

Abstract
Glioblastoma (GBM) is an aggressive and lethal disease that often results in a poor prognosis. Unlike most solid tumors, GBM is characterized by diffuse infiltrating margins, extensive angiogenesis, hypoxia, necrosis, and clonal heterogeneity. Recurrent disease is an unavoidable consequence for many patients as standard treatment options such as surgery, radiotherapy, and chemotherapy have proven to be insufficient in causing long-term survival benefits. Systemic delivery of promising drugs is hindered due to the blood-brain barrier and non-uniform perfusion within GBM tissue. In recent years, many investigations have highlighted the role of GBM stem cells (GSCs) and their microenvironment in the initiation and maintenance of tumor tissue. Preclinical and early clinical studies to target GSCs and microenvironmental components are currently underway. Of these strategies, immunotherapy using checkpoint inhibitors and redirected cytotoxic T cells have shown promising results in early investigations. But, GBM microenvironment is heterogenous and recent investigations have shown cell populations within this microenvironment to be plastic. These studies underline the importance of identifying the role of and targeting multiple cell populations within the GBM microenvironment which could have a synergistic effect when combined with novel therapies. Pericytes are multipotent perivascular cells that play a vital role within the GBM microenvironment by assisting in tumor initiation, survival, and progression. Due to their role in regulating the blood-brain barrier permeability, promoting angiogenesis, tumor growth, clearing extracellular matrix for infiltrating GBM cells and in helping GBM cells evade immune surveillance, pericytes could be ideal therapeutic targets for stymieing or exploiting their role within the GBM microenvironment. This chapter will introduce hallmarks of GBM and elaborate on the contributions of pericytes to these hallmarks by examining recent findings. In addition, the chapter also highlights the therapeutic value of targeting pericytes, while discussing conventional and novel GBM therapies and obstacles to their efficacy.
Keywords: Glioblastoma, GBM Pericytes, Microenvironment
Introduction
Glioblastoma: Incidence and Histological Characteristics
GBM is the most common and aggressive primary malignant brain cancer with a dismal prognosis (Sattiraju et al. 2017a; Van Meir et al. 2010). The median overall survival rate is currently 20.8 months for patients undergoing treatment with adjuvant chemotherapy (temozolomide) following maximum safe resection (Roh et al. 2017). It is estimated that about 13,000 patients are newly diagnosed with GBM annually with an estimated 5-year survival rate of about 10% for adults and about 40% for children (Rostomily et al. 2005; Song et al. 2010; Stupp et al. 2009).
Histopathological characteristics of GBM include anaplasia, macrophage and microglial infiltration, extensive angiogenesis and regions of severe hypoxia, resulting in pseudopalisading structures around necrosing neural tissue (Bissell and Radisky 2001; Van Meir et al. 2010). At a cellular level, GBM is characterized by the rapid proliferation of malignant cells which invade diffusely into the surrounding normal brain parenchyma accompanied by extensive proliferation of endothelial cells which assemble into highly torturous, disorganized and leaky blood vessels which often result in hypoxic microenvironments within tumor tissues (Van Meir et al. 2010; Zong et al. 2012). Invading GBM cells tend to displace preexisting astrocytes and pericytes that are otherwise tightly wrapped around endothelial cells, thereby disrupting the blood-brain barrier (BBB), resulting in leaky blood vessels (Dubois et al. 2014; Watkins et al. 2014). The cell of origin (COI) which is hypothesized to be the first malignant cell that sets the eventual formation of aberrant tissue into motion is thought to exist within the tumor mass in areas of severe hypoxia. GBM is a highly heterogenous disease indicated by the high degree of genetic variations and existence of multiple subclones within tumor tissues. This high degree of heterogeneity has also prompted some researchers to believe that there may be multiple cells of origin, giving rise to a mosaic of aberrant cells which we consider as the tumor tissue (Bradshaw et al. 2016; Cabrera et al. 2015; Dalerba et al. 2007; Fidoamore et al. 2016; Friedmann-Morvinski and Verma 2014; Heddleston et al. 2011).
Hypoxic Microenvironment Within GBM
GBM is an aggressive disease which requires abundant supply of oxygen and nutrients for the survival of highly proliferating cells. The high rate of cellular proliferation within the tumor tissue causes severe hypoxia (0.1–0.5%) within the tumor core and mild hypoxia (0.5–2.5%) in peripheral regions of the tumor (Evans et al. 2004). Within these hypoxic regions, cells residing far away from preexisting blood vessels that cannot adapt to their hypoxic microenvironment undergo apoptosis or coagulative necrosis (Brat and Van Meir 2004; Martínez-González et al. 2012). This results in the outward movement of cells in the surrounding areas towards the periphery of the hypoxic region. These outward moving cells form palisading structures called “Pseudopalisades” and these events are thought to enhance the invasiveness of GBM cells, which in turn stimulate endothelial proliferation and angiogenesis through secretion of VEGF and other factors. Pseudopalisades are a distinct feature of GBMs and a marker of aggressive disease, often distinguishing them from low-grade astrocytomas (Brat et al. 2004; Heddleston et al. 2009; Rong et al. 2006; Sattiraju et al. 2017a).
Hallmarks of GBM
Glioblastoma Initiation and Maintenance
Recent investigations have pointed towards progenitor cells present within the subventricular zone (SVZ) and in peripheral white matter of the cortex as potential COIs for GBM (Alcantara Llaguno et al. 2015). These investigations had employed cell population-specific knockdown of proto-oncogenes and in vivo transduction of progenitor cells expressing fluorescent labels in the cortex of the brain of transgenic mice to show that primitive progenitor cells give rise to aberrant populations of rapidly proliferating cells in a hierarchical fashion. Llaguno et al. pointed to the mutation of proto-oncogenes as a prerequisite for the transformation of otherwise normal progenitor cells and subsequent altered migration and production of aberrant glial cells. Assanah et al., on the other hand, showed that the overexpression of growth factors receptors without mutations to proto-oncogenes was sufficient to alter the function of progenitor cells and cause the growth of tumors within brains of mice (Assanah et al. 2006).
Even though the question of which cell population(s) initiate GBM is still being hotly debated (Safa et al. 2015; Soda et al. 2011), studies examining the effect of therapeutics on established GBM models have reported the existence of stem-like, plastic cells present within GBM tissue that tend to survive therapeutic exposure and later cause disease relapse (Bao et al. 2006; Baskar et al. 2012; Chen et al. 2012; Jackson et al. 2015; Mannino and Chalmers 2011; Murat et al. 2008; Weller et al. 2012). These tumor cells which show the genetic expression and functional characteristics of stem cells have been termed as glioblastoma stem cells (GSCs) (Bradshaw et al. 2016; Cabrera et al. 2015; Calabrese et al. 2007; Dalerba et al. 2007; Gilbertson and Rich 2007; Hanahan and Weinberg 2011; Heddleston et al. 2009; Lathia et al. 2015; Liebelt et al. 2016; Singh et al. 2004a, b). In recent years, further studies into the role of GSCs have indicated their importance in the maintenance of GBMs and reconstitution of tumors post therapy (Chou et al. 2012; Dewhirst et al. 2008; El Hallani et al. 2010; Heddleston et al. 2009; Jhaveri et al. 2016; Lathia et al. 2010; Nakada et al. 2013; Ogden et al. 2008; Ricci-Vitiani et al. 2010). Although several investigations have elucidated the role of GSCs and explored potential ways to target them to enhance the efficacy of current and future therapies, questions regarding the ontology of GSCs have not been conclusively answered (Brooks et al. 2013; Chen et al. 2014; Fan et al. 2010; Huang et al. 2012; Mendez et al. 2010; Persano et al. 2012; Sattiraju et al. 2017a). In addition, Segerman et al. Chaffer et al. and others showed that cancer cells are highly plastic and that terminally differentiated cancer cells dedifferentiate into cancer stem cells (CSCs) in response to stressors and therapy (Chaffer et al. 2013; Niklasson et al. 2017; Segerman et al. 2016). These studies highlighted the importance of tissue microenvironment in the regulation of cancer cell state, but further investigations are necessary to understand the importance of cellular plasticity in tumor maintenance and therapeutic resistance.
Angiogenesis and Perfusion with GBM
GBM is characterized by extensive angiogenesis to allow the growth and survival of rapidly proliferating cells (Das and Marsden 2013; Jain et al. 2007). Hypoxic microenvironments cause GBM cell invasion and stimulate vascular and perivascular cells to produce pro-angiogenic factors (Brat et al. 2004; McCord et al. 2009; Rong et al. 2006). Vascular endothelial growth factor (VEGF) and its receptors, platelet-derived growth factor (PDGF), PDGF receptor-beta (PDGFRβ), angiopoietins (Ang1 and Ang2), Tie2, matrix metalloproteinases (MMP-2 and MMP-9), bone morphogenic proteins (BMPs), etc. have been shown to be involved in this process (Jackson et al. 2017; Ribeiro and Okamoto 2015). The central nervous system (CNS) vasculature consists of tightly packed endothelial cells that are wrapped around by pericytes which provide structural support. Additionally, these vessels are further wrapped around by astrocytes that extend across endothelial cell tight junctions and by interneurons which together form the BBB. BBB is a protective vascular barrier that only allows the passive diffusion of water, oxygen, carbon dioxide, and highly lipophilic molecules. Glucose, amino acids, hormones, and larger molecules are actively transported across the BBB (Abbott 2002; Abbott et al. 2010). Co-opted vasculature and the newly formed angiogenic blood vessels within the GBM tissue show greater vascular permeability than normal CNS vasculature due to their disorganized architecture. This altered, often leaky vascular barrier within the tumor tissue is termed as the blood-tumor barrier (BTB) and results in regions of edema which hinder effective drug delivery (Agarwal et al. 2013; Dubois et al. 2014; Sattiraju et al. 2017b). The presence of BBB in peritumoral regions and a leaky BTB within the tumor results in ineffective perfusion of GBM tissue which further contributes to necrosis and hypoxia while certain parts of a tumor might escape exposure to systemic therapies, thereby resulting in recurrent disease (Pardridge 2005, 2012).
