

Abstract
Background
Single nucleotide polymorphisms (SNPs) in the α-synuclein (SNCA) gene are associated with differential risk and age at onset (AAO) of both idiopathic and Leucine-rich repeat kinase 2 (LRRK2)-associated Parkinson’s disease (PD). Yet potential combinatory or synergistic effects among several modulatory SNPs for PD risk or AAO remain largely underexplored.
Objectives
The mechanistic target of rapamycin (mTOR) signaling pathway is functionally impaired in PD. Here we explored whether SNPs in the mTOR pathway, alone or by epistatic interaction with known susceptibility factors, can modulate PD risk and AAO.
Methods
Based on functional relevance, we selected a total of 64 SNPs mapping to a total of 57 genes from the mTOR pathway and genotyped a discovery series cohort encompassing 898 PD patients and 921 controls. As a replication series, we screened 4170 PD and 3014 controls available from the International Parkinson’s Disease Genomics Consortium.
Results
In the discovery series cohort, we found a 4-loci interaction involving STK11 rs8111699, FCHSD1 rs456998, GSK3B rs1732170, and SNCA rs356219, which was associated with an increased risk of PD (odds ratio = 2.59, P < .001). In addition, we also found a 3-loci epistatic combination of RPTOR rs11868112 and RPS6KA2 rs6456121 with SNCA rs356219, which was associated (odds ratio = 2.89; P < .0001) with differential AAO. The latter was further validated (odds ratio = 1.56; P = 0.046–0.047) in the International Parkinson’s Disease Genomics Consortium cohort.
Conclusions
These findings indicate that genetic variability in the mTOR pathway contributes to SNCA effects in a nonlinear epistatic manner to modulate differential AAO in PD, unraveling the contribution of this cascade in the pathogenesis of the disease.
Keywords: age at onset, alpha-synuclein, epistasis, mTOR, Parkinson’s disease, SNP
Parkinson’s disease (PD) is a neurodegenerative disease that is characterized by α-synuclein (SNCA) aggregates and neural loss in several brain stem nuclei. Mainly, dopaminergic neurons loss in the substantia nigra leads to the related cardinal motor symptoms of bradykinesia, resting tremor, and rigidity.1 Intriguingly, the cause of this neural loss is still unknown. Whereas genetic mutations in PD causative genes represent 5% to 10% of total PD patients, the vast majority of cases are idiopathic PD (IPD).2 The age at onset (AAO) of the motor symptoms and the progression of PD are variable,3,4 and their modulatory factors remain largely unknown. Yet single nucleotide polymorphisms (SNPs) in the SNCA and the microtubule-associated protein Tau (MAPT) genes have shown top-hit association signals with PD risk in genomewide association studies5,6 and in candidate gene studies.7,8 For instance, the SNP rs356219 in the 3’ untranslated region (3’-UTR) of SNCA is a haplotype-tag SNP associated with a higher risk of PD9,10 and related to increased SNCA protein expression.11,12 This tag SNP also modulates AAO in both IPD and leucine-rich repeat kinase 2 (LRRK2)- associated PD (L2PD).13,14 Although IPD genetic risk factors such as the glucocerebrosidase gene and others have been identified,15–18 the overall heritability of IPD as a complex multifactorial disorder remains unclear.
An extended hypothesis in IPD is that multiple genetic susceptibility factors along with environmental cues19 and their potential interactions modulate disease presentation. In this sense, the genetic contribution to sporadic diseases such as PD can be additive when the effect of multiple genes is exerted in a linear fashion or nonadditive when the genetic effect is nonlinear and can include either dominance (intralocus interaction) or epistatic (interlocus interaction) effects. The latter, epistasis, is considered the complex effect of one gene upon other genes,20 and it is also suspected as an important genetic component of complex diseases such as IPD.21 Still, the potential epistatic effects among different susceptibility risk factors for IPD, despite their plausibility, remain largely neglected and underexplored in this disease.22
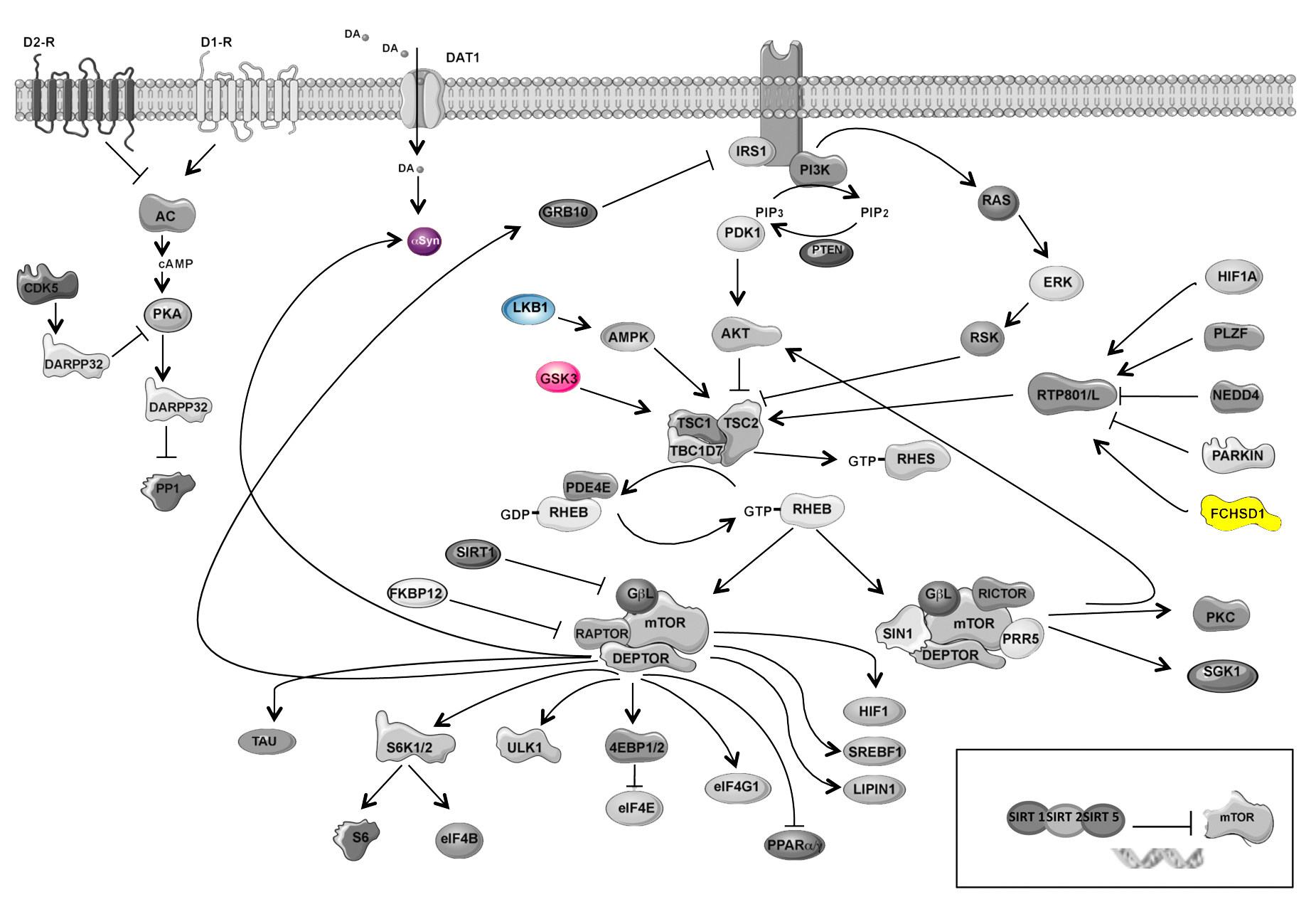
In this context, the signaling cascades regulating the neurodegenerative process in PD are not well established, but several pathways have been proposed.23 Recent studies have reported that deregulation of the mechanistic target of rapamycin (mTOR) pathway occurs in PD.24–27 For instance, mTOR and Akt activities are impaired in nigral neurons in human postmortem PD brains. Interestingly, restitution of mTOR or Akt function in cellular and animal models of PD prevents neuron cell death, pointing out a relevant contribution of this signaling cascade to PD pathophysiology.24,25,28 In basal conditions, the mTOR signaling integrates basic cellular functions such as growth, proliferation, and survival. Specifically in the brain, mTOR has a key role in neural development, neuron survival, synaptic plasticity, and memory formation.29 The central hub of the pathway is mTOR, a serine/threonine kinase that is present in 2 different multiprotein complexes, mTORC1 and mTORC2. When the multiprotein complex contains mTOR and Raptor, among other interactors, it is called mTORC1 and controls protein translation and autophagy.30,31 When mTOR multiprotein complex binds to Rictor, it is called mTORC2 and regulates actin polymerization and Akt trophic activity.30,32
Based on this biochemical evidence, we performed an mTOR genetic pathway candidate approach to explore whether common genetic variability in genes from the mTOR signaling cascade could modulate the risk or the AAO of PD. To this end, we genotyped 64 SNPs mapping to 57 genes of the mTOR pathway in a discovery series cohort of 898 PD patients and 921 healthy controls (total N = 1819). By classical allelic and genotypic association analyses, we investigated whether individual mTOR SNPs are associated with differential susceptibility and AAO of PD. By using the multifactor dimensionality reduction (MDR) method,33–35 we assessed potential multilocus epistatic interactions of SNPs in mTOR genes and classical PD-associated SNPs in SNCA and MAPT in modulating PD risk and AAO. Furthermore, we performed a validation analysis in a replication series cohort of 4170 PD and 3014 controls (total N = 7184 subjects) available from The International Parkinson’s Disease Genomics Consortium (IPDGC). Our study uncovers for the first time novel higher order genetic interactions influencing PD thus exploring a novel field in PD genetic research.
