

Abstract
A new family of structurally well-defined molybdenum alkylidyne catalysts for alkyne metathesis, which is distinguished by a tripodal trisilanolate ligand architecture, is presented. Complexes of type 1 combine the virtues of previous generations of silanolate-based catalysts with a significantly improved functional group tolerance. They are easy to prepare on scale; the modularity of the ligand synthesis allows the steric and electronic properties to be fine-tuned and hence the application profile of the catalysts to be optimized. This opportunity is manifested in the development of catalyst 1f, which is as reactive as the best ancestors but exhibits an unrivaled scope. The new catalysts work well in the presence of unprotected alcohols and various other protic groups. The chelate effect entails even a certain stability toward water, which marks a big leap forward in metal alkylidyne chemistry in general. At the same time, they tolerate many donor sites, including basic nitrogen and numerous heterocycles. This aspect is substantiated by applications to polyfunctional (natural) products. A combined spectroscopic, crystallographic, and computational study provides insights into structure and electronic character of complexes of type 1. Particularly informative are a density functional theory (DFT)-based chemical shift tensor analysis of the alkylidyne carbon atom and 95Mo NMR spectroscopy; this analytical tool had been rarely used in organometallic chemistry before but turns out to be a sensitive probe that deserves more attention. The data show that the podand ligands render a Mo-alkylidyne a priori more electrophilic than analogous monodentate triarylsilanols; proper ligand tuning, however, allows the Lewis acidity as well as the steric demand about the central atom to be adjusted to the point that excellent performance of the catalyst is ensured.
Introduction
In a recent Communication, we disclosed complex 1a as the prototype of a new generation of molybdenum alkylidyne complexes, termed “canopy catalysts”, for alkyne metathesis because of their distinguishing tripodal silanolate ligand framework (Figure 1).1 Shortly after our paper had been published, the Lee group presented its work that had incidentally pursued the same ligand design.2
Figure 1.

Prototype of the canopy catalysts and the parent triphenylsilanolate complexes; modulation of the donor properties of a silanolate ligand caused by the facile bending and stretching of the Mo–O–Si angle.
Even though the focus of these two parallel investigations had been somewhat different, both reached the conclusion that such complexes are more than just a tethered variant of catalyst 2a carrying triphenylsilanolate ligands, which had set new standards in the field of alkyne metathesis when it was introduced by our group a decade ago.3−6 Outlined below is a comprehensive study into this new family of canopy catalysts, including the optimization of their synthesis, a first ligand-tuning exercise, structural investigations, a portrayal of their electronic nature, and new insights into the elementary steps of the catalytic cycle. Most importantly, a focused evaluation of the catalytic performance revealed an unrivaled stability toward numerous basic and protic sites, including unprotected alcohols and even moisture. Overall, this investigation shows that 1a and relatives bear great potential and might well mark a new milestone in the evolution of alkyne metathesis at large.7−10
Results and Discussion
Design Principles
Complexes 2 and derived bench-stable adducts such as [2·(phen)] (phen = 1,10-phenanthroline) owe their excellent application profile to the synergy between the operative molybdenum alkylidyne unit11 and the ancillary silanolate ligands.3−5 Silanolates are weaker π- and σ-donors than ordinary alkoxides12 and have “adaptive” ligand properties (Figure 1):3−5 bending and stretching of a Mo–O–Si hinge comes with thermal motion at almost no cost. Any such change of the bond angle, however, alters the hybridization of oxygen and hence the degree of π-donation, which, in turn, gently modulates the energy of the catalyst’s frontier orbitals. For this very property, 2 is able to meet the opposing electronic optima of the elementary steps passed through during the catalytic cycle: substrate binding and metallacycle formation are favored by a more Lewis-acidic center, whereas the cycloreversion step and extrusion of the product as the chemically inverse operations have the exact opposite electronic demand.3−5,13,14 At the same time, (triaryl)silanolates15 are sufficiently bulky to preclude bimolecular decomposition and/or competing associative pathways, preventing alkyne polymerization from occurring. For the longer O–Si and Si–C bonds, however, the bulk is sufficiently remote from the molybdenum center not to impede substrate binding or product dissociation; only for very hindered alkynes is the size of the silanolates critical.16 In that silanolates lower the barriers of all elementary steps and, at the same time, protect the catalyst, they render the overall reaction efficient on electronic and steric grounds.3−7
The tempered Lewis acidity that 2a and relatives draw from the metal/ligand cooperativity also accounts for a remarkable compatibility with functional groups.3−5 Numerous applications to complex and/or sensitive target molecules bear ample witness of this fact;17,18 an instructive example is shown in Scheme 1. Even certain protic groups are tolerated,19,20 although important limitations still remain. This particular problem is challenging: the polarization of a Schrock alkylidyne renders the C atom of the [M≡CR] unit innately nucleophilic and basic and hence potentially prone to degradation in a protic environment.11,21 Ligand exchange, however, is equally obstructive: any in situ replacement of the silanolates of 2 jeopardizes the virtues of the catalyst and ultimately entails loss of activity. This may explain why 2 usually fails when primary alcohols or related protic sites are present; the comparison shown in Scheme 1 illustrates this paradox.
Scheme 1. Current State of the Art: Triarylsilanolate Catalysts That Tolerate Diverse Functionality and Complex Settings but Fail with Simple Unhindered Alcohols.

Complex 2 was released in situ from the corresponding ate complex chosen as precatalyst, cf. ref (17c).
Under the premise that ligand exchange is detrimental, it seemed reasonable to assume that a chelate structure might improve stability vis-à-vis alcohols and other protic substrates. Our first attempt to reduce this plan to practice met with only partial success (Scheme 2).22 Specifically, the trisilanol ligands 4 and 5 were designed but failed to afford distinct tripodal alkylidyne complexes on reaction with precatalyst 3. Rather, they lead to partial cross-linking with formation of ill-defined mixtures, which nonetheless show good activity and, as a matter of fact, an improved functional group tolerance compared to 2.22 It was with the help of these two-component systems that certain substrates containing primary −OH groups could be metathesized for the first time in appreciable yields.22
Scheme 2. Previous Attempts to Prepare Molybdenum Alkylidyne Complexes with a Podand Ligand Architecture22,23.

Cross-linking (partial) also seems to plague an alternative ligand scaffold of type 6 composed of tethered phenol units:23−25 the only fully characterized podand complex 7 derived from 3 and 6a proved inactive, whereas the composition of those mixtures that are catalytically competent is unclear.23 In consideration thereof, we were prompted to reassess the design and came up with 1 as the prototype of a new generation of catalysts for alkyne metathesis.1,26 In a formal sense, the three Ph3SiO– groups of the parent complex 2 are tied together via an additional phenyl ring that forms the basal plane of a podand ligand framework. In contrast to 4 and 5, the backbone of this new type of tridentate ligand (11) contains only sp2-hybridized C atoms, which reduces the degrees of conformational freedom and should render the formation of well-defined tripodal complexes more favorable; however, this comes at the cost of increased rigidity, even though we had hoped that the conformationally flexible Mo–O–Si angle would partly compensate the perceived stiffness of complexes of type 1. It is important to note that each alkyne metathesis catalyst must be able to accommodate different geometries, that is, the tetrahedral ligand environment of the alkylidyne and a trigonal-bipyramidal extreme at the stage of the metallacyclobutadiene intermediate formed upon [2 + 2] cycloaddition.7−10 Provided that this essential geometric boundary condition is met, the modularity of the design should allow the properties of this new catalyst family to be fine-tuned.
Ligand Synthesis and Variation
The preparation of the parent ligand 11a starts off with the cyclocondensation of 2-bromoacetophenone to afford tribromide 8a (Scheme 3).27,28 This reaction was originally performed with triflic acid at elevated temperature but was later found to be much higher yielding upon gentle release of HCl from SiCl4 and EtOH;29 under these conditions, the yield of 8a increased from 55% to 90% on a 20 g scale. Exhaustive metal/halogen exchange with excess tBuLi in Et2O followed by quenching of the resulting triorganolithium intermediate with Ph2Si(OMe)2 works nicely (75%, 4 g scale), provided that the metalation step is performed at very low temperature (−125 °C).30 The final hydrolysis proceeds quantitatively on treatment of 9a with aqueous HCl (5 g scale). Trisilanol 11a is thus available in multigram quantity in three high-yielding steps.
Scheme 3. Ligand Synthesis.

Reagents and conditions: (a) SiCl4, EtOH, 0 °C → RT, 90% (X = H, 20 g scale); (b) TfOH, 130 °C, 55% (X = F); (c) tBuLi, Et2O, R2Si(OMe)2, −125 °C → RT, 75% (9a), 86% (9b), 12% (9c); (d) tBuLi, Et2O, R2SiH(Cl), −125 °C → RT, 91% (10d), 81% (10e); (e) aq. HCl, 0 °C → RT, quant. (11a, 5 g scale), 78% (11b), quant. (11c); (f) mCPBA, 94% (11d in CH2Cl2), 81–87% (11e, in tetrahydrofuran (THF)); (g) tBuLi, Et2O, benzophenone, −125 °C → RT, 78% (2 g scale).
Because this route is modular, it provides ready access to analogues as necessary for catalyst screening and optimization. To this end, compound 11b(28) bearing a fluorine substituent on the three arenes forming the fence was prepared in good yield. Additional compounds were made to study the influence of the substituents on silicon: quenching of the triorganolithium species derived from 8a with (MeOC6H4)2Si(OMe)2 followed by hydrolysis gave 11c. Although this reaction was less clean,31 a sufficient amount was secured (ca. 400 mg) to study the properties of this particular ligand. For the preparation of 11d,e with two aliphatic substituents on silicon, it was best to use R2Si(H)Cl (R = Me, iPr) as the electrophilic partner; oxidative cleavage of the Si–H bond in 10 with meta-chloroperoxybenzoic acid (mCPBA) furnished the desired compounds. Even though silanols are privileged ligands for molybdenum alkylidynes,3−7 we also prepared the carbinol analogue 12 for comparison by lithiation of 8a followed by a benzophenone quench (Scheme 3).26
In the solid state, the trisilanol ligands 11a,111b,2811c,28 and 11e (Figure 2) invariably adopt a conformation in which the three Si–OH groups are upward/inward-oriented as a consequence of a cyclic array of hydrogen bonds between the individual −Si–OH units. This favorable approximate C3 symmetry, as necessary for the formation of the targeted podand complexes, is maintained in solution, as evident from the NMR spectra. The situation is very different for the carbinol analogue 12 (Figure 3): one pair of reciprocal H-bonds connects only two of the alcohol groups, whereas the third −OH is oriented to the opposite side of the basal plane. Although line broadening in the NMR spectra indicates that the system is dynamic at RT, the two-up/one-down geometry is the average conformation of 12 in solution that gets increasingly locked upon cooling (see the Supporting Information).
Figure 2.
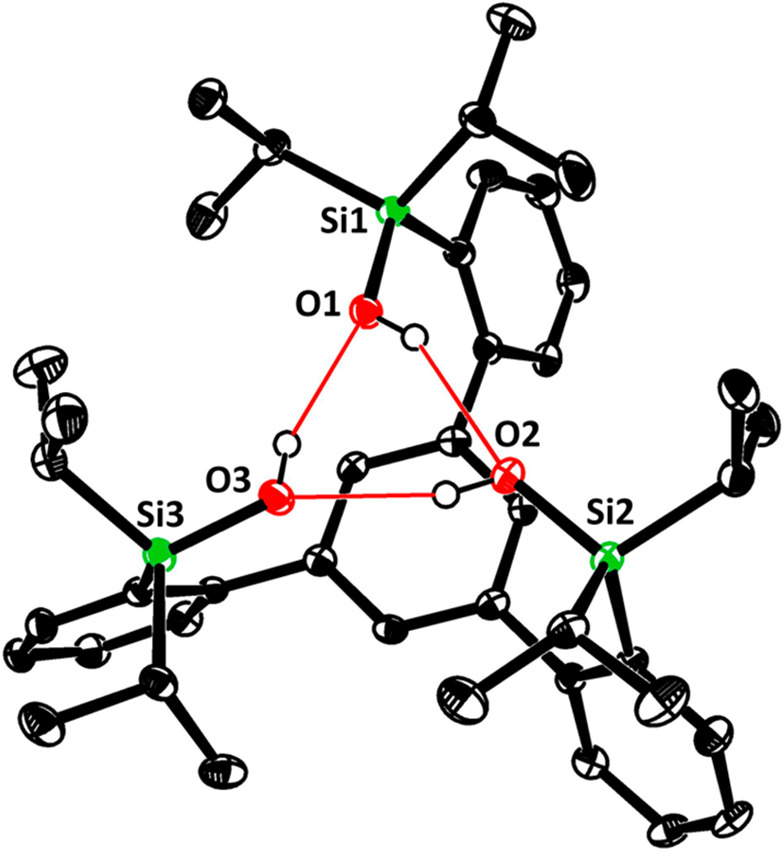
Structure of compound 11e in the solid state (only one of the two independent molecules in the unit cell is shown). Hydrogen atoms except for the −OH protons are not shown for clarity. The red lines indicate hydrogen bonds.
Figure 3.
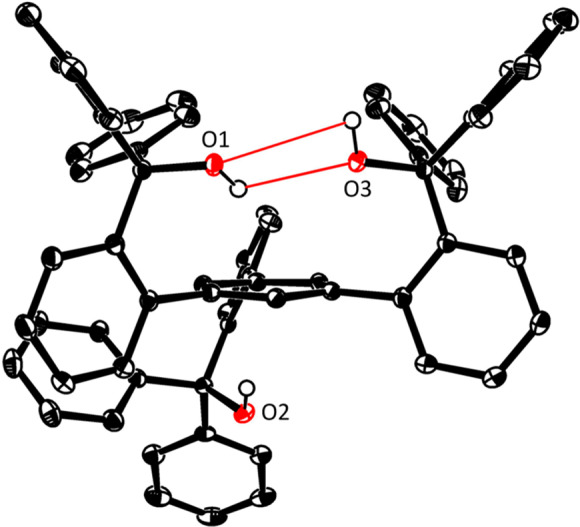
Structure of compound 12 in the solid state. Cocrystallized CH2Cl2 and hydrogen atoms (except for the −OH protons) are not shown for clarity. The red lines indicate hydrogen bonds.
Preparation of the Podand Complexes and the Aggregation Issue
The parent complex 2a is formed from the tribromoalkylidyne complex 13a by salt metathesis with Ph3SiOM (M = K, Na).1,3,4 However, this method was not deemed ideal for the preparation of the podand complexes of type 1 for the following reasons: (i) The conformation of trisilanols 11 is perfect for the envisaged podand complex formation; deprotonation almost certainly enforces an unfavorable change because charge repulsion destabilizes the upward/inward orientation of the Si–O vectors held together in 11 by hydrogen bonding. (ii) Any such conformational change, however, increases the likelihood of cross-linking. The resulting ill-defined products cannot convert to the targeted podand complex because salt metathesis is irreversible; a reduced yield of 1 is the likely consequence.32 (iii) The poor solubility of the trisodium (potassium) salts of 11 in aprotic organic media is a handicap in practical terms.33
The use of the molybdenum complex 3 bearing moderately basic amide ligands would probably allow these issues to be avoided. It has already been shown to undergo ligand exchange with our first-generation trisilanole ligands 4 and 5 (see Scheme 2).22 Yet, we sought a more practical solution because 3 is exceptionally sensitive, requiring rigorously anhydrous conditions and an Ar (not N2!) atmosphere.34 A convenient alternative was found in complexes 14, which themselves are catalytically inactive but fairly easy to make (Scheme 4). Because tert-butoxy groups are more basic than a silanole, it suffices to stir a solution of 11a and 14a in toluene at ambient temperature to achieve quantitative ligand exchange; moreover, the reaction is entropically favorable.35 The released tert-butanol can be evaporated in high vacuum, which makes the isolation of the resulting bright-yellow and only moderately air-sensitive complex [1a]2 straightforward. The analogous reaction of the all-carbon analogue 12 did not afford a podand complex (Scheme 5). Rather, the peculiar conformation of the ligand translates into the structure of the resulting complex 16: two tert-butoxide ligands of 14a were replaced by 12, which acts as a bidentate rather than tridentate ligand, and the third tert-alcohol unit is dangling and fails to substitute the remaining tert-butoxide even at elevated temperature (see the Supporting Information). Complex 16 showed only marginal activity in the test reaction chosen to benchmark the new catalysts (see below); however, it is reminiscent of a catalyst for applications in material science that had been deliberately designed to exhibit a tempered character.36,37
Scheme 4. Preparation of the Catalysts.

