

Abstract
Di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) is being developed as an iron chelator with selective anticancer activity. We investigated the mechanism whereby Dp44mT kills breast cancer cells, both as a single agent and in combination with doxorubicin. Dp44mT alone induced selective cell killing in the breast cancer cell line MDA-MB-231 when compared with healthy mammary epithelial cells (MCF-12A). It induces G1 cell cycle arrest and reduces cancer cell clonogenic growth at nanomolar concentrations. Dp44mT, but not the iron chelator desferal, induces DNA double-strand breaks quantified as S139 phosphorylated histone foci (γ-H2AX) and Comet tails induced in MDA-MB-231 cells. Doxorubicin-induced cytotoxicity and DNA damage were both enhanced significantly in the presence of low concentrations of Dp44mT. The chelator caused selective poisoning of DNA topoisomerase IIα (top2α) as measured by an in vitro DNA cleavage assay and cellular topoisomerase-DNA complex formation. Heterozygous Nalm-6 top2α knockout cells (top2α+/− ) were partially resistant to Dp44mT-induced cytotoxicity compared with isogenic top2α+/+ or top2β−/− cells. Specificity for top2α was confirmed using top2α and top2β small interfering BNA knockdown in HeLa cells. The results show that Dp44mT is cytotoxic to breast cancer cells, at least in part, due to selective inhibition of top2α. Thus, Dp44mT may serve as a mechanistically unique treatment for cancer due to its dual ability to chelate iron and inhibit top2α activity.
Introduction
Iron chelators have historically been studied for treatment of iron overload disease and for their potential to alleviate the cardotoxic side effects of anthracycline chemotherapy (1, 2). The essential nature of elemental iron is evident in a multitude of cellular processes including proliferation, DNA synthesis, mitochondrial electron transport, and oxygen sensing (3, 4). Iron is also critical for the functioning of ribonucleotide reductase, an essential enzyme that provides deoxynucleotide triphosphates for DNA synthesis and repair (5). Neoplastic cells have a higher requirement for iron than healthy cells because they rapidly proliferate. Exposure of cancer cells to some iron chelators has been shown to elicit a G1-S cell cycle arrest (6, 7). The R2 subunit of ribonucleotide reductase, a primarily S-phase protein, is dependent on iron supply for its stabilization with the R1 subunit and the subsequent activity of ribonucleotide reductase (8). Within serum, iron is transported by the protein transferrin and taken up by cells via binding of transferrin to transferrin receptor 1 (9). Cancer cells show increased uptake and utilization of iron by virtue of possessing significantly higher levels of transferrin receptor 1 than healthy cells (10). These unique characteristics of neoplastic cells have been exploited recently by the use of iron chelators that show significant antitumor activity in vitro and in animal models (11, 12). Targeting iron metabolism has resulted in compounds with some antitumor activity such as desferrioxamine (desferal or desferrioxamine; refs. 13, 14) and dexrazoxane (ICRF-187; refs. 15, 16) when used in combination with doxorubicin. The iron chelator dexrazoxane (ICRF-187), a cell-permeable cyclic derivative of EDTA, is Food and Drug Administration approved for reducing the incidence and severity of doxorubicin-induced cardiomyopathy in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2, but it has no appreciable antitumor activity of its own (17). Our previous work and that of others have shown desferrioxamine to be capable of inhibiting breast tumor growth without interfering with tumoricidal activity of doxorubicin (14). However, the short half-life and low membrane permeability of desferrioxamine limit its antitumor activity in clinical settings (18). Other newer iron chelators, tachypyridine and triapine, are currently being evaluated for antitumor activity in the clinic (19, 20).
A range of iron chelating di-2-pyridyl thiosemicarbazone analogues have been developed (14) with greater cytotoxic and iron chelating activities than desferrioxamine (21). One analogue, di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT), was proposed to have selective antitumor activity in vitro and in vivo (structure, Fig. 1A, inset; ref. 12). In previous studies, Dp44mT as a single agent was capable of inducing apoptosis in neuroepithelioma, melanoma, and breast cancer cells in contrast to healthy fibroblasts. It also showed activity in etoposide (VP-16)–resistant clones of MCF-7 breast and KB3–1 epidermoid carcinoma cells. Short-term studies with low-dose Dp44mT have inhibited lung, melanoma, and neuroepithelioma tumor growth in nude mice without systemic iron depletion, changes in organ weights, or differences in serum biochemical parameters, in contrast to triapine (12). We are interested in understanding the activity and mechanism of action of Dp44mT to determine whether it might serve as a therapeutic adjunct to doxorubicin treatment of breast cancer. Given the antitumor activity of Dp44mT by itself, we tested low doses of Dp44mT in vitro and in vivo, with and without doxorubicin, for potential cytotoxic activity and compared it to desferrioxamine and ICRF-187. The distinctive cytotoxic activity of Dp44mT led us to hypothesize that it may possess DNA-damaging activity.
Figure 1.

The growth inhibitory effect of Dp44mT in cancer cell lines and primary normal breast epithelial cells. A, cell proliferation was measured using a sulforhodamine B dye–based assay. MDA-MB-231 (breast cancer) and MCF-12A (healthy mammary epithelial) cells were incubated with increasing concentrations of Dp44mT, desferrioxamine (DFO), or ICRF-187 for 72 h. Results are expressed relative to control. Inset, chemical structure of Dp44mT. B, exponentially growing MDA-MB-231 cells were incubated for 48 or 72 h with 0.1 μmol/L doxorubicin (horizontally striped columns), 10 nmol/L Dp44mT, 15 μmol/L desferrioxamine, or 0.5 μmol/L ICRF-187 (vertically striped columns). When tested in combination with doxorubicin, cells were incubated with each iron chelator for 1 h before adding doxorubicin (crossed columns). Results are expressed relative to control cells (gray solid columns). C, colony formation assay was done on MDA-MB-231 cells incubated with Dp44mT (1 nmol/L–10 μmol/L), doxorubicin (10 nmol/L and 100 nmol/L), or the combination (10 nmol/L Dp44mT and 10 or 100 nmol/L doxorubicin). Cells were preincubated with Dp44mT for 1 h, exposed to doxorubicin for 24 h, washed, and then incubated for a period of 7 d. Representative of at least three independent experiments. Bars, SD. * P < 0.05, between treatment with combination and treatment with doxorubicin alone.
Members of the DNA damage response pathway are among the first responders to genomic alterations produced by DNA-targeting agents such as VP-16, doxorubicin, and camptothecin. The ataxia telangiectasia mutated (ATM) kinase, a member of the phosphatidylinositol 3-kinase-related kinase family, is autophosphorylated on Ser1981 and acts on downstream substrates responsible for DNA repair, cell cycle arrest, and apoptosis. Histone H2AX phosphorylated on Ser139, termed γ-H2AX, is one of the earliest markers of replication-associated DNA double-strand breaks that are induced by inhibition of DNA synthesis (22, 23). γ-H2AX is proposed to anchor broken chromosomes together and recruit DNA repair elements, including Mre11-Rad50-Nbs1. Other substrates of ATM are the cell cycle checkpoint-associated kinases Chk1 and Chk2 (24). The Chk2 kinase undergoes a cascade of autophosphorylation, including on Thr68, upon DNA damage induced by blockage of DNA replication (25).