Methods
Cohort of Study and Data Collection
Discovery Series
This study included 1819 subjects (898 PD cases and 921 unrelated healthy controls) of Spanish ancestry. The PD population ratio consisted of 76/100 women/ men (43.31/56.69%), with age at PD onset of 55.30± 13.12 (mean ± standard deviation) years in men and 57.75 ± 13.12 years in women and age at sample collection of 64.37 ± 12.25 years in men and 67.52 ± 11.31 years in women. Sex-matched, age-matched, and demographic-matched controls ratio consisted of 71/100 women/men (41.69/58.31%) with age at sample collection of 53.79 ± 12.16 years in men and 56.83 ± 14.30 years in women. In addition, we also genotyped an independent set of 127 L2PD patients carrying the LRRK2 p.G2019S mutation. This cohort consisted of a ratio of 84/100 men/women, with age at PD onset of 55.64 ± 14.20 years in men and 58.85 ± 13.56 years in women and age at sample collection of 58.30± 16.66 years in men and 62.22 ± 16.99 years in women.
All PD participants were residents in the northeastern region of the Iberian Peninsula (Catalonia) with European origin and were recruited at the Movement Disorders Unit of the Hospital Clinic de Barcelona. Patients had a clinical diagnosis of definite PD according to Unified Parkinson’s Disease Rating Scale (UPDRS) criteria36 except that family history was not used as exclusion criterion or a neuropathological diagnosis of definite PD according to proposed criteria.37 The control individuals (total N = 921) encompassed 75 healthy spouses of the patients recruited at the Hospital Clinic de Barcelona by expert neurologists specialized in movement disorders. The remaining 846 controls were population-based controls collected at the Spanish National DNA Bank of Salamanca. This DNA bank hosts a collection of DNA samples and their associated clinical, genealogical, and lifestyle data that are representative of the healthy Spanish population. Specifically, the population-based controls used in the current study passed a questionnaire on health status supervised by a physician and reported no signs, symptoms, or familial history of PD or of other neurological diseases. At recruitment, informed written consent was obtained and whole blood samples were obtained from each participant. Genomic DNA was isolated from peripheral blood lymphocytes as previously described38 and stored at −80°C until use. The study was approved by the ethics committee of the Hospital Clinic de Barcelona.
Replication Series
As a replication series, we used a large cohort consisting of 7184 subjects (4170 PD patients and 3014 controls) of Spanish ancestry from the IPDGC. The PD population consisted of 1861/2309 women/men (44.62/55.37%), with age at PD onset of 59.74 ± 12.69 years in men and 61.69 ± 12.57 years in women. Sex-matched, age-matched, and demographic-matched controls consisted of 1655/1359 women/men (54.92/45.08%) with age at sample collection of 62.51 ± 15.56 years in men and 62.68 ± 11.50 years in women.
SNP Selection Criteria and Genotyping
A total of 64 SNPs from 57 genes in the mTOR pathway and SNPs from genes involved in PD (SNCA, MAPT, LRRK2, or PRKN) were selected on the basis of the following criteria: (i) a minor allele frequency above 0.1 based on data from the HapMap project or in 1000Genomes39 and (ii) a published (Pubmed) association or functional deregulation of the SNP in human disease specifically including neurological and psychiatric disorders (Table S1). All of the SNPs were genotyped using TaqMan OpenArray Genotyping Plates (Madrid, Spain), Custom Format 64 QuantStudio TM 12K Flex (Madrid, Spain), in the Genomics Core facility (Universitat Pompeu Fabra, Parc de Recerca Biomedicaède Barcelona, Barcelona, Catalonia, Spain). SNPs that were not in Hardy-Weinberg equilibrium or did not surpass a genotyping call-rate threshold of 0.95 in all studied samples were filtered out. This quality control reduced the list to 54 SNPs, including 52-mTOR related genes along with SNCA and MAPT SNPs.
For the replication cohort, samples were genotyped using the customized NeuroChip Array v.1.0 or v.1.1 (Illumina, San Diego, California, USA).40 Quality control analysis was performed as previously described.5 Individuals related at the level of cousins or closer (sharing proportionally more than 18.5% of alleles) were dropped from the following analysis. Samples were clustered using principal component analysis to ensure European ancestry as compared to the HapMap3 Utah residents with northern and western european ancestry from the CEPH collection/Toscani in Italia (CEU/TSI) populations.39
Statistical Analyses
Hardy-Weinberg Equilibrium
We assessed each SNP for Hardy-Weinberg equilibrium separately in cases and controls using a Fisher exact test.
Allelic Association Analysis
We calculated the differences of SNP allelic frequencies between cases and controls using the Expectation- Maximization algorithm as implemented in the statistical package UNPHASED version 3.1.7,41 adjusting by potential confounders, including gender and age. We used a threshold of .05 for statistical significance. We corrected all P values for multiple testing by using the Benjamini and Hochberg method42 (n = 54 tests).
Genotypic Association Analysis
Statistically significant SNPs detected in the allelic analysis were further analyzed at the genotypic level under the different possible models of inheritance as computed in the software SNPstats (http://bioinfo.iconcologia.net/SNPstats),43 considering gender and age as covariates and adjusting P values by the Benjamini and Hochberg method.42 As implemented in SNPstats, among all possible models of inheritance for each SNP, the model best fitting the data was defined automatically as the model with the lowest Akaike information value and therefore minimized expected entropy.