Reagents and conditions: (a) see ref (4); (b) NaOtBu, THF, 83%; (c) 11, toluene, 95% ([1a]2), 50% ([1b]2, 76% ([1c]2); (d) toluene, 60 °C, see text; (e) MeCN, quant. (NMR), see text.
Scheme 5. Carbinol Variant.

As previously described, the new podand complex [1a]2 is a supramolecular aggregate that dissociates quantitatively in toluene solution at 60 °C to give the desired monomeric species 1a;1 once formed, the monomer persists for extended periods of time (≫7 days at RT (NMR); see the Supporting Information) and shows excellent thermal stability (≫12 h in [D8]-toluene at 60 °C; see the Supporting Information). However, monomeric 1a reverts to the dimeric aggregate [1a]2 upon evaporation of the solvent or on cooling; complexes 1b,c show the same behavior. The acetonitrile adduct [1a·MeCN], in contrast, remains monomeric in the solid state: it was in this format that we were originally able to prove the podand ligand architecture about the molybdenum alkylidyne unit by X-ray crystallography.1
Although this aggregation in the solid state does not preclude alkyne metathesis from occurring,1 the rather poor solubility of [1a]2 and the need to disassemble this aggregate via ligation and/or heating are not ideal for applications in catalysis. Because crystals of [1]2 suitable for X-ray diffraction could not be grown, indirect evidence had to guide our efforts to improve the design with the aim of avoiding such complications. To this end, we revisited the structure of the parent complex 2a, which is also a dimeric aggregate in the solid state as well as in concentrated solution. Upon dilution, however—that is, under conditions typically used for ring-closing alkyne metathesis (RCAM)38—[2a]2 fully dissociates to the monomeric complex 2a (DOSY; see the Supporting Information). The X-ray structure (Figure 4)4 shows that the ortho-H atoms of each benzylidyne unit of [2a]2 are engaged in tight C–H/π interactions with neighboring phenyl groups of the silanolate ligands; the latter, in turn, experience numerous intermolecular C–H/π and π/π interactions39 with the phenyl rings of the second independent molecule in the unit cell. We suppose that [1a]2 is the result of analogous noncovalent interactions:40 supramolecular aggregate formation is hence likely a consequence of the multitude of fairly preorganized arene rings in the first ligand sphere about the molybdenum center.
Figure 4.
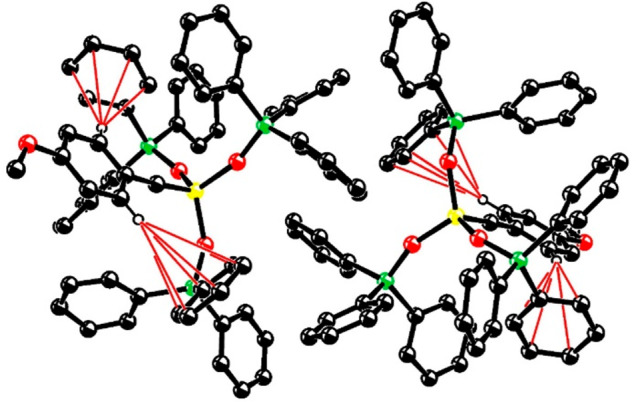
Structure of complex of [2a]2 in the unit cell. All H atoms except for the ortho-protons of the benzylidyne units are omitted for clarity. The red lines indicate the close interactions of these H atoms with neighboring phenyl rings of the silanolate ligands, indicative of C–H/π interactions. The packing reveals numerous intermolecular π/π and C–H/π interactions between the two independent molecules of this dimeric aggregate. Color code: Mo = yellow, O = red, Si = green, and C = black.
On the basis of this analysis, we envisaged two different ways to prevent supramolecular aggregate formation. Under the proviso that the C–H/π-contact of the ortho-benzylidyne protons instigates a network of tight intermolecular interactions, it may suffice to put ortho-substituents on the benzylidyne unit to break the contacts.41,42 The second conceivable design was the replacement of the phenyl groups responsible for the intermolecular contacts altogether by appropriate alkyl substituents.15
In pursuit of these ideas, the 2,6-dimethylbenzylidyne complex 1e was prepared from 14b(4,35) by the route outlined above (Scheme 6). In line with our expectation, 1e is indeed monomeric in the solid state (Figure 5) as well as in solution (DOSY; see the Supporting Information). The same is true for complexes 1d and 1f carrying two aliphatic substituents on the silicon bridges: both of them are monomeric in solution (see the Supporting Information), even though 1d retains the p-methoxybenzylidyne group. The X-ray structure of 1d (Figure 6) contains two independent molecules in the unit cell, but they do not entertain any short intermolecular contacts between them (see the Supporting Information).
Scheme 6. Additional Catalysts.