In this report, we present results that prove a unique DNA-damaging, topoisomerase IIa (top2α)–directed mechanism of action for Dp44mT that may be partly responsible for its antitumor activity.
Materials and Methods
Materials.
Dp44mT was obtained from Calbiochem (#412520). The Drug Synthesis and Chemistry Branch of the National Cancer Institute provided doxorubicin and camptothecin. VP-16 and propidium iodide were purchased from Sigma-Aldrich. MJ-III-65 (NSC 706744) was provided by Dr. Mark Cushman (Purdue University, Purdue, IN). Recombinant human top2 was provided by Dr. John Nitiss (St. Jude Children’s Research Hospital, Memphis, TN). Recombinant top2β was a gift from Dr. Neil Osheroff. T4 polynucleotide kinase, DNA polymerase I (Klenow fragment), deoxynucleotide triphosphates, φX174 DNA, agarose, and polyacrylamide/bis were purchased from Invitrogen or New England Biolabs. DNA Quick Spin columns were purchased from Roche Diagnostics. [γ-32P]dATP and [α-32P]dGTP 5′-triphosphate were purchased from Perkin-Elmer Life and Analytical Science. Oligonucleotides were synthesized by MWG Biotech.
Cell culture.
MDA-MB-231 breast cancer cells were obtained from American Type Culture Collection and maintained in DMEM:F12 (1:1) containing 5% heat-inactivated FCS, 2 mmol/L l-glycine, 1 mmol/L sodium pyruvate, and 50 μmol/L β-mercaptoethanol at 37°C in 5% CO2 in air. MCF-12A primary healthy epithelial cells were obtained from Biowhittaker Clonetics and maintained in mammary epithelial cell growth medium (Biowhittaker, Clonetics; ref. 14). The construction and characterization of top2α+/− and top2β−/− Nalm-6 leukemic cells are described elsewhere (26).
Colony formation assay.
Cell survival was determined using a colony formation assay after the indicated treatments as reported previously (27). Colonies were counted manually. Counts were obtained from at least three independent experiments. The number of colonies obtained from each experiment was normalized to the untreated sample (n = 100). For statistical analyses, the combination drug-treated values was compared with each single agent alone. Asterisks are used to denote statistically significant differences (P < 0.05) following an unpaired, two-tailed Student’s t test.
Assessment of antiproliferative activity.
Growth inhibition was assessed by the sulforhodamine B (Sigma-Aldrich) assay as described previously (27). IC50 was calculated using the software Prism 5 (GraphPad Software, Inc.). Data were obtained from at least three independent experiments. The absorbance values obtained from each experiment were normalized to the untreated sample (n = 100). For statistical analyses, the combination drug-treated values were compared with each single agent alone. Asterisks are used to denote statistically significant differences (P < 0.05) following an unpaired, two-tailed Student’s t test.
Flow cytometric analysis.
For cell cycle analyses, cells were harvested and fixed in 70% ice-cold ethanol for 30 min at 4°C (14). Cells were analyzed by FACScan (BD Biosciences) and cell cycle phases were quantitated using the CellQuest program (BD Biosciences). The percentage of cells with sub-G1 DNA content was used as a measure of apoptosis.
Small interfering RNA–mediated gene knockdown.
The top2α, top2β, and control small interfering RNAs (siRNA) were purchased from Invitrogen, and knockdown experiments were carried out as described previously (26). HeLa cells were transfected with siRNA using Lipofectamine (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were collected by trypsinization and cultured in 96-well plates for 72 h, followed by a sulforhodamine B–based growth inhibition assay.
Western blot analysis and antibodies.
Whole cell protein lysates were extracted and used for Western blot analysis as previously described (22). Antibodies for pChk1-S345, pChk2-T68, α-tubulin, and pATM-S1981 were obtained from Cell Signaling Technologies.
Top1- and top2-induced DNA cleavage assays.
Topoisomerase-mediated reactions have been described previously (27). The 161-bp fragment from pBluescript SK(−) phagemid DNA (Stratagene) was 3′-end labeled with [α-32P]dGTP. For topoisomerase I (top1) cleavage assays, labeled DNA (50 fmol/reaction) was incubated with 5 ng of recombinant topi with or without drug at 25°C in 10 μL of reaction buffer. Maxam-Gilbert loading buffer was then added to the reaction mixtures. Aliquots were separated in 16% denaturing polyacrylamide gels (7 mol/L urea) in 1× Tris-borate EDTA for 2 h at 40 V/cm at 50°C.
For top2 assays, the same pSK fragments used for top1 assays or single-stranded oligonucleotides were 5′-end labeled with [32P]ATP and T4 polynucleotide kinase. Labeling mixtures were subsequently centrifuged through Mini Quick Spin DNA columns (for pSK fragments) or Oligo columns (for oligonucleotides; Roche Diagnostics) to remove the unincorporated label. Annealing to the complementary strand of the oligonucleotides was done by heating the reaction mixture to 95o C and overnight cooling to room temperature in 10 mmol/L Tris-HCl (pH 7.8), 100 mmol/L NaCl, and 1 mmol/L EDTA. DNA substrates (10 μmol/reaction) were incubated with 500 ng of top2α or top2β in the presence or absence of drugs for the indicated times at 25°C in 10 μL of reaction buffer. Reactions were stopped by adding SDS (final concentration 0.5%). Samples were separated on 16% (for pSK DNA) or 20% (for the oligonucleotides) denaturing polyacrylamide gels (7 mol/L urea). Imaging and quantitation were done using a PhosphorImager (Molecular Dynamics).
Immunocomplex of enzyme assay.
Topoisomerase-DNA adducts were detected as described previously (27). Top1-DNA complexes were detected using the C21 top1 monoclonal antibody (gift from Yung-Chi Cheng, Yale University, New Haven, CT) following standard Western blotting procedures. Top2α and top2β antibodies were obtained from Abcam. The pan-caspase inhibitor z-VAD-fmk was obtained from Bachem.
Antibodies used for confocal microscopy of nuclear protein localization.
Cells grown in Nunc chamber slides (Nalgene) using 0.5 mL of tissue culture medium were fixed and permeabilized as described previously using 4% paraformaldehyde and ice-cold 70% ethanol (22, 28). The rabbit anti–γ-H2AX antibody was obtained from Dr. William Bonner (National Cancer Institute, Bethesda, MD). The phosphorylated S1981-ATM was purchased from Cell Signaling Technologies. Line scans were done to graphically represent the extent of colocalization using Image J software (NIH).
Alkaline single-cell gel electrophoresis (Comet) assay for evaluation of DNA damage.
