Hence, this minimal promoter represents a valid tool to investigate the interplay between EGFR-directed and TNF-α-driven signaling pathways on GM-CSF gene transcription.
We have also reported that EGFR activation promotes gene expression in keratinocytes through enhanced AP-1 whereas it does not affect NFκB, whose transcriptional activity is promoted by TNF-α (Pastore et al., 2005).
Basal activity of the minimal GM-CSF promoter was only slightly perturbed by TGF-α, whereas this growth factor synergized with TNF-α with a three-fold increase over TNF-α-driven reporter gene expression (Figure 3a). Of note, the selective pharmacological inhibition of EGFR by PD16 impaired GM-CSF promoter activity following induction by TNF-α. A similar inhibitory effect was exerted by the selective blockade of ERK1/2 by PD98059 (PD98), or JNK1/2 by SP600125 (SP600).
Figure 3. TGF-α upregulates GM-CSF transcription via increased c-Jun phosphorylation and transactivation.
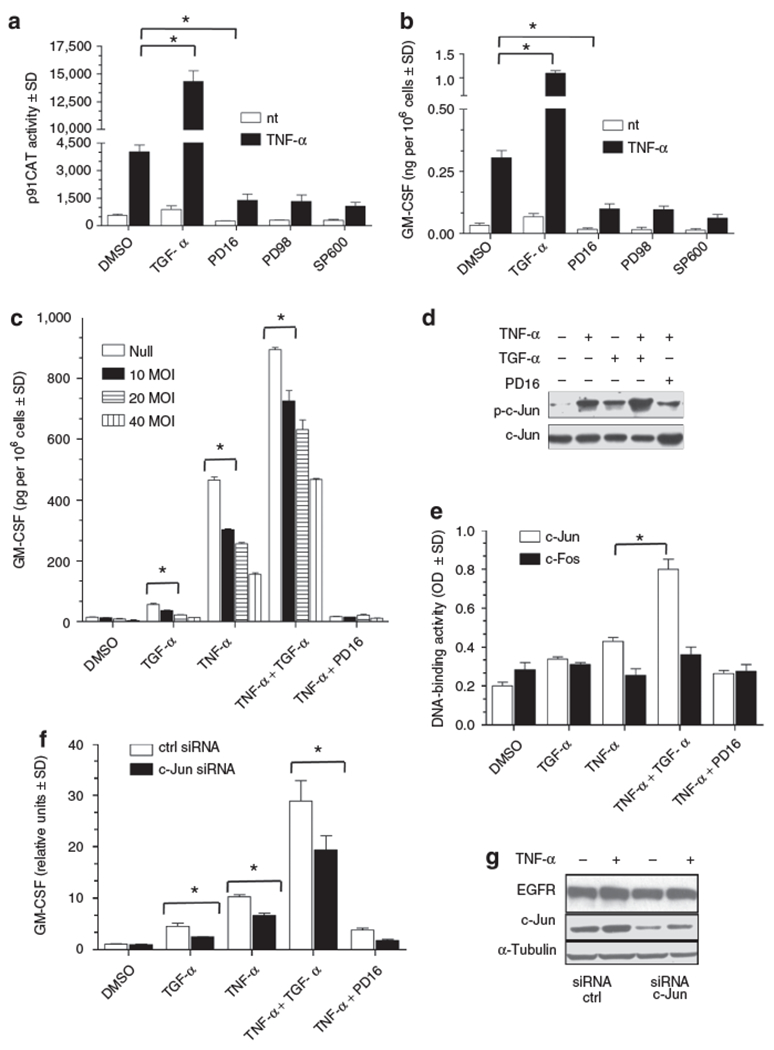
(a) CAT activity was determined in human keratinocytes transfected with the minimal GM-CSF promoter p91CAT following 6 hours stimulation with 50 ng/ml TNF-α and/or 50 ng/ml TGF-α directly, or following 30 minutes pre-incubation with 2 μ/M PD168393 (PD16), or 20 μm PD98059 (PD98) or SP600125 (SP600). (b) ELISA of GM-CSF levels in the supernatant of keratinocytes after 24 hours stimulation with same conditions as panel a. (c) GM-CSF ELISA detection of culture supernatants 48 hours after infection of human keratinocytes with increasing doses of A-FOS or null adenovirus. Cells were treated in the last 24 hours with TNF-α (50 ng/ml) and/or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). (d) Twenty μg of total cell lysates were run in an immunoblot to detect phospho c-Jun (ser 63) levels or used to evaluate the relative DNA binding activity versus an AP-1 binding site in (e) where c-Jun/c-Fos DNA binding was tested by TransAM assay. For these experiments, keratinocytes were treated for 15 minutes (panel d) or 30minutes (panel e) with TNF-α (50 ng/ml) and/or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). (f) GM-CSF mRNA levels were quantified by real-time RT-PCR with and without c-Jun silencing in human keratinocytes stimulated for 4 hours with TNF-α (50 ng/ml) and/or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). (g) Total levels of c-Jun and EGFR were detected via immunoblot as control for silencing experiment. Values in (a-c) and (f) are expressed as the mean of experimental duplicates± SD. *P<0.01. Panels d and g show one representative experiment. All experiments were repeated at least three times.
Endogenous GM-CSF protein release examined by ELISA in the supernatant of transfected keratinocytes closely paralleled GM-CSF promoter activity, confirming that the control of GM-CSF expression by EGFR signaling and downstream kinases was at the transcriptional level (Figure 3b). The dominant negative form of AP-1, A-FOS, is able to sequester AP-1 monomers and prevents their binding to DNA thereby suppressing AP-1 transcriptional activity (Olive et al., 1997). Human keratinocytes were stimulated for 24 hours with TNF-α in the presence (TGF-α) or absence (PD16) of EGFR activation and transduced with an adenovirus expressing A-FOS. A dose-dependent inhibition of GM-CSF release into culture supernatants was observed in all the conditions tested when AP-1 activity was reduced (Figure 3c). The same culture conditions were used to study proximal events upstream of GM-CSF release. To this aim lysates isolated from human keratinocytes stimulated for 15 and 30 minutes were analyzed by western blot and with an ELISA-based DNA binding assay. In this assay higher optical density (OD) values represent higher levels of a specific transcription factor bound to its cognate DNA sequence. Western blot analysis of human keratinocytes treated for 15 minutes showed that TNF-α induced an increase in phosphorylation levels of c-Jun in serine 63 (Figure 3d). EGFR activation by TGF-α or inhibition by PD16 was able to respectively potentiate or decrease TNF-α induced serine 63 phosphorylation of c-Jun. This residue has an important role in the transactivating function of AP-1 transcription factor (Smeal et al., 1991). In the same conditions, we observed that while c-Fos DNA binding was only slightly affected, c-Jun binding to AP-1 consensus was maximal when TNF-α and TGF-α were co-administered (Figure 3e). In addition, human keratinocytes were transfected with a siRNA targeting c-Jun or a control siRNA sequence, and cell lysates and RNAs were assayed for total c-Jun levels and GM-CSF mRNA expression (Figure 3f and g). In presence of lower levels of c-Jun protein, TNF-α and TGF-α failed to fully induce GM-CSF mRNA. c-Jun is an important upstream modulator of EGFR transcription and when its expression is completely abrogated in mice, EGFR levels are strongly reduced (Zenz et al., 2003). In our silencing condition, in human keratinocytes, in which c-Jun levels are reduced of about 70% of the original amount, EGFR transcription and expression still occurs at a normal rate. Overall, these data suggested that TNF-α induced GM-CSF transcription depended on EGFR and c-Jun activity.
