

Abstract
With the conceptual advance some four decades ago that type 1 diabetes (T1D) represents an autoimmune disease, hope emerged that immune-based therapies would quickly evolve as a means to prevent and reverse the disorder. However, despite dozens of clinical trials seeking to achieve that purpose, the goal remains unfulfilled, at least in a pragmatic form. With the benefit of hindsight, multiple reasons are likely to account for this unfortunate situation, and several stand out: failure to appreciate disease heterogeneity; inappropriate utilization of and insight from rodent models of disease; inadequacies in addressing the immunologic and metabolic contributions to the disease; suboptimal trial designs; and lack of a clear understanding of the disorder’s pathogenesis. This review conveys how recent knowledge gains in these areas, combined with efforts related to disease staging and emerging mechanistic data from clinical trials, provide cautious optimism that an immune-based means to prevent the loss of β-cells in T1D will emerge into clinical practice.
Introduction
Dozens of immune-based therapeutic efforts have been directed towards disrupting the type 1 diabetes (T1D) autoimmune process for the purpose of averting symptomatic onset of disease (hereafter, prevention), preserving the ability to produce C-peptide after diagnosis (hereafter, preservation), or achieving other clinically meaningful features (e.g., insulin independence, improved HbA1c, avoidance of hypoglycaemia) in those with a recent diagnosis. While advances have certainly been seen towards these therapeutic goals, no methodological achievements have been extended to a public health care (i.e., non-research based) setting1. Reasons for this shortcoming are multiple, with many resulting from views once held as true that are, in fact, erroneous or at least more complex in their reality (Table 1).
Table 1.
Roadblocks hampering advancement of therapies for type 1 diabetes from research into the public healthcare setting.
| Original Hypothesis | Current / Revised Hypothesis | Current Unknowns |
|---|---|---|
| Type 1 diabetes can be divided into two subsets (immune-mediated, Type 1A and β-cell-mediated Type 1B) | β-cell dysfunction and autoimmunity are thought to occur in all type 1 diabetes cases, though the extent to which each component contributes likely varies across individuals | Can type 1 diabetes be further subdivided based on different etiologies and/or natural histories? Can these sub-classifications be used to develop tailored therapies? |
| An environmental trigger is needed to initiate and/or perpetuate type 1 diabetes in at-risk individuals | A variety of environmental exposures may contribute toward risk for disease in susceptible individuals, but none have been directly associated with type 1 diabetes, to date | Can we identify specific environmental agents with clear contributions toward type 1 diabetes? What are the temporal factors and potential interactions involved? Do they vary by demographic group or geographic region? |
| β-cell mass declines in a linear fashion for months or years prior to diagnosis of type 1 diabetes. | β-cell loss may be attributable to both a decline in mass as well as dysfunction and may occur in a non-linear pattern with a sharp decline in the months immediately prior to type 1 diabetes diagnosis. | Is the pattern of β-cell loss truly relapsing/remitting? If so, how can we extend the remission periods? Are there biomarkers available to track β-cell mass and function in real time? |
| Loss of >85% of β-cell mass precipitates symptomatic onset of type 1 diabetes and after diagnosis, destruction continues until β-cells are no longer present | Appreciable heterogeneity exists regarding β-cell mass in healthy individuals and in those with type 1 diabetes; cells expressing insulin can often be detected in the type 1 diabetes pancreas for years, even decades, after diagnosis | How can we determine the β-cell mass in living patients (both functional and dysfunctional but potential amenable to restorative therapies)? Are dysfunctional β-cells “hiding” or masking themselves against the autoimmune attack? What factors render β-cell resistant to immune-mediated killing, and how can we provide similar protection to all β-cells in susceptible individuals? |
| C-peptide secretion and insulin production decline proportionally to β-cell mass. | β-cell mass/function may decline significantly prior to detectable deficits in C-peptide production and impairments in glycemic regulation | How long can residual β-cells compensate for decreasing β-cell mass and how does the increased insulin demand affect β-cell stress? |
| Glycemic control is normal until the onset of overt symptomatic disease and clinical diagnosis. | Stage 2 type 1 diabetes is defined by the presence of multiple disease-associated autoantibodies and impaired glycemic response to oral glucose tolerance test despite normal fasting blood glucose levels and HbA1c | How can we prevent progression from Stage 1 (multiple autoantibodies) to Stage 2 (dysglycemia)? |
Where have we journeyed in immune based therapy for T1D?