DNA damage was measured using the alkaline Comet assay based on the modification described by Singh and colleagues (29). For this assay, 5,000 MDA-MB-231 cells before and after the treatment were mixed with 40 μL of 0.5% low melting point agarose in PBS at 37°C. The mixture was layered onto a fully frosted microscope slide (Fisher Scientific) coated previously with 100 μL of 0.65% normal agarose in PBS, followed by a top layer of 80 μL of low melting point agarose. After solidification, the slides were left in a lysis solution (2.5 mol/L NaCl, 10 mmol/L Tris, 100 mmol/L EDTA, 10% DMSO, and 1% Triton X-100, pH 10) at 40c for 1 h. The slides were then placed in electrophoretic buffer (1 mmol/L EDTA, 300 mmol/L NaOH, pH 13) for 20 min at 40C to allow unwinding of DNA. Electrophoresis was conducted for the next 20 min at 35 V (0.7 V/cm) and 300 mA. After the electrophoresis, the slides were washed with neutralization buffer (0.4 mol/L Tris, pH 7.5) and stained with 25 μL of 20 μg/mL ethidium bromide. Cells, 100 per treatment condition, were analyzed under an epifluorescence microscope (Nikon). The raw images captured were quantitated using CometScore software by Tritek Corp. Cellular responses to DNA damage were expressed as the “tail moment,” which combines a measurement of the length of the DNA migration and the relative DNA content therein (30). Ionizing radiation (1 Gy) was used as a positive control.
Results
Inhibition of tumor cell growth by Dp44mT.
The antiproliferative activity of Dp44mT was first tested in various tumor cell lines compared with healthy cells using the sulforhodamine B dye–based assay and different concentrations of Dp44mT. The growth inhibition in the human breast cancer cell line MDA-MB-231 was compared with the effect on healthy human mammary epithelial MCF-12A cells. The biological activity of Dp44mT was compared with two other well-studied iron chelators, desferrioxamine and dexrazoxane (ICRF-187). As shown in Fig. 1, Dp44mT was highly toxic to MDA-MB-231, showing 50% growth inhibition (GI50 values) at ~ 100 nmol/L Dp44mT. Much greater concentrations of Dp44mT were required to inhibit the growth of MCF-12A, with GI50 values >10 μmol/L. Desferrioxamine and ICRF-187 had much less growth inhibitory activity on the aggressive tumor cells and were relatively nontoxic to the normal cells (Fig. 1A, middle and right), in agreement with our previous report (14).
The goal of adjuvant chemotherapy protocols is to maximize tumor cell killing while sparing healthy tissues from the toxic side effects. Iron chelators have been tested for their chemotherapeutic and cardioprotective potentials primarily in combination with anthracycline drugs such as doxorubicin. For this study, we tested the growth inhibitory activity of Dp44mT in combination with doxorubicin in MDA-MB-231 breast cancer cells. Pretreatment of MDA-MB-231 tumor cells with 10 nmol/L Dp44mT for 1 hour significantly enhanced the growth inhibition caused by doxorubicin (measured at 72 hours; Fig. 1B). In contrast, desferrioxamine did not significantly enhance the antitumor activity of doxorubicin at either 48 or 72 hours. Using ICRF-187 seemed to reduce doxorubicin-induced growth inhibition at 72 hours, although this effect was not statistically significant (P = 0.076). Overall, Dp44mT seemed to have the greatest effect on enhancing doxorubicin-induced growth inhibition in MDA-MB-231 cells.
One of the hallmark traits of cancer cells is their ability to undergo isolated clonal growth. The anticancer activity of Dp44mT was further shown using a colony formation assay with MDA-MB-231 cells. Dp44mT caused significant, dose-dependent inhibition of colony formation at concentrations of ≥10 nmol/L (Fig. 1C). Furthermore, the combination of 10 nmol/L Dp44mT with doxorubicin resulted in significantly fewer colonies than seen with either agent alone. Thus, Dp44mT has distinct anticancer cell killing activity in breast cancer cells and is only toxic to healthy mammary epithelial cells at higher concentrations.
Cell cycle arrest by Dp44mT as a mechanism leading to growth arrest.
The effects of Dp44mT on cell cycle progression and cell death in MDA-MB-231 cells were analyzed by flow cytometric analysis of DNA content using fixed, propidium iodide–stained cells. MDA-MB-231 cells were exposed to doxorubicin alone (0.1 μmol/L, 48 hours), Dp44mT alone (20–100 nmol/L, 48 hours), or a combination of the two drugs at 48 hours (Fig. 2A and B). The iron chelator desferrioxamine was tested for comparison. Doxorubicin (0.1 μmol/L) induced a G2-M arrest in MDA-MB-231 breast cancer cells (Fig. 2A), as shown in previous studies (14). The addition of Dp44mT (20 nmol/L) seemed to abrogate the G2-M arrest caused by doxorubicin and significantly increased the population of cells with sub-G1 (apoptotic) DNA content. Dp44mT alone caused a dose-dependent G1 cell cycle arrest in MDA-MB-231 cells and a 2% to 4% increase in cytoxicity (sub-G1 DNA) when tested at 20 to 100 nmol/L. The percentage of G1 cells increased from ~ 47% to 78% with 100 nmol/L Dp44mT after 48 hours. The ability of Dp44mT to induce G1-S cell cycle arrest in p53-mutant-containing MDA-MB-231 cells is also indicative of its p53-independent mechanism of action (31). The cell cycle modulating activity of Dp44mT was distinct from that of the iron chelator desferrioxamine (15 μmol/L; Fig. 2C), which, as shown previously, had minimal effect on the cell cycle profile of MDA-MB-231 cells. In addition, desferrioxamine did not inhibit the G2-M arrest induced by doxorubicin but slightly increased the percentage of cells with sub-G1 DNA content in the presence of doxorubicin. Overall, the cell cycle studies with Dp44mT are in agreement with the sulforhodamine B assay results and show that low concentrations of the iron chelator Dp44mT enhance the breast cancer cell killing activity of doxorubicin.
Figure 2.
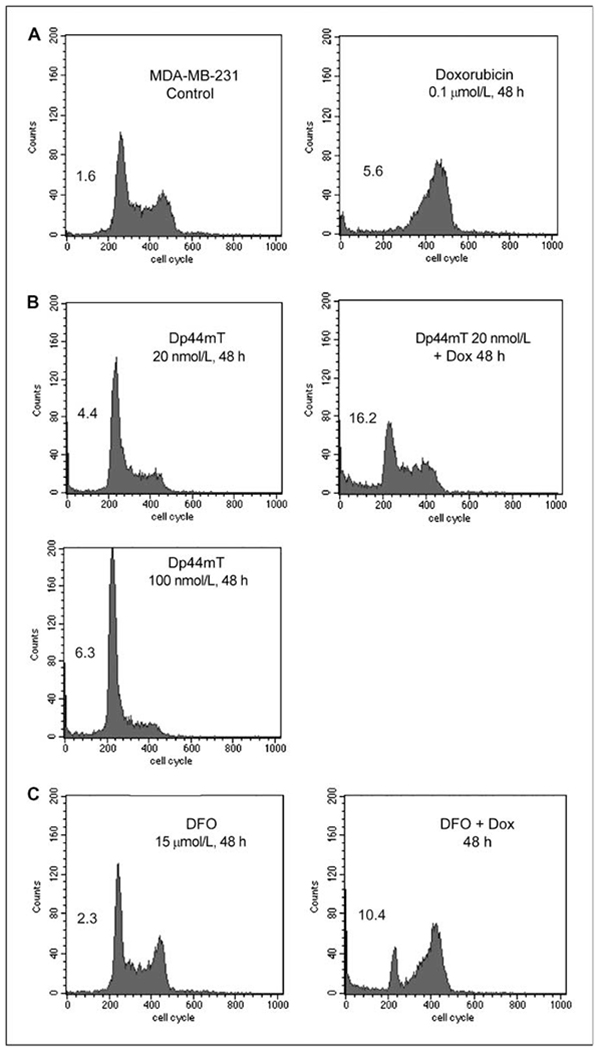
Cell cycle arrest and apoptosis by Dp44mT in MDA-MB-231 cells. A, for cell cycle analysis, proliferating MDA-MB-231 breast cancer cells were exposed to 0.1 μmol/L doxorubicin for 48 h and stained with propidium iodide. The numbers within each representative fluorescence-activated cell sorting profile indicate the percentage of cells with sub-G1 DNA content. B, MDA-MB-231 cells were exposed for 48 h to 20 or 100 nmol/L of Dp44mT or to a combination of 20 nmol/L Dp44mT (1 h pretreatment) and 0.1 μmol/L doxorubicin. C, cell cycle profile of MDA-MB-231 cells exposed to 15 μmol/L desferrioxamine with or without 0.1 μmol/L doxorubicin for 48 h. Fluorescence-activated cell sorting profiles are representative of at least three independent experiments.