Epistatic Association Analysis
We used the MDR software version 3.04 (http://www.multifactordimensionalityreduction.org/) to detect high-order SNP interactions associated with the risk or the AAO of PD. The MDR method is based on a data-mining strategy for detecting combinations of discrete attributes, such as SNPs, or those that are predictive of an outcome, such as case or control status.34 PD risk was considered a discrete outcome, whereas PD AAO was analyzed as continuous (quantitative) outcome. The MDR analyses were performed using 10-fold cross-validation, and the best model was selected based on balanced accuracy (for PD risk) or the build-in Student t-test (for PD AAO) and cross-validation consistency (CVC) scores. The CVC is the number of times a particular SNP combination is identified out of the 10 cross-validations. Statistical significance was evaluated by performing a 1000-time permutation test using the option “explicit test for epistasis,” which specifically tests for interactions and provides multiple testing correction.44 We used a threshold of .05 for significance.
Results
After filtering out SNPs that were not in Hardy- Weinberg equilibrium or SNPs that did not surpass the quality threshold of unambiguous genotypes above 0.95 in all studied samples, we obtained a set of 54 SNPs (Table S1) that were further analyzed for single or multiple associations with risk or AAO of PD in the discovery series.
Single Marker Association of SNCA and MAPT With the Risk of IPD
In the discovery series, we first performed allelic association analysis of single markers with the overall PD susceptibility. After adjusting by gender, age, and multiple testing (n = 54 tests), we found a statistically significant allelic association of SNP rs356219 in the SNCA gene with a differential PD risk in which the G risk allele had a frequency of 0.41 in cases and 0.33 in controls (odds ratio, OR [95% confidence interval, CI] = 1.35 [1.16–1.57], adjusted P = .0054). We also confirmed the previously described (45) significant association of SNP rs1800547 in the MAPT gene with PD risk (OR [95% CI] = 0.75 [0.64–0.88], adjusted P = .01; Table S2). At the genotypic level, we also detected significant association of SNCA rs356219 (OR [95% CI] = 1.36 [1.18–1.56], adjusted P = .0027) and also of MAPT rs1900547 (OR [95% CI] = 1.33 [1.15–1.54], adjusted P = .0027), both under a log-additive model of inheritance (Table 1). These data are in agreement with previous findings from our group7,8 and from others.13,45,46 On the contrary, we did not find a significant association with PD risk for any of the other 52 mTOR genetic markers, but top signals that did not reach statistical significance included DDIT4L rs1053227 (unadjusted P = .01) and EIF4EBP1 rs6605631 (unadjusted P = .01; Table 1).
TABLE 1.
Genotypic association of SNPs in SNCA and MAPT with PD risk adjusted by sex, age, and multiple testing adjustments of P values
| PD | Control | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | Alleles, M/m | Freq. 11 | Freq. 12 | Freq. 22 | Freq. 11 | Freq. 12 | Freq. 22 | OR (95% CI) | Model | Genotypic P value | Adj. Gen. P value |
| SNCA | rs356219 | A (1)/G (2) | 0.36 | 0.47 | 0.17 | 0.45 | 0.44 | 0.11 | 1.36 (1.18–1.56) | Log-additive | <.0001 | <.0027 |
| MAPT | rs1800547 | A (1)/G (2) | 0.56 | 0.38 | 0.06 | 0.49 | 0.41 | 0.11 | 1.33 (1.15–1.54) | Log-additive | .0001 | .0027 |
| DDIT4L | rs1053227 | G (1)/A (2) | 0.33 | 0.49 | 0.18 | 0.37 | 0.49 | 0.14 | 0.84 (0.73–0.96) | Log-additive | .0100 | 1350 |
| EIF4EBP1 | rs6605631 | T (1)/C (2) | 0.69 | 0.29 | 0.02 | 0.66 | 0.29 | 0.05 | 1.98 (1.16–3.38) | Recessive | .0100 | .1350 |
| LPIN1 | rs7595221 | A (1)/G (2) | 0.29 | 0.48 | 0.23 | 0.25 | 0.53 | 0.22 | 1.23 (1.02–1.49) | Overdominant | .0270 | .2916 |
| PRKCA | rs887797 | G (1)/A (2) | 0.48 | 0.40 | 0.12 | 0.43 | 0.45 | 0.12 | 1.22 (1.01–1.48) | Overdominant | .0360 | .2922 |
Statistically significant P values are bold. Genotypic test calculated in SNPstats software with sex and age adjustment of P values. P values were adjusted for 54 multiple testing by using FDR correction. The genotypic test was chosen as the genetic test model with lower Akaike information/lower P value. N = 898 PD cases; 921 controls (N = 1,819). CI, confidence interval; Freq., frequency; M, major allele; MAPT, microtubule-associated protein Tau; m, minor allele; OR, odds ratio; SNCA, α-synuclein; SNP, single nucleotide polymorphism.
Association of SNCA and mTOR Interactions With the Risk of IPD
Subsequently, in the discovery series we explored possible gene-gene interactions involving more than 2 loci by MDR analysis. Using this approach, we found that SNP rs356219 in SNCA synergistically interacts with mTOR markers, modulating the risk of PD, including the SNPs rs8111699 in SKT11, rs456998 in FCHSD1, and rs1732170 in GSK3B (OR [95% CI] = 2.59 [2.14–3.13], explicit epistasis test P < .001; Table 2 and Supporting Information Fig. 1). In addition, the CVC for this 4-marker model was 10, and both the training and testing balanced accuracies were around 60% (precision, specificity, and sensitivity; Table S3). However, we could not replicate these results in the IPDGC cohort (Table S4), and thus they are likely to be attributable to a main effect of the SNCA marker.
TABLE 2.
SNP rs356219 in SNCA interacts with rs8111699 in SKT11, rs456998 in FCHSD1, and rs1732170 in GSK3B in modulating PD risk
| Gene | SNP | Bal. acc. CV training | Bal. acc. CV testing | CVC | OR (95% CI) | Association P valuea | Epistatic association P valueb |
|---|---|---|---|---|---|---|---|
| SNCA | rs356219 | 0.5378 | 0.5245 | 7/10 | 1.37 (1.13–1.66) | .306–.307 | .925–.926 |
| STK11 | rs8111699 | 0.5529 | 0.5283 | 5/10 | 1.52 (1.26–1.83) | .214–.215 | .874–.875 |
| GSK3B | rs1732170 | ||||||
| SNCA | rs356219 | 0.5729 | 0.5247 | 5/10 | 1.79 (1.48–2.16) | .300–.301 | .924–.925 |
| STK11 | rs8111699 | ||||||
| GSK3B | rs1732170 | ||||||
| SNCA | rs356219 | 0.6181 | 0.5875 | 10/10 | 2.59 (2.14–3.13) | <.001 | <.001 |
| STK11 | rs8111699 | ||||||
| FCHSD1 | rs456998 | ||||||
| GSK3B | rs1732170 | ||||||
Statistically significant P values are bold. Test of association for SNP combinations with PD risk. N = 898 PD cases and N = 921 unrelated controls. Random seed = 10; CVC = 10. CI, confidence interval; CV, cross-validation; CVC, cross-validation count; OR, odds ratio; SNCA, α-synuclein; SNP, single nucleotide polymorphism.
Normal P value.
P value of explicit test of epistasis obtained with 1000 permutations.