The earliest attempts at immune intervention involved generalized immunosuppression (e.g., cyclosporine, azathioprine, cyclosporine plus methotrexate)2–5. While partially successful in improving metabolic outcomes in recent-onset cases of T1D, these interventions were neither durable, nor were the complications associated with their use considered demonstrative of adequate equipoise. Hence, clinical trialists largely turned their attention towards therapeutics targeting specific signalling pathways, molecules, and cell populations deemed to underlie the disorder’s development.
In the last ten years alone, nearly 70 phase I-III studies (prevention or preservation) tested agents with mechanisms largely thought to be directed at immune modulation (Appendix Page 1), cytokines and inflammation (Appendix Page 2), vitamin D (Appendix Page 3), as well as immune cell-based strategies (Appendix Page 4–5). Beyond these, numerous antigen-specific immunotherapies have also been attempted (reviewed separately in Roep et al. Lancet Diabetes & Endocrinology), and several other T1D trials remain in progress (Tables 2–3). Past efforts have not always been well “focused” in their approach, and significant shifts in emphasis clearly occurred during this 10-year period (Figure 1), largely influenced by issues of drug availability, follow-on from pre-clinical studies, perceived safety, or enthusiasm regarding an agent’s ability to target a particular immune process deemed critical to the disorder’s pathogenesis. However, in contrast with other autoimmune diseases (Appendix Page 6–8), the risk/benefit bar for T1D has been set so high that a number of drugs (e.g., Alemtuzumab [anti-CD52]) shown to be effective in other disease areas have not been trialled in T1D for reasons related to safety concerns in pediatric patients and inflated expectations to prevent or cure the disease.
Table 2.
Type 1 Diabetes Mellitus immunotherapy trials (non-antigen specific) that are awaiting, or currently recruiting participants.
| Sponsor | Clinical Trial Identifier | Title | Estimated Enrolment | Intervention | Primary Outcome | Estimated Completion Date |
|---|---|---|---|---|---|---|
| Nanjing Medical University | NCT02307695 | The Effect of Saxagliptin on Glucose Fluctuation and Immune Regulation in Patients with Type 1 Diabetes | 184 | Saxagliptin | MAGE at 24 weeks | Mar-17 |
| Federal University of São Paulo | NCT01559025 | Evaluation of Vildagliptin (Galvus®) as add-on to Insulin in Residual β-cell Function and Inflammatory Markers in New-onset Type 1 Diabetes Mellitus | 44 | Vildagliptin | MMTT C-peptide at 3, 6, 9 and 12 month | Mar-17; UNKNOWN STATUS* |
| University of Calgary | NCT02442544 | Effect of Prebiotic Fibre on Gut Microbiota, Intestinal Permeability and Glycaemic Control in Children with Type 1 Diabetes: A Pilot Randomized, Double Blind, Placebo Controlled Study | 30 | Prebiotic 1:1 Oligofructose:Inulin | HbA1C at 3 months | Jun-17 |
| Vanilloid Genetics Inc. | NCT02820558 | A Phase I Study of Safety and Pharmacological Activity of Substance P in the Reversal of Recent-Onset Type 1 Diabetes | 12 | Substance P | Safety at 20–27 days | Sep-17 |