DNA damage and checkpoint activation induced by Dp44mT.
We tested the hypothesis that Dp44mT might be inducing its cell cycle and growth inhibitory activity by causing DNA damage. First, DNA damage was measured by confocal microscopy. The phosphorylation of Ser139 (S139) in histone H2AX and the resultant formation of nuclear foci of phospho-histone γ-H2AX were used as a measure of DNA double strand breaks, as previously described (23). The autophosphorylation of the upstream kinase ATM at Ser1981 (S1981) was used to ascertain if the ATM kinase was colocalized with γ-H2AX. Such colocalization would confirm the activation of a DNA damage checkpoint at the site of damage (25). As expected, doxorubicin treatment of MDA-MB-231 cells resulted in a significant increase in nuclear foci for both γ-H2AX and pS1981-ATM (Fig. 3A). For γ-H2AX, doxorubicin produced on average of 9.2 foci per nucleus with 46% of cells having at least one focus. Interestingly, 20 nmol/L Dp44mT as a single agent caused a significant increase in both the number of foci per nucleus and the percentage of MDA-MB-231 cells with detectable foci after exposure for 6 hours (quantitation, Fig. 3B), with an average of 4.6 foci per nucleus (compared with 0.5 focus per nucleus in control cells) and with 28% of the nuclei with at least 1 focus (compared with <7% in control cells). The combination of Dp44mT and doxorubicin resulted in an increased percentage of cells (68%) with DNA damage. The number of foci per nucleus also increased to 19.1. Desferrioxamine alone at 15 μmol/L did not cause any appreciable foci formation, but it, too, modestly enhanced the level of DNA damage caused by doxorubicin. The signal for γ-H2AX and pS1981-ATM was found to be colocalized, as confirmed by line scan analysis (Fig. 3A, bottom). The induction of DNA strand breaks by Dp44mT alone in MDA-MB-231 cells was confirmed by an alkaline Comet assay (Fig. 3C). Appreciable DNA damage was measured with 100 nmol/L Dp44mT after a 24-hour exposure.
Figure 3.
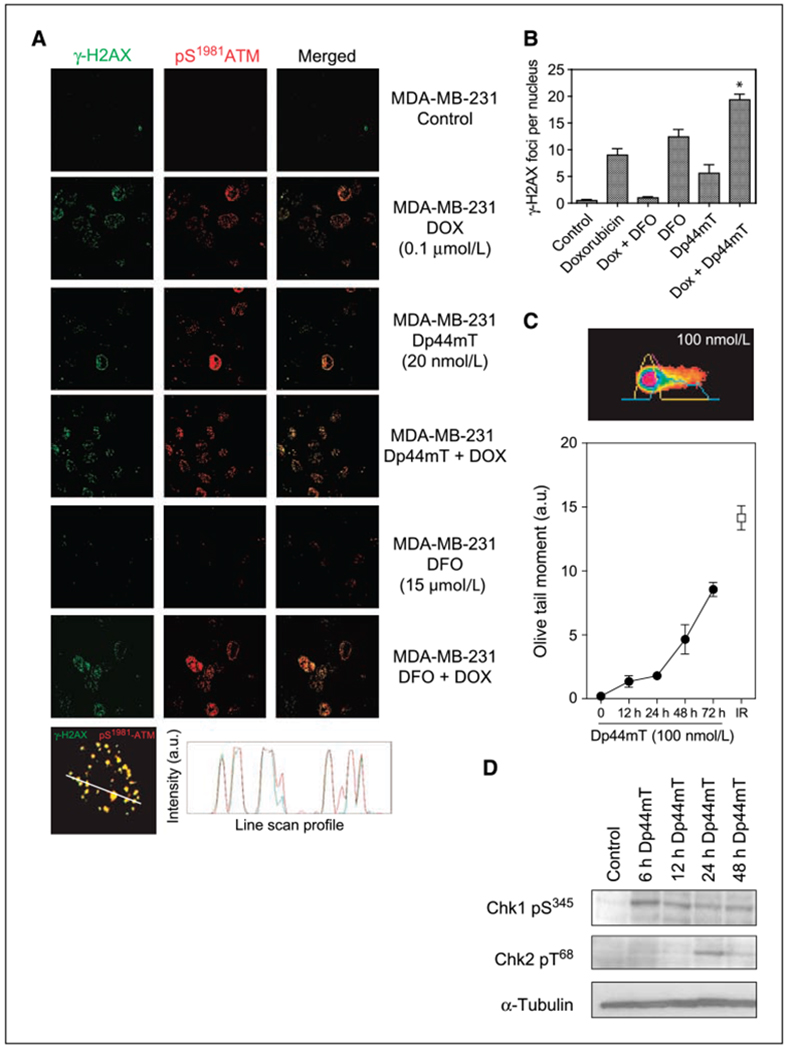
DNA damage and checkpoint activation by Dp44mT in MDA-MB-231 cells. A, immunofluorescence of the S139 phosphorylated form of histone H2A (γ-H2AX) and the S1981 phosphorylated form of ATM kinase assessed as a measure of DNA damage. Cells were exposed to doxorubicin, iron chelator (desferrioxamine or Dp44mT), or the combination for 6 h followed by measurement of γ-H2AX (green fluorescence) and pATM (red fluorescence). Representative confocal microscopy images for MDA-MB-231 show nuclear foci of phosphorylated protein. Colocalization between γ-H2AX and pATM following Dp44mT treatment was confirmed using qualitative line scan analysis (bottom). B, quantitation of γ-H2AX foci formation by Dp44mT and doxorubicin in MDA-MB-231 cells. At least 100 nuclei from three independent experimental slides were used for the quantitation. Asterisk, statistically significant values of the combination of Dp44mT and doxorubicin versus either single agent. C, alkaline Comet assay with MDA-MB-231 cells was used to measure DNA strand breaks induced by Dp44mT (100 nmol/L) at increasing incubation times (closed circles). Tritek Comet measurement software was used for analyzing Olive tail moments from at least 100 nuclei. Cells exposed to 1-Gy ionizing radiation were used as a positive control (open square). Inset, representative image from Dp44mT-treated single nuclei pseudocolored by CometScore program. Representative of at least three independent experiments. D, checkpoint activation by Dp44mT was analyzed by measurement of phosphorylated forms of Chk1 (S345) and Chk2 (T68) in MDA-MB-231 cells. Cells were exposed to 20 nmol/L Dp44mT for 6, 12, 24, or 48 h. α-Tubulin was used as loading control. Representative Western blot images are shown.