Single-Marker Association of SNCA With the AAO of IPD
We further performed a single-marker association analysis of the studied SNPs with the AAO of IPD in the discovery series. We analyzed whether those SNPs significantly associated with PD risk could also modulate the AAO at a single-marker level. For this reason, only the SNP in MAPT and SNCA were subjected to the analysis. In the single-marker genotypic analysis, adjusting by gender, age, and multiple testing (n = 2 tests), we found that SNP rs356219 in SNCA was the only marker associated with differential AAO of PD in our sample. Specifically, we observed a mean IPD AAO ± Standard Error of the Mean (S.E.M). of 55.6 ± 0.8 years for GG carriers, 55.8 ± 0.5 for AG, and 58.0 ± 0.5 for AA (adjusted P = .0034) following a log-additive model, that is, an overall IPD AAO difference of approximately 2.5 years attributable to this SNP. These data are consistent with previous results reported in IPD13,47 or in L2PD,7,14 although in the latter, SNCA rs356219 seemed to have a stronger effect on the L2PD AAO with a difference of up to 11 years (58 years for GG carriers vs 69 years for AA).14 In addition, we did not find an association of MAPT rs1800547 with the AAO of PD (Table 3).
TABLE 3.
Genotypic association tests of SNCA rs356219 and MAPT rs1800547 SNPs with AAO of Parkinson’s disease with sex, age, and multiple testing adjustments of P values
| Gene | SNP | Alleles, M/m | Freq. 11 | Freq. 12 | Freq. 22 | AAO 11, y | AAO 12, y | AAO 22, y | Difference, y | Model | P value | Adjusted P value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNCA | rs356219 | A (1)/G (2) | 0.36 | 0.47 | 0.17 | 58.03 ± 0.5 | 55.84 ± 0.48 | 55.64 ± 0.85 | −1.35 (−2.19 to −0.51) | Log-additive | .0017 | .0034 |
| MAPT | rs1800547 | A (1)/G (2) | 0.56 | 0.38 | 0.06 | 56.46 ± 0.61 | 56.15 ± 0.75 | 58.36 ± 1.73 | 0.30 (−0.66 to 1.26) | Log-additive | .54 | .54 |
Statistically significant P values are bold. Genotypic test calculated in the SNPstats software with sex and age adjustment of P values. The model of inheritance best fitting the data was automatically selected as the model with the lowest Akaike information value. Two SNPs were considered for P value multiple test adjustment. N = 748 PD cases with AAO available. AAO, age at onset; Freq., frequency; M, major allele; MAPT, microtubule-associated protein Tau; m, minor allele; SNCA, α-synuclein; SNP, single nucleotide polymorphism; y, years.
SNCA and mTOR Epistatic Interactions Modify the AAO of IPD
We then explored whether epistatic combinations of mTOR SNPs were associated with the AAO of idiopathic PD in the discovery series. Using the MDR analysis, we found that SNCA SNP rs356219 interacts with rs11868112 in RPTOR and rs6456121 in RPS6KA2 in modulating the AAO of IPD with the maximum CVC of 10/10 (OR [95% CI] = 2.89 [2.90–4.00], explicit epistasis test P < .001; Table 4 and Supporting Information Fig. 2). Interestingly, in the IPDGC replication series, we confirmed the epistatic interaction of this 3-loci and their association with differential AAO of IPD with the maximum CVC of 10/10, although with a slighter effect magnitude (OR [95% CI] = 1.56 [1.34–1.81], explicit epistasis test P = .046-.047; Table 5 and Supporting Information Fig. 2), thus validating findings from the discovery series.
TABLE 4.
SNP rs356219 in SNCA interacts with rs11868112 in RPTOR and rs6456121 in RPS6KA2 in modulating the AAO of idiopathic PD
| Gene | SNP | t statistic CV training | t statistic CV testing | CVC | OR (95% CI) | Association P valuea | Epistatic association P valueb |
|---|---|---|---|---|---|---|---|
| SNCA | rs356219 | 2.6757 | 1.0612 | 5/10 | 1.31 (0.96–1.79) | .477–.478 | .640–.641 |
| RPTOR | rs11868112 | 4.1806 | 3.1885 | 8/10 | 1.63 (1.20–2.21) | .0260–.0270 | .0460–.0470 |
| RPS6KA2 | rs6456121 | ||||||
| RPTOR | rs11868112 | 7.6501 | 6.0162 | 10/10 | 2.89 (2.90–4.00) | <.001 | <.001 |
| SNCA | rs356219 | ||||||
| RPS6KA2 | rs6456121 | ||||||
Statistically significant P values are bold. Test of association for SNP combinations with PD AAO. AAO was considered a continuous variable. N = 748 PD cases; random seed = 10; CVC = 10. OR were obtained generating a dichotomous dataset comparing early PD AAO (before 57 years) and late PD AAO (after 58 years) based on the average of AAO found with the multifactor dimensionality reduction (56.66 years). AAO, age at onset; CI, confidence interval; CV, cross-validation; CVC, cross-validation count; OR, odds ratio; PD, Parkinson’s disease; SNCA, α-synuclein; SNP, single nucleotide polymorphism.
Normal P value.
P value of explicit test of epistasis obtained with 1000 permutations.
TABLE 5.
SNP rs356219 in SNCA interacts with rs11868112 in RPTOR and rs6456121 in RPS6KA2 in modulating the AAO of idiooathic PD in the International Parkinson’s Disease Genomics Consortium cohort
| Gene | SNP | t statistic CV training | t statistic CV testing | CVC | OR (95% CI) | Association P valuea | Epistatic association P valueb |
|---|---|---|---|---|---|---|---|
| RPTOR | rs11868112 | 5.7190 | 4.2753 | 10/10 | 1.56(1.34–1.81) | 0.001–0.002 | 0.046–0.047 |
| SNCA | rs356219 | ||||||
| RPS6KA2 | rs6456121 | ||||||
Statistically significant P values are bold. Forced Interaction of SNPs rs11868112 in RPTOR, rs356219 in SNCA, and rs6456121 in RPS6KA2 with PD AAO in the International Parkinson’s Disease Genomics Consortium cohort. AAO was considered a continuous variable. N = 2798 PD cases; random seed = 10; CVC = 10. OR was obtained generating a dichotomous dataset comparing early PD AAO (before 60 years) and late PD AAO (after 61 years) based on the average of AAO found with the multifactor dimensionality reduction (60.86 years). AAO, age at onset; CI, confidence interval; CV, cross-validation; CVC, cross-validation count; OR, odds ratio; PD, Parkinson’s disease; SNCA, α-synuclein; SNP, single nucleotide polymorphism.
Normal P value.
P value of explicit test of epistasis obtained with 1000 permutations.
We also explored whether this 3-loci epistatic interaction modulates AAO in our cohort of 127 L2PD patients whose mean AAO was 56.74 years, similar to the mean AAO of 56.66 years observed in IPD. Despite the relative limited number of participants, we exploratively performed a forced MDR analysis for this 3-loci interaction and observed a similar association trend in L2PD as in IPD that did not reach statistical significance (OR [95% CI] = 3.99 [1.89–8.40], explicit epistasis test P = .1170-.1180; Table S5). However, when we oversampled this L2PD cohort to the number of IPD patients (N = 748), we could again observe the epistatic association of these 3 SNPs with L2PD AAO, thus suggesting a similar effect in L2PD as in IPD, which should be further validated in larger L2PD cohorts (data not shown; OR [95% CI] = 4.68 [2.56–8.58], explicit epistasis test P < .001).