Reagents and conditions: (a) 11e, toluene, 99%; (b) 11d, toluene; (c) 11a, toluene, 65%; (d) 11d, toluene, 84%.
Figure 5.
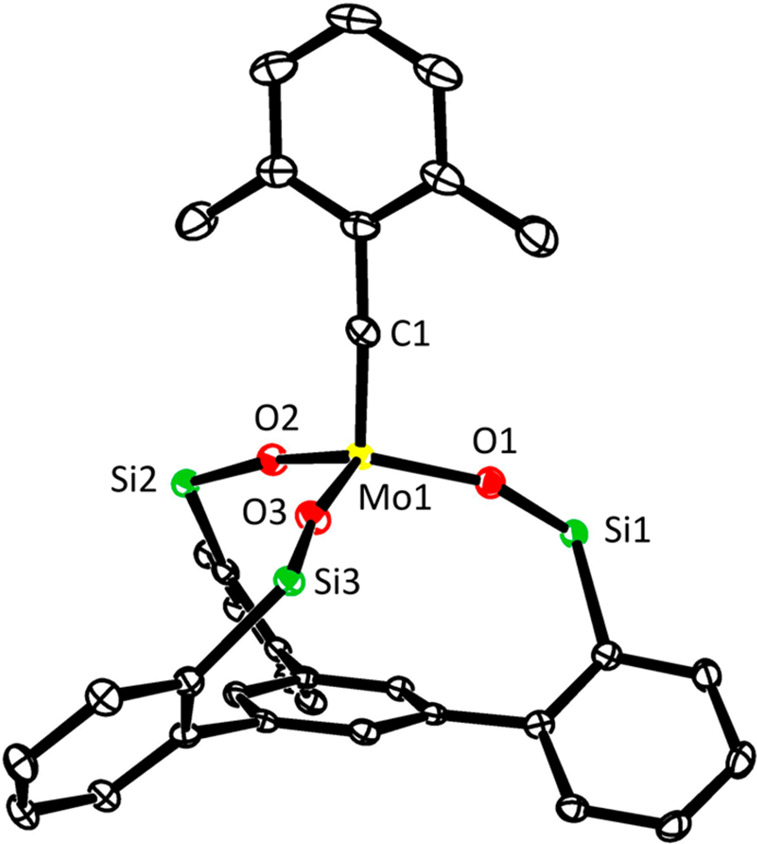
Representation of the truncated structure of complex 1e in the solid state, in which the lateral phenyl rings on silicon were removed to unveil the almost linear benzylidyne unit and the compressed array of the core, which clearly deviates from an ideal tetrahedral geometry; see text. Hydrogen atoms are omitted for clarity. For the full structure, see the Supporting Information.
Figure 6.
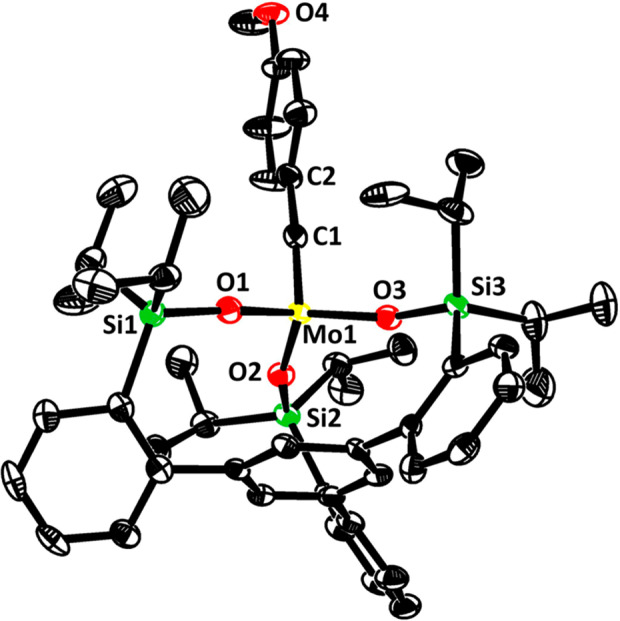
Structure of complex 1d in the solid state. Hydrogen atoms are omitted for clarity.
It is important to note that the reaction of the slimmest ligand 11d with 14b carrying an encumbered 2,6-dimethylbenzylidyne unit afforded the expected podand complex 1f together with a small amount of an organometallic impurity, which also contains an alkylidyne (see the Supporting Information). The analogous reaction of 11d with 14a bearing a p-methoxybenzylidyne, in contrast, furnished an ill-defined mixture that was not analyzed any further (Scheme 6). These observations suggest that an adequate steric balance between the size of the alkylidyne and the steric demand of the periphery is necessary to prevent (partial) cross-linking from occurring.
Structural Aspects and Electronic Implications
The structures of 1d, 1e, and [1a·MeCN]1 in the solid state verify the tripodal silanolate ligand architecture capping the coordination site trans to the alkylidyne. While the lengths of the Mo1≡C1 bonds (1.742(2) Å in 1e; 1.741(5) Å in 1d) fall into the expected range,3,4 the alkylidyne units Mo1≡C1–C2 of these complexes are almost linear, with bond angles of 177.1(2)° and 176.2(4)°, respectively. For comparison, the alkylidyne of 2b has a bond angle of only 171.4(2)°.4 Therefore, we believe that the linearity in 1d and 1e is significant rather than incidental.43 Such a linear array leads to optimal orbital overlap between the Mo1≡C1 unit and the arene and explains the rather short C1–C2 bond (1.448(3) Å in 1e). In line with this notion, the computed lowest unoccupied molecular orbital (LUMO) is indeed delocalized over the entire benzylidyne unit, with strong lobes on the 2,6-dimethylphenyl ring (Figure 7).
Figure 7.
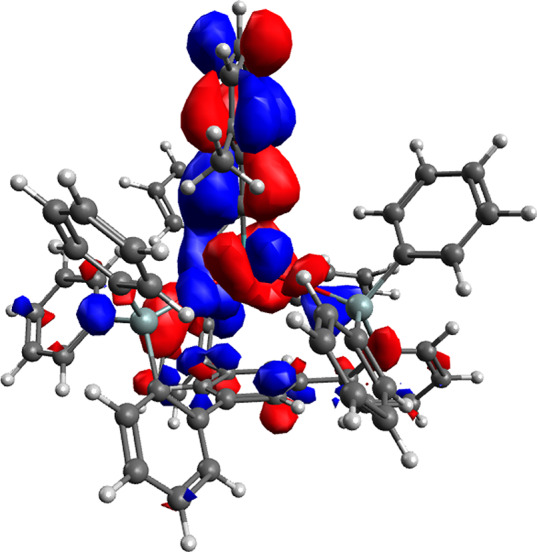
LUMO of complex 1e.
The bond angles merit detailed consideration. For the canopy catalyst 1e, the Mo–O–Si bond angles are 156.2(1)°, 164.4(1)°, and 170.6(2)°, whereas its monodentate cousin 2b shows 146.4(1)°, 154.1(1)°, and 169.4(1)°.4 The corresponding bond angles in 1d are fairly uniform (160.9(2)°, 161.3(2)°, 165(2)°) but again more obtuse on average than that of 2b. As a consequence of this stretching in the periphery, the ligand sphere about the Mo center in 1d and 1e notably deviates from an ideal tetrahedral geometry (Figure 5). This fact is also manifested in the significantly reduced C1–Mo1–O bond angles (102.6(1)°, 103.2(1)°, 103.4(1)° in 1e; ideal tetrahedron: 109.47°). Although the Mo center resides above the plane defined by the three O atoms (0.42 Å), the elevation is smaller than in the parent complex 2b (0.49 Å).
The Mo atom of 1e is located 3.52 Å above the centroid of the phenyl ring that forms the basal plane of the tripodal framework. At first sight, this situation resembles a largely electrostatic cation/π interaction39 but is likely more involved; the fact that the LUMO shows small but distinct lobes on the basal phenyl group (Figure 7) indicates a weak but non-negligible through-space orbital interaction.44 This aspect is subject to further investigations.
95Mo NMR Study
The structures of 1d, 1e, and [1a·MeCN]1 in the solid state provide only a static picture, but the observed geometric attributes of the complexes have important electronic implications as they impact on the hydridization of the O atoms and on the orbital overlap. Therefore, a more detailed investigation into the electronic character of the new catalysts in solution seemed warranted.
As a spin 5/2 nucleus with low natural abundance (ca. 15.9%), a low gyromagnetic ratio, but a rather low quadrupole moment, the 95Mo isotope has only rarely been used for analytical purposes in organometallic chemistry.45 We are aware of a single investigation into molybdenum alkylidynes,46 which reports the 95Mo NMR shifts of [(Me3SiCH2)3Mo≡CSiMe3] and [(Me3CCH2)3Mo≡CCMe3]47 but failed to record the signal of [(tBuO)3Mo≡CPh]; all three complexes are catalytically incompetent.
Despite this only partly encouraging precedent, we found this technique very useful in the present context. Good 95Mo NMR spectra of a representative set of complexes were obtained in [D8]-toluene at 60 °C. Quadrupolar relaxation is slower at higher temperature and hence the lines are sharper;48 at the same time, all complexes are monomeric in solution under these conditions. A well-resolved signal at δMo = 79.6 ppm was recorded for [(tBuO)3Mo≡CAr] (14a, Ar = p-MeOC6H4) (see the Supporting Information); this complex is a close relative of [(tBuO)3Mo≡CPh], which had defied detection in the only prior study.46 While 14a itself is catalytically inactive, it serves as the starting point for the preparation of the canopy catalysts (see Schemes 4 and 6).
For the lack of pertinent literature data, it is currently impossible to construe the recorded shifts by comparison with reference compounds. However, we are inclined to believe that the 95Mo shifts reflect changes in the electronic character of these complexes quite closely. Figure 8 compares the spectra of 1a and 1e, which carry the identical tripodal ligand but differ in the substitution of the benzylidyne. Although quite remote, the change from the p-MeO group in 1a to the 2,6-dimethyl substitution pattern in 1e entails notable deshielding. This fact proves that the Mo-alkylidyne is effectively coupled to the π-system of the arene: as a good donor substituent, the MeO– group imparts higher electron density onto the Mo center, which the spectral response seems to mirror.
Figure 8.

95Mo NMR spectra of the monomeric canopy complexes 1a and 1e differing only in the aryl substituent on the alkylidyne. The spectra were recorded at 60 °C.
To further probe this aspect, an additional series of complexes was analyzed. For the sake of direct comparison, all of them carried the p-methoxybenzylidyne group (Figure 9) but differ in the silanolate ligands; any electronic communication with the Mo center is now mediated via the O bridge. The electron-withdrawing fluorides in 1b cause deshielding relative to the parent compound 1a, whereas the two MeO groups in 1c entail a small but significant shift to higher frequencies. As expected, the more drastic electronic change upon formal replacement of the aryl groups on silicon by more electron-releasing isopropyl substituents in 1d causes a more pronounced effect in the same direction.49 Equally indicative is the finding that [(tBuO)3Mo≡CAr] (14a, Ar = p-MeOC6H4) (δMo = 79.6 ppm) resonates at much higher field (see the Supporting Information), reflecting the fact that tertiary carbinol ligands are better donors than silanols.12
Figure 9.

95Mo NMR spectra of different complexes (all in monomeric form) bearing the same aryl substituent on the alkylidyne. All spectra were recorded at 60 °C; Ar = p-MeOC6H4.
The trend observed in the 95Mo spectra finds correspondence in the 13C as well as 29Si NMR shifts (Table 1); the only exception is the alkylidyne C signal of 2a, but this complex has a different ligand set. Although caution has to be exerted, it is tempting to see a qualitative correlation between these spectral data with the electron density and hence Lewis acidity of the metal center. Under this proviso, the new complex 1a is more Lewis acidic than the parent complex 2a, even though the rather obtuse Mo–O–Si angles would suggest otherwise. The influence of the bond angles seems to be outweighed by the distortion of the first coordination sphere about the Mo(+6) center and the particular structural features of the tripodal ligand framework.
Table 1. Relevant 95Mo, 29Si, and 13C NMR Shifts ([D8]-Toluene, ppm) of Molybdenum p-Methoxybenzylidyne Complexes Bearing Different Silanolate Ligands.
| complex | 95Mo (δ) | 29Si (δ) | 13C (δ) |
|---|---|---|---|
| 1d | 358 | +10.2 | 303.3 |
| 2a | 397 | –8.0 | 300.5 |
| 1c | 414 | –9.1 | 309.3 |
| 1a | 419 | –9.9 | 310.4 |
| 1b | 434 | –9.8 | 311.4 |
Chemical Shift Tensor Analysis
The qualitative conclusion that the molybdenum alkylidyne unit of the canopy catalyst 1e is slightly more Lewis acidic than that of its cousins 2b containing monodentate triarylsilanolate ligands can be probed in greater detail by chemical shift tensor (CST) analysis of the alkylidyne carbon atom.50 This approach is particularly appealing as the relation between the 13C chemical shift and the molecular electronic structure of transition metal complexes is fairly well-explored51 and has been used to gain information on the electronic properties of metal alkylidynes and related complexes.13,52
Chemical shift is an anisotropic property that can be described by the three principal components of the chemical shift tensor ((δiso = (δ11 + δ22 + δ33)/3). The property calculated by ab initio methods is the shielding tensor σ (δii ≈ σiso,ref – σii), which can be deconvoluted into diamagnetic and paramagnetic terms (σ = σdia + σpara): while diamagnetic terms mainly arise from core orbitals and are hence quite insensitive to the electronic environment, the paramagnetic contributions are sensitive to frontier orbitals as they arise from the magnetically induced admixture of electronically excited states into the electronic ground state via the angular momentum operator Li. This relation between chemical shift and molecular electronic structure makes chemical shift one of the few molecular descriptors that are sensitive to the anisotropy of the electronic structure around a nucleus. In a pictorial view, strong deshielding of a given nucleus occurs along a direction i if a high-lying occupied orbital on this nucleus can be superimposed onto a low-lying vacant orbital on the same nucleus by rotation along the axis i. Importantly, this deshielding is expected to increase for a decreasing energy gap between the involved orbitals and is hence often indicative of reactive frontier orbitals (i.e., high-lying highest occupied molecular orbital (HOMO) and/or low-lying LUMO).
The three most relevant orbital couplings for a metal alkylidyne are schematically shown in Figure 10: a previous investigation into metal alkylidynes showed that the σ-symmetric orbitals contribute to deshielding by a magnetic coupling with the low-lying vacant π*(M–C) orbital, whereas the bonding π(M–C) orbital causes deshielding by interaction with the vacant σ*(M–C) orbital.13 The computed geometries of 2b and 1e were found to reproduce the crystal structures well (for computational details, see the Supporting Information). The calculated isotropic shifts of 2b (δC = 303 ppm) and its podand cousin 1e (δC = 321 ppm) show good agreement with the experimental values (307 and 312 ppm, respectively). Consistent with the experiment, the podand complex 1e is more deshielded. Interestingly, all three principal components of the shielding tensor make small contributions that sum up to this net outcome (Figure 11). The larger deshielding of 1e suggests that the π*(Mo–C) orbital as the LUMO of 1e is lower-lying than that of 2b, which indicates a higher electrophilicity of the canopy variant. This conclusion is consistent with the strongly delocalized π*(Mo–C) orbital (Figure 7): the lobes on the basal phenyl group of 1e suggest that this remote substituent assists in lowering the energy of the LUMO.
Figure 10.
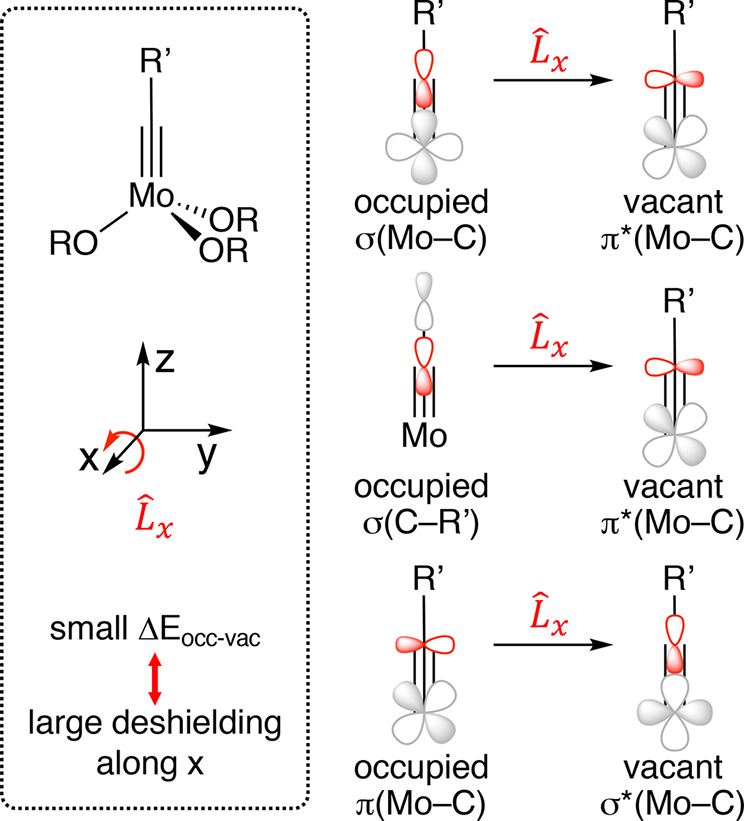
Three relevant orbital couplings.
Figure 11.
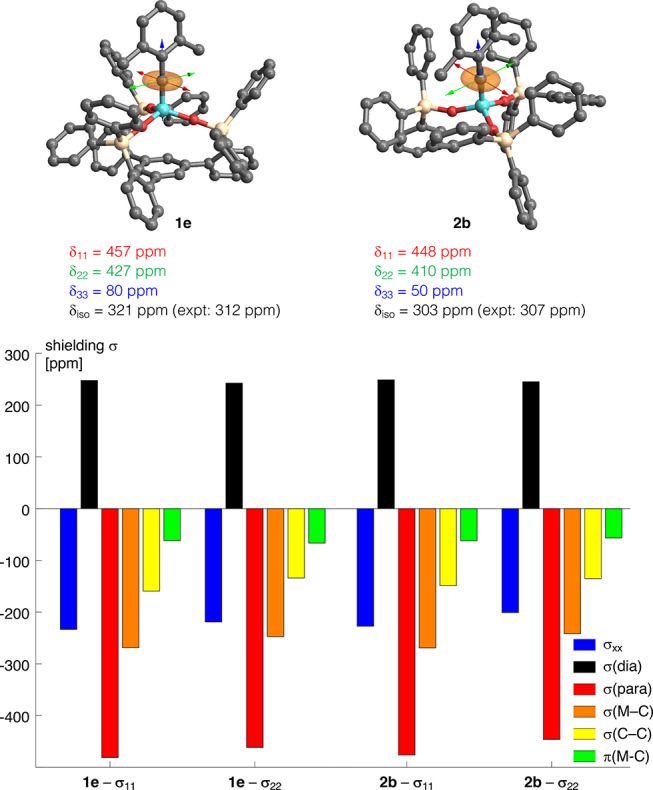
CST analysis of complexes 1e and 2b.
Reactive Intermediates
It is not clear, a priori, whether the lower-lying LUMO of complexes of type 1 is an advantage for catalysis. As mentioned in the Introduction, it takes a well-balanced electrophilicity management for a complex to become an efficient alkyne metathesis catalyst: a certain Lewis acidity is necessary for the [2 + 2] cycloaddition to happen,7−10,53 but if the electrophilic character is too pronounced, the resulting metallacyclobutadiene is (over)stabilized and cycloreversion is disfavored (not to speak of the functional group tolerance that gets lost).13,14
It was therefore deemed relevant to check whether the more electrophilic canopy catalysts 1 might already enter the regime of (meta)stable metallacyclobutadienes. When 1a was treated with 3-hexyne (5 equiv) in [D8]-toluene in a temperature range between +23 and +40 °C, a new complex 18, which turned out to be a metallatetrahedrane, was detected rather than the expected metallacyclobutadiene 17 (Scheme 7). The Lee group had been the first to observe the formation of this unusual complex; because they managed to characterize 18 in detail by spectroscopic means and even X-ray crystallography, we refrained from repeating the entire exercise.2 Our observations fully confirm their lead finding; the fact that 18 is formed so easily implies that the metallatetrahedrane is slightly more stable than (or at least isoenergetic with) the starting molybdenum alkylidyne and the presumed metallacyclobutadiene 17 from which it is thought to derive according to theory.54 Because 1a is a competent catalyst (see below), the question arises about which role complex 18 plays in the catalytic process. On the basis of probability arguments, metallatetrahedranes had been considered as possible on-cycle intermediates,55 but computational studies and the isolation of a few unreactive representatives advocated against this early assumption.56−59 Since then, it is generally believed that metallatetrahedranes are unreactive sinks and/or a gateway to decomposition.60
Scheme 7. Discrete Intermediates Downstream from the Molybdenum Alkylidynes.