Genetic ablation of EGFR prevents full induction of GM-CSF, and maximal phosphorylation of ERK 1/2, JNK 1/2 and c-Jun in vitro and in vivo
To determine whether genetic ablation of EGFR impacted GM-CSF regulation, neonatal mouse keratinocytes were isolated from EGFR null mice (Threadgill et al., 1995) and stimulated with TNF-α in the presence (TGF-α) or absence (PD16) of EGFR activation (Figure 4a). After 1 hour stimulation of wild type cells, TGF-α or TNF-α elicited a small increase of GM-CSF mRNA expression. As seen for human keratinocytes, the combination of TGF-α and TNF-α gave a synergistic response, while the pharmacologic inhibition of EGFR reduced TNF-α induced GM-CSF levels. A similar response was detected at the protein level after 24 hours stimulation (Figure 4b). Interestingly, keratinocytes isolated from EGFR null pups showed a decreased capacity to fully upregulate GM-CSF expression after TNF-α stimulation (Figure 4a and Figure 4b). In parallel, if compared to keratinocytes from wild type littermates, ERK1/2, JNK1/2 and c-Jun phosphorylation levels were reduced in keratinocytes of EGFR null mice (Figure 4c lanes 9 versus 3 and lanes 11 versus 5). Figure 4d shows the densitometric analysis of the phosphorylated form of c-Jun normalized by the total c-Jun band. In wild-type keratinocytes the phosphorylation of c-Jun increased over the basal level at 10 minutes and returned to baseline level after 1 hour of stimulation by TNF-α. At all timepoints, EGFR ablated keratinocytes showed lower levels of the phosphorylated form of c-Jun. Keratinocytes from EGFR null mice displayed low levels of an alternatively spliced variant detected with the EGFR antibody at 140-kDa, as previously described (Dlugosz et al., 1997). This alternatively spliced form could account for the faint induction of ERK1/2 and JNK 1/2 after TGF-α stimulation and the small reduction of ERK1/2 and JNK 1/2 phosphorylation in the presence of the EGFR inhibitor PD16.
Figure 4. Genetic ablation of EGFR impairs TNF-α-induced GM-CSF expression and ERK1/2, JNK1/2, and c-Jun phosphorylation in murine keratinocytes.
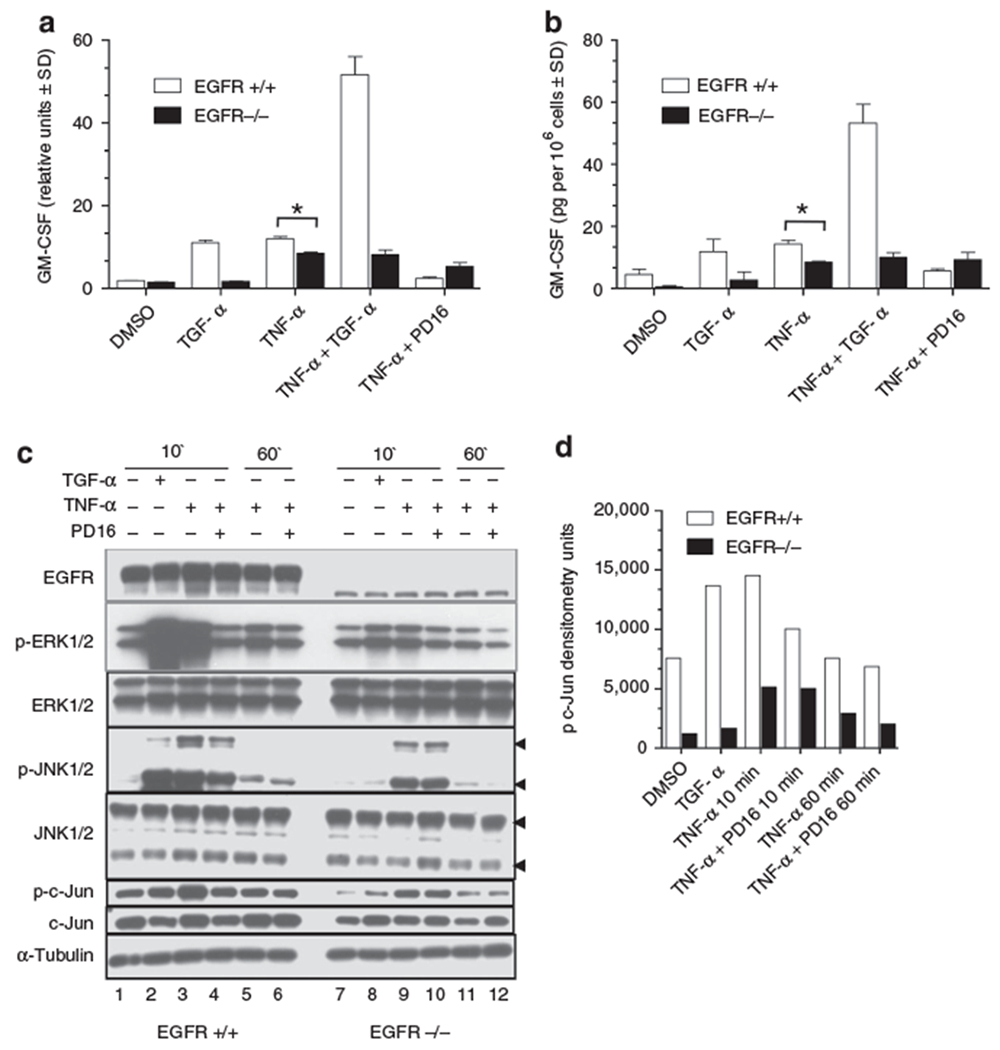
(a) real-time RT-PCR for murine GM-CSF on wild-type and EGFR null keratinocytes. Cells were stimulated for 1 hour with TNF-α (50 ng/ml) and/or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). (b) ELISA of cell supernatant after 24 hours stimulation as panel a. (c) Keratinocytes from wild-type (lanes 1–6) and EGFR null mice (lanes 7–12) were stimulated for 10 minutes and 1 hour with TNF-α (50 ng/ml) or TGF-α (50 ng/ml), directly or following 30 minutes incubation with PD168393 (1 μm). Twenty μg of cell lysates were subjected to immunoblotting. (d) Densitometric analysis of phospho c-Jun bands normalized over total c-Jun bands of immunoblot from panel c. Values in (a) and (b) are expressed as the mean of experimental duplicates ± SD. *P<0.01. Experiments were repeated at least three times with one representative experiment shown in the figure.