We further investigated the molecular events associated with Dp44mT-induced cell cycle arrest by measuring the phosphorylation/activation of Chk1 and Chk2, which are known regulators of cell cycle checkpoints. The phosphorylation of Chk1 at Ser345 and Chk2 at Thr68 in MDA-MB-231 cells was measured by Western blot immunoassay. A rapid activation of Chk1 and a delayed, temporal activation of Chk2 were observed with increasing incubation times following addition of 20 nmol/L Dp44mT to the cells (Fig. 3D). Thus, Dp44mT activated a DNA damage and cell cycle checkpoint marked by phosphorylation of H2AX, ATM, and Chk1.
Top2α as a target for Dp44mT.
Topoisomerase targeting is one of the mechanisms by which DNA-damaging agents manifest their cytotoxicity (32). DNA supercoiling during replication and repair is relaxed by the topoisomerase enzymes (33), which can then form interfacial covalent inhibitory complexes with the DNA and topoisomerase inhibitors and cause DNA strand breaks when completion of their catalytic activity is inhibited. Whereas top1 induces single-stranded nicks, top2 is associated with double-strand breaks. Topoisomerases are targeted by several established clinical anticancer agents such as VP-16 and camptothecins. To determine whether the mechanism of induction of DNA strand breaks by Dp44mT involves the targeting of nuclear DNA topoisomerases, we first performed an in vitro DNA cleavage assay. Radiolabeled DNA substrates were incubated with top1, top2α, or top2β in the presence or absence of drug. Dp44mT at concentrations of ≥100 nmol/L specifically targeted top2α (Fig. 4A). The DNA cleavage pattern of Dp44mT was distinct from that of VP-16, which was used as a positive control. Dp44mT had no appreciable effect on 2β activity. Dp44mT also did not show any topi-directed activity (Fig. 4A), for which camptothecin (1 μmol/L) and MJ-III-65 (1 μmol/L) were used as positive controls (32).
Figure 4.
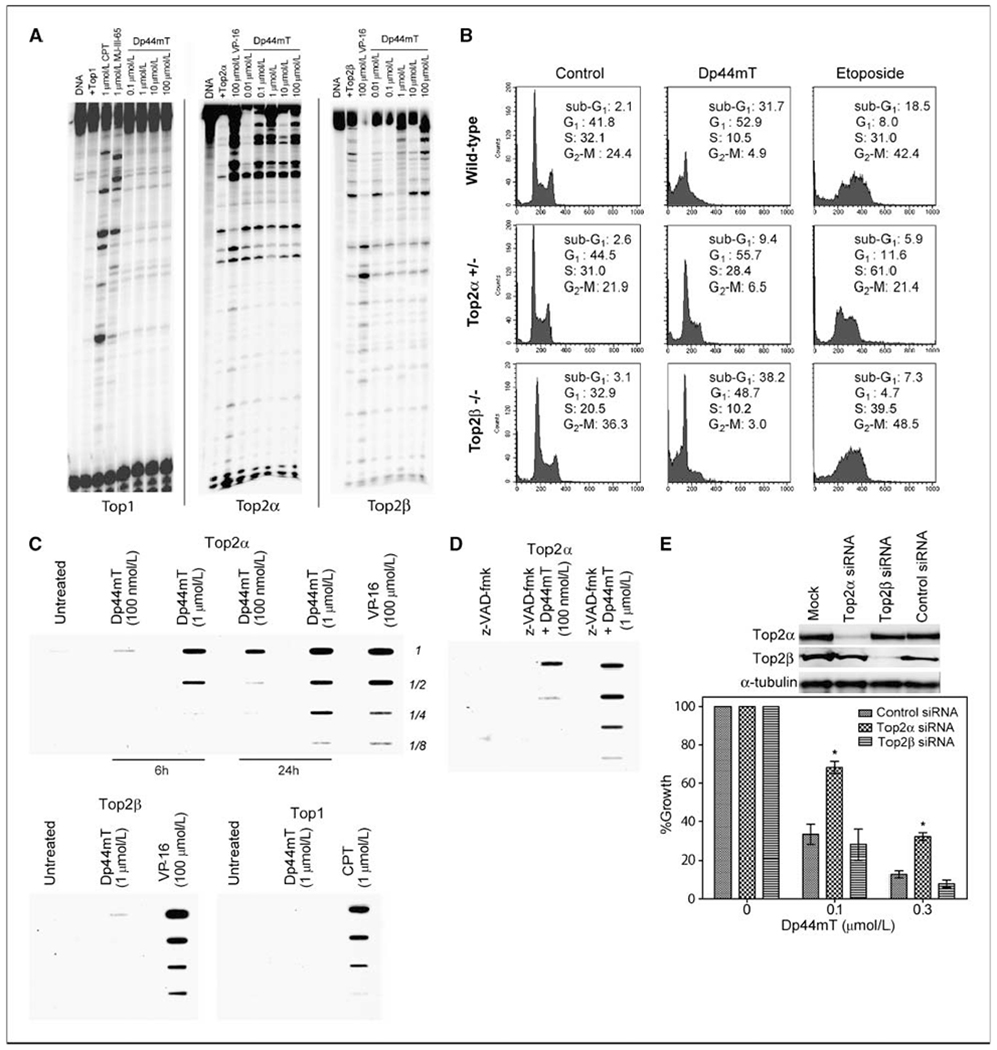
Topoisomerase IIα–mediated activity of Dp44mT. A, for topi activity, DNA corresponding to the 3′-end-labeled PvuII/HindIII fragment of pBluescript SK(−) phagemid DNA (pSK) was incubated with recombinant top1 in the absence of drug (Top1) or in the presence of camptothecin, MJ-III-65, or increasing concentrations of Dp44mT (0.1–100 μmol/L). Reactions were carried out at 25°C for 20 min and stopped by addition of 0.5% SDS. To probe top2α-mediated cleavage activity of Dp44mT, the PvuII/HindIII fragment of pBluescript SK(−) phagemid DNA (pSK) was labeled at the 5′ ends, and reactions were carried out with recombinant top2α. VP-16 (100 μmol/L for 30 min) was included as a positive control. For testing top2β-mediated activity, recombinant top2β was used in the reactions as above with increasing concentrations of Dp44mT or with VP-16. B, apoptotic analysis of isogenic Nalm-6 leukemia cells (top2α/β+/+, top2α+/−, and top2β−/−) by propidium iodide staining and flow cytometry. The population of cells with sub-G1 DNA content was compared at Dp44mT (0.1 μmol/L) or VP-16 (10 μmol/L) at 48 h. Representative of at least four independent experiments. The percentage of cells in each phase of cell cycle is provided. C, cellular topoisomerase cleavage complexes by top1, top2α, and top2β were determined using the immunocomplex of enzyme formation bioassay at 6 or 24 h as indicated in top2α/β wild-type Nalm-6 leukemic cells. Camptothecin (at 1 μmol/L) or VP-16 (at 100 μmol/L) was used as a positive control. Similar data were obtained in at least four independent experiments. D, Nalm-6 cells were also pretreated with pan-caspase inhibitor z-VAD-fmk (100 μmol/L for 30 min) and assayed for formation of nonapoptotic top2α cleavage complexes. E, Dp44mT sensitivity in siRNA-mediated top2α or top2β knockdown HeLa cells. Western blots indicate protein levels 48 h after siRNA transfection. Asterisks, statistically significant difference between the response in top2α+/− compared with top2α+/+ or top2β−/− cells. Bars, SD from two independent experiments.