Immune Microenvironment with GBM
A bulk of GBM tissue consists of infiltrated microglia and macrophages, but their response to tumor cells is often suppressed. Immune suppressive factors such as interleukin-10 (IL-10), IL6, transforming growth factor-beta (TGF-β), prostaglandin E2 (PGE2) suppress immune response against GBM cells, promote transformation of dendritic cells (DCs) into a regulatory phenotype and promote the activation of FOXP3+ regulatory T cells (Tregs). Hypoxia and subsequent expression of HIF-1a and VEGF production have also been reported to cause Treg activation and immune suppression. Macrophages isolated from GBMs tend to polarize towards M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes. Tumor-activated macrophages (TAMs) which are similar to M2 macrophages in function and cell surface marker presentation have been shown to play a vital role within the GBM microenvironment by promoting invasion and growth of tumor cells. TAMs have been reported to cause matrix degradation and enhance survival of GBM cells and GSCs. Hypoxic microenvironments within GBM have been reported to stimulate activation of microglia into TAMs (Jackson et al. 2017; Nduom et al. 2015; Razavi et al. 2016).
Activation of naïve T cells within the GBM microenvironment not only requires contact with major histocompatibility complexes (MHCs) on antigen-presenting cells (APCs) but also activation of co-stimulatory receptors (Driessens et al. 2009; Sharpe and Abbas 2006). Based on significant findings in the past decade that shined the spotlight on the role of immune cells and immune-related mechanisms within tumor microenvironment that effect tumor evasion and clearance, a lot of efforts are currently being made to exploit these mechanisms to deliver therapeutics which could suppress inhibitory signals and allow immune cells to identify and clear tumor cells (Rosenberg and Restifo 2015; Wang et al. 2014; Yang 2015). Inhibiting immune checkpoints is currently the most commonly exploited mechanism for therapeutic purposes. Immune checkpoints prevent naïve T cells from causing autoimmune responses during infections by producing inhibitory signals. Programmed death-1 (PD-1) is an immune checkpoint co-stimulatory receptor expressed on the cell surface of T cells. Normal cells of the body express ligands for PD-1, namely PDL-L1 and PDL-L2, to prevent activation of naïve T cells and subsequent cytotoxicity. GBM cells exploit this mechanism and overexpress PDL-L1 on their cell surface to evade immune recognition and attack. Monoclonal antibodies that inhibit PD-1-PDL-L1 binding have shown to boost immune response against tumor cells and enhance survival in pre-clinical studies (Iwai et al. 2017). Promising results in clinical trials resulted in the approval of immune checkpoint inhibitors (ICIs) such as Nivolumab (approved for NSCLC, melanoma and renal cell carcinoma), Pembrolizumab (approved for melanoma, lung cancer and head and neck cancer), and Azetolizumab (approved for Urothelial and lung cancers) for the treatment of cancer patients (Alsaab et al. 2017; Hamanishi et al. 2016; Kang et al. 2016).
Cytotoxic T cell antigen-4 (CTLA4) is a co-stimulatory receptor, similar to PD-1, which negatively regulates T-cell activation. Inhibiting the binding of CTLA4 to its ligand had been shown to cause increased immune response against tumors. Monoclonal antibodies that inhibit CTLA4 binding such as Ipilimumab were approved for use in the clinic for melanoma patients after they showed promising results in preclinical and clinical trials (Alsaab et al. 2017; Hamanishi et al. 2016; Kang et al. 2016). The delivery of ICIs to GBMs is hindered, however, by the presence of BBB and the ineffective perfusion within tumor tissue due to angiogenic blood vessels (Hodges et al. 2016; Lyon et al. 2017; Rolle et al. 2010).
A recent phase-I study showed the potential for using interleukin-13 receptor alpha 2 (IL13RA2) directed chimeric antigen receptor (CAR) T cells against recurrent GBM. The IL13RA2 redirected CAR T cells in this study were delivered into the resection cavity of a 50-year-old patient using a neurosurgical technique called convection-enhanced delivery (CED) (Brown et al. 2016). CED provides the opportunity to bypass the BBB and enhance the efficacy of drugs by delivering them directly into the bed of the tumor (Bobo et al. 1994; Pardridge 2005; Lidar et al. 2004). Even though results from the study were promising, more efforts are needed to enhance the efficacy of this strategy.
Failure of Conventional Therapies Against GBM and Trends for the Future
Recurrent disease is a major contributor to GBM patient mortality and is almost an eventual outcome for newly diagnosed patients whose tumors regress following adjuvant chemotherapy using temozolomide. Invasive GBM cells residing outside the area of resection that escape surgical debulking, therapy-resistant GBM cells that either attain stem cell-like characteristics through de-differentiation or retain their stem cell state after tumor initiation (COIs), insufficient diffusion due to the BBB, and resulting accumulation of ineffective concentrations of systemic therapies within GBM tissue are thought to be major reasons for therapeutic failure and disease relapse (Chaichana 2014; Eyupoglu et al. 2013; Lathia et al. 2015; Pardridge 2005; Persidsky et al. 2006; Wolburg and Lippoldt 2002; Wolburg et al. 2012).
In recent years, a lot of attention has been placed on stymieing angiogenic processes using anti-angiogenic therapies such as Bevacizumab (which binds to and inhibits VEGF function) and to target GBM-specific cell surface receptors, often overexpressed by tumors such as the mutated EGFRvIII (Gilbert et al. 2014). These therapies have proven to be ineffective in causing a long-term effect on the progression of tumors and have not dramatically increased the median survival rate. Tumor heterogeneity, barriers to effective drug delivery, and additional factors involved in the promotion and maintenance of angiogenic vessels are thought to have caused the failure of these therapies in the clinic. Latest strategies to modulate the immune activity within GBM microenvironment, redirecting T cells using chimeric antigen receptors (CARs) towards GBM cells and oncolytic viruses have shown promise in preclinical investigations and phase-I clinical trials, but further research is needed to increase their effectiveness in the future. Additionally, preclinical investigations evaluating the efficacy of targeting GSCs, strategies to transiently enhance BBB permeability, therapeutic stem cells-based drug delivery and targeted molecular irradiation are currently underway to deliver systemic therapies more effectively to GBM tissue (Sattiraju et al. 2017b, c) (Fig. 2.1).
Fig. 2.1.
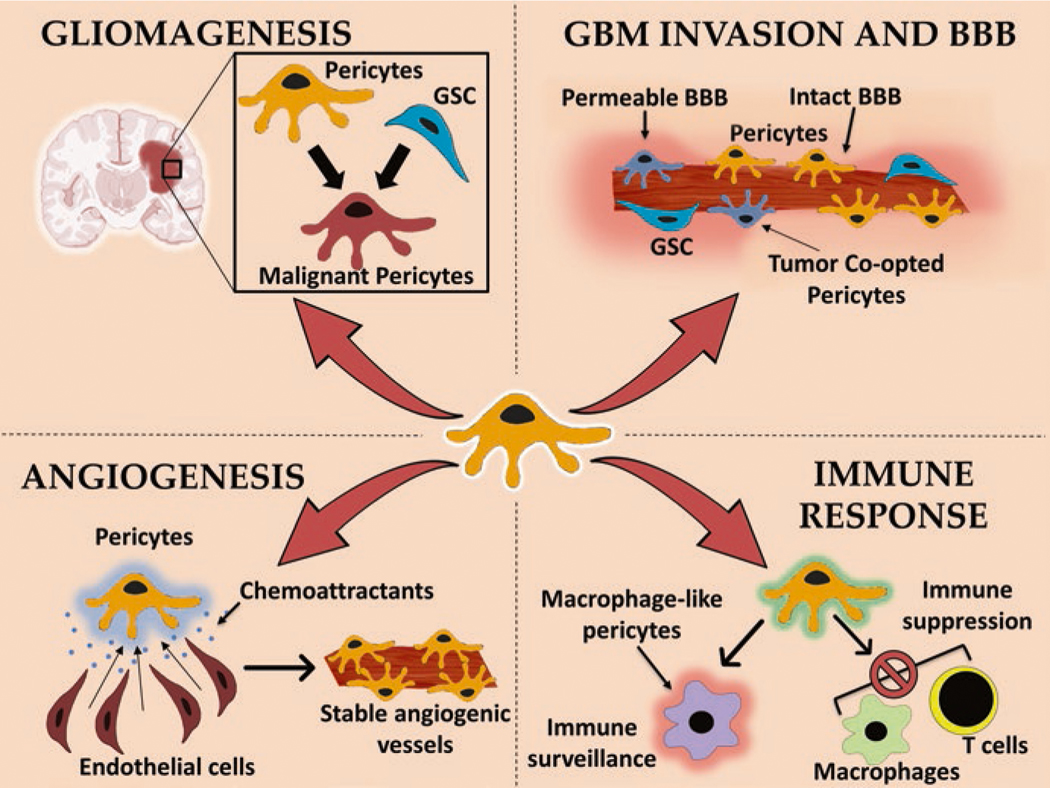
Diagram showing the multifaceted role of pericytes in various critical events of GBM initiation, establishment, maintenance, and progression. GSC glioblastoma stem cells, BBB blood-brain barrier
Multifaceted Role of Pericytes Within GBM
Stemness and Tumor Initiation
Pericytes were previously thought to mainly play a role in supporting vascular architectures within the brain as part of the neurovascular unit (NVU) and to regulate blood flow within capillaries, but recent investigations have shed light on their role in tissue homeostasis and disease pathologies (Jackson et al. 2017; Sweeney et al. 2016). Using transgenic mouse models and cell surface receptor expression analysis, pericytes have been shown to be plastic multipotent perivascular cells which have the capability to differentiate into vascular smooth muscle cells, adipocytes, primary osteocytes, chondrocytes, fibroblasts, myofibroblasts, and neural cell lineages (Birbrair et al. 2014a, 2017a; Ribeiro and Okamoto 2015). Even though studies have shown pericytes to be stem cell-like, not all pericytes are plastic. Birbrair et al. had previously discovered two distinct subpopulations of pericytes in Nestin-GFP/NG2-DsRed double-transgenic mice, of which only Nestin+/NG2+ “Type-2” pericytes were observed to be involved in tumor angiogenesis while Nestin-/NG2+ “Type-1” pericytes were not, indicating that pericytes are heterogenous in their function (Birbrair et al. 2011, 2013a, b, 2014b). Birbrair et al. and others had discovered that pericytes (Type-2) can also be differentiated into neural and myeloid lineages to give rise to neural-like stem cells (NLSCs) and macrophages (Armulik et al. 2011; Birbrair and Frenette 2016; Birbrair et al. 2015).