To confirm our in vitro observations in vivo, we examined the level of activation of ERK1/2, JNK1/2, c-Jun and the levels of GM-CSF in skin tissue lysates and tissue sections. To this aim, we exploited a second mouse model with a skin targeted deletion of EGFR. These mice were created by crossing Keratin 5 promoter-driven Cre recombinase transgenics (Ramirez et al., 2004) with mice containing loxP sites flanking exon 3 of the EGFR gene (Lee and Threadgill, 2009). Double transgenic deleted mice, Tg(K5Cre)/Egfr(f/f) did not display a specific defect at birth but, at the end of the first week of life, showed a strong skin phenotype that resembled that of the EGFR null mice (Threadgill et al., 1995). In contrast to EGFR null mice, skin targeted mutants survive for months and maintain a skin inflammatory phenotype that is currently under investigation.
TPA painting of mouse skin induces the upregulation of GM-CSF expression in total skin extracts (Koury et al., 1983). We treated the back skin of double Tg(K5Cre)/Egfr(f/+): wild type for EGFR and Tg(K5Cre)/Egfr(f/f): EGFR deleted) transgenic mice with 10 μg TPA for 2 hours and 7 hours and harvested the whole skin for tissue lysates and histology respectively. Immunohistochemical staining for total leukocytes (blue staining = CD45 positive cells) showed a highly abundant infiltrate in EGFR ablated skin while sparse cells were detected in untreated skin of littermate controls. TPA treatment increased the number of CD45 infiltrating cells in EGFR expressing skin but not in the highly infiltrated skin of EGFR ablated mice. TPA administration caused an increase in GM-CSF levels in EGFR competent double transgenics both in the dermal and in the epidermal compartment while it did not affect the basal level in the epidermis of EGFR ablated double transgenics. In spite of the highly abundant leukocyte presence, EGFR ablated mice did not express higher levels of GM-CSF in the epidermis than wild type controls. Thus epidermal GM-CSF expression was not associated with the degree of leukocyte infiltration in EGFR ablated skin. Cultured keratinocytes from EGFR null skin were less responsive than wild type cells to TPA stimulation in terms of GM-CSF mRNA expression (Figure 5b). These data showed that keratinocytes lacking EGFR failed to fully upregulate GM-CSF expression both in vitro and in vivo during an inflammatory event. The profile of phosphorylation of ERK1/2, JNK1/2 and c-Jun in skin samples mirrored the levels that we observed in isolated keratinocytes lysates. The skin of EGFR ablated mice showed overall lower levels of phospho-ERK1/2, phospho-JNK1/2 and lower total and phospho-c-Jun levels when compared to correspondent lysates from EGFR competent skin (Figure 5c).
Figure 5. Skin-targeted deletion of EGFR impairs TPA-induced epithelial GM-CSF expression in vivo.
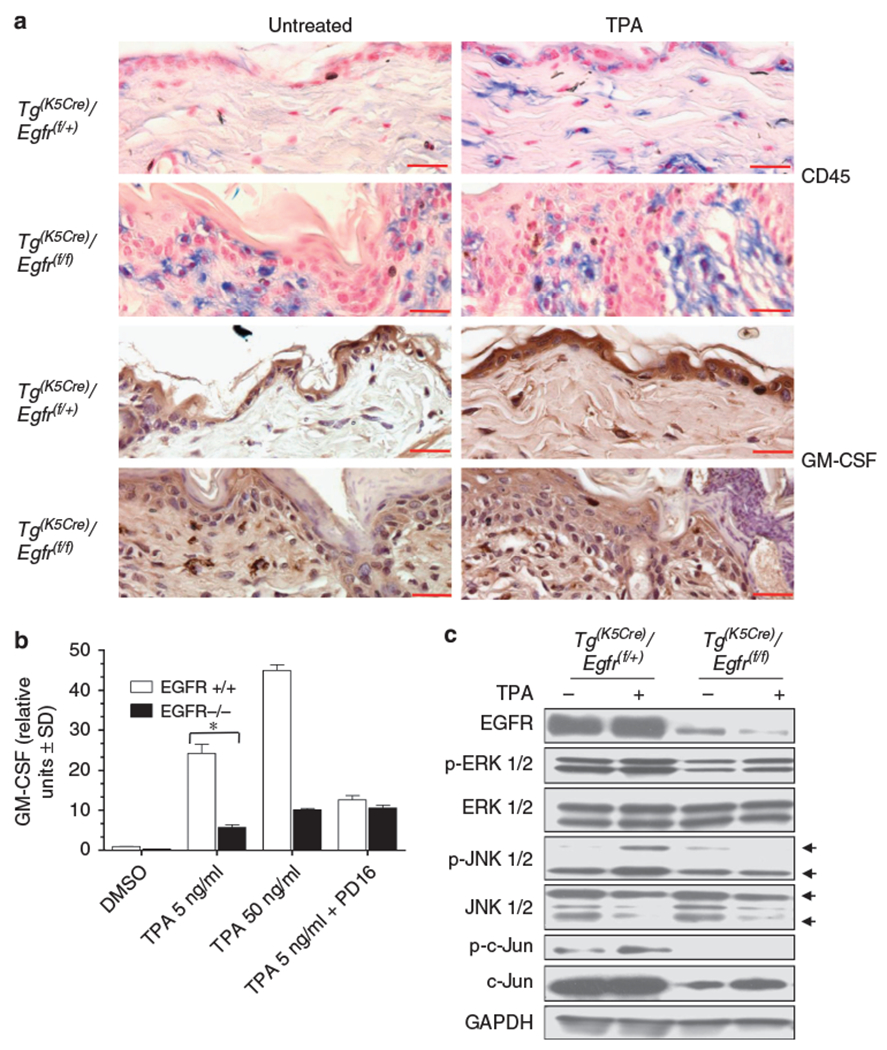
(a) Immunohistochemical detection of CD45 in skin sections from double transgenic wild-type (Tg(K5Cre)/Egft(f/+)) and double transgenic EGFR deleted (Tg(K5Cre)/Egfr(f/f)) mice, untreated and treated with 10 μg of TPA for 7 hours. Immunohistochemical detection of GM-CSF in skin sections from double transgenic wild-type and double transgenic EGFR deleted mice, untreated and treated with 10 μg of TPA for 7 hours. Normalization bar = 50 μm. (b) real-time RT-PCR for murine GM-CSF on wild-type and EGFR null keratinocytes. Cells were stimulated for 1 hour with 5 and 50 ng/ml of TPA directly or following 30 minutes incubation with PD168393 1 μm. (c) Double transgenic wild-type (Tg(K5Cre)/Egft(f/+)) and double transgenic EGFR deleted (Tg(K5Cre)/Egfr(f/f)) mice were treated for 2 hours with vehicle alone or 10 μg of TPA and 50 μg of tissue lysates were immunoblotted. Experiments were repeated at least three times with one representative experiment showed in the figure. Values in (b) are expressed as the mean of experimental duplicates ± SD. *P<0.01.