To determine whether top2α is associated with the cytotoxic activity of Dp44mT, we used a set of isogenic Nalm-6 leukemic cell lines heterozygous for top2α (denoted top2α+/+ or top2α+/−) or with a homozygous deletion of top2β (denoted top2β−/− ref. 26). Because top2α is an essential gene (34), these top2α+/− isogenic cells provided a useful model for testing the role of top2α in cytotoxic signaling. In control experiments, the top2α+/− cells were found to express ~ 57% as much top2α enzyme levels as the wild-type counterpart cells (see ref. 26 for extensive characterization data). The cytotoxicity of Dp44mT (at 100 nmol/L) was compared in the three cell lines by flow cytometric analysis of propidium iodide staining for sub-G1 DNA–containing cells (Fig. 4B). The results shows that the top2α+/− cells are partially resistant to the cytotoxic effects of Dp44mT compared with the wild-type (top2α+/+) or top2β−/− cells (Fig. 4B; Supplementary Fig. S1A). After exposure to Dp44mT (100 nmol/L), the top2α+/+ and top2β−/− cells showed 31.7% and 38.2% sub-G1 containing cells, respectively. In contrast, the top2α+/~ cells only had 9.4%. VP-16 (10 μmol/L) was used as a positive control. The reduced cytotoxicity of Dp44mT selectively in top2α/− Nalm-6 cells was also confirmed using a direct morphologic assessment following Hoescht-33342/propidium iodide staining (Supplementary Fig. S1B). We then performed an in vivo assay to measure cellular complex formation between top1, top2α, or top2β and DNA following Dp44mT treatment of the top2α/β wild-type Nalm-6 cells. The immunocomplex of enzyme formation assay was done 6 and 24 hours after treatment with 0.1 and 1 μmol/L of Dp44mT. As shown in Fig. 4C, Dp44mT caused covalent complex formation between top2α and the DNA. VP-16 (at 100 μmol/L) was used as a positive control. No complex formation was observed when probed for top1 or top2β. To confirm that the top2α-DNA complexes formed were not secondary to apoptosis (35), we preincubated Nalm-6 cells with the pan-caspase inhibitor z-VAD-fmk (100 μmol/L for 30 minutes) and tested for top2α complex formation after 24 hours (Fig. 4D). Pretreatment with caspase inhibitor failed to rescue the top2α complex formation, confirming top2α targeting by Dp44mT. An orthogonal approach was then taken to establish the association between top2α and Dp44mT-mediated cytotoxicity. Transient siRNA-mediated knockdown of top2α and top2β was carried out in HeLa cells (26). A protein level reduction of ~ 78% and 85% was observed for top2α and top2β, respectively (Fig. 4E). Using the sulforhodamine B–based growth inhibition assay, top2α siRNA cells showed a partial resistance to Dp44mT (0.1 and 0.3 μmol/L) when compared with the top2β siRNA– or control siRNA–treated cells at 72 hours. Taken together, these results show that top2α mediates, at least in part, the in vivo biological activity of Dp44mT.
Discussion
Use of iron chelators for cancer therapy.
Iron chelators are being explored as anticancer agents largely because cancer cells have an increased requirement for iron due to their increased levels of metabolism as compared with normal cells. However, iron chelators themselves have not met with clear success in the clinic, although they have shown promise in some nonclinical models (16). For example, the short half-life, low membrane permeability, and lack of activity following oral administration of desferrioxamine have limited its antitumor activity in clinical settings (18). Recently, triapine, a tridentate iron chelator, produced dose-limiting toxicities in phase II trials in patients with non–small-cell lung and prostrate cancer without clear therapeutic benefit (36, 37). The hexadentate iron chelator tachypyridine, by arresting cancer cells in the G2 phase of the cell cycle, showed antitumor and radiation-sensitizing activity that remains to be extended to the clinic (11). ICRF-I87 has no appreciable antitumor activity in animals (38) or in vitro unless it is used at very high concentrations that are not pharmacologically achievable (e.g., see Fig. 1). The reduction in cytotoxicity we observed with ICRF-I87 in combination with doxorubicin is probably due to the intact bisdioxopiperazine acting as a catalytic inhibitor of top2 (both isozymes) and likely preventing doxorubicin-induced poisoning of top2 (both isozymes) as previously shown (39). Dp44mT is a newer, more potent iron chelator that has been shown to have antitumor activity in mice xenografted with a range of human tumors, including tumors resistant to VP-I6 (12). Here we show that low concentrations of Dp44mT are selectively toxic to breast cancer cells compared with normal breast epithelial cells. Whereas Dp44mT is highly lipophilic and antiproliferative, its exact mechanism of action has been unclear. We show that Dp44Mt may be able to serve as an anticancer agent by virtue of its ability to inhibit cancer cell metabolism through iron chelation and its novel ability to cause cytotoxic chromatin damage.
Inhibition of top2α activity by Dp44mT.
In this report, we show that Dp44mT causes DNA damage and inhibition of top2α activity. Top2 alters DNA topology by catalyzing the passage of the intact DNA double helix through a transient double-stranded break made on another DNA double helix. This step is critical for relieving torsional stress that occurs during DNA replication, repair, and transcription (33). When top2 is prevented from completing its catalytic cycle, DNA strand breaks ensue. Dp44mT induces top2α-mediated DNA cleavage in vitro and has no apparent effect on top2β or topi (Fig. 4). The top2α-mediated activity of Dp44mT was shown using Nalm-6 leukemia cells that were heterozygous deficient for top2α, in which Dp44mT had only half of the cytotoxic activity compared with wild-type cells. This association was confirmed using a siRNA knockdown approach in HeLa cells, in which top2α siRNA cells, but not top2β or control siRNA cells, were partially resistant to Dp44mT. In addition, Dp44mT is able to induce the formation of stable cellular top2α-DNA complexes, indicative of top2α poisoning in cancer cells. To our knowledge, this is the first report of a specific top2α-directed activity for an iron chelator. In early studies, ICRF-187 (dexrazoxane) was shown to have the ability to inhibit topoisomerases, but not top2α specifically and not at such low concentrations (16). Dp44mT inhibits top2α activity at concentrations (100 nmol/L) that are 10- to 100-fold lower than those used to show top2 inhibition by ICRF-187 (39). ICRF-187 is a prodrug that is hydrolyzed or converted to an EDTA-like compound. Further studies will be needed to ascertain the conversion of Dp44mT in vivo and the activity of the metabolites for better comparison with bisdioxopiperazines (ICRF-187/ICRF-193) activity (40, 41). Other iron chelating analogues of di-2-pyridylketone isonicotinoyl hydrazone have been tested for DNA cleavage activity and do not inhibit top1 (42). No reports have provided data on top2 activity for these analogues.