| Ospedale San Raffaele | NCT02505893 | A Monocentric, Open-label Pilot Study to Assess the Safety and Efficacy of Minimal Islet Transplantation in Patients with New-onset Type 1 Diabetes | 6 | ATG + pG-CSF + Rapamycin + Human Pancreatic Islet | Safety and MMTT C-peptide at 12 months | May-18 |
| University of Jordan | NCT02940418 | Use of Stem Cells in Diabetes Mellitus Type 1 | 20 | AD-MSCs + BM-MNCs | Safety at 6 months | Jul-18 |
| ITN | NCT02293837 | EXTEND | 177 | Tocilizumab | MMTT C-peptide at 12 months | Jul-18 |
| Uppsala University Hospital | NCT02617654 | A Randomized, Double-blinded Placebo-controlled, Paralleled Designed, Investigator Sponsored Study of the Effect of the GLP-1 Receptor Agonist Liraglutide on Β-cell Function in C-peptide Positive Type 1 Diabetic Patients | 50 | Liraglutide | MMTT C-peptide at 12 months | Sep-18 |
| Merck Sharp & Dohme Corp | NCT03170544 | A Single Ascending Dose Clinical Trial to Study the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of MK-1092 in Healthy Subjects and in Subjects With Type 1 Diabetes Mellitus | 76 | MK-1092 | Safety and Maximal Glucose Infusion Rate at 33 days | Sep-18 |
| Dompé Farmaceutici | NCT02814838 | A Phase 2, Multicentre, Randomized, Double-blind, Placebo-controlled Study in Patients with New-onset Type 1 Diabetes | 72 | Ladarixin | MMTT C-peptide at 13±1 weeks | Nov-18 |
| Assistance Publique - Hôpitaux de Paris | NCT02411253 | DIABIL-2 | 138 | rhIL-2 | MMTT C-peptide at 12 months | Dec-18 |
| Piemonti Lorenzo | NCT02803892 | MONORAPA | 60 | Rapamycin /+ Vildagliptin | MMTT C-peptide at 4±1, 12±2 weeks | Dec-18 |
| Columbia University | NCT02218619 | Clinical Investigation of Efficacy of Tauroursodeoxycholic Acid (TUDCA) to Enhance Pancreatic Β-cell Survival in Type 1 Diabetes by Reducing Endoplasmic Reticulum Stress | 20 | Tauroursodeoxycholic Acid (TUDCA) | MMTT C-peptide at 6, 12 and 18 months | Dec-18 |
| ImCyse SA | NCT03272269 | A Phase I Placebo-controlled, Double-blind, Dose Escalation Clinical Trial to Evaluate the Safety and Immune Responses of Imcyse’s IMCY-0098 in Patients With Recent Onset Type 1 Diabetes | 40 | IMCY-0098 | Safety at 24 weeks | Dec-18 |
| Medical University of Warsaw | NCT03032354 | Effect of Lactobacillus Rhamnosus GG and Bifidobacterium Lactis BB 12 on Beta-cell Function in Children With Newly Diagnosed Type 1 Diabetes - a Randomized Controlled Trial | 96 | Probiotics | MMTT C-peptide at 6 and 12 months | Dec-18 |
| Stem Cells Arabia | NCT02644759 | Transplantation of Autologous Stem Cells for the Treatment of Type 1 Diabetes Mellitus | 100 | aLD-SCs / CB-MSCs + G-CSF | Insulin requirement at 1 month | Jan-19 |
| DiaVacs, Inc. | NCT02354911 | A Randomized, Double-Blind, Placebo-Controlled, Cross-Over Study of the Safety and Efficacy of Autologous Immunoregulatory Dendritic Cells in Patients With Type 1 Diabetes | 24 | Immunoregulatory Dendritic Cells | MMTT C-peptide at 12 and 24 months | Jan-19 |
| Hackensack University Medical Center | NCT02624804 | A Pilot Study of the Therapeutic Potential of Stem Cell Educator Therapy in Type 1 Diabetes | 10 | Stem Cell Educator Therapy | Safety at 12 months | Jun-19 |