Epidermal GM-CSF levels in cetuximab-associated skin rash are highly variable and correlate with ERK1/2 and c-Jun phosphorylation levels but not the inflammatory infiltrate
A papulo-pustular rash is a common adverse effect of several anti EGFR drugs. Noteworthy, the degree of severity and intensity of the eruption is variable within patients, and it is reversible when the treatment is withdrawn. By immunohistochemistry we detected GM-CSF, phospho-ERK1/2, phospho-c-Jun and CD45 expression in human skin samples obtained from inflamed skin of patients undergoing chemotherapy with cetuximab and displaying grade II lesions. Serial sections of the skin of 3 representative patients (#1, #2, #3) are shown in Figure 6 compared with normal control skin. Even though all the patients showed a high levels of CD45 staining they displayed variable expression levels of epidermal GM-CSF. Thus, we could not correlate epidermal GM-CSF expression with the abundance of the local inflammatory infiltrate. The variable degree of GM-CSF expression in different patients was associated with the phosphorylation levels of ERK1/2 and c-Jun.
Figure 6. Epidermal GM-CSF expression levels in the inflamed skin of cetuximab treated patients are associated with phosphorylation levels of ERK1/2 and c-Jun and not with CD45 staining.
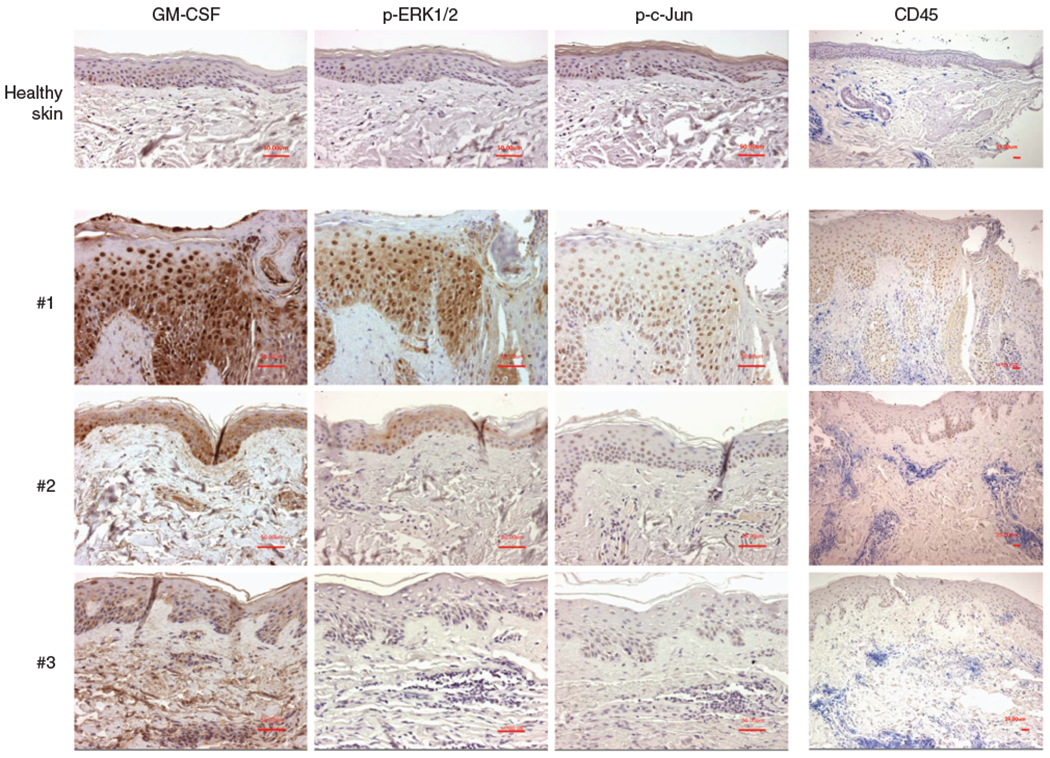
Skin sections of three representative patients and one control are shown. Patients were affected by cetuximab-induced skin rash (grade II). GM-CSF, phospho ERK1/2, phospho c-Jun, and CD45 immunohistochemistry were performed on serial sections from the same patient. Normalization bar = 50 μm for × 200 magnification of GM-CSF, phospho ERK1/2, and phospho c-Jun staining. Normalization bar = 25 μm for × 100 magnification of CD45 staining.
DISCUSSION
In the current study we broaden the concept of EGFR as a skin immunomodulator by adding GM-CSF to the pool of inflammatory mediators that are regulated by EGFR activity. The EGFR ligand TGF-α, potentiates keratinocyte response to the pro-inflammatory cytokines TNF-α and IFN-γ with an enhanced expression of GM-CSF. This synergy is likely active in the lesional skin of chronic inflammatory diseases, and helps to explain the intense GM-CSF staining that we found in the skin lesions from patients with atopic dermatitis (Pastore et al., 1997), but also in allergic contact dermatitis and psoriasis. Strong TGF-α expression characterizes the whole epidermis of lesional skin from patients with these disorders (Mascia et al., 2003), whereas TNF-α and IFN-γ are locally released by a variety of infiltrating leukocyte populations, which include dendritic cells, monocytes, mast cells and T cells. In turn, these cytokines not only induce the release of the mature form of TGF-α from its membrane precursor (Mascia et al., 2003), but they also effectively stimulate its de novo expression by keratinocytes (Valyi-Nagy et al., 1992; Matsuura et al., 1999). The enhancement of cytokine-induced GM-CSF expression by TGF-α closely resembled what we previously described for CXCL8 in cultured keratinocytes (Mascia et al., 2003), and suggests that both GM-CSF and CXCL8 are part of a TGF-α-driven amplification circuit during keratinocyte response to skin inflammation.