The top2α directed activity by Dp44mT is novel and may be clinically advantageous for several reasons. First, proliferating cells such as cancer cells express highly elevated levels of top2α compared with healthy cells, making this isozyme a suitable target for anticancer therapy (43). Second, the targeting of top2β for cancer chemotherapy has been implicated in development of secondary malignancies (44), thereby diminishing the utility of top2β inhibition in the treatment of some cancers. Finally, one key goal of iron chelation therapy is to use it in conjunction with other chemotherapy drugs to limit the toxic side effects of drugs that induce reactive oxygen species, as occurs in heart tissue exposed to anthracycline drugs such as doxorubicin (45). ICRF-187 does not itself inhibit the growth of tumors in animal models and thus is viewed as a cardioprotective drug to be used combination with doxorubicin (17, 39). Because cardiac cells do not express top2α (46), Dp44mT may be able to serve as a cardioprotective agent while also enhancing cancer cell cytotoxicity when administered in combination with doxorubicin.
DNA-damaging activity of Dp44mT.
To our knowledge, this is also the first report of a DNA-damaging activity for Dp44mT. Dp44mT caused a clear increase in number of γ-H2AX foci along with an increased colocalized signal for phosphorylated ATM when MDA-MB-231 cells were treated with nanomolar chelator concentrations. Induction of DNA breaks by Dp44mT was confirmed in breast cancer cells by measuring Comet tails under alkaline conditions. The concentrations that caused “y-H2AX formation are within the same range as those that showed antiproliferative activity by Dp44mT. In contrast, ICRF-187 has been shown to enhance the formation of y-H2AX by VP-16 or ionizing radiation, and there is no clear evidence that it can itself induce y-H2AX formation (38).
Effects of Dp44mT on the cell cycle.
The cytotoxic activity of Dp44mT was associated with an ability to induce growth inhibition and G1-S cell cycle arrest in breast cancer cell lines. The G1-S phase arrest by an iron chelator is expected based on previous findings with other agents that interfere with iron metabolism (6, 47). The G1-S cell cycle arrest by Dp44mT may also indicate that the iron chelating activity of Dp44mT is, in fact, its primary mechanism of cancer cell growth inhibition because classic top2 inhibitors such as doxorubicin and VP-16 induce a G2-M arrest in cancer cells (43). Although we cannot rule out that Dp44mT can extend part of its effect by interfering with DNA condensation during mitosis (as with ICRF-187), this is unlikely because we observe a G1-S arrest and no polyploidization by Dp44mT (41). We also found that Dp44mT activates a cell cycle checkpoint via phosphorylation of ATM and Chkl/Chk2. Following its autophosphorylation, ATM acts as the upstream kinase for sensing and transducing the damage signal to a range of substrates including Chkl and Chk2. The appearance of phosphorylated forms of both Chkl and Chk2 is possibly indicative of their redundant effect for downstream inhibitory kinase activity on the Cdc25A phosphatase, which normally allows cellular progression through the G1-S phase (48). The early appearance of Chkl and late temporal appearance of Chk2 may indicate that Chkl is the primary kinase responsible for cell cycle arrest induced by Dp44mT. Recent work suggesting that iron chelation can induce proteasome-mediated degradation of cell cycle proteins underlines the more complicated nature of downstream signaling following cell cycle arrest by an iron chelator (7). Ubiquitin-independent proteasomal degradation of cyclin D1 and p21waf1 has been described in response to iron chelation by desferrioxamine and 311 (49, 50). The G1-S cell cycle arrest by Dp44mT (and likely the escape from a G2-M arrest) could possibly be explained by iron-mediated proteolysis of these and other unknown cell cycle checkpoint proteins. Further investigations will be needed to characterize the degradation of proteins that could signal for a G1-S cell cycle arrest by Dp44mT, despite targeting top2α.
Combination of Dp44mT and doxorubicin.
The combined treatment of MDA-MB-231 breast cancer cells with both Dp44mT and doxorubicin enhanced all of the cytotoxic effects induced by doxorubicin alone. This included growth inhibition and induction of sub-G1 DNA–containing cells. Importantly for cancer treatment, the combination of doxorubicin with Dp44mT completely inhibited the clonogenic growth of MDA-MB-231 cells. In the presence of Dp44mT, doxorubicin was also unable to induce a G2-M cell cycle arrest. The abrogation of a G2-M checkpoint is likely the mechanism by which Dp44mT exacerbates the cytotoxicity of doxorubicin because checkpoint activity is required for the DNA repair steps that permit cells to survive otherwise toxic DNA damage. The DNA-damaging activity of doxorubicin was also enhanced in the presence of Dp44mT as measured by induction of S139 phosphorylated histone γ-H2AX. The presence of γ-H2AX nuclear foci is currently being developed as a clinical biomarker by the National Cancer Institute and might be of clinical utility for evaluating Dp44mT (27). The increased γ-H2AX–associated DNA double-strand breaks induced by the drug combination may derive from the combined inhibition of top2α by Dp44mT and of top2β by doxorubicin (39).
In summary, the novel and multifaceted modes of action for Dp44mT present a mechanistically advantageous agent for cancer therapy, especially for cancers resistant to conventional chemotherapy. Further preclinical studies on Dp44mT are therefore warranted to evaluate its clinical potential.
Supplementary Material
Acknowledgments
Grant support: Intramural Research Program, Center for Cancer Research, National Cancer Institute, NIH.