In later studies, Birbrair et al. also observed that NLSCs derived from type-2 pericytes tend to migrate to the subventricular zone (SVZ) of healthy mice when implanted intracranially (Birbrair et al. 2013a). Importantly, Birbrair et al. also observed that in mice bearing orthotopic GBMs, NLSCs migrate to regions of orthotropic tumors and invade along infiltrating margins when intracranially implanted in the ipsilateral hemisphere. These results indicate NLSCs to behave similarly to mesenchymal stem cells (MSCs) with regard to their migration to GBM sites when implanted intracranially but unlike MSCs which are co-opted by the tumor cells and transform into tumor-associated fibroblasts (TAFs), which assist in tumor growth and expansion, NLSCs did not differentiate into a TAF-like phenotype when co-cultured with GBM cells. In addition, unlike MSCs, NLSCs did not promote angiogenesis when in contact with GBM cells (Birbrair et al. 2017b). These results further contribute to the hypothesis that pericytes could be closely related to MSCs either as their precursors or as a specialized subpopulation of MSCs within the brain. Further adding fuel to this line of thought are reports showing that pericytes and MSCs share cell surface marker expression such as NG2, CD44, αSMA, pDGFRβ, CD90, CD73, CD105, and Sca-1 (Crisan et al. 2008; Ribeiro and Okamoto 2015). The ability of pericytes to differentiate into neural and myeloid lineages could point to their role of pericytes in responding to injury and neurological diseases (Dore-Duffy et al. 2000; ElAli et al. 2014).
In GBM, GSCs have been reported to transdifferentiate into tumor pericytes, which form the majority of pericytes found within tumor tissue and assist in GBM cell proliferation and GSC self-renewal (Caspani et al. 2014; Cheng et al. 2013; Jackson et al. 2017). Neural stem cells (NSCs) have previously been shown to harbor the capacity to transdifferentiate into pericytes in normal brain tissue, indicating that GSCs could exploit such mechanisms to give rise to perivascular niche components (Cheng et al. 2013; Goldberg and Hirschi 2009). Due to the tendency of GSCs to localize near vasculature and due to their ability to give rise to malignant multipotent pericytes, researchers have suggested the possibility for such GSC-derived CD133+ “malignant pericytes” to drive tumor progression. Appaix et al. suggested an alternative “Cancer Pericytes” model for GBM initiation and progression where they hypothesized malignant CD133+ pericytes to act as COIs (Appaix et al. 2014). According to this hypothesis, existing pericytes behaving as MSCs could attain a neural stem cell-like phenotype by detaching from basement membrane, thereby forming a cancer stem-cell pool. These malignant pericyte-derived CSCs could then proliferate extensively, giving rise to a tumor mass which results in a hypoxic microenvironment. Subsequently, endothelial cells recruited through the CXCR4/ CXCL12 pathway could initiate angiogenesis. Malignant pericyte-derived CSCs could then differentiate into an aggressive mesenchymal phenotype, transdifferentiate into other cell types and drive tumor heterogeneity within the tumor mass or migrate to co-opted blood vessels to initiate another cycle of tumor formation. The authors speculated that such malignant pericytes could gain and lose CD133 expression during different stages of tumor formation and could thus explain the existence of CD133- stem cells within GBMs.
Additionally, Zhang et al. have reported that overexpression of cytoplasmic GT198 (a DNA repair gene that activates VEGF) within pericytes gives rise to tumors. The authors suggested that malignant pericytes could be derived from GT198 expressing GSCs or from normal pericytes that undergo somatic mutations upon microenvironmental stimuli. GT198+ malignant pericytes could also be resistant to radiotherapy and cause the failure of anti-VEGF therapies (Zhang et al. 2017).
Endothelial cells on co-opted blood vessels within the brain are thought to stimulate the migration of GSCs towards them through SDF-1/CXCR4 pathway and later induce their transdifferentiation into tumor pericytes by secreting TGF-β (Cheng et al. 2013). This has been suggested as a mechanism to allow for the proliferation of endothelial cells, as tumor pericytes present within the perivascular niche secrete VEGF and other paracrine factors. In addition, enhanced pericyte coverage of co-opted and angiogenic blood vessels is thought to render resistance to anti-angiogenic therapies (Gabriele Bergers and Hanahan 2008; Ribeiro and Okamoto 2015). The ability of GSCs to give rise to tumor pericytes would also indicate their independence from relying on perivascular cells and their progenitors within the peri-tumoral region for engineering a suitable microenvironment.
Caspani et al. have suggested a “Dual Cell of Origin” hypothesis where pericytes drive tumor diversification upon direct contact with GSCs. In their study, the authors reported that pericytes attain a stem cell-like state upon transfer of cytoplasm from GSCs giving rise to GSC-pericyte cell fusions. The authors reported the existence of GBM cells that expressed labels for both pericytes and GBM cells within orthotopically implanted xenografts, suggesting that aneuploid cells derived from multipolar division of these GSC-pericyte cell fusions could drive GBM diversification (Caspani et al. 2014). The findings mentioned in the above section indicate that pericytes within NVU are plastic and play a critical role in GBM initiation and progression.
Pericytes Are Involved in GBM Invasion
Pericytes are thought to play a protective role against tumor invasion by acting as physical barriers but poor and disorganized pericyte coverage is often observed within the tumor microenvironments which allows tumor to spread (Xian et al. 2006). Invading GBM cells tend to incorporate existing blood vessels into tumor tissue by a process termed “Vascular Co-option.” These co-opted blood vessels undergo necrosis upon angiopoietin-2 secretion by GBM cells, repeating events of hypoxia and subsequent invasion into surrounding areas of normal brain parenchyma (Liebelt et al. 2016; Reiss et al. 2005). Alternatively, edema caused due to incomplete coverage of angiogenic blood vessels by pericytes within tumor tissue has also been hypothesized to enhance tumor invasion by increasing intratumoral fluid pressure. This increased intratumoral fluid pressure is further thought to suppress surrounding blood circulation, resulting in hypoxic microenvironments, thereby stimulating the invasion of GBM cells into surrounding normal brain parenchyma (Cooke et al. 2012).
Cytoplasmic extensions of invading GBM cells around pericytes that they encounter, termed “Flectopodia,” have been suggested to facilitate the co-option of existing blood vessels by trafficking GBM cell cytoplasm into the cellular cortex of pericytes. GTPase Cdc42 was shown to play a critical role in the formation of such cytoplasmic extensions and pericyte activation (Caspani et al. 2014).
Poor pericyte coverage allows spreading of GBM, as incomplete coverage of angiogenic vessels allows vascular invasion by GBM cells. In addition, poor pericyte coverage also enhances the metastatic potential of other solid tumors that tend to migrate to the brain. Brain metastatic lung and melanoma cells that extravasate through capillaries were shown to survive and proliferate for long periods of time only when in contact with the abluminal endothelial cells of capillaries in a pericyte-like position. This mechanism where tumor cells position themselves similar to pericytes when in contact with the abluminal endothelial cells has been extensively studied in melanomas and is termed as pericyte mimicry or angiotropism (Bentolila et al. 2016; Lugassy et al. 2014; Scott et al. 2015). Pericyte mimicry not only allows for the survival and proliferation of tumor cells but has also been reported to be exploited by tumor cells for extravascular migratory metastasis. This mechanism allows tumor cells to spread to local and distant sites by avoiding vascular invasion. GBM cells have been well documented in in vivo and in vitro studies to invade surrounding normal brain parenchyma along abluminal side of capillaries through the mechanism of pericyte mimicry (Scott et al. 2015).
As mentioned in the previous section, the ability of GSCs to attain a pericyte-like phenotype and their similarities to pericytes within the GBM microenvironment, with regard to cell surface marker expression, could be a result of pericyte mimicry of tumor cells and could support the hypothesis that pericyte-like cells within the GBM microenvironment could also act as initiating cells.
Pericytes and the BBB
Pericytes have been shown to play a vital role in regulating BBB permeability by controlling the expression and alignment of tight and adherens junction proteins along with the transcytosis of molecules across the BBB (Sweeney et al. 2016). The importance of pericytes for the structural stability of CNS vessels and for maintaining the BBB was observed in PDGFβ knockout mice where pericyte depletion resulted in enhanced CNS vascular permeability due to BBB disruption (Armulik et al. 2011; Bell et al. 2010; Daneman et al. 2010; Sweeney et al. 2016). PDGF-BB(ligand)-PDGFRβ signalling pathway is critical for pericyte-endothelial cell interactions to maintain BBB stability and to regulate extravasation across the barrier into brain parenchyma. Transgenic mice with both PDGF and PDGFRβ null mutations have shown embryonic lethality due to the development of microvascular instability and hyperplasia, aneurysms, and microhemorrhages (Sweeney et al. 2016). In studies where PDGF-BB-PDGFRβ signalling pathway was partially disrupted, age-dependent BBB disruption was observed (Armulik et al. 2011; Bell et al. 2010; Daneman et al. 2010; Sweeney et al. 2016). Similarly, Abramsson et al. showed that the lack of PDGFRβ expression on pericytes resulted in their poor vascular coverage and enlargement of blood vessel diameter, resulting in increased leakiness (Abramsson et al. 2002). In addition, TGFβ, Ang1, and Notch signaling have been reported to play an important role in maintaining the integrity of the BBB (Bruna et al. 2007; Cheng et al. 2013; Liebner and Plate 2010; Reis and Liebner 2013; Sweeney et al. 2016). TGFβ-TGFβR2 signaling promotes pericyte maturation, proliferation, and attachment to endothelial cells. Aberrant signaling of downstream effectors of TGFβ-TGFβR2 pathway such as Smad1, Smad2, and Smad4 results in vascular destabilization and brain hemorrhages (Goumans and Mummery 2000; Li et al. 2011; Maddaluno et al. 2013; Sweeney et al. 2016). Additionally, forkhead transcription factor (Foxf2) which affects TGFβ signaling also plays a role in maintaining BBB integrity as Foxf2 knockout transgenic mice show BBB disruption, hemorrhages, perivascular edema, increase in luminal endothelial caveolae, and thinning of basal lamina of capillaries (Reyahi et al. 2015).