GM-CSF is considered a pro-inflammatory cytokine, since it contributes to the establishment of a favorable milieu for the recruitment, functional differentiation and survival of a variety of leukocyte cell populations, including monocytes and dendritic cells (Hamilton, 2008). High expression of GM-CSF in the lesional skin of atopic patients has been implicated in the facilitated recruitment and maturation of dendritic cell progenitors in this disease (Hamid et al., 1994;Pastore et al., 1997). In addition, we reported that keratinocytes cultured from nonlesional skin of atopic dermatitis patients release abnormal levels of GM-CSF in response to proinflammatory stimuli, and reasonably contribute to GM-CSF accumulation in atopic lesions in vivo (Pastore et al., 1997, 2000). Analogously, the direct correlation between GM-CSF and TNF-α levels in the suction blister fluids from psoriatic lesions suggested the contribution of GM-CSF in the pathogenesis of this disease (Bonifati et al., 1994). However, a direct evidence for a pro-inflammatory effect associated with high levels of GM-CSF in the epidermis has not been provided. Transgenic mice overexpressing GM-CSF in the basal layer of the epidermis display accelerated re-epithelialization of fullthickness skin wounds, characterized by higher numbers of proliferating keratinocytes at the wound edges and increased formation of granulation tissue (Mann et al., 2001). A selective accumulation of Langerhans cells and mast cells could be observed in the epidermis, without evident signs of active inflammation (Breuhahn et al., 2000). When injected intradermally into the human skin, GM-CSF induces epidermal thickening, with accumulation of dermal CD1+, Birbeck granule-positive Langerhans cells, but no evidence of T cell, monocyte or granulocyte recruitment or changes in cell-mediated immune responses (Kaplan et al., 1992; Braunstein et al., 1994). Finally, intradermal injection of recombinant human GM-CSF around chronic venous ulcers can be successfully employed to accelerate wound closure (Stagno et al., 1999; Cianfarani et al., 2006). We speculate that during an inflammatory response in the skin, as already suggested in inflammatory conditions of the lungs and colon (Abdul Rhaman et al., 2004; Korzenik et al., 2005), GM-CSF may not necessarily contribute to the aggravation of the flogistic response but its enhanced expression could participate to the clearance of the inflammatory condition through a promotion of the wound healing response. In this study we demonstrated that functional EGFR signaling was involved in TNF-α-induced GM-CSF expression in human and murine keratinocytes. The genetic deletion of the EGFR pathway is associated with impaired wound healing response (Repertinger et al., 2004). Our data showed a strong association between EGFR activity and GM-CSF expression and it is possible that the delayed wound healing response observed in EGFR null skin could depend on the insufficient expression of this cytokine in the epidermis.
We demonstrated that the abrogation of EGFR signaling by specific EGFR tyrosine kinase inhibition caused a marked decrease of GM-CSF transcription associated with suppression of ERK1/2 activity. In contrast, the blockade of Her-2 did not affect GM-CSF expression. Previous papers have already established that functional ERK1/2 activity plays a relevant role in GM-CSF gene expression in human epithelial cells, including keratinocytes (Reibman et al., 2000; Kanda and Watanabe, 2004; Matsubara et al., 2004).
With our study we identified EGFR and c-Jun as important regulators of TNF-α induced GM-CSF promoter activity. In particular we showed that c-Jun phosphorylation and c-Jun DNA binding activity is maximally activated by combined treatment with TGF-α and TNF-α. In parallel, inhibitors of EGFR or genetic abrogation of this pathway prevented the full induction of c-Jun phosphorylation both in vitro and in vivo. Due to the lack of higher number of samples we could not do an extensive study to correlate GM-CSF levels, ERK1/2 and c-Jun phosphorylation to the extent of the skin rash, the amount or the quality of skin infiltrating cells or the tumor response to the drug. In the small subset of patients on anti-EGFR therapy that we analyzed, the amount of skin inflammatory infiltrate was not directly related to the levels of GM-CSF expressed in the epidermis. Nevertheless in all patients studied, GM-CSF levels in the epidermis were consistently associated with ERK1/2 and c-Jun phosphorylation levels, confirming the relevance for the human system of data obtained in vitro and in the mouse models. We speculate that GM-CSF might not have a pathogenetic role in the lesions formed after treatment with cetuximab. Further studies are needed to confirm if epidermal GM-CSF, can be used as a surrogate marker for drug efficacy evaluation.
GM-CSF stimulates proliferation and migration of transformed epithelial cells suggests a possible role of this factor in tumor progression and invasion (Mueller and Fusenig, 1999; Gutschalk et al., 2006). Since GM-CSF expression is among the transcription-controlled events downstream of the EGFR/ERK1/2 pathway in epithelial cells, it is possible that suppression of GM-CSF may play a relevant role in the anti-cancer mechanisms triggered by the EGFR inhibitors presently employed for the therapy of EGFR-dependent epithelial cancers. These hypotheses are currently under investigation. Previous work associated the expression levels of an epithelial cell derived chemokine with tumor progression (Pivarcsi et al., 2007). These authors highlighted the role of EGFR in CCL27 expression both in vitro in Ras transformed cells, in skin with epithelial tumors and in lesions from Erlotinib treated patients. Altogether, these and our independent observations underline the need of further study on the role of EGFR and EGFR blockade into the creation of a selective environment that shapes the immune system response within the skin and possibly within the tumor.
MATERIALS AND METHODS
Subjects
Four-mm punch biopsies were taken from lesional skin of adult patients with chronic plaque psoriasis (n = 5; two females and three males; age 30–48), chronic allergic contact dermatitis (n = 3; two females and one male; age 30–40), cetuximab-associated skin rash grade II, according to Melosky et al., 2009 (n = 9, three females and six males, age 50–66) and normal skin of healthy subjects (n = 7; two females and two males; age 24–59). Epidermal sheets for keratinocyte cultures were obtained from healthy individuals undergoing plastic surgery (mammoplasty or abdominoplasty) (n = 4, two females and two males; age 25–40). None of the patients had received oral corticosteroids within 1 month of skin explant, and topical corticosteroids were not allowed for a period of at least 2 weeks prior to enrolment. All subjects gave written informed consent prior to enrolment. These studies were conducted according to the Declaration of Helsinki Guidelines and were approved by the institutional review boards of the Istituto Dermopatico dell’Immacolata (healthy donors and patients affected by psoriasis and allergic contact dermatitis) and the Universita’ di Chieti and Pescara (patients treated with anti EGFR therapy).