| Janssen Research & Development, LLC | NCT02846545 | T1GER | 81 | Golimumab | MMTT C-peptide at 12 months | Oct-19 |
| Second Xiangya Hospital of Central South University | NCT03011021 | Safety and Efficacy of Umbilical Cord Blood Regulatory T Cells Plus Liraglutide on Autoimmune Diabetes | 40 | UCB - Tregs + Liraglutide | Safety at 24 months | Nov-19 |
| Second Xiangya Hospital of Central South University | NCT02932826 | Safety Study and Therapeutic Effects of Umbilical Cord Blood Treg Cells on Autoimmune Diabetes | 40 | UCB - Tregs | Safety at 24 months | Nov-19 |
| Indiana University | NCT02384889 | Targeting Polyamines Using DFMO in Persons with Type 1 Diabetes: A Randomized, Double-Masked, Placebo-Controlled Phase I Study to Evaluate the Safety, Tolerability, and Initial Pharmacodynamics of Multiple Ascending Doses | 42 | Difluoromethylornithine | Safety with dose escalation at 6 months | Dec-19 |
| University of Massachusetts, Worcester | NCT03046927 | Vitamin D and Residual Beta-Cell Function in Type 1 Diabetes | 40 | Ergocalciferol | MMTT C-peptide at 12 months | Jul-20 |
| TrialNet | NCT01773707 | CTLA4-Ig (Abatacept)for Prevention of Abnormal Glucose Tolerance and Diabetes in Relatives At -Risk for Type 1 | 206 | Abatacept | Abnormal Glucose Tolerance | Nov-20 |
| Janssen Research & Development, LLC | NCT03298542 | A Phase 1b Study to Evaluate SIMPONI (Golimumab) Therapy in Children, Adolescents and Young Adults With Pre-Symptomatic Type 1 Diabetes | 30 | Golimumab | Safety at 26, 52, and 78 weeks | Jul-21 |
| University of Alberta | NCT03182426 | Autologous Hematopoietic Stem Cell Mobilization (Plerixafor) and Immunologic Reset in New Onset Type 1 Diabetes Mellitus | 60 | Alemtuzumab + Anakinra + Etanercept + Liraglutide + Plerixafor | Safety and MMTT C-peptide at 3, 6, 9, 12, 18 and 24 months | Dec-21 |
| Yale University | NCT02772679 | TILT | 16 | Tregs + IL-2 | Safety and Treg proportion at 3 years | Dec-21 |
| Hospices Civils de Lyon | NCT02804165 | Gene-virus Interactions Implicated in Type 1 Diabetes | 1100 | Enterovirus vaccination | T1D Diagnosis | Jul-22 |
| University of Miami | NCT03243058 | A Randomized, Double Blind, Phase I/II Trial of Low-Dose Interlekin-2 Immunotherapy in Established Type 1 Diabetes | 54 | Proleukin (IL-2) | MMTT C-peptide at 12 months | Jan-23 |
| Massachusetts General Hospital | NCT02081326 | Repeat BCG Vaccinations for the Treatment of Established Type 1 Diabetes | 150 | BCG | HbA1C at 1,2,3,4 and 5 years | Jul-23 |
| Medical University of Gdansk | EudraCT: 2014-004319-35 | TregVac2.0 | N/A | Tregs + anti-CD20 antibody | (Phase II Trial assessing expanded polytTregs in patients with recent onset Type 1 Diabetes Mellitus) | N/A |
| Riita Veijola, Finland | EudraCT 2014-004760-37 | Incretin-based therapy in non-symptomatic, early diagnosed Type 1 Diabetics | 15 | Liraglutide | MMTT C-peptide at 3, 6, 9 and 12 months | N/A |
denotes change in status of trial to ‘unknown’ over the course of producing this review, as the completion date has passed and trial status has not been verified for two years or more, according to Clinicaltrials.gov