Pericytes have been observed to attach loosely and improperly cover angiogenic vessels within GBMs, yet proper pericyte coverage has been shown to result in stabilization of tumor blood vessels, thereby accelerating GBM cell proliferation and invasion. High TGF-β-Smad activity was shown to result in highly aggressive and proliferative GBMs in patients which conferred a poor prognosis. TGFβ-Smad pathway has also been shown to activate PDGFB gene expression in primary GBM cells in vitro (Bruna et al. 2007). Majority of pericytes found in GBMs are thought to arise from GSCs that transdifferentiate upon migration to endothelial cells through SDF-1-CXCR4 signaling pathway. TGFβ has been shown to play a vital role in inducing this transdifferentiation of GSCs into pericyte-like cells which help promote angiogenesis, stabilize tumor vessels, and contribute to GBM growth (Cheng et al. 2013).
Notch3, expressed by brain pericytes, has been shown to play an important role in interactions with endothelial cells and in maintaining the integrity of BBB. Abnormal TGFβ-Notch signaling has been reported to cause cerebral cavernous malformation. Transgenic mice with dysfunctional Notch signaling caused by knockout of RBP-Jκ transcriptional factor, showed brain hemorrhages (Li et al. 2011; Maddaluno et al. 2013; Sweeney et al. 2016). Like TGFβ, Notch1 signaling has also been shown to induce transdifferentiation of GSCs into pericyte-like cells in vitro which promote angiogenesis (Guichet et al. 2015). Angiopoietin-1 (Ang1) expressed by pericytes, which binds to Tie2 receptor tyrosine kinase on endothelial cells, maintains BBB integrity. Like TGFβ and Notch1, knockout of Ang1 in transgenic mice resulted in BBB disruption and promotion of angiogenesis (Suri et al. 1996). The major facilitator superfamily domain-containing 2a (MFSD2A), a BBB transporter whose expression is thought to depend on presence of pericytes, has also been reported to play a role in the formation and maintenance of BBB integrity (Ben-Zvi et al. 2014).
Pericytes interact with astrocytes through apolipoprotein E (APOE4)-LRP1 interaction, resulting in the activation of MMP-9 activity by signaling through cyclophilin A (CypA)-NFKB pathway, which promotes inflammation. Increased APOE4-LRP1 signaling and resulting increase in the activity of MMP-9 causes BBB disruption due to degradation of endothelial tight junctions and basement membrane (Bell et al. 2012; Sweeney et al. 2016). Pericytes also regulate perfusion through brain capillaries and regulate BBB permeability through their contractile nature which is facilitated by synthesis of vimentin, actin and myosin microfilaments, tropomyosin and desmin. As part of the NVU, pericytes are also in contact with interneurons and receive communications from the nervous system as the average distance between a neuron and a brain capillary is 8–23 μm. Conditioned media from cultured human brain pericytes has shown the presence of neurotrophic factors and pericytes have been shown to regulate capillary diameter and cerebral blood flow upon signaling from neurons (Hawkes et al. 2011; Lovick et al. 1999). Studies have shown that norepinephrine leads to pericyte contraction (reduction of capillary diameter) while GABA, dopamine, glutamate, and adenosine cause pericyte relaxation (increase in capillary diameter) (Sweeney et al. 2016).
The BBB provides a formidable challenge for delivering drugs into the CNS, especially to brain tumors. In addition to intact BBB in areas around brain tumors, leaky angiogenic vessels which cause edema in certain areas of tumors result in the ineffective and non-uniform delivery of systemically delivered therapies (Sattiraju et al. 2017a, b). Ineffective drug delivery is thought to be one of the major reasons for GBM relapse, as cells that are not exposed to effective concentrations of systemic therapies reduce therapeutic efficacy. Therefore, efforts are being made to either transiently disrupt the BBB or to target cells involved in regulating BBB permeability, in order to enhance the extravasation of systemic therapies into GBM. As pericyte recruitment and stabilization of blood vessels within brain tumors have been shown to regulate vascular permeability, strategies to disrupt pericyte recruitment by angiogenic endothelial cells are currently being investigated.
In a recent study by Behling et al. by targeting monomeric vascular endothelial cadherin, which is expressed on angiogenic vessels and endothelial progenitor cells (EPCs), using a monoclonal antibody E4G10 that was labeled to α particles, the authors reported enhanced survival in Ntva transgenic mice bearing GBMs. As extensive proliferation of pericytes is usually observed in this GBM model, their higher density was suspected to protect endothelial cells from radiotherapy. In addition to mitigating tumor growth, the authors also observed normalization of the morphology of angiogenic vessels, reduction of edema, and a decrease in pericyte coverage of these vessels. In addition, depletion of regulatory T cells (Tregs) and EPCs was also observed (Behling et al. 2016). In another recent study, Sattiraju et al. showed enhancement in BBB permeability in brains of mice bearing orthotopic GBMs by targeting integrin alpha-V beta-3 (αvβ3) using an αvβ3 targeted antagonist conjugated to partially polymerized liposomes that was labeled to α particles. Apart from observing enhanced BBB permeability, the authors also reported tumor cytotoxicity evidenced by nuclear accumulations of γH2Ax double-strand DNA break repair protein within tumor mass. Overexpression of αvβ3 by GBM cells, especially at invasive ends and the presence of proliferating malignant cells in perivascular regions could explain this effect. The authors in this study also observed enhanced BBB permeability in peritumoral and normal regions of the brain surrounding tumors, indicating that enhanced vascular permeability in these distal regions might not have been caused due to direct a particle-induced cellular effects (Sattiraju et al. 2017c).
Studies by both Behling et al. and Sattiraju et al. show the feasibility of targeting vascular and perivascular components within the NVU using short-ranged, targeted molecular irradiation to either enhance vascular perfusion or BBB permeability, to better deliver systemic drugs to GBMs and to reduce radio-resistance conferred by perivascular cells within GBM microenvironment. But, as evidenced in previous studies where pericyte ablation caused enhanced invasion and metastasis, further long-term evaluations to assess tumor resistance and remission in abovementioned strategies are necessary (Bentolila et al. 2016; Lugassy et al. 2014; Scott et al. 2015). Xiong et al. reported an alternative strategy of enhancing BBB permeability by remotely activating intracranially implanted genetically engineered MSCs using high-frequency focused ultrasound (HIFU) to secrete TNF-α. This study highlights the ability to locally deliver factors which can transiently influence vascular and perivascular cells that regulate BBB integrity and perfusion within normal brain parenchyma and GBMs, thus enhancing the delivery of systemic drugs to GBMs and CNS (Sattiraju et al. 2017b; Xiong et al. 2015).
Pericytes Drive Angiogenesis
Angiogenesis is a crucial multi-stage process resulting in the transformation of GBM into an aggressive disease (Jain et al. 2007). Highly proliferating cells within GBM tissue drive up nutrient and oxygen demand and the resulting hypoxic microenvironment stimulates the production of new and often-disorganized angiogenic blood vessels. Pericytes play a major role in this process by forming a scaffold for newly proliferating endothelial cells to form blood vessels and secrete factors that stabilize these newly formed angiogenic vessels (Mancuso et al. 2006; Ribeiro and Okamoto 2015). The process of angiogenesis is tightly regulated and requires direct contact and crosstalk between endothelial cells and pericytes. Pericytes are involved in secreting factors that stimulate endothelial tip sprouting from existing blood vessels in their vicinity, recruit endothelial cells, and aid in their proliferation. Upon endothelial recruitment and formation of a vessel structure, pericytes within GBM microenvironment wrap endothelial cells which results in vessel stabilization and maturation (Caspani et al. 2014).
Endothelial cells are thought to produce PDGFβ which recruits PDGFRβ expressing pericytes, which in turn secrete VEGF and Ang-1 to stabilize angiogenic blood vessels (Hellstrom et al. 1999). Studies examining the effect of a pan tyrosine kinase inhibitor (SU6668), which preferentially targets PDGFRβ in RIP1-Tag2 transgenic mice that generate pancreatic islet carcinomas showed that ablating pericytes from tumor blood vessels caused vascular regression (Bergers et al. 2003). GSC-derived pericyte-like cells within the GBM microenvironment have also been reported to express PDGFRβ and aid in vessel stabilization and maturation (Cheng et al. 2013). Inhibiting PDGF-BB-PDGFRβ signaling resulted in regression of tumor vessels, indicating that it affects the survival of endothelial cells, GSC-derived pericyte-like cells and co-opted pericytes (Hellstrom et al. 2001). Overexpression of PDGFβ by endothelial cells has been reported to increase pericyte coverage and accelerate tumor growth (Furuhashi et al. 2004).
TGF-β, expressed by pericytes in a latent form, also plays a critical role in angiogenesis and GBM progression. TGF-β-TGFβR2 signaling has been reported to result in the stabilization of angiogenic vessels, and high expression of TGF-β-Smad has been reported to result in a poor prognosis for GBM patients. TGF-β-Smad pathway has been shown to induce expression of PDGF-B in GBM cells and the transdifferentiation of GSCs into pericyte-like cells, which aid in angiogenic vessels stabilization and maturation (Goumans and Mummery 2000; Maddaluno et al. 2013). TGF-β signaling alone has been reported to be able to induce genetic and phenotypic changes, often seen in altered vasculature within GBMs.