Cell cultures
Primary cultures of normal human keratinocytes were obtained as previously described (Pastore et al., 1997), and routinely grown in serum-free Epilife Medium from Invitrogen (Carlsbad, CA), prepared from an essential nutrient solution supplemented with HKGS growth supplement kit that contains: 0.2 ng/ml EGF, 0.18 μg/ml hydrocortisone, 5 μg/ml bovine transferrin, 0.2% bovine pituitary extract, 5 μg/ml bovine insulin and antibiotic solution of Gentamicin/Amphotericin. In the 24 hours preceding the experiments, 80% confluent cell cultures were switched to EGF and hydrocortisone-depleted medium. All assays were performed on human keratinocytes obtained from at least three distinct healthy donors. Newborn mouse keratinocytes were cultured as described in Lichti et al., 2008.
Mice and treatment
Mouse studies were performed under a protocol approved by the National Cancer Institute and the NIH Animal Care and Use Committee. Homozygous or heterozygous EGFR null mice and wild-type siblings on a CD-1 background were developed using homologous recombination in 129/Sv-derived D3 embryonic stem cells. Mice were genotyped as described elsewhere. (Threadgill et al., 1995). The construction and characterization of K5Cre mice were previously described (Ramirez et al., 2004). K5Cre mice express Cre Recombinase under the promoter of bovine Keratin 5 that targets the transgene to the basal layer of the epidermis and the outer root sheath of the hair follicle. EGFR floxed mice containing loxP sites flanking exon 3 of the EGFR gene (Lee and Threadgill, 2009) were crossed with K5Cre mice to obtain double transgenic mice with EGFR deletion in the epidermis (Tg(K5Cre)/Egfr(f/f)). The skin of the above mentioned mice was compared with double transgenic littermates that were heterozygous for the EGFR floxed allele (Tg(K5Cre)/Egfr(f/+)). In vivo experiments were performed by painting the back skin of shaved mice with 10 μg of TPA from Axxora (San Diego CA).
Cytokines and chemicals
Recombinant human TGF-α, Amphiregulin, EGF, HB-EGF, TNF-α and IFN-γ were purchased from R&D Systems (Minneapolis, MN). The small molecule, cell-permeant protein kinase inhibitors PD168393 and AG1478 (EGFR inhibitor), PD98059 (ERK1/2 inhibitor), and SP600125 (JNK1/2 inhibitor) were from Calbiochem (La Jolla, CA). Herceptin was purchased from Genentech Inc., (San Francisco, CA). Human IgG was purchased from Sigma (St Louis, MI).
RNA isolation, RNAse protection assay and real-time RT-PCR
Total RNA was extracted from keratinocytes using the TRIzol Reagent, according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). The templates of human GM-CSF and the housekeeping molecule L32, and the kit for RNase protection assay were purchased from BD Biosciences (San Diego, CA). Total RNA (10 μg) was hybridized overnight with α-32ATP-labeled cDNA templates, and reactions were performed per the manufacturer’s instructions.
For cDNA synthesis, 1 μg of total RNA was reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen). PCR were performed in a volume of 20 μl using iQ SYBR Green Supermix from BIORAD (Hercules, CA) and 1:100 dilution of cDNA. Primers used for this analysis were as follows: human GAPDH from Gene Link (Hawthorne NY), human GM-CSF 5′: GAAGTCATCTCAGAAATGTTTGACCTC, 3′: GGTCTGTAGGCAGGTCGGC, mouse GM-CSF 5′: GGAGGATGTGGCTGCAGAAT, 3′: GGCTGTAGACCACAATGCCC and mouse GAPDH 5′: CATCGCCTTCCGTGTTCCTA, 3′: GCGGCACGTCAGAATCCA. Real Time analysis was performed using a Bio-Rad iCycler iQ and Gene Expression Macro (version 1.1). Results are presented as relative quantity ±SD from at least three independent experiments.
Cell lysates and western blot
Total cell lysates were prepared from human and murine keratinocytes cultures with cold RIPA buffer freshly additioned with antiprotease cocktail from Roche (Mannheim, Germany), 1 mm sodium orthovanadate, 1 mm sodium fluoride. Cell lysates were vortexed and incubated on ice for 10 minutes and then centrifuged at 14,000 rpm for 15 minutes. Supernatants were quantified with Bradford method and used for western blot or TransAM assays (Active Motif, Carlsbad, CA). Total lysates from skin samples were treated as follows: skin was excised and snap frozen in liquid nitrogen and cold processed at 2,000 rpm for 2 minutes in a Mikro-Dismembrator S from Sartorius (Goettingen, Germany). Powderized skin was then lysed with cold RIPA buffer as previously described. Twenty mg of protein lysates were run on poliacrylamide gels under denaturing conditions and then transferred on PVDF membranes for protein detection. All primary and HRP secondary antibodies were from Cell Signaling (Boston, MA).
Enzyme-Linked Immunosorbent Assay (ELISA) and TransAM assay
Quantitation of GM-CSF was performed on cell-free supernatants using ELISA kits from R&D Systems (Abingdon, United Kingdom). Data are expressed as nanograms or picograms per 106 cells ± SD from three independent experiments. TransAM is an ELISA based assay kit to detect and quantify transcription factor activation. Twenty mg of total cell lysates from human keratinocytes were used for each condition on the AP-1 family kit (44296) from Active Motif (Carlsbad, CA). Results were read on a spectrophotometer and data are expressed as O.D. optical density ± SD. Experiments were performed in duplicates with three independent repeats.
Plasmid construct and keratinocyte transfection
The chloranphenicol-acetyltransferase (CAT) reporter construct carrying the sequence of the minimal GM-CSF promoter, from 91 nucleotides upstream to 23 nucleotides downstream of the GM-CSF cap site (p91 GM-CSF CAT), has been previously described (Kaushansky, 1989). For transfection experiments, we followed the procedure previously described (Pastore et al., 2000). CAT activity in cell lysates was measured using CAT ELISA kit from Roche (Mannheim, Germany). Results are plotted p91CAT activity ± SD from duplicates values of three independent experiments.
Adenovirus infection and siRNA transfection
A dominant negative AP-1 vector, A-FOS, or empty vector control, Null, were introduced into human keratinocytes using an adenoviral construct driven by a cytomegalovirus promoter (Olive et al., 1997). Sixty percent confluent keratinocytes were infected with increasing amounts of A-FOS adenovirus in 0.3 ml/6 well plates in order to enhance viral uptake. Ten to forty MOI (multiplicity of infection from 10 viral particles/cell to 40 viral particles/cell) were used in this experiment. After 90 minutes. of infection, additional 2 ml of starvation medium were added to the cells. After 24 hours cells were stimulated for additional 24 hours before collecting supernatants. Human keratinocytes were treated with c-Jun siRNA (Cat. No. SI00300580) or non targeting siRNA (Cat No. D-001210–02–20) from Qiagen (Valencia, CA) and Dharmacon (Lafayette, CO) respectively. Cells were transfected with 100 nm siRNA and RNAiFect transfection reagent from Qiagen according to manufacturer’s instruction. Effective silencing was verified by western blot.