The recorded NMR data (EXSY, ROESY) show beyond a doubt that 18 exchanges with 3-hexyne as well as with the propylidyne complex 1a′, which proves that formation of the metallatetrahedrane is reversible (see the Supporting Information). However, the data gathered so far do not allow us to decide if 18 is a true on-cycle intermediate or just an off-cycle reservoir, which would be more in line with previous views.56−58 If it is off the cycle, the high concentration of 18 in the mixture is a serious handicap because it keeps much of the active molybdenum species away from doing its job. This question is highly relevant for further optimization and hence subject to ongoing investigations in this laboratory.
Benchmarking of the Catalytic Activity
With a series of well-characterized tripodal alkylidyne complexes of type 1 in hand, we were in the position to study how steric and electronic changes impact on the catalytic properties. The homometathesis of 1-methoxy-4-(prop-1-yn-1-yl)benzene (19) to form 1,2-bis(4-methoxyphenyl)ethyne (20a) was chosen to benchmark the catalysts, each in monomeric form (Figure 12). To monitor their activity by NMR, the reaction was performed at 27 °C in the absence of molecular sieves as a 2-butyne-sequestering agent3 to avoid any interference.61 Therefore the reaction leads only to an equilibrium comprising ca. 45% of unreacted starting material.
Figure 12.
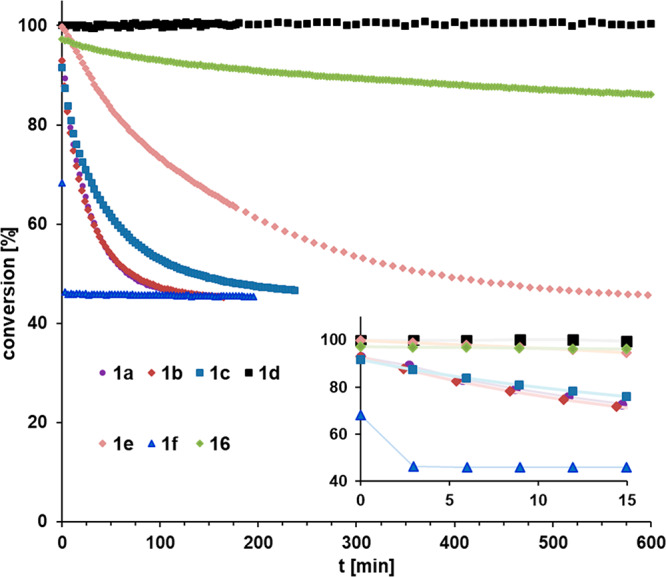
Benchmarking experiment (1H NMR): consumption of 1-methoxy-4-(prop-1-yn-1-yl)benzene (19) ([D8]-toluene, 0.1 M, 27 °C, 5 mol % catalyst loading). The inset shows that the reaction catalyzed by 1f had reached equilibrium in <5 min, when the second data point was recorded.
1a as the prototype complex of the new series shows appreciable activity in that the equilibrium is reached in ≤100 min.1 The reaction rate of 1b bearing fluorine substituents on the fence of the podand ligand is identical, within experimental error, whereas 1c containing MeO groups on the silanolate propellers is slightly less active. The comparison of 1a and 1e, which differ from each other only in the substitution pattern of the benzylidyne, shows that the 2,6-dimethyl-substituted variant results in markedly slower conversion even though equilibrium is eventually reached. This result must also be seen in light of the report by the Lee group, who found that their complex bearing a 2,4,6-trimethylphenyl group induced scrambling of 1-methoxy-4-(phenylethynyl)benzene only upon heating.2 Taken together, these observations indicate that 2,6-disubstitution of the benzylidyne group causes slow(er) and/or incomplete initiation. Figure 12 also shows that complex 16 with the carbinol ligand set exhibits much lower activity than 1a as the closest relative of the trisilanol series. This finding is in line with the notion that silanols are privileged ligands for molybdenum alkylidynes.3,4
Although 1a is a competent catalyst, it does not rival the parent complex 2a carrying three simple monodentate Ph3SiO groups, which effects the model reaction in ≤10 min. In our preliminary Communication, we conjectured that the higher rigidity of the tripodal scaffold might be the reason why the canopy variant 1a is ∼10 times slower than 2a.1 A stiffer backbone retards the changes back and forth between tetrahedral and trigonal-bipyramidal coordination geometries of the reactive intermediates passed through during a catalytic cycle. While this argument is certainly valid, the improved understanding of the nature of 1 makes us believe that additional factors might come into play. The accumulation of metallatetrahedrane 18 (see Scheme 7) is arguably relevant in this context: unless 18 itself is kinetically competitive, its formation in high concentration is a serious handicap as it siphons much of the active species into an off-cycle reservoir.
In any case, it seems that the ligand design must somehow counteract the build-up of Lewis acidity resulting from the distorted ligand sphere about the metal. Encouraged by the 95Mo NMR data, we tested the activity of 1d and 1f bearing more donating alkyl substituents on the Si bridges. The effect is massive, and the outcome is extreme. 1d endowed with isopropyl groups failed to catalyze the model reaction at ambient temperature (but does so in refluxing toluene), whereas its sibling 1f bearing methyl substituents is by far the most active precatalyst of the entire new series; it basically regains the activity of the parent complex 2a (Figure 12). We tentatively ascribe the striking difference between 1d and 1f to steric effects, supposing that the bulky isopropyl moieties disfavor or even prevent substrate binding (compare Figure 6). In any case, the performance of 1f is gratifying: this “turbo catalyst” is as reactive as its best ancestors but outperforms them by far in terms of functional group tolerance.
Scope and Applications
A truly comprehensive investigation into the functional group compatibility of the canopy complexes is beyond the scope of this Article, but the examples compiled below clearly show their promise.
The direct comparison of the parent triphenylsilanolate complex 2a with the new catalysts 1a and 1f is particularly instructive (Table 2): while 2a invariably failed to metathesize alkynes carrying unhindered primary alcohol groups, both canopy complexes afforded the desired products in good to excellent yields. As expected, catalyst 1f reacted much faster than 1a (ca. 2 versus 16 h), but their productivity is comparable, suggesting that different members of this family likely cover a similar substrate scope. The superior performance of the new catalysts is further illustrated by the formation of phenol 20b and product 22 carrying an ordinary secondary alcohol. The RCAM reaction leading to 23 containing two different propargylic substituents showcases two advantageous attributes at the same time: 2a is unable to cope with the unprotected −OH group, whereas both new catalysts are able to do so. However, only the slim turbo catalyst 1f provides the targeted cyclic monomer, whereas 1a gave mostly dimeric products; this aspect becomes highly relevant in the context of the much more demanding cycloalkyne 51 as the key precursor for the marine macrolide amphidinolide F (see below).
Table 2. Homo-Metathesis and Ring-Closing Alkyne Metathesis Reactions in the Presence of Unprotected −OH Groupsa.

Isolated yields of reactions performed with 5 mol % of catalyst loading in toluene at RT in the presence of MS 5 Å, unless stated otherwise.
At 60 °C.
With 10 mol % of 2a.
Mostly formation of dimers (NMR).
With 10 mol % of catalyst at 110 °C.
Yield determined by high-performance liquid chromatography (HPLC); in addition, ca. 20% of what appeared to be dimeric products were detected.
With 20 mol % of catalyst at 80 °C.
Using 30 mol % of catalyst at reflux temperature; yield over two steps (metathetic ring closure and reductive cleavage of the 2,2,6,6-tetramethylpiperidinyl group).
Compatibility with a propargylic −OH group is quintessential for the formation of 24. It is also of note that two of the three alkyne groups in the substrate reacted selectively.22 Once again, the superiority of 1f is striking because the standard catalyst 2a gave a poor yield and a bad mass balance. Cycloalkyne 25 as the key intermediate of the first total synthesis of the marine nor-cembranoid sinulariadiolide further illustrates the excellent application profile,62 in that the new catalyst tolerates the free propargylic −OH but also an electron-rich enol ester as well as a hydroxylamine.
In line with our initial design concept, it is the chelate effect that largely accounts for the much improved compatibility with unprotected −OH groups; it even entails a certain meta-stability of the canopy catalysts toward water (Scheme 8). Whereas addition of either a primary alcohol or H2O (10 equiv) to a solution of 2b in [D8]-toluene results in almost instantaneous solvolysis (hydrolysis) with quantitative release of Ph3SiOH (≤5 min, NMR), it takes ∼9 h until 1e is fully decomposed by water under otherwise identical conditions. Importantly, the spectra do not provide any indication for protonation of the alkylidyne: ligand exchange rather than destruction of the operative Mo≡CR unit is (mainly) accountable for the loss of activity. This observation is in line with recent experimental as well as computational data from the literature.21,63 Although water remains ultimately detrimental, a half-life on the order of 1 h is a chemical virtue without precedent in metal alkylidyne chemistry in general; it is fully appreciated if one considers the extreme sensitivity of earlier generations of alkyne metathesis catalysts, not least the otherwise very powerful complex 3 and its precursors.7−10,34,64 In this context, we reiterate that it has become common practice to supplement the reaction mixtures with molecular sieves.3,4 Although this additive primarily serves as a sequestering agent for the released 2-butyne in reactions conducted at RT, it ensures a level of dryness and hence a lifespan of a canopy catalyst that allow certain applications to be carried out in technical-grade solvents that need not be rigorously dried and purified prior to use. Indeed, under these standard conditions, the test reaction used to benchmark the catalyst performance proceeded quantitatively in technical-grade toluene (87 ppm water) as well as in toluene containing residual EtOH (5300 ppm) in the presence of MS 5 Å. Although there is certainly very much room for improvement, these preliminary data are encouraging and show that the podand ligand structure pays a valuable dividend in practical terms too.
Scheme 8. Positive Impact of the Chelate Effect.