In summary, these results indicate that there is a higher order complex epistatic effect of SNCA rs356219 with markers RPTOR rs11868112 and RPS6KA2 rs6456121 from the mTOR pathway in modulating the AAO of IPD. This finding illustrates the proof of principle that epistatic interactions acting on known PD genetic risk factors can further modulate disease presentation.
Discussion
Here, we report for the first time that common genetic variability in the mTOR genetic pathway synergistically interacts with known PD risk SNPs in SNCA to modulate the AAO of IPD. The association of genetic polymorphisms such as SNCA rs356219 or of MAPT rs1800547 with the risk of PD was previously reported in our cohort7,48 and in other cohorts45,46 as well as by genomewide association studies.5,6 Here we also observed significant association of these markers with PD risk. At the multilocus level for the MAPT polymorphism, we did not find any epistatic interaction with other SNPs. However, for SNCA, our data set established the grounds to perform further epistatic analyses involving the candidate mTOR pathway. Indeed, at a multilocus level, we did detect a higher order loci epistatic interaction comprising SNCA rs356219, SKT11 rs8111699, FCHSD1 rs456998, and GSK3B rs1732170, which was associated with a differential risk for PD in the discovery series. However, we did not replicate this finding in the IPDGC cohort. This negative result suggests that the SNCA SNP could be driving the main effect in PD risk or, alternatively, that other factors such as genetic heterogeneity, linkage disequilibrium differences, population allele frequency difference, or even additional environmental effects could be affecting the power to corroborate epistatic associations in populations other than the discovery series.49
As for the AAO of IPD, we found a significant association of SNCA rs356219 located in the 3’UTR with a 2.4 mean AAO difference of IPD with 55.6 years for carriers of the GG risk genotype and 58.0 years for AA carriers, but no association for MAPT. These findings are largely consistent with the most recent meta-analysis of PD AAO to date.50 Previously, the SNCA rs356219 was shown to modulate the AAO of IPD in the German population (3 years difference; 55.7 years for GG vs 58.7 years for AA)13 and also in another Northern Spain cohort genotyping of the SNCA 3’UTR neighboring marker rs356165, which is in absolute linkage disequilibrium with rs356219 (3.5 years difference; 60.1 years for GG vs 56.6 for AA).47 The most recent meta-analysis of PD AAO to date has reported a similar effect for the SNCA 3’UTR rs356203 marker, yet with a slighter AAO effect of 0.6 years difference, probably because of the larger diversity of populations screened in this study.50 In addition, in our Spanish sample we also found that SNCA rs356219 specifically influences the AAO of monogenic L2PD (11 years difference; 58 years for GG vs 69 for AA),14 overall suggesting a greater effect of SNCA rs356165 on AAO in monogenic L2PD than in IPD. At the multilocus level, we found a 3-loci high-order epistatic interaction involving SNCA rs356219, RPTOR rs11868112, and RPS6KA2 rs6456121, which was associated with IPD AAO. Indeed, the markers RPTOR rs11868112 and RPS6KA2 rs6456121 were not individually associated with IPD AAO but only in conjunction with SNCA rs356165 and in an epistatic manner. Most important, we validated the association of this epistatic interaction with the AAO of IPD in the IPDGC replication series.49,51 Overall, these findings indicate that common genetic variability in genes from the mTOR pathway, by genetic epistatic interaction with SNCA, can modulate the pathophysiology of PD and contribute to the AAO of IPD by influencing, either potentiating or diminishing, the effects of well-known risk factors of disease.
SNCA implications in the PD pathogenesis has been widely described at the genetic but also at the protein level (SNCA). In fact, the variant rs356219 of SNCA has been associated with enhanced transcription of SNCA and increased levels of SNCA.11,12 Furthermore, we have found that SNCA, RPTOR, and RPS6KA2 interact epistatically in modulating PD AAO. At the biological and biochemical levels, epistasis can be related with the presence of a physical interaction between proteins that participate in the same cellular pathway.20,21 The RPTOR gene codifies for the Raptor protein that is the main component of the mTORC1 complex. This complex regulates important cellular functions such as protein synthesis and autophagy.52 The third interacting marker is located in the RPS6KA2 gene, which encodes ribosomal S6 kinase RSK3. This protein is a serine/threonine kinase that stimulates mTOR signaling activation and modulates protein synthesis initiation via the phosphorylation of the eukaryotic translation initiation factor B.52 Increased protein synthesis and reduced autophagy in PD have been both related to SNCA accumulation and oligomerization and, thus, to dopaminergic neurodegeneration.53–55 Hence, dysfunctional activation of the mTOR pathway can contribute to SNCA aggregation and the spreading of PD pathology in the brain.
Given the large number of genetic polymorphisms in genes from the mTOR pathway, in the order of thousands, the number of SNPs screened in our study is limited and could be scaled, most specially in selected promising candidates pinpointed in this study. In fact, it is conceivable that the genetic interactions identified here involve physical and functional interactions between the different proteins, a hypothesis that should be tested in further studies. These results also should be validated in other genetically similar populations, to dismiss not only the possibility of false positive association but also any potential population-specific effects. Overall, we found that genetic variability in the mTOR pathway interacts with SNCA risk variants modulating the effect of SNCA rs356219 and determining AAO of IPD. Our findings indicate that the individual effects of classical susceptibility loci associated with PD risk such as SNCA could be additionally influenced by genetic variability in other genes, as here shown with loci from the mTOR pathway, thus further contributing to elucidating the genetic contribution to IPD as a complex disease. Moreover, a relevant implication of our findings is that classical association of individual markers reported in PD, especially in large genomewide association studies, could be revisited in light of potential epistatic complex interactions among markers that until the present have been overlooked and poorly explored in the disease.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Peter Andrews for this kind assessment with the multifactor dimensionality reduction (MDR) software use and helpful discussion. We also thank Dr. Roger Anglada from the Genomics Core Facility from the Universitat Pompeu Fabra (Barcelona) for his work and helpful assessment with sample analysis. We acknowledge the Centres de Recerca de Catalunya (CERCA) Program from the Generalitat de Catalunya and the Fondo Europeo de Desarrollo Regional (FEDER) Program from the European Union to Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS). RF-S was supported by a Jóvenes Investigadores grant (SAF2015-73508-JIN) through the Programa Estatal de Investigación, Desarrollo e Innovación Orientada a los Retos de la Sociedad (Plan Estatal de Investigacion Científica, tecnica y de innovacion (ICDCI) 2013-2016) of the Spanish Ministry of Economy and Competitiveness Ministerio de Economia (MINECO), and the Agencia Estatal de Investigación, which is cofunded by FEDER Agencia Europea de Investigacion/Fondo Europeo de Desarrollo Regional/Union Europea (AEI/FEDER/UE).
Funding agency: MichaelJ. Fox Foundation.