Immunohistochemistry
Paraffin embedded sections of biopsies fixed with 4% paraformaldehyde, were incubated with citrate buffer pH 6 and microwaved for 3 minutes at high power. Slides were allowed to cool down for 20 minutes. at room temperature, treated with 3% hydrogen peroxide, and then incubated for 1 hour with 10% goat serum. After overnight incubation at 4 °C with the appropriate primary dilution of each antibody, slides were treated for 1 hour at room temperature with the corresponding secondary antibody. For the detection of GM-CSF, phospho ERK1/2, c-Jun and CD45 in human tissues, we employed: sc-13101 (1:10) from Santa Cruz Biotech (Santa Cruz, CA), 9101 and 2361 (1:50) from Cell Signaling (Boston MA) clone 2B11 + PD7/26 from DAKO (Glostrup, Denmark); for the detection of GM-CSF in mouse tissue we used AF-415-NA (1:15) from R&D Systems (Abingdon, United Kingdom)and for mouse CD45 we used (1:100) CD45-AF 114 from R&D Systems (Minneapolis, MN). Secondary biotinylated antibodies and staining kits were from Vector Laboratories (Burlingame Laboratories, CA). Immunoreactivity was revealed using avidin-biotin-peroxidase system and AEC (Figure 1) or DAB (Figure 5 and Figure 6) as chromogen or alkaline phosphatase system and AP-blue as substrate, all from Vector Laboratories (Burlingame Laboratories, CA). CD45 staining (AP-blue, blue staining) for human tissue was performed as a second staining after phospho c-Jun staining (DAB, brown staining). Sections were counterstained with Mayer’s hematoxylin or Vector Red.
Statistical analysis
The Wilcoxon signed-rank test (GraphPad Prism software, La Jolla, CA) was applied to compare differences between groups of data. Significance was assumed at a P-value of 0.05 or less.
Supplementary Material
ACKNOWLEDGMENTS
The authors thanks Marta Custer for her technical assistance with the mouse colony and the summer students Susan Shen and Rachel Packer for their help in genotyping the mice. The authors also acknowledge Professor Michele De Tursi (Oncology Department of University of Chieti and Pescara) for providing skin samples from cetuximab-treated patients. This work was supported by grants from the Ministero della Salute, Ministero dell’Istruzione, dell’Università e della Ricerca (Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale (PRIN), and Fondo 60%) and from the Intramural Research Program of the Center for Cancer Research, NCI, NIH.
Abbreviations
- ACD
allergic contact dermatitis
- AD
atopic dermatitis
- AP-1
activator protein 1
- AR
amphiregulin
- CAT
chloranphenicol-acetyltransferase
- CCL
(CC ligand), chemokine of the CC subfamily
- CCR
receptor for CCL
- CXCL
(CXC ligand), chemokine of the CXC subfamily
- CXCR
receptor for CXCL
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- GM-CSF
granulocyte/macrophage-colony stimulating factor
- HB-EGF
heparin-binding EGF-like protein
- HER
Herceptin
- IFN-γ
interferon-gamma
- JNK
Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MOI
multiplicity of infection
- NFκB
nuclear factor kappa B
- RANTES
regulated upon activation normal T cell expressed and secreted
- RPA
RNAse protection assay
- TGF-α
transforming growth factor-alpha
- TNF-α
tumor necrosis factor-alpha
Footnotes
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
CONFLICT OF INTEREST
The author states no conflict of interest.
REFERENCES
- Abdul Rhaman JA, Moodley YP, Phillips MJ (2004) Pulmonary alveolar proteinosis associated with psoriasis and complicated by mycobacterial infection: successful treatment with granulocyte-macrophage colony stimulating factor after a partial response to whole lavage. Respirology 9:419–22 [DOI] [PubMed] [Google Scholar]
- Bonifati C, Carducci M, Cordiali Fei P, Trento E, Sacerdoti G, Fazio M et al. (1994) Correlated increases of TNF-α, IL-6 and GM-CSF levels in suction blister fluids and sera of psoriatic patients-relation with disease severity. Clin Exp Dermatol 19:383–7 [DOI] [PubMed] [Google Scholar]
- Braunstein S, Kaplan G, Gottlieb AB, Schwartz M, Walsh G, Abalos RM et al. (1994) GM-CSF activates regenerative epidermal growth and stimulates proliferation in human skin in vivo. J Invest Dermatol 103:601–4 [DOI] [PubMed] [Google Scholar]
- Breuhahn K, Mann A, Müller G, Wilhelmi A, Schirmacher P, Enk A et al. (2000) Epidermal overexpression of granulocyte-macrophage colony-stimulating factor induces keratinocyte proliferation and apoptosis. Cell Growth Differ 11:111–21 [PubMed] [Google Scholar]
- Cianfarani F, Tommasi R, Failla CM, Viviano MT, Annessi G, Papi M et al. (2006) Granulocyte/macrophage colony-stimulating factor treatment of human chronic ulcers promotes angiogenesis associated with de novo vascular endothelial growth factor transcription in the ulcer bed. Br J Dermatol 154:34–41 [DOI] [PubMed] [Google Scholar]
- Dlugosz AA, Hansen L, Cheng C, Alexander N, Denning MF, Threadgill DW et al. (1997) Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-RasHA oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res 57:3180–8 [PubMed] [Google Scholar]
- Florin L, Knebel G, Zigrino P, Wonderstrass B, Mauch C, Shorpp-Kistner M et al. (2006) Delayed wound healing and epidermal hyperproliferation in mice lacking Jun B in the skin. J Invest Dermatol 126:902–11 [DOI] [PubMed] [Google Scholar]
- Gutschalk CM, Herold-Mende CC, Fusenig NE, Mueller MM (2006) Granulocyte colony-stimulating factor promote malignant growth of cells from head and neck squamous cell carcinomas in vivo. Cancer Res 66:8026–36 [DOI] [PubMed] [Google Scholar]
- Hamid Q, Boguniewicz M, Leung DY (1994) Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest 94:870–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA (2008) Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 7:533–44 [DOI] [PubMed] [Google Scholar]
- Hudis CA (2007) Trastuzumab mechanism of action and use in clinic. NEJM 357:39–51 [DOI] [PubMed] [Google Scholar]
- Jost M, Kari C, Rodeck U (2000) The EGF receptor -an essential regulator of multiple epidermal functions. Eur J Dermatol 10:505–10 [PubMed] [Google Scholar]
- Kanda N, Watanabe S (2004) Histamine enhances the production of granulocyte-macrophage colony-stimulating factor via proten kinase C-α and extracellular signal-regulated kinase in human keratinocytes. J Invest Dermatol 122:863–72 [DOI] [PubMed] [Google Scholar]
- Kaplan G, Walsh G, Guido LS, Meyn P, Burkhardt RA, Abalos RM et al. (1992) Novel responses of human skin to intradermal recombinant granulocyte/macrophage colony-stimulating factor: langerhans cell recruitment, keratinocyte growth, and enhanced wound healing. J Exp Med 175:1717–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushansky K (1989) Control of GM-CSF production in normal endothelial cells by positive and negative regulatory elements. J Immunol 143:2525–9 [PubMed] [Google Scholar]
- Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ (2005) Sargramostim for active Chrohn’s disease. N Engl J Med 352: 2193–201 [DOI] [PubMed] [Google Scholar]
- Koury MJ, Balmain A, Pragnell IB (1983) Induction of granulocyte-macrophage colony-stimulating activity in mouse skin by proinflammatory agents and tumor promoters. EMBO J 2:1877–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TC, Threadgill DW (2009) Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47:85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichti U, Anders J, Yuspa SH (2008) Isolation and short term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc 3:799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann A, Breuhahn K, Schirmacher P, Blessing M (2001) Keratinocyte-derived GM-CSF accelerates wound healing: stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol 117:1382–90 [DOI] [PubMed] [Google Scholar]
- Mascia F, Mariani V, Girolomoni G, Pastore S (2003) Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 163:303–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara M, Harada D, Manabe H, Hasegawa K (2004) Staphylococcus aureus peptidoglycan stimulates granulocyte macrophage colony-stimulating factor production from human epidermal keratinocytes via mitogen-activated protein kinases. FEBS Lett 566:195–200 [DOI] [PubMed] [Google Scholar]
- Matsuura H, Sakaue M, Subbaramaiah K, Kamitani H, Eling TE, Dannenberg AJ et al. (1999) Regulation of cyclooxygenase-2 by interferon-γ and transforming growth factor α in normal human epidermal keratinocytes and squamous carcinoma cells. J Biol Chem 274:29138–48 [DOI] [PubMed] [Google Scholar]
- Melosky B, Burkes R, Rayson D, Alcinor T, Shear N, Lacouture M (2009) Management of skin rash during EGFR-targeted monoclonal antibody treatment for gastrointestinal malignancies: canadian recommendations. Curr Oncol 16:16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LS, Sørensen OE, Liu PT, Jalian HR, Eshttiaghpour D, Behmanesh BE et al. (2005) TGF-α regulates TLR expression and function on epidermal keratinocytes. J Immunol 174:6137–43 [DOI] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE (1999) Constitutive expression of G-CSF and GM-CSF in human skin carcinoma cells with functional consequence for tumor progression. Int J Cancer 83:780–9 [DOI] [PubMed] [Google Scholar]
- Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C (1997) A dominant negative to activation protein-1 (AP-1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem 272:18586–94 [DOI] [PubMed] [Google Scholar]
- Pastore S, Fanales-Belasio E, Albanesi C, Chinni LM, Giannetti A, Girolomoni G (1997) Granulocyte macrophage-colony stimulating factor is overproduced by keratinocytes in atopic dermatitis. Implications for sustained dendritic cell activation in the skin. J Clin Invest 99: 3009–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore S, Giustizieri ML, Mascia F, Giannetti A, Kaushansky K, Girolomoni G (2000) Dysregulated activator of activating protein 1 in keratinocytes of atopic dermatitis patients with enhanced expression of granulocyte/macrophage-colony stimulating factor. J Invest Dermatol 115:1134–43 [DOI] [PubMed] [Google Scholar]
- Pastore S, Mascia F, Mariotti F, Dattilo C, Mariani V, Girolomoni G (2005) ERK1/2 regulates epidermal chemokine expression and skin inflammation. J Immunol 174:5047–56 [DOI] [PubMed] [Google Scholar]
- Pérez-Soler R, Delord JP, Halpern A, Kelly K, Krueger J, Massutì Surreda B et al. (2005) HER1/EGFR inhibitor-associated rash: future directions form management and investigation outcomes from the HER1/EGFR inhibitor Rash Management Forum. The Oncologist 10:345–56 [DOI] [PubMed] [Google Scholar]
- Pivarcsi A, Muller A, Hippe A, Rieker J, van Lierop A, Steinhoff M et al. (2007) Tumor immune escape by the loss of homeostatic chemokine expression. Proc Natl Acad Sci 104:19055–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M et al. (2004) A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 39:52–7 [DOI] [PubMed] [Google Scholar]
- Reibman J, Talbot AT, Hsu Y, Ou G, Jover J, Nilsen D (2000) Regulation of expression of granulocyte-macrophage colony-stimulating factor in human bronchial epithelial cells: roles of protein kinase C and mitogen-activated protein kinases. J Immunol 165:1618–25 [DOI] [PubMed] [Google Scholar]
- Repertinger SK, Campagnaro E, Fuhrman J, El-Abaseri T, Yuspa SH, Hansen L (2004) EGFR enhances early healing after cutaneous incisional wounding. J Invest Dermatol 123:982–9 [DOI] [PubMed] [Google Scholar]
- Robert C, Soria JC, Spatz A, Le Cesne A, Malka D, Pautier P et al. (2005) Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol 6:491–500 [DOI] [PubMed] [Google Scholar]
- Roberts RB, Arteaga CL, Threadgill DW (2004) Modeling the cancer patient with genetically engineered mice: prediction of toxicity from molecule-targeted therapies. Cancer Cell 5:115–20 [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M (1991) Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 354:494–6 [DOI] [PubMed] [Google Scholar]
- Sørensen OE, Cowland JB, Theilgaard-Mönch K, Liu L, Ganz T, Borrregaard N (2003) Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol 170:5583–9 [DOI] [PubMed] [Google Scholar]
- Stagno F, Guglielmo P, Consoli U, Russo M, Giustolisi R (1999) Successful healing of hydroxyurea-related leg ulcers with topical granulocyte/macrophage colony-stimulating factor. Blood 94:1479–80 [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D et al. (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutated phenotype. Science 269:230–4 [DOI] [PubMed] [Google Scholar]
- Valdembri D, Serini G, Vacca A, Ribatti D, Bussolino F (2002) In vivo activation of JAK/STAT-3 pathway during angiogenesis induced by GM-CSF. FASEB J 16:225–7 [DOI] [PubMed] [Google Scholar]
- Valyi-Nagy I, Jensen PJ, Albelda SM, Rodeck U (1992) Cytokine-induced expression of transforming growth factor-α and the epidermal growth factor receptor in neonatal skin explants. J Invest Dermatol 99:350–6 [DOI] [PubMed] [Google Scholar]
- Wetzker R, Böhmer FD (2003) Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol 4:651–57 [DOI] [PubMed] [Google Scholar]
- Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L et al. (2003) C-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell 6:879–89 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.