We thank Drs. Eugene Herman and James Doroshow for helpful discussions; Drs. Neil Osheroff and Jo Ann Byl (Department of Biochemistry, Vanderbilt University, Nashville, TN) for generously providing the top2α. and top2β enzymes; Dr. Stephanie Zdanov whose technical assistance is greatly appreciated; and Drs. Fabio Klamt, Kathy Lee, and Melanie Simpson for critical reading of the manuscript.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the U.S. Food and Drug Administration.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Barry E, Alvarez JA, Scully RE, Miller TL, Lipshultz SE. Anthracycline-induced cardiotoxicity: course, pathophysiology, prevention and management. Expert Opin Pharmacother 2007;8:1039–58. [DOI] [PubMed] [Google Scholar]
- 2.Bernhardt PV. Coordination chemistry and biology of chelators for the treatment of iron overload disorders. Dalton Trans 2007;30:3214–20. [DOI] [PubMed] [Google Scholar]
- 3.De Domenico I, McVey Ward D, Kaplan J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol 2008;9:72–81. [DOI] [PubMed] [Google Scholar]
- 4.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2006;2:406–14. [DOI] [PubMed] [Google Scholar]
- 5.Thelander L, Graslund A, Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem Biophys Res Commun 1983;110:859–65. [DOI] [PubMed] [Google Scholar]
- 6.Brodie C, Siriwardana G, Lucas J, et al. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: evidence for a block in Gx phase of the cell cycle. Cancer Res 1993;53:3968–75. [PubMed] [Google Scholar]
- 7.Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle 2007;6:1982–94. [DOI] [PubMed] [Google Scholar]
- 8.Green DA, Antholine WE, Wong SJ, Richardson DR, Chitambar CR. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: effect on the R2 subunit of ribonucleotide reductase. Clin Cancer Res 2001;7:3574–9. [PubMed] [Google Scholar]
- 9.Sargent PJ, Famaud S, Evans RW. Structure/function overview of proteins involved in iron storage and transport. Curr Med Chem 2005;12:2683–93. [DOI] [PubMed] [Google Scholar]
- 10.Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet 1980;2:390–2. [DOI] [PubMed] [Google Scholar]
- 11.Turner J, Koumenis C, Kute TE, et al. Tachpyridine, a metal chelator, induces G2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation. Blood 2005;106:3191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci USA 2006;103:14901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovejoy DB, Richardson DR. Novel “hybrid” iron chelators derived from aroylhydrazones and thiosemi-carbazones demonstrate selective antiproliferative activity against tumor cells. Blood 2002;100:666–76. [DOI] [PubMed] [Google Scholar]
- 14.Hoke EM, Maylock CA, Shacter E. Desferal inhibits breast tumor growth and does not interfere with the tumoricidal activity of doxorubicin. Free Radic Biol Med 2005;39:403–11. [DOI] [PubMed] [Google Scholar]
- 15.Hasinoff BB, Herman EH. Dexrazoxane: how it works in cardiac and tumor cells. Is it a prodrug or is it a drug? Cardiovasc Toxicol 2007;7:140–4. [DOI] [PubMed] [Google Scholar]
- 16.Hasinoff BB, Kuschak TI, Yalowich JC, Creighton AM. A QSAR study comparing the cytotoxicity and DNA topoisomerase II inhibitory effects of bisdioxopiperazine analogs of ICRF-187 (dexrazoxane). Biochem Pharmacol 1995;50:953–8. [DOI] [PubMed] [Google Scholar]
- 17.Imondi AR. Preclinical models of cardiac protection and testing for effects of dexrazoxane on doxorubicin antitumor effects. Semin Oncol 1998;25:22–30. [PubMed] [Google Scholar]
- 18.Selig RA, White L, Gramacho C, et al. Failure of iron chelators to reduce tumor growth in human neuroblastoma xenografts. Cancer Res 1998;58:473–8. [PubMed] [Google Scholar]
- 19.Finch RA, Liu M, Grill SP, et al. Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): a potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol 2000;59:983–91. [DOI] [PubMed] [Google Scholar]
- 20.Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr Top Med Chem 2004;4:1623–35. [DOI] [PubMed] [Google Scholar]
- 21.Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood 2004;104:1450–8. [DOI] [PubMed] [Google Scholar]
- 22.Rao VA, Fan AM, Meng L, et al. Phosphorylation of BLM, dissociation from topoisomerase IIIα, and colocalization with γ-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol 2005;25:8925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilch DR, Sedelnikova OA, Redon C, et al. Characteristics of γ-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 2003;81:123–9. [DOI] [PubMed] [Google Scholar]
- 24.Shiloh Y ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003;3:155–68. [DOI] [PubMed] [Google Scholar]
- 25.Takemura H, Rao VA, Sordet O, et al. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J Biol Chem 2006;281:30814–23. [DOI] [PubMed] [Google Scholar]
- 26.Toyoda E, Kagaya S, Cowell IG, et al. NK314, a topoisomerase II inhibitor that specifically targets the α isoform. J Biol Chem 2008;283:23711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao VA, Agama K, Holbeck S, Pommier Y. Batracylin (NSC 320846), a dual inhibitor of DNA topoisomerases I and II induces histone γ-H2AX as a biomarker of DNA damage. Cancer Res 2007;67:9971–9. [DOI] [PubMed] [Google Scholar]
- 28.Rao VA, Conti C, Guirouilh-Barbat J, et al. Endogenous γ-H2AX-ATM-Chk2 checkpoint activation in Bloom’s syndrome helicase deficient cells is related to DNA replication arrested forks. Mol Cancer Res 2007;5: 713–24. [DOI] [PubMed] [Google Scholar]
- 29.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175:184–91. [DOI] [PubMed] [Google Scholar]
- 30.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res 1990;122:86–94. [PubMed] [Google Scholar]
- 31.Bartek J, Iggo R, Gannon J, Lane DP. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene 1990;5:893–9. [PubMed] [Google Scholar]
- 32.Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006;6:789–802. [DOI] [PubMed] [Google Scholar]
- 33.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001; 70:369–413. [DOI] [PubMed] [Google Scholar]
- 34.Akimitsu N, Adachi N, Hirai H, et al. Enforced cytokinesis without complete nuclear division in embryonic cells depleting the activity of DNA topoisomerase IIα. Genes Cells 2003;8:393–402. [DOI] [PubMed] [Google Scholar]
- 35.Sordet O, Khan QA, Plo I, et al. Apoptotic topoisomerase I-DNA complexes induced by staurosporine-mediated oxygen radicals. J Biol Chem 2004; 279:50499–504. [DOI] [PubMed] [Google Scholar]
- 36.Ma B, Goh BC, Tan EH, et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine®) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest New Drugs 2008;26:169–73. [DOI] [PubMed] [Google Scholar]
- 37.Attia S, Kolesar J, Mahoney MR, et al. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Invest New Drugs 2008;26: 369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofland KF, Thougaard AV, Dejligbjerg M, et al. Combining etoposide and dexrazoxane synergizes with radiotherapy and improves survival in mice with central nervous system tumors. Clin Cancer Res 2005;11:6722–9. [DOI] [PubMed] [Google Scholar]
- 39.Lyu YL, Kerrigan JE, Lin CP, et al. Topoisomerase IIβ mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 2007;67:8839–46. [DOI] [PubMed] [Google Scholar]
- 40.Hajji N, Pastor N, Mateos S, Dominguez I, Cortes F. DNA strand breaks induced by the anti-topoisomerase II bis-dioxopiperazine ICRF-193. Mutat Res 2003;530: 35–46. [DOI] [PubMed] [Google Scholar]
- 41.Hasinoff BB, Abram ME, Barnabe N, et al. The catalytic DNA topoisomerase II inhibitor dexrazoxane (ICRF-187) induces differentiation and apoptosis in human leukemia K562 cells. Mol Pharmacol 2001;59: 453–61. [DOI] [PubMed] [Google Scholar]
- 42.Chaston TB, Watts RN, Yuan J, Richardson DR. Potent antitumor activity of novel iron chelators derived from di-2-pyridylketone isonicotinoyl hydrazone involves fenton-derived free radical generation. Clin Cancer Res 2004;10:7365–74. [DOI] [PubMed] [Google Scholar]
- 43.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002;3: 430–40. [DOI] [PubMed] [Google Scholar]
- 44.Azarova AM, Lyu YL, Lin CP, et al. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci U S A 2007;104: 11014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Wong J, Lovejoy DB, Kalinowski DS, Richardson DR. Chelators at the cancer coalface: desferrioxamine to Triapine and beyond. Clin Cancer Res 2006;12:6876–83. [DOI] [PubMed] [Google Scholar]
- 46.Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta 1992;1132:43–8. [DOI] [PubMed] [Google Scholar]
- 47.Szuts D, Krude T. Cell cycle arrest at the initiation step of human chromosomal DNA replication causes DNA damage. J Cell Sci 2004;117:4897–908. [DOI] [PubMed] [Google Scholar]
- 48.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007;28:739–45. [DOI] [PubMed] [Google Scholar]
- 49.Fu D, Richardson DR. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood 2007; 110:752–61. [DOI] [PubMed] [Google Scholar]
- 50.Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood 2007;109:4045–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.