In view of the foregoing, it will not come as a surprise that various other protic sites are also well-tolerated by the new catalysts, including a free phenol, amides, carbamates, malonates, β-ketoesters, sulfones, and the fluorenyl group (Table 2 and Schemes 9, 10, 11, and 12). β-Ketoesters in particular caused problems in the past for the parent complexes 2,5 whereas the new catalyst 1a furnished product 26 containing this common motif in good yield.
Scheme 9. Homo-Metathesis Reactions of Substrates Comprising C–H and/or N–H Acidic Sites.

The reactions were performed with 1a (5 mol %) in toluene at RT in the presence of MS 5 Å.
At 90 °C with 10 mol % of catalyst.
Scheme 10. Survey of the Functional Group Tolerance: Homo-Metathesis Reactions.

The choice of catalyst is color-coded; at 60 °C.
At 90 °C; NR = no reaction
Scheme 11. Ring-Closing Alkyne Metathesis (RCAM): Test Reactions.

Unless stated otherwise, all reactions were performed with 1a (5 mol %) in toluene in the presence of powdered MS 5 Å at ambient temperature.
At 60 °C.
Scheme 12. Formation of Key Intermediates of Previous Natural Product Total Syntheses.

Reagents and conditions: (a) 1a (5 mol %), toluene, MS 5 Å, RT, quant.; (b) 1a (5 mol %), toluene, MS 5 Å, 60 °C, 75%; (c) 1a (5 mol %), toluene, MS 5 Å, RT, 85%; (d) 1f (30 mol %), toluene, MS 5 Å, 80 °C, 81%.
Because our spectral and computational data had suggested that the podand ligand architecture upregulates the Lewis acidity, it was deemed equally important to prove that the new catalysts remain active in the presence of various donor sites. Although gentle heating was necessary in several cases, it is rewarding to find that basic nitrogen in the form of a pyridine (31), a secondary or tertiary amine (34 and 35), or a hydroxylamine (25) does not bring metathesis to a halt. The same is true for a nitrile-containing compound (20d), even though the structure of the adduct [1a·MeCN] had shown that cyano groups exhibit appreciable affinity to the molybdenum center.1 The good results with thioethers and various sulfur-containing heterocycles (30, 32, 36, and 47) are equally remarkable, as is the compatibility with a Weinreb amide (37), which is a potential chelate ligand for a high-valent transition metal center.
Auspicious results were also obtained with yet other exigent functionality. An elimination-prone primary tosylate and a primary iodide went uncompromised (38 and 39). The ring-closing alkyne metathesis (RCAM) reaction leading to cycloalkyne 43 proves that a nitro group, as such, is compatible (Scheme 11); however, attempted formation of the nitro-substituted tolane derivative 20f failed even at elevated temperature. This limitation shows that electron-deficient alkynes in general are quite refractory substrates.65 The observation that other products bearing electron-withdrawing substituents required more forcing conditions corroborates this aspect (20c,d and 31). Attempted formation of the aldehyde derivative 20g, however, resulted in decomposition. Aldehydes have previously been found incompatible, a limitation that obviously persists;3−5 ketones, in contrast, pose no problem (20e, 26, and 47).
The rewarding outcome of the model reactions made us confident that the new catalysts qualify for more advanced applications. Material from previous total synthesis campaigns from our laboratory provided us with the opportunity to confirm this expectation. The two demanding cases 24 and 25 (Table 2) have already been discussed earlier in the context of the tolerance of the new catalysts toward unprotected −OH groups. Next, we reprepared product 45, which is a key precursor for the synthesis of homoepilachnene, an ingredient of the defense secretions of the pupae of a Mexican beetle (Scheme 12);66 although rather simple in structural terms, this example shows the compatibility of 1a with a base-sensitive C–H acidic fluorenyl group. Clearly more challenging is the formation of cycloalkyne 47, which leads to the potent anticancer agent epothilone C upon Lindlar reduction of the triple bond;67 the thiazole ring of 47 adds an important entry to the list of heterocycles that do not quench the catalyst’s activity. Equally noteworthy is the fact that the elimination-prone aldol substructure and the ketone are not affected. Even more compelling evidence for the mildness of the method is found in the successful formation of 49, an immediate precursor of amphidinolide V:68 the vinyl epoxide and the allyl ether subunits are both sensitive to acid and/or base yet remain intact. As expected, the double bonds present in the substrate are tolerated, in line with observations already made with previous generations of alkyne metathesis catalysts.3−10
The successful formation of 51 as the key intermediate en route to the marine toxin amphidinolide F69 illustrates yet another relevant aspect: only the slim turbo catalyst 1f allowed this densely decorated macrocycle to be formed in good yield, whereas 1a afforded acyclic dimers by homo-metathesis; complex 2a with Ph3SiO– ligands had also largely caused homodimerization, most likely as a result of steric hindrance about one of the double bonds in diyne 50.69 The leaner ligand sphere of 1f obviously helps to overcome yet another of the few limitations that siloxide-based alkyne metathesis catalysts had faced in the past (see also entry 7 in Table 2).19
Overall, the functional group tolerance of catalysts of type 1 is deemed remarkable and certainly unrivaled by any other molecularly defined alkyne metathesis catalyst known to date. From the conceptual viewpoint, this excellent profile may help to correct the common misperception that high-valent early transition metal catalysts in general and MoVI-based alkylidyne and alkylidene catalysts in particular provide only limited opportunities when working with polyfunctionalized compounds.70
Conclusions
The new family of well-defined molybdenum alkylidyne complexes endowed with a distinctive podand topology is distinguished by good to excellent catalytic activity for alkyne metathesis and a truly remarkable compatibility with exigent functionality, including various free −OH groups and several other protic sites. It is largely due to the chelate effect that even a certain stability toward water was noticed, which is without parallel in the literature. Because the preparation of the required ligands is straightforward on a multigram scale, their design is inherently modular, and the complexes themselves are readily prepared; they might very well mark an important milestone in the development of ever more efficient alkyne metathesis catalysts. Ongoing studies in our laboratory intend to gain additional insights into the factors relevant for catalysis, study the pertinent reactive intermediates, improve activity and stability even further, map the functional group tolerance in greater detail, extend the coverage beyond molybdenum, and showcase the virtues of the new catalysts by advanced applications.
Acknowledgments
Generous financial support by the MPG, the Leverhulme Trust (studentship to E.Y.), and Scholarship Fund of the Swiss Chemical Industry (SSCI fellowship to C.P.G.) is gratefully acknowledged. We thank all analytical departments of our Institute for valuable support, especially Mr. J. Rust and Prof. C. W. Lehmann for the excellent X-ray service, Dr. R. Goddard for valuable discussions, and Dr. C. Farès and M.Sc. J. B. Lingnau for assistance with various NMR analyses.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04742.
The authors declare no competing financial interest.
Supplementary Material
References
- Hillenbrand J.; Leutzsch M.; Fürstner A. Molybdenum Alkylidyne Complexes with Tripodal Silanolate Ligands: The Next Generation of Alkyne Metathesis Catalysts. Angew. Chem., Int. Ed. 2019, 58, 15690–15696. 10.1002/anie.201908571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. R.; Rotella M. E.; Du P.; Zhou X.; Fronczek F. R.; Kumar R.; Gutierrez O.; Lee S. Siloxide Podand Ligand Scaffold for Molybdenum-Catalyzed Alkyne Metathesis and Isolation of a Dynamic Metallatetrahedrane Intermediate. Organometallics 2019, 38, 4054–4059. 10.1021/acs.organomet.9b00430. [DOI] [Google Scholar]
- a Heppekausen J.; Stade R.; Goddard R.; Fürstner A. Practical New Silyloxy-based Alkyne Metathesis Catalysts with Optimized Activity and Selectivity Profiles. J. Am. Chem. Soc. 2010, 132, 11045–11057. 10.1021/ja104800w. [DOI] [PubMed] [Google Scholar]; For the lead finding that siloxides are outstanding ligands for Mo-based catalysts, see:; b Bindl M.; Stade R.; Heilmann E. K.; Picot A.; Goddard R.; Fürstner A. Molybdenum Nitride Complexes with Ph3SiO- Ligands are Exceedingly Practical and Tolerant Precatalysts for Alkyne Metathesis and Efficient Nitrogen Transfer Agents. J. Am. Chem. Soc. 2009, 131, 9468–9470. 10.1021/ja903259g. [DOI] [PubMed] [Google Scholar]
- Heppekausen J.; Stade R.; Kondoh A.; Seidel G.; Goddard R.; Fürstner A. Optimized Synthesis, Structural Investigations, Ligand Tuning and Synthetic Evaluation of Silyloxy-based Alkyne Metathesis Catalysts. Chem. - Eur. J. 2012, 18, 10281–10299. 10.1002/chem.201200621. [DOI] [PubMed] [Google Scholar]
- a Persich P.; Llaveria J.; Lhermet R.; de Haro T.; Stade R.; Kondoh A.; Fürstner A. Increasing the Structural Span of Alkyne Metathesis. Chem. - Eur. J. 2013, 19, 13047–13058. 10.1002/chem.201302320. [DOI] [PubMed] [Google Scholar]; b Lhermet R.; Fürstner A. Cross-Metathesis of Terminal Alkynes. Chem. - Eur. J. 2014, 20, 13188–13193. 10.1002/chem.201404166. [DOI] [PubMed] [Google Scholar]
- Lackner A.; Fürstner A. The Triple-Bond Metathesis of Aryldiazonium Salts: A Prospect for Dinitrogen Cleavage. Angew. Chem., Int. Ed. 2015, 54, 12814–12818. 10.1002/anie.201506546. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Alkyne Metathesis on the Rise. Angew. Chem., Int. Ed. 2013, 52, 2794–2819. 10.1002/anie.201204513. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Teaching Metathesis “Simple” Stereochemistry. Science 2013, 341, 1229713. 10.1126/science.1229713. [DOI] [PubMed] [Google Scholar]
- Ehrhorn H.; Tamm M. Well-Defined Alkyne Metathesis Catalysts: Developments and Recent Applications. Chem. - Eur. J. 2018, 25, 3190–3208. 10.1002/chem.201804511. [DOI] [PubMed] [Google Scholar]