Appendix: International Parkinson’s Disease Genomics Consortium Members and Affiliations
United Kingdom: Alastair J Noyce (Preventive Neurology Unit, Wolfson Institute of Preventive Medicine, QMUL, London, UK and Department of Molecular Neuroscience, UCL, London, UK), Rauan Kaiyrzhanov (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Ben Middlehurst (Institute of Translational Medicine, University of Liverpool, Liverpool, UK), Demis A Kia (UCL Genetics Institute; and Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Manuela Tan (Department of Clinical Neuroscience, University College London, London, UK), Henry Houlden (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Huw R Morris (Department of Clinical Neuroscience, University College London, London, UK), Helene Plun-Favreau (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Peter Holmans (Biostatistics & Bioinformatics Unit, Institute of Psychological Medicine and Clinical Neuroscience, MRC Centre for Neuropsychiatric Genetics & Genomics, Cardiff, UK), John Hardy (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Daniah Trabzuni (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK; Department of Genetics, King Faisal Specialist Hospital and Research Centre, Riyadh, 11211 Saudi Arabia), Jose Bras (UK Dementia Research Institute at UCL and Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), John Quinn PhD (Institute of Translational Medicine, University of Liverpool, Liverpool, UK), Kin Y. Mok (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Kerri J. Kinghorn (Institute of Healthy Ageing, University College London, London, UK), Kimberley Billingsley (Institute of Translational Medicine, University of Liverpool, Liverpool, UK), Nicholas W. Wood (UCL Genetics Institute; and Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Patrick Lewis (University of Reading, Reading, UK), Sebastian Schreglmann (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Rita Guerreiro (UK Dementia Research Institute at UCL and Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Ruth Lovering (University College London, London, UK), Lea R’Bibo (Department of Molecular Neuroscience, UCL Institute of Neurology,London, UK), Claudia Manzoni (University of Reading, Reading, UK), Mie Rizig (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Mina Ryten (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Sebastian Guelfi (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Valentina Escott-Price (MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University School of Medicine, Cardiff, UK), Viorica Chelban (Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Thomas Foltynie (UCL Institute of Neurology, London, UK), Nigel Williams (MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff, UK), Karen E. Morrison (Faculty of Medicine, University of Southampton, UK), Carl Clarke (University of Birmingham, Birmingham, UK and Sandwell and West Birmingham Hospitals NHS Trust, Birmingham, UK).
France: Alexis Brice (Institut du Cerveau et de la Moelle épinière, ICM, Inserm U 1127, CNRS, UMR 7225, Sorbonne Universités, UPMC University Paris 06, UMR S 1127, AP-HP, Pitié-Salpêtrière Hospital, Paris, France), Fabrice Danjou (Institut du Cerveau et de la Moelle épinière, ICM, Inserm U 1127, CNRS, UMR 7225, Sorbonne Universités, UPMC University Paris 06, UMR S 1127, AP-HP, Pitié-Salpêtrière Hospital, Paris, France), Suzanne Lesage (Institut du Cerveau et de la Moelle épinière, ICM, Inserm U 1127, CNRS, UMR 7225, Sorbonne Universités, UPMC University Paris 06, UMR S 1127, AP-HP, Pitié-Salpêtrière Hospital, Paris, France), Jean-Christophe Corvol (Institut du Cerveau et de la Moelle épinière, ICM, Inserm U 1127, CNRS, UMR 7225, Sorbonne Universités, UPMC University Paris 06, UMR S 1127, Centre d’Investigation Clinique Pitié Neurosciences CIC-1422, AP-HP, Pitié-Salpêtrière Hospital, Paris, France), Maria Martinez (INSERM UMR 1220; and Paul Sabatier University, Toulouse, France).
Germany: Claudia Schulte (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, and Deutsches Zentrum fur Neurodegenerative Erkrankungen (DZNE), German Center for Neurodegenerative Diseases, Tübingen, Germany), Kathrin Brockmann (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, and DZNE, German Center for Neurodegenerative Diseases, Tübingen, Germany), Javier Simon-Sanchez (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, and DZNE, German Center for Neurodegenerative Diseases, Tübingen, Germany), Peter Heutink (DZNE, German Center for Neurodegenerative Diseases and Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany), Patrizia Rizzu (DZNE, German Center for Neurodegenerative Diseases), Manu Sharma (Centre for Genetic Epidemiology, Institute for Clinical Epidemiology and Applied Biometry, University of Tubingen, Germany), Thomas Gasser (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, and DZNE, German Center for Neurodegenerative Diseases, Tübingen, Germany).
United States of America: Aude Nicolas (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Mark R. Cookson (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, USA), Sara Bandres-Ciga (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Cornelis Blauwendraat (National Institute on Aging and National Institute of Neurological Disorders and Stroke, USA), David W. Craig (Department of Translational Genomics, Keck School of Medicine, University of Southern California, Los Angeles, USA), Faraz Faghri (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, USA; Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, IL, USA), J. Raphael Gibbs (Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA), Dena G. Hernandez (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Kendall Van Keuren-Jensen (Neurogenomics Division, TGen, Phoenix, AZ USA), Joshua M. Shulman (Departments of Neurology, Neuroscience, and Molecular & Human Genetics, Baylor College of Medicine, Houston, Texas, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, Texas, USA), Hirotaka Iwaki (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Hampton L. Leonard (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Mike A. Nalls (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, USA; CEO/Consultant Data Tecnica International, Glen Echo, MD, USA), Laurie Robak (Baylor College of Medicine, Houston, Texas, USA), Steven Lubbe (Ken and Ruth Davee Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA), Steven Finkbeiner (Departments of Neurology and Physiology, University of California, San Francisco; Gladstone Institute of Neurological Disease; Taube/ Koret Center for Neurodegenerative Disease Research, San Francisco, CA, USA), Niccolo E. Mencacci (Northwestern University Feinberg School of Medicine, Chicago, IL, USA), Codrin Lungu (National Institutes of Health Division of Clinical Research, NINDS, National Institutes of Health, Bethesda, MD, USA), Andrew B. Singleton (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Sonja W. Scholz (Neurodegenerative Diseases Research Unit, National Institute of Neurological Disorders and Stroke, Bethesda, MD, USA), Xylena Reed (Laboratory of Neurogenetics, National Institute on Aging, Bethesda, MD, USA), Roy N. Alcalay (Department of Neurology, College of Physicians and Surgeons, Columbia University Medical Center, New York, NY, USA, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, College of Physicians and Surgeons, Columbia University Medical Center, New York, NY, USA).
Canada: Ziv Gan-Or (Montreal Neurological Institute and Hospital, Department of Neurology & Neurosurgery, Department of Human Genetics, McGill University, Montréal, QC, H3A 0G4, Canada), Guy A. Rouleau (Montreal Neurological Institute and Hospital, Department of Neurology & Neurosurgery, Department of Human Genetics, McGill University, Montréal, QC, H3A 0G4, Canada), Lynne Krohn (Montreal Neurological Institute and Hospital, Department of Neurology & Neurosurgery, Department of Human Genetics, McGill University, Montréal, QC, H3A 0G4, Canada).
The Netherlands: Jacobus J. van Hilten (Department of Neurology, Leiden University Medical Center, Leiden, Netherlands) and Johan Marinus (Department of Neurology, Leiden University Medical Center, Leiden, Netherlands).