Notch1-DII4 signaling has been reported to be critical in regulating sprouting angiogenesis. Inactivation of Notch1 or DII4 genes have shown to cause increased endothelial tip sprouting and tip-cell numbers, indicating that Notch1-DII4 signaling at endothelial sprout restricts tip sprouting towards VEGF-A gradients, thereby ensuring proper sprouting and branching patterns (Gerhardt et al. 2003). Pericytes have been reported to enhance the survival of endothelial cells and promote angiogenesis by secreting VEGF-A, which binds to VEGFR2 expressed on endothelial cells. VEGFR2 stimulates the upregulation of survival genes such as Bcl-2, survivin and X-linked inhibitor of apoptosis protein (XIAP) in endothelial cells (Franco et al. 2011). VEFG-A-VEGFR2 autocrine signaling within endothelial cells also promotes their survival. Vitronectin secreted by pericytes also causes an increase of VEGF-A expression in endothelial cells through integrin αv-NFκB signaling, which in turn causes intracellular stimulation of VEGFR2 to promote endothelial cell survival (Franco et al. 2011; Sweeney et al. 2016). Inhibiting angiogenesis by interfering with VEGF-A-VEGFR2 signaling was thought to provide significant benefit to GBM patients as it would result in the collapse of existing angiogenic vessels and prevent further tumor vascularization and growth. Bevacizumab (Avastin), a monoclonal antibody against VEGF-A showed very promising results in preclinical studies and was tested in two phase-III clinical trials (RTOG and AVAGlio) as a stand-alone and combinational therapeutic for patients with recurrent GBM (Friedman et al. 2009; Gilbert et al. 2014). Although patients initially showed decreased tumor growth, their tumors eventually grew resistant to the drug, resulting in a progression-free survival of about 10.6 months in Phase-III clinical trials with no added benefit to overall survival when compared to placebo arm (Friedman et al. 2009; Hamza et al. 2014).
Ephrin receptor EphB4, which controls vascular morphogenesis within the NVU during developmental angiogenesis, and its ligand ephrin-B2, which is expressed on brain pericytes and endothelial cells, play a vital role in pericyte-endothelial cell interactions. Ephrin-B2-EphB4 signaling has been reported to regulate pericyte migration and interaction with maturing blood vessels and could be involved in the vascular remodeling within the GBM microenvironment (Augustin and Reiss 2003). Vascular cell adhesion molecule-1 (VCAM-1), N-cadherin and integrin α4β1 play critical roles in regulating pericyte migration, vessel maturation, and survival of endothelial and mural cells during angiogenesis (Gerhardt et al. 2000).
Overexpression of endosialin (CD248) has also been linked to pro-angiogenic role of pericytes within the GBM microenvironment (Brady et al. 2004). Angiogenic vessels within GBMs were reported to be composed of two layers of pericytes, an abluminal layer of proliferating cells and an adluminal layer of cells surrounded by basal lamina. By restricting the recruitment of pericytes and their signaling with endothelial cells during angiogenesis, newly formed GBM blood vessels could be disrupted causing reduced GBM invasion and growth.
In a study by Svensson et al. using a RGS5-GFP transgenic mouse model, the authors showed that PDGFRβ and neuroregulin-2 (NG2)-expressing pericytes were activated and recruited to blood vessels within orthotopically implanted GBMs from peritumoral and distal regions within ipsilateral and contralateral hemispheres, including from the rostral SVZ. Activation and recruitment of pericytes from distant regions of the brain towards tumor tissue has been attributed to parenchymal diffusion of paracrine factors such as hypoxia inducible factor-1a (HIF-1α) through cerebrospinal fluid or cerebral edema. These tumor trophic pericytes showed CD13 MSC cell surface marker expression in areas surrounding tumor tissue, but did not show CD13 expression within tumor tissues, indicating a phenotypic shift as pericytes enter paracrine signaling networks within the GBM microenvironment. The results from this study indicate that pericytes migrate and integrate into the vasculature within GBMs, stabilize angiogenic blood vessels, and promote further angiogenesis (Svensson et al. 2015). In another study by Huang et al., NG2 was identified to be critical in integrin β1-dependent pericyte-endothelial cell interactions and ablation of NG2 resulted in two-fold reduction in vessel ensheathment by pericytes. NG2 ablation was also reported to cause reduced collagen IV deposition due to loss of collagen VI anchorage. Impaired vessel maturation and stabilization within GBM tissues of mice lacking NG2 expression resulted in decreased tumor progression, highlighting the importance of NG2 for successful angiogenesis (Huang et al. 2010).
Pericytes Contribute to Immune Microenvironment
GBM cell survival, proliferation, and invasion into peritumoral regions are thought to be facilitated by their evasion of immune surveillance by using multiple immunosuppressive mechanisms. Pericytes have been shown to regulate immune cell activity by secreting chemokines and other factors in addition to differentiating into macrophages, thus playing a critical role in immune surveillance and tumor clearance within the GBM microenvironment (Valdor et al. 2017). Brain pericytes have been reported to exhibit phagocytic properties and express macrophages cell surface markers such as CD11b, CD68, and MHC class II. Stimulation by IL-1β has been reported to result in the upregulation of iNOS and COX-2 within porcine brain pericytes (Balabanov et al. 1996). In addition, Pieper et al. reported that stimulation by IFN- γ or TNF-α resulted in an antigen-presenting activity within brain pericytes (Pieper et al. 2014). Pieper et al. also showed that stimulation by LPS, TNF-α, and IL-1β resulted in chemotactic recruitment and transmigration of neutrophils into the brain parenchyma (Pieper et al. 2013).
During GBM establishment and expansion, pericytes have also been reported to be co-opted by tumor cells through direct cell-cell contact to facilitate immunosuppression and eventual immune evasion of GBM cells (Caspani et al. 2014). Upregulation of PD-L1, CD90, PDGFRβ, CD248, and Rgs5 which inhibit CD4+ and CD8+ cytotoxic T cell activity was reported in pericytes derived from within-tumor microenvironments (Bose et al. 2013; Ochs et al. 2013; Ribeiro and Okamoto 2015). Valdor et al. recently reported that pericytes conditioned in vitro by GBM cells are characterized by high levels of anti-inflammatory cytokines, suppress the function of cytotoxic T cells and reduce antigen presentation within perivascular regions of orthotopically implanted GBMs. GBM conditioned pericytes (GBM-PCs) showed high levels of IL-10 and TGFβ anti-inflammatory cytokine expression (100–400 pg/mL) and low levels of TNFα expression (45 pg/mL) in vitro when compared to control native pericytes, indicating an immunosuppressive phenotype. GBM-PCs also showed upregulation of Il4ra (encoding interleukin-4 receptor alpha), Il1rn (encoding interleukin-1 receptor inhibitor), and increased expression of angiogenic cytokine IL-6. However, expression of CD80 and CD86 co-stimulatory molecules was reported to be reduced (Valdor et al. 2017).
The exact mechanisms through which pericytes influence immune activity within the tumor microenvironment are not yet clearly understood and further research is required to gain a pristine understanding of the multifaceted role that pericytes play within tumor microenvironments and the way in which their role changes during tumor progression and in response to therapy (Fig. 2.2).
Fig. 2.2.
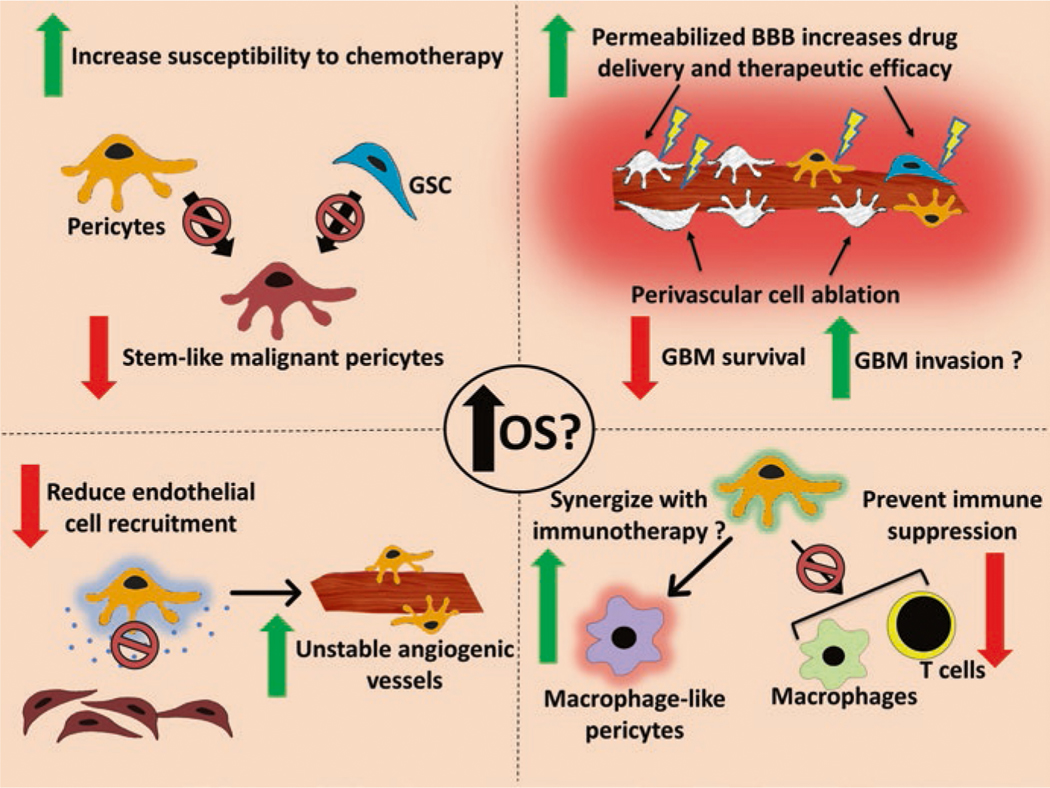
Diagram showing potential therapeutic interventions which could limit the contribution of pericytes to GBM overall survival and progression. OS overall survival, GSC glioblastoma stem cells, GBM glioblastoma, BBB blood-brain barrier
Conclusions: Opportunities for Therapeutic Intervention
The median overall survival of GBM patients has been improved to ~16 months using surgical resection followed by adjuvant temozolomide, but recurrent disease remains a major contributor to patient mortality (Roh et al. 2017). Novel therapeutic strategies such as immune checkpoint inhibitors, alternating electric fields, CAR T cells, stem cell-based drug and oncolytic viral delivery and strategies to enhance delivery of systemic therapies show the potential to increase patient survival and to reduce GBM recurrence in the future. As highlighted in this chapter, microenvironmental components play a vital role in the survival and proliferation of GBM cells. It is therefore critical to appreciate the complex relationship between GBM and their microenvironment and to design therapeutic strategies that would not just target one component of their microenvironment, as it would only serve to partially impede GBM survival and progression. The studies and their finding mentioned in this chapter highlight the critical role that pericytes play at various stages of GBM development.