- a Schrock R. R.; Czekelius C. Recent Advances in the Synthesis and Applications of Molybdenum and Tungsten Alkylidene and Alkylidyne Catalysts for the Metathesis of Alkenes and Alkynes. Adv. Synth. Catal. 2007, 349, 55–77. 10.1002/adsc.200600459. [DOI] [Google Scholar]; b Zhang W.; Moore J. S. Alkyne Metathesis: Catalysts and Synthetic Applications. Adv. Synth. Catal. 2007, 349, 93–120. 10.1002/adsc.200600476. [DOI] [Google Scholar]; c Fürstner A.; Davies P. W. Alkyne Metathesis. Chem. Commun. 2005, 2307–2320. 10.1039/b419143a. [DOI] [PubMed] [Google Scholar]; d Mortreux A.; Coutelier O. Alkyne Metathesis Catalysts: Scope and Future. J. Mol. Catal. A: Chem. 2006, 254, 96–104. 10.1016/j.molcata.2006.03.054. [DOI] [Google Scholar]; e Yang H.; Jin Y.; Du Y.; Zhang W. Application of Alkyne Metathesis in Polymer Synthesis. J. Mater. Chem. A 2014, 2, 5986–5993. 10.1039/c3ta14227b. [DOI] [Google Scholar]
- a Schrock R. R. Multiple Metal–Carbon Bonds for Catalytic Metathesis Reactions. Angew. Chem., Int. Ed. 2006, 45, 3748–3759. 10.1002/anie.200600085. [DOI] [PubMed] [Google Scholar]; b Schrock R. R. High Oxidation State Multiple Metal-Carbon Bonds. Chem. Rev. 2002, 102, 145–180. 10.1021/cr0103726. [DOI] [PubMed] [Google Scholar]
- Krempner C. Role of Siloxides in Transition Metal Chemistry and Homogeneous Catalysis. Eur. J. Inorg. Chem. 2011, 2011, 1689–1698. 10.1002/ejic.201100044. [DOI] [Google Scholar]
- An instructive illustration is manifest in a volcano-type correlation: increasing electrophilicity of the alkylidyne leads to higher activity up to a certain point, at which the metallacyclobutadiene intermediate becomes (too) stable and the retro-cycloaddition (too) slow; see:Estes D. P.; Gordon C. P.; Fedorov A.; Liao W.-C.; Ehrhorn H.; Bittner C.; Zier M. L.; Bockfeld D.; Chan K. W.; Eisenstein O.; Raynaud C.; Tamm M.; Copéret C. Molecular and Silica-Supported Molybdenum Alkyne Metathesis Catalysts: Influence of Electrons and Dynamics on Activity Revealed by Kinetics, Solid-State NMR, and Chemical Shift Analysis. J. Am. Chem. Soc. 2017, 139, 17597–17607. 10.1021/jacs.7b09934. [DOI] [PubMed] [Google Scholar]
- Yet another illustration that electrophilicity management is critical for success can be seen in the fact that unfluorinated and perfluorinated tert-butoxy ligands entail no or very low activity, whereas trifluoro- and hexafluoro-tert-butoxy groups give competent molybdenum alkylidyne catalysts; see:; a McCullough L. G.; Schrock R. R. Metathesis of Acetylenes by Molybdenum(VI) Alkylidyne Complexes. J. Am. Chem. Soc. 1984, 106, 4067–4068. 10.1021/ja00326a051. [DOI] [Google Scholar]; b McCullough L. G.; Schrock R. R.; Dewan J. C.; Murdzek J. C. Preparation of Trialkoxymolybdenum(VI) Alkylidyne Complexes, Their Reactions with Acetylenes, and the X-ray Structure of Mo[C3(CMe3)2][OCH(CF3)2]2(C5H5N)2. J. Am. Chem. Soc. 1985, 107, 5987–5998. 10.1021/ja00307a025. [DOI] [Google Scholar]; c Bittner C.; Ehrhorn H.; Bockfeld D.; Brandhorst K.; Tamm M. Tuning the Catalytic Alkyne Metathesis Activity of Molybdenum and Tungsten 2,4,6-Trimethylbenzylidyne Complexes with Fluoroalkoxide Ligands OC(CF3)nMe3–n. Organometallics 2017, 36, 3398–3406. 10.1021/acs.organomet.7b00519. [DOI] [Google Scholar]
- Certain alkylsilanolates also lead to active catalysts but have been studied less systematically; see refs (4) and (22).
- See, for example:; a Willwacher J.; Kausch-Busies N.; Fürstner A. Divergent Total Synthesis of the Antimitotic Agent Leiodermatolide. Angew. Chem., Int. Ed. 2012, 51, 12041–12046. 10.1002/anie.201206670. [DOI] [PubMed] [Google Scholar]; b Kwon Y.; Schulthoff S.; Dao Q. M.; Wirtz C.; Fürstner A. Total Synthesis of Disciformycin A and B: Unusually Exigent Targets of Biological Significance. Chem. - Eur. J. 2018, 24, 109–114. 10.1002/chem.201705550. [DOI] [PubMed] [Google Scholar]
- a Micoine K.; Fürstner A. Concise Total Synthesis of the Potent Translation and Cell Migration Inhibitor Lactimidomycin. J. Am. Chem. Soc. 2010, 132, 14064–14066. 10.1021/ja107141p. [DOI] [PubMed] [Google Scholar]; b Hickmann V.; Kondoh A.; Gabor B.; Alcarazo M.; Fürstner A. Catalysis-based and Protecting-Group-free Total Syntheses of the Marine Oxylipins Hybridalactone and the Ecklonialactones A, B, and C. J. Am. Chem. Soc. 2011, 133, 13471–13480. 10.1021/ja204027a. [DOI] [PubMed] [Google Scholar]; c Benson S.; Collin M.-P.; Arlt A.; Gabor B.; Goddard R.; Fürstner A. Second-Generation Total Synthesis of Spirastrellolide F Methyl Ester: The Alkyne Route. Angew. Chem., Int. Ed. 2011, 50, 8739–8744. 10.1002/anie.201103270. [DOI] [PubMed] [Google Scholar]; d Chaladaj W.; Corbet M.; Fürstner A. Total Synthesis of Neurymenolide A Based on a Gold-Catalyzed Synthesis of 4-Hydroxy-2-pyrones. Angew. Chem., Int. Ed. 2012, 51, 6929–6933. 10.1002/anie.201203180. [DOI] [PubMed] [Google Scholar]; e Willwacher J.; Fürstner A. Catalysis-Based Total Synthesis of Putative Mandelalide A. Angew. Chem., Int. Ed. 2014, 53, 4217–4221. 10.1002/anie.201400605. [DOI] [PubMed] [Google Scholar]; f Hoffmeister L.; Persich P.; Fürstner A. Formal Total Synthesis of Kendomycin by Way of Alkyne Metathesis/Gold Catalysis. Chem. - Eur. J. 2014, 20, 4396–4402. 10.1002/chem.201304580. [DOI] [PubMed] [Google Scholar]; g Lehr K.; Schulthoff S.; Ueda Y.; Mariz R.; Leseurre L.; Gabor B.; Fürstner A. A New Method for the Preparation of Non-Terminal Alkynes: Application to the Total Synthesis of Tulearin A and C. Chem. - Eur. J. 2015, 21, 219–227. 10.1002/chem.201404873. [DOI] [PubMed] [Google Scholar]; h Hoffmeister L.; Fukuda T.; Pototschnig G.; Fürstner A. Total Synthesis of an Exceptional Brominated 4-Pyrone Derivative of Algal Origin: An Exercise in Gold Catalysis and Alkyne Metathesis. Chem. - Eur. J. 2015, 21, 4529–4533. 10.1002/chem.201500437. [DOI] [PubMed] [Google Scholar]; i Ungeheuer F.; Fürstner A. Concise Total Synthesis of Ivorenolide B. Chem. - Eur. J. 2015, 21, 11387–11392. 10.1002/chem.201501765. [DOI] [PubMed] [Google Scholar]; j Rummelt S. M.; Preindl J.; Sommer H.; Fürstner A. Selective Formation of a Trisubstituted Alkene Motif by trans-Hydrostannation/Stille Coupling: Application to the Total Synthesis and Late-Stage Modification of 5,6-Dihydrocineromycin B. Angew. Chem., Int. Ed. 2015, 54, 6241–6245. 10.1002/anie.201501608. [DOI] [PubMed] [Google Scholar]; k Fuchs M.; Fürstner A. trans-Hydrogenation: Application to a Concise and Scalable Synthesis of Brefeldin A. Angew. Chem., Int. Ed. 2015, 54, 3978–3982. 10.1002/anie.201411618. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Ahlers A.; de Haro T.; Gabor B.; Fürstner A. Concise Total Synthesis of Enigmazole A. Angew. Chem., Int. Ed. 2016, 55, 1406–1411. 10.1002/anie.201510026. [DOI] [PubMed] [Google Scholar]; m Meng Z.; Souillart L.; Monks B.; Huwyler N.; Herrmann J.; Müller R.; Fürstner A. A, ,Motif-Oriented“ Total Synthesis of Nannocystin Ax. Preparation and Biological Evaluation of Analogues. J. Org. Chem. 2018, 83, 6977–6994. 10.1021/acs.joc.7b02871. [DOI] [PubMed] [Google Scholar]
- a Neuhaus C. M.; Liniger M.; Stieger M.; Altmann K.-H. Total Synthesis of the Tubulin Inhibitor WF-1360F Based on Macrocycle Formation through Ring-Closing Alkyne Metathesis. Angew. Chem., Int. Ed. 2013, 52, 5866–5870. 10.1002/anie.201300576. [DOI] [PubMed] [Google Scholar]; b Ralston K. J.; Ramstadius H. C.; Brewster R. C.; Niblock H. S.; Hulme A. N. Self-Assembly of Disorazole C1 Through a One-Pot Alkyne Metathesis Homodimerization Strategy. Angew. Chem., Int. Ed. 2015, 54, 7086–7090. 10.1002/anie.201501922. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burnley J.; Jackson W. R.; Robinson A. J. One-Pot Selective Homodimerization/Hydrogenation Strategy for Sequential Dicarba Bridge Formation. J. Org. Chem. 2015, 80, 9057–9063. 10.1021/acs.joc.5b01312. [DOI] [PubMed] [Google Scholar]; d Guo L.-D.; Huang X.-Z.; Luo S.-P.; Cao W.-S.; Ruan Y.-P.; Ye J.-L.; Huang P.-Q. Organocatalytic, Asymmetric Total Synthesis of (−)-Haliclonin A. Angew. Chem., Int. Ed. 2016, 55, 4064–4068. 10.1002/anie.201512005. [DOI] [PubMed] [Google Scholar]; e Boeckman R. K. Jr.; Wang H.; Rugg K. W.; Genung N. E.; Chen K.; Ryder T. R. A Scalable Total Synthesis of (−)-Nakadomarin A. Org. Lett. 2016, 18, 6136–6139. 10.1021/acs.orglett.6b03137. [DOI] [PubMed] [Google Scholar]; f Herstad G.; Molesworth P. P.; Miller C.; Benneche T.; Tius M. A. Ring-Closing Metathesis in an Enantioselective Synthesis of the Macrocyclic Core of Crassin Acetate. Tetrahedron 2016, 72, 2084–2093. 10.1016/j.tet.2016.02.050. [DOI] [Google Scholar]; g Vendeville J.-B.; Matters R. E.; Chen A.; Light M. E.; Tizzard G. J.; Chai C. L. L.; Harrowven D. C. A Synthetic Approach to Chrysophaerntin F. Chem. Commun. 2019, 55, 4837–4840. 10.1039/C9CC01666J. [DOI] [PubMed] [Google Scholar]; For applications in which the catalysts were generated in situ from nitride precursors as described in ref (3b), see:; h Fouché M.; Rooney L.; Barrett A. G. M. Biomimetic Total Synthesis of Cruentaren A via Aromatizaiton of Diketodioxinones. J. Org. Chem. 2012, 77, 3060–3070. 10.1021/jo300225z. [DOI] [PubMed] [Google Scholar]; i Smith B. J.; Sulikowski G. A. Total Synthesis of (±)-Haliclonacylamine C. Angew. Chem., Int. Ed. 2010, 49, 1599–1602. 10.1002/anie.200905732. [DOI] [PubMed] [Google Scholar]; j Nilson M. G.; Funk R. L. Total Synthesis of (−)-Nakadomarin A. Org. Lett. 2010, 12, 4912–4915. 10.1021/ol102079z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For pertinent examples, see:; a Gebauer K.; Fürstner A. Total Synthesis of the Biphenyl Alkaloid (−)-Lythranidine. Angew. Chem., Int. Ed. 2014, 53, 6393–6396. 10.1002/anie.201402550. [DOI] [PubMed] [Google Scholar]; b Mailhol D.; Willwacher J.; Kausch-Busies N.; Rubitski E. E.; Sobol Z.; Schuler M.; Lam M.-H.; Musto S.; Loganzo F.; Maderna A.; Fürstner A. Synthesis, Molecular Editing, and Biological Assessment of the Potent Cytotoxin Leiodermatolide. J. Am. Chem. Soc. 2014, 136, 15719–15729. 10.1021/ja508846g. [DOI] [PubMed] [Google Scholar]; c Karier P.; Ungeheuer F.; Ahlers A.; Anderl F.; Wille C.; Fürstner A. Metathesis at an Implausible Site: A Formal Total Synthesis of Rhizoxin D. Angew. Chem., Int. Ed. 2019, 58, 248–253. 10.1002/anie.201812096. [DOI] [PubMed] [Google Scholar]