Spain: Astrid D. Adarmes-Gómez (Instituto de Biome- dicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), iquel Aguilar (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona.), Ignacio Alvarez (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona.), Victoria Alvarez (Hospital Universitario Central de Asturias, Oviedo), Francisco Javier Barrero (Hospital Universitario San Cecilio de Granada, Universidad de Granada), Jesús Alberto Bergareche Yarza (Instituto de Investigación Sanitaria Biodonostia, San Sebastián), Inmaculada Bernal-Bernal (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Marta Blazquez (Hospital Universitario Central de Asturias, Oviedo), Marta Bonilla-Toribio (Instituto de Biome- dicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Juan A. Botía (Universidad de Murcia, Murcia), María Teresa Boungiorno (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona), Dolores Buiza-Rueda (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Ana Cámara (Hospital Clinic de Barcelona), Fátima Carrillo (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/ Universidad de Sevilla, Seville), Mario Carrión-Claro (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Debora Cerdan (Hospital General de Segovia, Segovia), Jordi Clarimón (Memory Unit, Department of Neurology, IIB Sant Pau, Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [Madrid]), Yaroslau Compta (Hospital Clinic de Barcelona), Beatriz de la Casa (Hospital Gregorio Marañon, Madrid), Monica Diez-Fairen (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona), Oriol Dols-Icardo (Memory Unit, Department of Neurology, IIB Sant Pau, Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Barcelona, and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [CIBERNED], Madrid), Jacinto Duarte (Hospital General de Segovia, Segovia), Raquel Duran (Centro de Investigacion Biomedica, Universidad de Granada, Granada), Francisco Escamilla-Sevilla (Hospital Universitario Virgen de las Nieves, Instituto de Investigación Biosanitaria de Granada, Granada), Mario Ezquerra (Hospital Clinic de Barcelona), Cici Feliz (Departmento de Neurologia, Instituto de Investigación Sanitaria Fundación Jiménez Díaz, Madrid, Spain), Manel Fernández (Hospital Clinic de Barcelona), Rubén Fernández- Santiago (Hospital Clinic de Barcelona), Ciara Garcia (Hospital Universitario Central de Asturias, Oviedo), Pedro García-Ruiz (Instituto de Investigación Sanitaria Fundación Jiménez Díaz, Madrid), Pilar Gómez-Garre (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Maria Jose Gomez Heredia (Hospital Universitario Virgen de la Victoria, Malaga), Isabel Gonzalez-Aramburu (Hospital Universitario Marqués de Valdecilla-IDIVAL, Santander), Ana Gorostidi Pagola (Instituto de Investigación Sanitaria Bio- donostia, San Sebastián), Janet Hoenicka (Institut de Recerca Sant Joan de Déu, Barcelona), Jon Infante (Hospital Universitario Marqués de Valdecilla-IDIVAL and University of Cantabria, Santander, and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [CIBERNED]), Silvia Jesús (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Adriano Jimenez-Escrig (Hospital Universitario Ramón y Cajal, Madrid), Jaime Kulisevsky (Movement Disorders Unit, Department of Neurology, IIB Sant Pau, Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Barcelona, and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [CIBERNED]), Miguel A. Labrador-Espinosa (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Jose Luis Lopez-Sendon (Hospital Universitario Ramón y Cajal, Madrid), Adolfo López de Munain Arregui (Instituto de Investigación Sanitaria Biodonostia, San Sebastián), Daniel Macias (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Irene Martínez Torres (Department of Neurology, Instituto de Investigación Sanitaria La Fe, Hospital Universitario y Politécnico La Fe, Valencia), Juan Marín (Movement Disorders Unit, Department of Neurology, IIB Sant Pau, Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Barcelona, and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [CIBERNED]), Maria Jose Marti (Hospital Clinic Barcelona), Juan Carlos Mar- tínez-Castrillo (Instituto Ramón y Cajal de Investigación Sanitaria, Hospital Universitario Ramón y Cajal, Madrid), Carlota Méndez-del-Barrio (Instituto de Biome- dicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Manuel Menéndez González (Hospital Universitario Central de Asturias, Oviedo), Marina Mata (Department of Neurology, Hospital Universitario Infanta Sofía, Madrid, Spain), Adolfo Mínguez (Hospital Universitario Virgen de las Nieves, Granada, Instituto de Investigación Biosanitaria de Granada), Pablo Mir (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/ CSIC/Universidad de Sevilla, Seville), Elisabet Mondragon Rezola (Instituto de Investigación Sanitaria Biodonostia, San Sebastián), Esteban Muñoz (Hospital Clinic Barcelona), Javier Pagonabarraga (Movement Disorders Unit, Department of Neurology, IIB Sant Pau, Hospital de la Santa Creu i Sant Pau, Universitat Autónoma de Barcelona, Barcelona, and Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas [CIBERNED]), Berta Pascual-Sedano (Unidad de Trastornos del Movimiento, Hospital Sant Pau, Barcelona), Pau Pastor (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona), Francisco Perez Errazquin (Hospital Universitario Virgen de la Victoria, Malaga), Teresa Periñán-Tocino (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Javier Ruiz- Martínez (Hospital Universitario Donostia, Instituto de Investigación Sanitaria Biodonostia, San Sebastián), Clara Ruz (Centro de Investigacion Biomedica, Universidad de Granada, Granada), Antonio Sanchez Rodriguez (Hospital Universitario Marqués de Valdecilla-IDIVAL, Santander), María Sierra (Hospital Universitario Marqués de Valdecilla-Instituto de Investigacion Valdecilla (IDIVAL), Santander), Esther Suarez-Sanmartin (Hospital Universitario Central de Asturias, Oviedo), Cesar Tabernero (Hospital General de Segovia, Segovia), Juan Pablo Tartari (Fundació Docencia i Recerca Mútua de Terrassa and Movement Disorders Unit, Department of Neurology, University Hospital Mutua de Terrassa, Terrassa, Barcelona), Cristina Tejera-Parrado (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Eduard Tolosa (Hospital Clinic Barcelona), Francesc Valldeoriola (Hospital Clinic Barcelona), Laura Vargas- González (Instituto de Biomedicina de Sevilla [IBiS], Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville), Lydia Vela (Department of Neurology, Hospital Universitario Fundación Alcorcón, Madrid), Francisco Vives (Centro de Investigacion Biomedica, Universidad de Granada, Granada).
Austria: Alexander Zimprich (Department of Neurology, Medical University of Vienna, Austria).
Norway: Lasse Pihlstrom (Department of Neurology, Oslo University Hospital, Oslo, Norway), Mathias Toft (Department of Neurology and Institute of Clinical Medicine, Oslo University Hospital, Oslo, Norway).
Estonia: Sulev Koks (Department of Pathophysiology, University of Tartu, Tartu, Estonia; Department of Reproductive Biology, Estonian University of Life Sciences, Tartu, Estonia), Pille Taba (Department of Neurology and Neurosurgery, University of Tartu, Tartu, Estonia).
Israel: Sharon Hassin-Baer (The Movement Disorders Institute, Department of Neurology and Sagol Neuroscience Center, Chaim Sheba Medical Center, Tel- Hashomer, 5262101, Ramat Gan, Israel, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel).