The failure of multiple anti-GBM therapeutics in phase-III clinical trials has been attributed to tumor heterogeneity, ineffective drug delivery, invasive GBM cells, and therapy-resistant GSCs. As this chapter details, pericytes have been reported to significantly contribute to stemness, angiogenesis and altered perfusion, altered BBB permeability, GBM cell invasion and suppression of immune activity within the GBM microenvironment. It is therefore important to further elucidate the role of GSC-derived pericytes and native brain pericytes that are either present within or recruited into the tumor microenvironment. As previous studies have shown, targeting and ablating pericytes might not always stymie the growth and survival of GBM cells. This could be due to the multifaceted role of pericytes within the tumor microenvironments, their cellular plasticity and the existence of different subtypes of pericytes that contribute to various events at various stages of GBM establishment and expansion.
Knowledge of the role that pericytes play within the GBM microenvironment would therefore allow us to design therapies in the future such that they can circumvent cellular events which could otherwise compromise their efficacy and to also possibly design strategies to mitigate the role played by pericytes and other perivascular components co-opted by GBMs in treatment resistance.
References
- Abbott NJ. (2002). Astrocyte-endothelial interactions and blood-brain barrier permeability. Journal of Anatomy, 200(6), 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, & Begley DJ. (2010). Structure and function of the blood-brain barrier. Neurobiology of Disease, 37(1), 13–25. https://doi.org/10.1016Zj.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abramsson A, Berlin O, Papayan H, Paulin D, Shani M, & Betsholtz C. (2002). Analysis of mural cell recruitment to tumor vessels. Circulation, 105(1), 112. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Manchanda P, Vogelbaum MA, Ohlfest JR, & Elmquist WF. (2013). Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: Findings in an orthotopic rat xenograft model of glioma. Drug Metabolism and Disposition, 41(1), 33–39. 10.1124/dmd.112.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, et al. (2015). Adult lineage restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell, 28(4), 429–440. 10.1016/jxcell.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, & Iyer AK. (2017). PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Frontiers in Pharmacology, 8, 561 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appaix F, Nissou MF, van der Sanden B, Dreyfus M, Berger F, Issartel JP, & Wion D. (2014). Brain mesenchymal stem cells: The other stem cells of the brain? World Journal of Stem Cells, 6(2), 134–143. 10.4252/wjsc.v6.i2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, & Betsholtz C. (2011). Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell, 21(2), 193–215. 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, & Canoll P. (2006). Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. The Journal of Neuroscience, 26(25), 6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin HG, & Reiss Y. (2003). EphB receptors and ephrinB ligands: Regulators of vascular assembly and homeostasis. Cell and Tissue Research, 314(1), 25–31. 10.1007/s00441-003-0770-9. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Washington R, Wagnerova J, & Dore-Duffy P. (1996). CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvascular Research, 52(2), 127–142. 10.1006/mvre.1996.0049. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444(7120), 756–760. 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Baskar R, Lee KA, Yeo R, & Yeoh K-W (2012). Cancer and radiation therapy: Current advances and future directions. International Journal of Medical Sciences, 9(3), 193–199. 10.7150/ijms.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behling K, Maguire WF, Di Gialleonardo V, Heeb LEM, Hassan IF, Veach DR, et al. (2016). Remodeling the vascular microenvironment of glioblastoma with a-particles. Journal of Nuclear Medicine, 57(11), 1771–1777. 10.2967/jnumed.116.173559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, & Zlokovic BV. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron, 68(3), 409–427. https://doi.org/10.1016Zj.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature, 485(7399), 512–516. 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, & Gu C. (2014). Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature, 509, 507 10.1038/nature13324 https://www.nature.com/articles/nature13324#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentolila LA, Prakash R, Mihic-Probst D, Wadehra M, Kleinman HK, Carmichael TS, et al. (2016). Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: Implications for melanoma progression along extravascular pathways. Scientific Reports, 6, 23834 10.1038/srep23834 https://www.nature.com/articles/srep23834#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, & Hanahan D. (2008). Modes of resistance to anti-angiogenic therapy. Nature Reviews Cancer, 8(8), 592–603. 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, & Hanahan D. (2003). Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. The Journal of Clinical Investigation, 111(9), 1287–1295. 10.1172/jci17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Borges IDT, Gilson Sena IF, Almeida GG, da Silva Meirelles L, Goncalves R, et al. (2017a). How plastic are pericytes? Stem Cells and Development, 26(14), 1013–1019. 10.1089/scd.2017.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, & Frenette PS. (2016). Niche heterogeneity in the bone marrow. Annals of the New York Academy of Sciences, 1370(1), 82–96. 10.1111/nyas.13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Sattiraju A, Zhu D, Zulato G, Batista I, Nguyen VT, et al. (2017b). Novel peripherally derived neural-like stem cells as therapeutic carriers for treating glioblastomas. Stem Cells Translational Medicine, 6(2), 471–481. 10.5966/sctm.2016-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Wang Z-M, Messi ML, Enikolopov GN, & Delbono O. (2011). Nestin-GFP transgene reveals neural precursor cells in adult skeletal muscle. PLoS One, 6(2), e16816 10.1371/journal.pone.0016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang Z-M, Messi ML, Enikolopov GN, Mintz A, & Delbono O. (2013a). Skeletal muscle neural progenitor cells exhibit properties of NG2-glia. Experimental Cell Research, 319(1), 45–63. 10.1016/j.yexcr.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang Z-M, Messi ML, Mintz A, & Delbono O. (2013b). Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. American Journal of Physiology-Cell Physiology, 505(11), C1098–C1113. 10.1152/ajpcell.00171.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang Z-M, Messi ML, Mintz A, & Delbono O. (2014a). Pericytes: Multitasking cells in the regeneration of injured, diseased, and aged skeletal muscle. Frontiers in Aging Neuroscience, 6, 245 10.3389/fnagi.2014.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang Z-M, Messi ML, Olson JD, Mintz A, & Delbono O. (2014b). Type-2 pericytes participate in normal and tumoral angiogenesis. American Journal of Physiology-Cell Physiology, 307(1), C25–C38. 10.1152/ajpcell.00084.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, & Delbono O. (2015). Pericytes at the intersection between tissue regeneration and pathology. Clinical Science (London, England), 128(2), 81–93. 10.1042/cs20140278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, & Radisky D. (2001). Putting tumours in context. Nature Reviews Cancer, 1(1), 46–54. 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, & Oldfield EH. (1994). Convection-enhanced delivery of macromolecules in the brain. Proceedings of the National Academy of Sciences, 91(6), 2076–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A, Barik S, Banerjee S, Ghosh T, Mallick A, Bhattacharyya Majumdar S, et al. (2013). Tumor-derived vascular pericytes anergize Th cells. The Journal of Immunology, 191, 971–981. [DOI] [PubMed] [Google Scholar]
- Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, & Itinteang T. (2016). Cancer stem cell hierarchy in glioblastoma multiforme. Frontiers in Surgery, 3, 21 10.3389/fsurg.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady J, Neal J, Sadakar N, & Gasque P. (2004). Human endosialin (tumor endothelial marker 1) is abundantly expressed in highly malignant and invasive brain tumors. Journal of Neuropathology and Experimental Neurology, 63(12), 1274–1283. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. (2004). Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Research, 64(3), 920–927. 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- Brat DJ, & Van Meir EG. (2004). Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Laboratory Investigation, 84, 397 10.1038/labinvest.3700070. [DOI] [PubMed] [Google Scholar]
- Brooks MD, Sengupta R, Snyder SC, & Rubin JB. (2013). Hitting them where they live: Targeting the glioblastoma perivascular stem cell niche. Current Pathobiology Reports, 1(2), 101–110. 10.1007/s40139-013-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. (2016). Regression of glioblastoma after chimeric antigen receptor T-cell therapy. The New England Journal of Medicine, 375(26), 2561–2569. 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, et al. (2007). High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell, 11(2), 147–160. 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Cabrera MC, Hollingsworth RE, & Hurt EM. (2015). Cancer stem cell plasticity and tumor hierarchy. World Journal of Stem Cells, 7(1), 27–36. 10.4252/wjsc.v7.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell, 11(1), 69–82. 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Caspani EM, Crossley PH, Redondo-Garcia C, & Martinez S. (2014). Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS One, 9(7), e101402 10.1371/journal.pone.0101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorige-nicity. Cell, 154(1), 61–74. 10.1016/jxell.2013.00.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaichana KL. (2014). The need to continually redefine the goals of surgery for glioblastoma. Neuro-Oncology, 16, 611–612. 10.1093/neuonc/not326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ding Z, Peng Y, Pan F, Li J, Zou L, et al. (2014). HIF-1a inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS One, 9(6), e98882 10.1371/journal.pone.0098882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, & Parada LF. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature, 488(7412), 522–526. 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. (2013). Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell, 153(1), 139–152. 10.1016/jxell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C-W, Wang C-C, Wu C-P, Lin Y-J, Lee Y-C, Cheng Y-W, & Hsieh C-H (2012). Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro-Oncology, 14(10), 1227–1238. 10.1093/neuonc/nos195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. (2012). Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell, 21(1), 66–81. 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. (2008). A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell, 3(3), 301–313. 