- a Cromm P. M.; Schaubach S.; Spiegel J.; Fürstner A.; Grossmann T. N.; Waldmann H. Orthogonal Ring-Closing Alkyne and Olefin Metathesis for the Synthesis of Small GTPase-targeting Bicyclic Peptides. Nat. Commun. 2016, 7, 11300. 10.1038/ncomms11300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cromm P. M.; Wallraven K.; Glas A.; Bier D.; Fürstner A.; Ottmann C.; Grossmann T. N. Contraining an Irregular Peptide Secondary Structure through Ring-Closing Alkyne Metathesis. ChemBioChem 2016, 17, 1915–1919. 10.1002/cbic.201600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For illustrations how protonation of the alkylidyne can be used to advantage, see:; a Bukhryakov K. V.; Schrock R. R.; Hoveyda A. H.; Tsay C.; Müller P. Syntheses of Molybdenum Oxo Alkylidene Complexes through Addition of Water to an Alkylidyne Complex. J. Am. Chem. Soc. 2018, 140, 2797–2800. 10.1021/jacs.8b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhai F.; Bukhryakov K. V.; Schrock R. R.; Hoveyda A. H.; Tsay C.; Müller P. Syntheses of Molybdenum Oxo Benzylidene Complexes. J. Am. Chem. Soc. 2018, 140, 13609–13613. 10.1021/jacs.8b09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaubach S.; Gebauer K.; Ungeheuer F.; Hoffmeister L.; Ilg M. K.; Wirtz C.; Fürstner A. A Two-Component Alkyne Metathesis Catalyst System with an Improved Substrate Scope and Functional Group Tolerance: Development and Applications to Natural Product Synthesis. Chem. - Eur. J. 2016, 22, 8494–8507. 10.1002/chem.201601163. [DOI] [PubMed] [Google Scholar]
- a Jyothish K.; Zhang W. Introducing a Podand Motif to Alkyne Metathesis Catalyst Design: A Highly Active Multidentate Molybdenum(VI) Catalyst that Resists Alkyne Polymerization. Angew. Chem., Int. Ed. 2011, 50, 3435–3438. 10.1002/anie.201007559. [DOI] [PubMed] [Google Scholar]; b Jyothish K.; Wang Q.; Zhang W. Highly Active Multidentate Alkyne Metathesis Catalysts: Ligand-Activity Relationship and Their Applications in Efficient Synthesis of Porphyrin-Based Aryleneethynylene Polymers. Adv. Synth. Catal. 2012, 354, 2073–2078. 10.1002/adsc.201200243. [DOI] [Google Scholar]; c Yang H.; Liu Z.; Zhang W. Multidentate Triphenolsilane-Based Alkyne Metathesis Catalysts. Adv. Synth. Catal. 2013, 355, 885–890. 10.1002/adsc.201201105. [DOI] [Google Scholar]; d Du Y.; Yang H.; Zhu C.; Ortiz M.; Okochi K. D.; Shoemaker R.; Jin Y.; Zhang W. Highly Active Multidentate Ligand-Based Alkyne Metathesis Catalysts. Chem. - Eur. J. 2016, 22, 7959–7963. 10.1002/chem.201505174. [DOI] [PubMed] [Google Scholar]; e Paley D. W.; Sedbrook D. F.; Decatur J.; Fischer F. R.; Steigerwald M. L.; Nuckolls C. Alcohol-Promoted Ring-Opening Alkyne Metathesis Polymerization. Angew. Chem., Int. Ed. 2013, 52, 4591–4594. 10.1002/anie.201300758. [DOI] [PubMed] [Google Scholar]
- For other alkylidyne complexes containing pincer-type ligands, see:; a O’Reilly M. E.; Ghiviriga I.; Abboud K. A.; Veige A. S. A New ONO3– Trianionic Pincer-Type Ligand for Generating Highly Nucleophilic Metal-Carbon Multiple Bonds. J. Am. Chem. Soc. 2012, 134, 11185–11195. 10.1021/ja302222s. [DOI] [PubMed] [Google Scholar]; b O’Reilly M. E.; Nadif S. S.; Ghiviriga I.; Abboud K. A.; Veige A. S. Synthesis and Characterization of Tungsten Alkylidene and Alkylidyne Complexes Supported by a New Pyrrolide-Centered Trianionic ONO3– Pincer Type Ligand. Organometallics 2014, 33, 836–839. 10.1021/om4009422. [DOI] [Google Scholar]; c Roland C. D.; VenkatRamani S.; Jakhar V. K.; Ghiviriga J.; Abboud K. A.; Veige A. S. Synthesis and Characterization of a Molybdenum Alkylidyne Supported by a Trianionic OCO3– Pincer Ligand. Organometallics 2018, 37, 4500–4505. 10.1021/acs.organomet.8b00677. [DOI] [Google Scholar]; d Bellone D. E.; Bours J.; Menke E. H.; Fischer F. R. Highly Selective Molybdenum ONO Pincer Complex Initiates the Living Ring-Opening Metathesis Polymerization of Strained Alkynes with Exceptionally Low Polydispersity Indices. J. Am. Chem. Soc. 2015, 137, 850–856. 10.1021/ja510919v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Elser I.; Groos J.; Hauser P. M.; Koy M.; van der Ende M.; Wang D.; Frey W.; Wurst K.; Meisner J.; Ziegler F.; Kästner J.; Buchmeiser M. R. Molybdenum and Tungsten Alkylidyne Complexes Containing Mono-, Bi-, and Tridentate N-Heterocyclic Carbenes. Organometallics 2019, 38, 4133–4146. 10.1021/acs.organomet.9b00481. [DOI] [Google Scholar]
- For other advances in catalyst design, see:; a Hauser P. M.; Hunger M.; Buchmeiser M. R. Silica-Supported Molybdenum Alkylidyne N-Heterocyclic Carbene Catalysts: Relevance of Site Isolation to Catalytic Performance. ChemCatChem 2018, 10, 1829–1834. 10.1002/cctc.201701654. [DOI] [Google Scholar]; b Koy M.; Elser I.; Meisner J.; Frey W.; Wurst K.; Kästner J.; Buchmeiser M. R. High Oxidation State Molybdenum N-Heterocyclic Carbene Alkylidyne Complexes: Synthesis, Mechanistic Studies, and Reactivity. Chem. - Eur. J. 2017, 23, 15484–15490. 10.1002/chem.201703313. [DOI] [PubMed] [Google Scholar]; c Bai W.; Wei W.; Sung H. H. Y.; Williams I. D.; Lin Z.; Jia G. Syntheses of Re(V) Alkylidyne Complexes and Ligand Effect on the Reactivity of Re(V) Alkylidyne Complexes toward Alkynes. Organometallics 2018, 37, 559–569. 10.1021/acs.organomet.7b00877. [DOI] [Google Scholar]; d Estes P. D.; Bittner C.; Arias O.; Casey M.; Fedorov A.; Tamm M.; Copéret C. Alkyne Metathesis with Silica-Supported and Molecular Catalysts at Parts-per-Million Loadings. Angew. Chem., Int. Ed. 2016, 55, 13960–13964. 10.1002/anie.201605129. [DOI] [PubMed] [Google Scholar]
- For inspiring precedent on the use of related ligand platforms, see:; a Kanady J. S.; Mendoza-Cortes J. L.; Tsui E. Y.; Nielsen R. J.; Goddard W. A.; Agapie T. Oxygen Atom Transfer and Oxidative Water Incorporation in Cuboidal Mn3MOn Complexes Based on Synthetic, isotope Labeling, and Computational Studies. J. Am. Chem. Soc. 2013, 135, 1073–1082. 10.1021/ja310022p. [DOI] [PubMed] [Google Scholar]; b Tsui E. Y.; Day M. W.; Agapie T. Trinuclear Copper: Synthesis and Magnetostructural Characterizatioin of Complexes Supported by a Hexapyridyl 1,3,5-Triarylbenzene Ligand. Angew. Chem., Int. Ed. 2011, 50, 1668–1672. 10.1002/anie.201005232. [DOI] [PubMed] [Google Scholar]
- Feng X.; Wu J.; Enkelmann V.; Müllen K. Hexa-peri-hexabenzocoronenes by Efficient Oxidative Cyclodehydrogenation. The Role of the Oligophenylene Precursors. Org. Lett. 2006, 8, 1145–1148. 10.1021/ol053043z. [DOI] [PubMed] [Google Scholar]
- The X-ray structures of compounds 8a, 11b, 11c, 15b, and 1,2-bis(2,6-dimethylphenyl)ethyne (formed as a byproduct during the preparation of 14b) are included in the Supporting Information.
- a Elmorsy S. S.; Pelter A.; Smith K. The Direct Production of Tri- and Hexa-Substituted Benzenes from Ketones under Mild Conditions. Tetrahedron Lett. 1991, 32, 4175–4176. 10.1016/S0040-4039(00)79896-0. [DOI] [Google Scholar]; b Trawny D.; Quennet M.; Rades N.; Lentz D.; Paulus B.; Reissig H.-U. Syntheses, Structures and Conformational Dynamics of 1,3,5-Tris-(3″-ethynylbiphenyl-2′-yl)benzene Derivatives. Eur. J. Org. Chem. 2015, 2015, 4667–4674. 10.1002/ejoc.201500615. [DOI] [Google Scholar]
- The reaction can be performed at dry ice temperature, but the yield is lower (ca. 40%) due to the formation of isomers that result from scrambling of the lithiated sites.
- The MeO– substituents favor scrambling at the stage of the organolithium intermediate, which results in the formation of isomers that are difficult to separate; for details, see the Supporting Information.
- The Lee group relied on salt metathesis to form their podand complexes. The two reported examples were obtained in modest yields of 30% and 50%; see ref (2).
- The Lee group had to use benzylpotassium for the deprotonation of the trisilanole; see ref (2).
- Zhang W.; Lu Y.; Moore J. S. Preparation of a Trisamidomolybdenum(VI) Propylidyne Complex. Org. Synth. 2007, 84, 163–176. 10.1002/0471264229.os084.17. [DOI] [Google Scholar]
- In the case of 14b, the crude product contains complex 15b (Ar = 2,6-(Me)2C6H3) and 1,2-bis(2,6-dimethylphenyl)ethyne as byproducts, the formation of which is not entirely clear. These compounds can be removed by careful recrystallization, and their structure was secured by X-ray diffraction; see the Supporting Information.
- von Kugelgen S.; Piskun I.; Griffin J. H.; Eckdahl C. T.; Jarenwattananon N. N.; Fischer F. R. Templated Synthesis of End-Functionalized Graphene Nanoribbons through Living Ring-Opening Alkyne Metathesis Polymerization. J. Am. Chem. Soc. 2019, 141, 11050–11058. 10.1021/jacs.9b01805. [DOI] [PubMed] [Google Scholar]
- The 95Mo NMR signal (δMo = 118 ppm) of 16 is broad (see the Supporting Information), in line with the lower symmetry of the complex that causes faster quadrupolar relaxation. The shift is considerably closer to that of the catalytically incompetent complex [(tBuO)3Mo≡CAr] (14a, Ar = p-MeOC6H4) (δMo = 79.6 ppm) than to that of the canopy catalysts with tripodal silanolate ligands (359–434 ppm).
- Fürstner A.; Seidel G. Ring-closing Metathesis of Functinalized Acetylene Derivatives: A New Entry into Cycloalkynes. Angew. Chem., Int. Ed. 1998, 37, 1734–1736. . [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Neel A. J.; Hilton M. J.; Sigman M. S.; Toste F. D. Exploiting Non-Covalent π-Interactions for Catalyst Design. Nature 2017, 543, 637–646. 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meyer E. A.; Castellano R. K.; Diederich F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- The ortho-benzylidyne protons of [2a]2 (δH ([D8]-toluene) = 6.06 (d), 5.49 ppm (d), ref (4)) and [1a]2 (δH = 5.91, 4.93 ppm) are strongly shielded. The X-ray structure of [2a]2 (Figure 4) shows that these protons reside in the anisotropy cone of the adjacent phenyl ring. Therefore, it is likely that a similar effect and hence a similar overall arrangement is present in [1a]2. We like to emphasize, however, that aggregation via reversible covalent cross-linking in [1a]2 cannot be rigorously excluded as long as no X-ray structure is available.
- This anticipation was also based on the fact that complex [(Ph3SiO)3Mo≡CAr] (2b, Ar = 2,6-(Me)2C6H3) featuring a 2,6-disubstituted phenyl ring in lieu of the p-methoxyphenyl group of 2a is monomeric in the solid state (see ref (4)) as well as in solution (DOSY; see the Supporting Information).