Footnotes
Relevant conflicts of interests/financial disclosures: Nothing to report.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Fahn S Medical treatment of Parkinson’s disease. J Neurol 1998; 245(11 suppl 3):P15–P24. [DOI] [PubMed] [Google Scholar]
- 2.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39(6):889–909. [DOI] [PubMed] [Google Scholar]
- 3.Marras C, Lang A, van de Warrenburg BP, Sue CM, Tabrizi SJ, Bertram L, et al. Nomenclature of genetic movement disorders: Recommendations of the International Parkinson and Movement Disorder Society task force. Mov Disord 2017;32(5):724–725. [DOI] [PubMed] [Google Scholar]
- 4.Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The Onset of Nonmotor Symptoms in Parkinson’s disease (The ONSET PDStudy). Mov Disord 2015;30(2):229–237. [DOI] [PubMed] [Google Scholar]
- 5.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014; 46(9):989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41(12):1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botta-Orfila T, Ezquerra M, Ríos J, Fernández-Santiago R, Cervantes S, Samaranch L, et al. Lack of interaction of SNCA and MAPT genotypes in Parkinson’s disease. Eur J Neurol 2011;18(2):e32. [DOI] [PubMed] [Google Scholar]
- 8.Ezquerra M, Compta Y, Marti MJ. Identifying the genetic components underlying the pathophysiology of movement disorders. Appl Clin Genet 2011;4:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N-N, Mao X-Y, Chang X-L, et al. SNCA rs356219 variant increases risk of sporadic Parkinson’s disease in ethnic Chinese. Am J Med Genet Part B Neuropsychiatr Genet 2013;162(5): 452–456. [DOI] [PubMed] [Google Scholar]
- 10.Campelo CL, Cagni FC, de Siqueira Figueredo D, et al. Variants in SNCA Gene Are Associated with Parkinson’s Disease Risk and Cognitive Symptoms in a Brazilian Sample . Front Aging Neurosci 2017; 9:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuchs J, Tichopad A, Golub Y, et al. Genetic variability in the SNCA gene influences α-synuclein levels in the blood and brain. FASEB J 2008;22(5):1327–1334. [DOI] [PubMed] [Google Scholar]
- 12.Mata IF, Shi M, Agarwal P, et al. SNCA variant associated with Parkinson disease and plasma α-synuclein level. Arch Neurol 2010; 67(11):1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brockmann K, Schulte C, Hauser AK, et al. SNCA: Major genetic modifier of age at onset of Parkinson’s disease. Mov Disord 2013; 28(9):1217–1221. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Santiago R, Garrido A, Infante J, et al. α-synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine-rich repeat kinase 2 (LRRK2) Parkinson’s disease in Spain. Mov Disord 2018;33(4):637–641. [DOI] [PubMed] [Google Scholar]
- 15.Kyratzi E, Pavlaki M, Stefanis L. The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum Mol Genet 2008;17(14):2160–2171. [DOI] [PubMed] [Google Scholar]
- 16.Golab-Janowska M, Honczarenko K, Gawronska-Szklarz B, Potemkowski A. CYP2D6 gene polymorphism as a probable risk factor for Alzheimer’s disease and Parkinson’s disease with dementia. Neurol Neurochir Pol 2007;41(2):113–121. [PubMed] [Google Scholar]
- 17.Lill CM. Genetics of Parkinson’s disease. Mol Cell Probes 2016;30 (6):386–396. [DOI] [PubMed] [Google Scholar]
- 18.Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004;81(1):70–73. [DOI] [PubMed] [Google Scholar]
- 19.Rajput AH, Uitti RJ, Stern W, et al. Geography, drinking water chemistry, pesticides and herbicides and the etiology of Parkinson’s disease. Can J Neurol Sci 1987;14(3 suppl):414–418. [DOI] [PubMed] [Google Scholar]
- 20.Campbell RF, McGrath PT, Paaby AB. Analysis of epistasis in natural traits using model organisms. Trends Genet 2018;34(11): 883–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattin KA, White BC, Barney N, et al. A computationally efficient hypothesis testing method for epistasis analysis using multifactor dimensionality reduction. Genet Epidemiol 2009;33(1):87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turton JC, Bullock J, Medway C, et al. Investigating Statistical Epis tasis in Complex Disorders. J Alzheimer’s Dis 2011;25(4):635–644. [DOI] [PubMed] [Google Scholar]
- 23.Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis 2009;14:478–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci 2006;26(39):9996–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci 2008;28(53):14363–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci 2009;12(9):1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci 2010;30(3):1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Q, Liu C, Liu W, et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol Sci 2015;143(1):81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calabresi P, Galletti F, Saggese E, Ghiglieri V, Picconi B . Neuronal networks and synaptic plasticity in Parkinson’s disease: beyond motor deficits. Park Relat Disord 2007;13(suppl 3):S259–S262. [DOI] [PubMed] [Google Scholar]
- 30.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptorindependent pathway that regulates the cytoskeleton. Curr Biol 2004;14(14):1296–1302. [DOI] [PubMed] [Google Scholar]
- 31.Kim D-H, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002;110(2):163–175. [DOI] [PubMed] [Google Scholar]
- 32.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004;6(11):1122–1128. [DOI] [PubMed] [Google Scholar]
- 33.Moore JH, Andrews PC. Epistasis analysis using multifactor dimensionality reduction In: Moore Jason H., Williams Scott M., eds Epistasis: Methods and Protocols. Switzerland AG: Springer Nature, 2014:301–314. [DOI] [PubMed] [Google Scholar]
- 34.Ritchie M, Hahn L, Roodi N, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet 2001; 69(1):138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Motsinger AA, Ritchie MD. The effect of reduction in cross-validation intervals on the performance of multifactor dimensionality reduction. Genet Epidemiol 2006;30(6):546–555. [DOI] [PubMed] [Google Scholar]
- 36.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 1993;50(2): 140–148. [DOI] [PubMed] [Google Scholar]
- 37.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 2009;8:50–57. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez-Santiago R, Iranzo A, Gaig C, et al. Absence of LRRK2 mutations in a cohort of patients with idiopathic REM sleep behavior disorder. Neurology 2016;86(11):1072–1073. [DOI] [PubMed] [Google Scholar]
- 39.Gibbs RA, Belmont JW, Hardenbol P, et al. The International HapMap Project. Nature 2003;426(6968):789–796. [DOI] [PubMed] [Google Scholar]
- 40.Blauwendraat C, Bandrés-Ciga S, Singleton AB. Predicting progression in patients with Parkinson’s disease. Lancet Neurol 2017;16 (11):860–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudbridge F Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol 2003;25(2):115–121. [DOI] [PubMed] [Google Scholar]
- 42.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995;57(1):289–300. [Google Scholar]
- 43.Sole X, Guino E, Valls J, Iniesta R, Moreno V. SNPStats: a web tool for the analysis of association studies. Bioinformatics 2006;22(15): 1928–1929. [DOI] [PubMed] [Google Scholar]
- 44.Greene CS, Himmelstein DS, Nelson HH, et al. Enabling personal genomics with an explicit test of epistasis In: Biocomputing 2010. Singapore: World Scientific; 2009:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mata IF, Yearout D, Alvarez V, et al. Replication of MAPT and SNCA, but not PARK16–18, as susceptibility genes for Parkinson’s disease. Mov Disord 2011;26(5):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elbaz A, Ross OA, Ioannidis JPA, et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann Neurol 2011;69(5):778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardo LF, Coto E, de Mena L, et al. A aearch for SNCA 3′ UTR variants identified SNP rs356165 as a determinant of disease risk and onset age in Parkinson’s disease. J Mol Neurosci 2012;47(3): 425–430. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez-Santiago R, Iranzo A, Gaig C, et al. MAPT association with REM sleep behavior disorder. Neurol Genet 2017. 13;3(1): e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greene CS, Penrod NM, Williams SM, Moore JH . Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS One 2009:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blauwendraat C, Heilbron K, Vallerga CL, et al. Parkinson disease age of onset GWAS: defining heritability, genetic loci and α- synuclein mechanisms. Cold Spring Harbor Laboratory; 2019. 10.1002/mds.27659. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verma SS, Cooke Bailey JN, Lucas A, et al. Epistatic gene-based interaction analyses for glaucoma in eMERGE and NEIGHBOR Consortium. PLoS Genet 2016;12(9):e1006186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carrière A, Cargnello M, Julien L-A, et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol 2008;18(17):1269–1277. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Esparcia P, Hernandez-Ortega K, Koneti A, et al. Altered machinery of protein synthesis is region- and stage-dependent and is associated with α-synuclein oligomers in Parkinson’s disease. Acta Neuropathol Commun 2015;3(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from -synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110(19):E1817–E1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dehay B, Bove J, Rodriguez-Muela N, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 2010;30(37):12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009;122(Pt 20):3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.