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Cho RW, & Clarke MF. (2007). Cancer stem cells: Models and concepts. Annual Review of Medicine, 58, 267–284. 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, & Barres BA. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature, 468(7323), 562–566. 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, & Marsden PA. (2013). Angiogenesis in Glioblastoma. New England Journal of Medicine, 369(16), 1561–1563. 10.1056/NEJMcibr1309402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst MW, Cao Y, & Moeller B. (2008). Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nature Reviews Cancer, 8(6), 425–437. http://www.nature.com/nrc/journal/v8/n6/suppinfo/nrc2397_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, & Rafols JA. (2000). Pericyte migration from the vascular wall in response to traumatic brain injury. Microvascular Research, 60(1), 55–69. 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- Driessens G, Kline J, & Gajewski TF. (2009). Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunological Reviews, 229(1), 126–144. 10.1111/j.1600-065X.2009.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois LG, Campanati L, Righy C, D’Andrea-Meira I, Spohr T. C. L. d. S. e., Porto-Carreiro I, et al. (2014). Gliomas and the vascular fragility of the blood brain barrier. Frontiers in Cellular Neuroscience, 8, 418 10.3389/fncel.2014.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. (2010). A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain, 133(Pt 4), 973–982. 10.1093/brain/awq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A, Thériault P, & Rivest S. (2014). The role of pericytes in neurovascular unit remodeling in brain disorders. International Journal of Molecular Sciences, 15(4), 6453–6474. 10.3390/ijms15046453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SM, Judy KD, Dunphy I, Jenkins WT, Nelson PT, Collins R, et al. (2004). Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Research, 64(5), 1886–1892. [DOI] [PubMed] [Google Scholar]
- Eyupoglu IY, Buchfelder M, & Savaskan NE. (2013). Surgical resection of malignant gliomas-role in optimizing patient outcome. Nature Reviews Neurology, 9(3), 141–151. [DOI] [PubMed] [Google Scholar]
- Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. (2010). NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells, 28(1), 5–16. 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidoamore A, Cristiano L, Antonosante A, d’Angelo M, Di Giacomo E, Astarita C, et al. (2016). Glioblastoma stem cells microenvironment: The paracrine roles of the niche in drug and radioresistance. Stem Cells International, 2016, 6809105 10.1155/2016/6809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco M, Roswall P, Cortez E, Hanahan D, & Pietras K. (2011). Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood, 118(10), 2906–2917. 10.1182/blood-2011-01-331694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. (2009). Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Journal of Clinical Oncology, 27(28), 4733–4740. 10.1200/jco.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, & Verma IM. (2014). Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Reports, 15(3), 244–253. 10.1002/embr.201338254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Sjoblom T, Abramsson A, Ellingsen J, Micke P, Li H, et al. (2004). Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Research, 64(8), 2725–2733. [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. (2003). VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. The Journal of Cell Biology, 161(6), 1163–1177. 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Wolburg H, & Redies C. (2000). N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Developmental Dynamics, 218(3), 472–479. 10.1002/1097-0177(200007)218:3<472::aid-dvdy1008>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. (2014). A randomized trial of bevacizumab for newly diagnosed glioblastoma. New England Journal of Medicine, 370(8), 699–708. 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, & Rich JN. (2007). Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nature Reviews Cancer, 7(10), 733–736. 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- Goldberg JS, & Hirschi KK. (2009). Diverse roles of the vasculature within the neural stem cell niche. Regenerative Medicine, 4(6), 879–897. 10.2217/rme.09.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, & Mummery C. (2000). Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. The International Journal of Developmental Biology, 44(3), 253–265. [PubMed] [Google Scholar]
- Guichet PO, Guelfi S, Teigell M, Hoppe L, Bakalara N, Bauchet L, et al. (2015). Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells, 33(1), 21–34. 10.1002/stem.1767. [DOI] [PubMed] [Google Scholar]
- Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, & Konishi I. (2016). PD-1/ PD-L1 blockade in cancer treatment: Perspectives and issues. International Journal of Clinical Oncology, 21(3), 462–473. 10.1007/s10147-016-0959-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza MA, Mandel JJ, Conrad CA, Gilbert MR, Yung WK, Puduvalli VK, & DeGroot JF. (2014). Survival outcome of early versus delayed bevacizumab treatment in patients with recurrent glioblastoma. Journal of Neuro-Oncology, 119(1), 135–140. 10.1007/s11060-014-1460-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, & Weinberg RA. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. 10.1016/jxell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, & Carare RO. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathologica, 121(4), 431–443. 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yang K, Kim Y, et al. (2011). Glioma stem cell maintenance: The role of the microenvironment. Current Pharmaceutical Design, 17(23), 2386–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddleston JM, Li Z, Hjelmeland AB, & Rich JN. (2009). The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle, 8(20), 3274–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, & Betsholtz C. (2001). Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. The Journal of Cell Biology, 153(3), 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, & Betsholtz C. (1999). Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development, 126(14), 3047–3055. [DOI] [PubMed] [Google Scholar]
- Hodges TR, Ferguson SD, & Heimberger AB. (2016). Immunotherapy in glioblastoma: Emerging options in precision medicine. CNS Oncology, 5(3), 175–186. 10.2217/cns-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FJ, You WK, Bonaldo P, Seyfried TN, Pasquale EB, & Stallcup WB. (2010). Pericyte deficiencies lead to aberrant tumor vascularizaton in the brain of the NG2 null mouse. Developmental Biology, 344(2), 1035–1046. https://doi.org/10.1016Zj.ydbio.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Rani MR, Ahluwalia MS, Bae E, Prayson RA, Weil RJ, et al. (2012). Endothelial expression of TNF receptor-1 generates a proapoptotic signal inhibited by integrin alpha6beta1 in glioblastoma. Cancer Research, 72(6), 1428–1437. 10.1158/0008-5472.can-11-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Hamanishi J, Chamoto K, & Honjo T. (2017). Cancer immunotherapies targeting the PD-1 signaling pathway. Journal of Biomedical Science, 24(1), 26 10.1186/s12929-017-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Hassiotou F, & Nowak A. (2015). Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis, 36(2), 177–185. 10.1093/carcin/bgu243. [DOI] [PubMed] [Google Scholar]
- Jackson S, ElAli A, Virgintino D, & Gilbert MR. (2017). Blood-brain barrier pericyte importance in malignant gliomas: What we can learn from stroke and Alzheimer’s disease. Neuro-Oncology, 19(9), 1173–1182. 10.1093/neuonc/nox058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, & Batchelor TT. (2007). Angiogenesis in brain tumours. Nature Reviews Neuroscience, 8(8), 610–622. http://www.nature.com/nrn/journal/v8/n8/suppinfo/nrn2175_S1.html. [DOI] [PubMed] [Google Scholar]
- Jhaveri N, Chen TC, & Hofman FM. (2016). Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Letters, 380(2), 545–551. 10.1016/j.canlet.2014.12.028. [DOI] [PubMed] [Google Scholar]
- Kang J, Demaria S, & Formenti S. (2016). Current clinical trials testing the combination of immunotherapy with radiotherapy. Journal for Immunotherapy of Cancer, 4, 51 10.1186/s40425-016-0156-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. (2010). Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell, 6(5), 421–432. 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, & Rich JN. (2015). Cancer stem cells in glioblastoma. Genes & Development, 29(12), 1203–1217. 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, et al. (2011). Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Developmental Cell, 20(3), 291–302. 10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, & Hu J. (2016). Glioma stem cells: Signaling, microenvironment, and therapy. Stem Cells International, 2016, 7849890 10.1155/2016/7849890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, & Plate KH. (2010). Differentiation of the brain vasculature: The answer came blowing by the Wnt. Journal of Angiogenesis Research, 2(1), 1 10.1186/2040-2384-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovick TA, Brown LA, & Key BJ. (1999). Neurovascular relationships in hippocampal slices: Physiological and anatomical studies of mechanisms underlying flow-metabolism coupling in intraparenchymal microvessels. Neuroscience, 92(1), 47–60. [DOI] [PubMed] [Google Scholar]
- Lugassy C, Zadran S, Bentolila LA, Wadehra M, Prakash R, Carmichael ST, et al. (2014). Angiotropism, pericytic mimicry and extravascular migratory metastasis in melanoma: An alternative to intravascular cancer dissemination. Cancer Microenvironment, 7(3), 139–152. 10.1007/s12307-014-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon JG, Mokarram N, Saxena T, Carroll SL, & Bellamkonda RV. (2017). Engineering challenges for brain tumor immunotherapy. Advanced Drug Delivery Reviews, 114, 19–32. https://doi.org/10.1016Zj.addr.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. (2013). EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature, 498(7455), 492–496. 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, et al. (2006). Rapid vascular regrowth in tumors after reversal of VEGF inhibition. The Journal of Clinical Investigation, 116(10), 2610–2621. 10.1172/jci24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannino M, & Chalmers AJ. (2011). Radioresistance of glioma stem cells: Intrinsic characteristic or property of the ‘microenvironment-stem cell unit’? Molecular Oncology, 5(4), 374–386. 10.1016/j.molonc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gonzâlez A, Calvo GF, Pérez Romasanta LA, & Pérez-Garcia VM. (2012). Hypoxic cell waves around necrotic cores in glioblastoma: A biomathematical model and its therapeutic implications. Bulletin of Mathematical Biology, 74(12), 2875–2896. 10.1007/s11538-012-9786-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, & Tofilon PJ. (2009). Physiologic oxygen concentration enhances the stem-like properties of CD 133+ human glioblastoma cells in vitro. Molecular Cancer Research, 7(4), 489–497. 10.1158/1541-7786.mcr-08-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, et al. (2010). Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Molecular Cancer, 9, 133 10.1186/1476-4598-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, et al. (2008). Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. Journal of Clinical Oncology, 26(18), 3015–3024. 10.1200/jco.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- Nakada M, Nambu E, Furuyama N, Yoshida Y, Takino T, Hayashi Y, et al. (2013). Integrin a3 is overexpressed in glioma stem-like cells and promotes invasion. British Journal of Cancer, 108(12), 2516–2524. 10.1038/bjc.2013.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nduom EK, Weller M, & Heimberger AB. (2015). Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology, 17(suppl_7), vii9–vii14. 10.1093/neuonc/nov151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklasson M, Jarvius M, Haglund C, Chantzi E, Bergstrom T, Nyberg F, et al. (2017). Abstract 4175: Targeting of a mesenchymal profile in order to sensitize multitherapy resistant glioblastoma clones. Cancer Research, 77(13 Supplement), 4175. [Google Scholar]