- A complex prepared by the Lee group carrying a mesityl group on the alkylidyne is monomeric (but failed to initiate the alkyne metathesis model reaction chosen by this team); see ref (2).
- The alkylidyne of the adduct [1a·MeCN], however, shows a significant kink (161.4(2)°); cf. ref (1).
- For weak bonding between the metal and a basal phenyl ring in tripodal uranium complexes, see:; a Bart S. C.; Heinemann F.; Anthon C.; Hauser C.; Meyer K. A New Tripodal Ligand System with Steric and Electronic Modularity for Uranium Coordination Chemistry. Inorg. Chem. 2009, 48, 9419–9426. 10.1021/ic9012697. [DOI] [PubMed] [Google Scholar]; b La Pierre H. S.; Kameo H.; Halter D. P.; Heinemann F. W.; Meyer K. Coordination and Redox Isomerization in the Reduction of a Uranium(III) Monoarene Complex. Angew. Chem., Int. Ed. 2014, 53, 7154–7157. 10.1002/anie.201402048. [DOI] [PubMed] [Google Scholar]
- a Malito J. Molybdenum-95 NMR Spectroscopy. Annu. Rep. NMR Spectrosc. 1996, 33, 151–206. 10.1016/S0066-4103(08)60050-2. [DOI] [Google Scholar]; b Mann B. E. The Cinderella Nuclei. Annu. Rep. NMR Spectrosc. 1991, 23, 141–207. 10.1016/S0066-4103(08)60277-X. [DOI] [Google Scholar]
- Young C. G.; Kober E. M.; Enemark J. H. 95Mo and 183W NMR Studies of Triply Bonded Dinuclear M(III) and Related M≡C (M = Mo or W) Complexes. Polyhedron 1987, 6, 255–259. 10.1016/S0277-5387(00)80794-9. [DOI] [Google Scholar]
- The extremely low-field 95Mo resonances of the alkylidynes bearing alkyl groups as ancillary ligands reported in ref (46) ([(Me3SiCH2)3Mo≡CSiMe3] (1845 ppm) and [(Me3CCH2)3Mo≡CCMe3] (1400 ppm)) are puzzling and should be rerecorded.
- For a detailed discussion, see:; Petrakis L. Quadrupolar Relaxation of Aluminum-27 Nuclear Magnetic Resonance in Aluminum Alkyls. J. Phys. Chem. 1968, 72, 4182–4188. 10.1021/j100858a041. [DOI] [Google Scholar]
- The same effect was recorded for 1f comprising −SiMe2– linkers (δMo = 422 ppm), which also resonates upfield from its otherwise identical sibling 1e with −SiPh2– bridges (δMo = 434 ppm).
- a Bohmann J. A.; Weinhold F.; Farrar T. C. Natural Chemical Shielding Analysis of Nuclear Magnetic Resonance Shielding Tensors from Gauge-including Atomic Orbital Calculations. J. Chem. Phys. 1997, 107, 1173–1184. 10.1063/1.474464. [DOI] [Google Scholar]; b Autschbach J. Analyzing NMR Shielding Tensors Calculated with Two-component Relativistic Methods using Sspin-free Localized Molecular Orbitals. J. Chem. Phys. 2008, 128, 164112. 10.1063/1.2905235. [DOI] [PubMed] [Google Scholar]; c Widdifield C. M.; Schurko R. Understanding Chemical Shielding Tensors using Group Theory, MO Analysis, and Modern Density-Functional Theory. Concepts Magn. Reson., Part A 2009, 34A, 91–123. 10.1002/cmr.a.20136. [DOI] [Google Scholar]; d Kaupp M.Interpretation of NMR Chemical Shifts. In Calculation of NMR and EPR Parameters; Kaupp M., Bühl M., Malkin V. G., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 293–306. [Google Scholar]
- Gordon C. P.; Raynaud C.; Andersen R. A.; Copéret C.; Eisenstein O. Carbon-13 NMR Chemical Shifts: A Descriptor for Electronic Structure and Reactivity of Organometallic Compounds. Acc. Chem. Res. 2019, 52, 2278–2289. 10.1021/acs.accounts.9b00225. [DOI] [PubMed] [Google Scholar]
- For CST analyses of metal alkylidenes and carbenes, see:; a Blanc F.; Basset J.-M.; Copéret C.; Sinha A.; Tonzetich Z. J.; Schrock R. R.; Solans-Monfort X.; Clot E.; Eisenstein O.; Lesage A.; Emsley L. Dynamics of Silica-Supported Catalysts Determined by Combining Solid-State NMR Spectroscopy and DFT Calculations. J. Am. Chem. Soc. 2008, 130, 5886–5900. 10.1021/ja077749v. [DOI] [PubMed] [Google Scholar]; b Halbert S.; Copéret C.; Raynaud C.; Eisenstein O. Elucidating the Link between NMR Chemical Shifts and Electronic Structure in d0 Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2016, 138, 2261–2272. 10.1021/jacs.5b12597. [DOI] [PubMed] [Google Scholar]; c Yamamoto K.; Gordon C. P.; Liao W.-C.; Copéret C.; Raynaud C.; Eisenstein O. Orbital Analysis of Carbon-13 Chemical Shift Tensors Reveals Patterns to Distinguish Fischer and Schrock Carbenes. Angew. Chem., Int. Ed. 2017, 56, 10127–10131. 10.1002/anie.201701537. [DOI] [PubMed] [Google Scholar]; d Biberger T.; Gordon C. P.; Leutzsch M.; Peil S.; Guthertz A.; Copéret C.; Fürstner A. Alkyne gem-Hydrogenation: Formation of Pianostool Ruthenium Carbene Complexes and Analysis of Their Chemical Character. Angew. Chem., Int. Ed. 2019, 58, 8845–8850. 10.1002/anie.201904255. [DOI] [PubMed] [Google Scholar]
- a Suresh C. H.; Frenking G. 1,3-Metal-Carbon Bonding and Alkyne Metathesis: DFT Investigations on Model Complexes of Group 4, 5, and 6 Transition Metals. Organometallics 2012, 31, 7171–7180. 10.1021/om3007097. [DOI] [Google Scholar]; b Zhu J.; Jia G.; Lin Z. Theoretical Investigation of Alkyne Metathesis Catalyzed by W/Mo Alkylidyne Complexes. Organometallics 2006, 25, 1812–1819. 10.1021/om060116p. [DOI] [Google Scholar]
- Computations suggest that the direct formation of a metallatetrahedrane from a molybdenum alkylidyne and an alkyne substrate is symmetry-forbidden. Rather, it is thought to be a derivative of a metallacyclobutadiene precursor; see ref (58).
- Sancho J.; Schrock R. R. Acetylene Metathesis by Tungsten(VI) Alkylidyne Complexes. J. Mol. Catal. 1982, 15, 75–79. 10.1016/0304-5102(82)80006-0. [DOI] [Google Scholar]
- Churchill M. R.; Fettinger J. C.; McCullough L. G.; Schrock R. R. Transformation of a Tungstenacyclobutadiene Complex into a Nonfluxional η3-Cyclopropenyl Complex by Addition of a Donor Ligand. The X-ray Structure of W(η5-C5H5)[C3(CMe3)2Me](PMe3)Cl2. J. Am. Chem. Soc. 1984, 106, 3356–3357. 10.1021/ja00323a051. [DOI] [Google Scholar]
- Schrock R. R.; Murdzek J. S.; Freudenberger J. H.; Churchill M. R.; Ziller J. W. Preparation of Molybdenum and Tungsten Neopentylidyne Complexes of the Type M(CCMe3)(O2CR)3, Their Reactions with Acetylenes, and the X-ray Structure of the η3-Cyclopropenyl Complex W[C3(CMe3)Et2](O2CCH3)3. Organometallics 1986, 5, 25–33. 10.1021/om00132a005. [DOI] [Google Scholar]
- Woo T.; Folga E.; Ziegler T. Density Functional Study of Acetylene Metathesis Catalyzed by High Oxidation State Molybdenum and Tungsten Carbyne Complexes. Organometallics 1993, 12, 1289–1298. 10.1021/om00028a052. [DOI] [Google Scholar]
- Lin Z.; Hall M. B. Stabilities of Metallacyclobutadiene and Metallatetrahedrane Complexes. Organometallics 1994, 13, 2878–2884. 10.1021/om00019a050. [DOI] [Google Scholar]
- Schrock R. R. High-Oxidation-State Molybdenum and Tungsten Alkylidyne Complexes. Acc. Chem. Res. 1986, 19, 342–348. 10.1021/ar00131a003. [DOI] [Google Scholar]
- At 60 °C with added MS 5Å, the fast rates make a clear ranking difficult.
- Meng Z.; Fürstner A. Total Synthesis of (−)-Sinulariadiolide. A Transannular Approach. J. Am. Chem. Soc. 2019, 141, 805–809. 10.1021/jacs.8b12185. [DOI] [PubMed] [Google Scholar]
- Chen P.; Zhang L.; Xue Z.-L.; Wu Y.-D.; Zhang X. Density Functional Theory Study of the Reaction between d0 Tungsten Alkylidyne Complexes and H2O: Addition versus Hydrolysis. Inorg. Chem. 2017, 56, 7111–7119. 10.1021/acs.inorgchem.7b00713. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Mathes C.; Lehmann C. W. Mo[N(tBu)(Ar)]3 Complexes as Catalyst Precursors: In Situ Activation and Application to Metathesis Reactions of Alkynes and Diynes. J. Am. Chem. Soc. 1999, 121, 9453–9454. 10.1021/ja991340r. [DOI] [Google Scholar]
- For pertinent examples, see:; a Wölfl B.; Mata G.; Fürstner A. Total Synthesis of Callyspongiolide, Part 2: The Ynoate Metathesis/cis-Reduction Strategy. Chem. - Eur. J. 2019, 25, 255–259. 10.1002/chem.201804988. [DOI] [PubMed] [Google Scholar]; b Schaubach S.; Michigami K.; Fürstner A. Hydroxyl-Assisted trans-Reduction of 1,3-Enynes: Applicatio to the Foraml Syntehis of (+)-Aspicillin. Synthesis 2016, 49, 202–208. 10.1055/s-0035-1562381. [DOI] [Google Scholar]
- Fürstner A.; Guth O.; Rumbo A.; Seidel G. Ring Closing Alkyne Metathesis. Comparative Investigation of Two Different Catalyst Systems and Application to the Stereoselective Synthesis of Olfactory Lactones, Azamacrolides, and the Macrocyclic Perimeter of the Marine Alkaloid Nakadomarin A. J. Am. Chem. Soc. 1999, 121, 11108–11113. 10.1021/ja992074k. [DOI] [Google Scholar]
- a Fürstner A.; Mathes C.; Lehmann C. W. Alkyne Metathesis: Development of a Novel Molybdenum-Based Catalyst System and its Application to the Total Synthesis of Epothilone A and C. Chem. - Eur. J. 2001, 7, 5299–5315. . [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Mathes C.; Grela K. Concise Total Syntheses of Epothilone A and C Based on Alkyne Metathesis. Chem. Commun. 2001, 1057–1059. 10.1039/b101669p. [DOI] [Google Scholar]
- a Fürstner A.; Larionov O.; Flügge S. What is Amphidinolide V? Report on a Likely Conquest. Angew. Chem., Int. Ed. 2007, 46, 5545–5548. 10.1002/anie.200701640. [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Flügge S.; Larionov O.; Takahashi Y.; Kubota T.; Kobayashi J. Total Synthesis and Biological Evaluation of Amphidinolide V and Analogues. Chem. - Eur. J. 2009, 15, 4011–4029. 10.1002/chem.200802068. [DOI] [PubMed] [Google Scholar]
- a Valot G.; Regens C. S.; O’Malley D. P.; Godineau E.; Takikawa H.; Fürstner A. Total Synthesis of Amphidinolide F. Angew. Chem., Int. Ed. 2013, 52, 9534–9538. 10.1002/anie.201301700. [DOI] [PubMed] [Google Scholar]; b Valot G.; Mailhol D.; Regens C. S.; O’Malley D. P.; Godineau E.; Takikawa H.; Philipps P.; Fürstner A. Concise Total Syntheses of Amphidinolide C and F. Chem. - Eur. J. 2015, 21, 2398–2408. 10.1002/chem.201405790. [DOI] [PubMed] [Google Scholar]
- For other examples, see the following and literature cited therein:; a Yu M.; Wang C.; Kyle A. f.; Jakubec P.; Dixon D. J.; Schrock R. R.; Hoveyda A. H. Synthesis of Macrocyclic Natural Products by Catalyst-Controlled Stereoselective Ring-Closing Metathesis. Nature 2011, 479, 88–93. 10.1038/nature10563. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mu Y.; Nguyen T. T.; Koh M. J.; Schrock R. R.; Hoveyda A. H. E- and Z-, Di- and Tri-Substituted Alkenyl Nitriles through Catalytic Cross-Metathesis. Nat. Chem. 2019, 11, 478–487. 10.1038/s41557-019-0233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a general treatise, see:; c Catalysis Without Precious Metals; Bullock R. M., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.