- Ochs K, Sahm F, Opitz CA, Lanz TV, Oezen I, Couraud PO, et al. (2013). Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. Journal of Neuroimmunology, 265(1–2), 106–116. 10.1016/j.jneuroim.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, et al. (2008). Identification of A2B5+CD133-tumor-initiating cells in adult human gliomas. Neurosurgery, 62(2), 505–514.; discussion 514–505 10.1227/01.neu.0000316019.28421.95. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. (2005). The blood-brain barrier: Bottleneck in brain drug development. NeuroRx, 2(1), 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. (2012). Drug transport across the blood-brain barrier. Journal of Cerebral Blood Flow & Metabolism, 32(11), 1959–1972. 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, et al. (2012). BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1a stability and MGMT expression. Cell Death & Disease, 3, e412 http://www.nature.com/cddis/journal/v3/n10/suppinfo/cddis2012153s1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, & Kanmogne GD. (2006). Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. Journal of Neuroimmune Pharmacology, 1(3), 223–236. 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Pieper C, Marek JJ, Unterberg M, Schwerdtle T, & Galla HJ. (2014). Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Research, 1550, 1–8. https://doi.org/10.1016Zj.brainres.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Pieper C, Pieloch P, & Galla HJ. (2013). Pericytes support neutrophil transmigration via interleukin-8 across a porcine co-culture model of the blood-brain barrier. Brain Research, 1524, 1–11. 10.1016/j.brainres.2013.05.047. [DOI] [PubMed] [Google Scholar]
- Razavi S-M, Lee KE, Jin BE, Aujla PS, Gholamin S, & Li G. (2016). Immune evasion strategies of glioblastoma. Frontiers in Surgery, 3, 11 10.3389/fsurg.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis M, & Liebner S. (2013). Wnt signaling in the vasculature. Experimental Cell Research, 319(9), 1317–1323. 10.1016/j.yexcr.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Reiss Y, Machein MR, & Plate KH. (2005). The role of angiopoietins during angiogenesis in gliomas. Brain Pathology, 15(4), 311–317. 10.1111/j.1750-3639.2005.tb00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyahi A, Nik AM, Ghiami M, Gritli-Linde A, Pontén F, Johansson BR, & Carlsson P. (2015). Foxf2 is required for brain pericyte differentiation and development and maintenance of the blood-brain barrier. Developmental Cell, 34(1), 19–32. 10.1016/j.devcel.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Ribeiro AL, & Okamoto OK. (2015). Combined effects of pericytes in the tumor microenvironment. Stem Cells International, 2015, 8 10.1155/2015/868475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. (2010). Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature, 468(7325), 824–828. 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Roh TH, Park HH, Kang S-G, Moon JH, Kim EH, Hong C-K, et al. (2017). Long-term outcomes of concomitant chemoradiotherapy with temozolomide for newly diagnosed glioblastoma patients: A single-center analysis. Medicine, 96(27), e7422 10.1097/md.0000000000007422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolle CE, Sengupta S, & Lesniak MS. (2010). Challenges in clinical design of immunotherapy trials for malignant glioma. Neurosurgery Clinics of North America, 21(1), 201–214. 10.1016/j.nec.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Durden DL, Van Meir EG, & Brat DJ. (2006). ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. Journal of Neuropathology & Experimental Neurology, 65(6), 529. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, & Restifo NP. (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science, 348(6230), 62–68. 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostomily RC, Spence AM, & Silbergeld DL. (2005). Neurosurgical management of high-grade gliomas In Moore AJ. & Newell DW. (Eds.), Neurosurgery: Principles and practice (pp. 167–186). London: Springer London. [Google Scholar]
- Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, & Bijangi-Vishehsaraei K. (2015). Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes & Diseases, 2(2), 152–163. 10.1016/j.gendis.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattiraju A, Sai KKS, & Mintz A. (2017a). Glioblastoma stem cells and their microenvironment. Advances in Experimental Medicine and Biology, 1041, 119–140. 10.1007/978-3-319-69194-7_7. [DOI] [PubMed] [Google Scholar]
- Sattiraju A, Sun Y, Solingapuram Sai KK, Li KCP, & Mintz A. (2017b). Maximizing local access to therapeutic deliveries in glioblastoma. Part IV: Image-guided, remote-controlled opening of the blood-brain barrier for systemic brain tumor therapy In De Vleeschouwer S. (Ed.), Glioblastoma. Brisbane (AU: ): Codon Publications; Copyright: The Authors. [PubMed] [Google Scholar]
- Sattiraju A, Xiong X, Pandya DN, Wadas TJ, Xuan A, Sun Y, et al. (2017c). Alpha particle enhanced blood brain/tumor barrier permeabilization in glioblastomas using integrin alpha-v beta-3-targeted liposomes. Molecular Cancer Therapeutics, 16(10), 2191–2200. 10.1158/1535-7163.mct-16-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MA, Shen J, Lam K, Yen YH, Shrestha S, Mravic M, Asatrian G, Chung CG, Lugassy C, Barnhill RL, Dry SM, Peault B, & James AW. (2015). Review of pericytes in tumor biology. International Journal of Orthopaedics, 2(3), 301–306. [Google Scholar]
- Segerman A, Niklasson M, Haglund C, Bergstrom T, Jarvius M, Xie Y, et al. (2016). Clonal variation in drug and radiation response among glioma-initiating cells is linked to proneural-mesenchymal transition. Cell Reports, 17(11), 2994–3009. 10.1016/j.celrep.2016.11.056. [DOI] [PubMed] [Google Scholar]
- Sharpe AH, & Abbas AK. (2006). T-cell costimulation—biology, therapeutic potential, and challenges. New England Journal of Medicine, 355(10), 973–975. 10.1056/NEJMp068087. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Hide T, & Dirks PB. (2004a). Cancer stem cells in nervous system tumors. Oncogene, 23(43), 7267–7273. 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. (2004b). Identification of human brain tumour initiating cells. Nature, 432, 396–401. 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. (2011). Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proceedings of the National Academy of Sciences of the United States of America, 108(11), 4274–4280. 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KS, Phi JH, Cho B-K, Wang K-C, Lee JY, Kim DG, et al. (2010). Long-term outcomes in children with glioblastoma. Journal of Neurosurgery: Pediatrics, 6(2), 145–149. 10.3171/2010.5.PEDS09558. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology, 10(5), 459–466. 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. (1996). Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell, 87(7), 1171–1180. [DOI] [PubMed] [Google Scholar]
- Svensson A, Ozen I, Genové G, Paul G, & Bengzon J. (2015). Endogenous brain pericytes are widely activated and contribute to mouse glioma microvasculature. PLoS One, 10(4), e0123553 10.1371/journal.pone.0123553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Ayyadurai S, & Zlokovic BV. (2016). Pericytes of the neurovascular unit: Key functions and signaling pathways. Nature Neuroscience, 19, 771 10.1038/nn.4288 https://www.nature.com/articles/nn.4288#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R, Garcia-Bernal D, Bueno C, Rodenas M, Moraleda JM, Macian F, & Martinez S. (2017). Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget, 8(40), 68614–68626. https://doiorg/10.18632/oncotarget.19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, & Olson JJ. (2010). Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA: A Cancer Journal for Clinicians, 60(3), 166–193. 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yin B, Wang HY, & Wang RF. (2014). Current advances in T-cell-based cancer immunotherapy. Immunotherapy, 6(12), 1265–1278. 10.2217/imt.14.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, & Sontheimer H. (2014). Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nature Communications, 5, 4196–4196. 10.1038/ncomms5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M, Cloughesy T, Perry JR, & Wick W. (2012). Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology, 15, 4–27. 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, & Lippoldt A. (2002). Tight junctions of the blood-brain barrier: Development, composition and regulation. Vascular Pharmacology, 38(6), 323–337. 10.1016/S1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Fallier-Becker P, Mack AF, & Wolburg-Buchholz K. (2012). The disturbed blood-brain barrier in human glioblastoma. Molecular Aspects of Medicine, 33(5–6), 579–589. 10.1016/j.mam.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Xian X, Hâkansson J, Stâhlberg A, Lindblom P, Betsholtz C, Gerhardt H, & Semb H. (2006). Pericytes limit tumor cell metastasis. The Journal of Clinical Investigation, 116(3), 642–651. 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Sun Y, Sattiraju A, Jung Y, Mintz A, Hayasaka S, & Li KCP (2015). Remote spatiotemporally controlled and biologically selective permeabilization of blood-brain barrier. Journal of Controlled Release: Official Journal of the Controlled Release Society, 217, 113–120. https://doi.org/10.1016Zj.jconrel.2015.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. (2015). Cancer immunotherapy: Harnessing the immune system to battle cancer. The Journal of Clinical Investigation, 125(9), 3335–3337. 10.1172/jci83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang Y, Rashid MH, Liu M, Angara K, Mivechi NF, et al. (2017). Malignant pericytes expressing GT198 give rise to tumor cells through angiogenesis. Oncotarget, 8(31), 51591–51607. 10.18632/oncotarget.18196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Verhaak RGW, & Canoll P. (2012). The cellular origin for malignant glioma and prospects for clinical advancements. Expert Review of Molecular Diagnostics, 12(4), 383–394. 10.1586/erm.12.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidar Z, Mardor Y, Jonas T, Pfeffer R, Faibel M, Nass D, et al. (2004). Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: A phase I/II clinical study. Journal of Neurosurgery, 100(3), 472–479. 10.3171/jns.2004.1003.0472. [DOI] [PubMed] [Google Scholar]