AD-MSC=Adipose Tissue-derived - Mesenchymal Stromal Cell, aLD-SC=autologous Leukapheresis-Derived – Stem Cell, ATG=Antithymocyte Globulin, BCG=Bacillus Calmette-Guérin, BM-MNC=Bone Marrow Mononuclear Cell, CB-MSC=Cord Blood–derived - Mesenchymal Stem Cell, CD(4+, 127-, 25+)=Cluster of Differentiation(4 positive, 127 negative, 25 positive), CTLA4-Ig=Cytotoxic T-Lymphocyte-associated Protein Immunoglobulin, DFMO=Difluoromethylornithine, DIABIL-2=Low-dose rhIL-2 in Patients With Recently-diagnosed Type 1 Diabetes, EXTEND=Toclizumab in New-onset Type 1 Diabetes, (p)G-CSF=(pegylated)Granulocyte Colony Stimulating Factor, GLP-1=Glucagon-like peptide-1, HbA1C=Haemoglobin A1C, IL-2=Interleukin-2, IMCY-0098=ImCyse Cytotoxic T lymphocyte stimulant, ITN=Immune Tolerance Network, LLC=Limited Liability Company, MAGE=Mean Amplitude of Glycaemic Excursion, MMTT=Mixed Meal Tolerance Test, MK-1092 Merck Sharp and Dohme Prostaglandin D2 receptor antagonist, MONORAPA=Monotherapy With Rapamycin in Long-standing Type 1 Diabetes, rhIL-2=recombinant human Interleukin-2, T1D=type 1 Diabetes, T1DM=Type 1 Diabetes Mellitus, T1GER=A Study of SIMPONI® to Arrest Β-cell Loss in Type 1 Diabetes, TILT=T1DM Immunotherapy Using Polyclonal Tregs + IL-2, Treg=Regulatory T cell, TregVac2.0=Cell therapy of type 1 diabetes based on the amplified artificially Regulatory T Cell CD4+ CD25+ CD127-, and anti-CD20 antibody - randomized study, TUDCA=Tauroursodeoxycholic Acid, UCB=Umbilical Cord Blood.
Table 3.
Type 1 Diabetes immunotherapy trials (non-antigen specific) that are currently active but not recruiting participants.
| Sponsor | Clinical Trial Identifier | Title | Estimated Enrolment | Intervention | Primary Outcome | Estimated Completion Date |
|---|---|---|---|---|---|---|
| Cambridge University Hospitals NHS Foundation Trust | NCT02265809 | DILfrequency | 41 | Aldesleukin | Treg proportion at 14 weeks | Oct-16 |
| Kamada | NCT02005848 | Phase II Study to Evaluate the Efficacy and Safety of Human, Alpha-1 Antitrypsin (AAT) [Glassia®] in the Treatment of New Onset Type-1 Diabetes | 71 | AAT | MMTT C-peptide at 12 months | Jan-17; COMPLETED* |
| Assistance Publique - Hôpitaux de Paris | NCT01862120 | DFIL2-Child | 24 | IL-2 | Treg proportion at 5 days | Mar-17 |
| Universidad del Desarrollo | NCT02893306 | DMT1-MSC | 10 | MSCs | Challenge test Insulin at 1, 6 and 24 months | Mar-17 |
| University Hospital, Basel | NCT02127047 | Effects of Exercise and Inhibition of Dipeptidyl Peptidase-4 on Insulin Secretion in Subjects with Type 1 Diabetes | 24 | Sitagliptin | MMTT C-peptide at 90 days | Jun-17; COMPLETED* |
| University of British Columbia | NCT02117765 | UST1D | 20 | Ustekinumab | Safety at 12 months | Jun-17 |
| GlaxoSmithKline | NCT02284009 | Study 110933: Albiglutide Versus Placebo in Insulin-treated Subjects with New-onset Type 1 Diabetes Mellitus | 68 | Albiglutide + Insulin | MMTT C-peptide at 12 months | Oct-17; COMPLETED* |
| Jewish General Hospital | NCT02204397 | Study of Tolerability and Safety of Adding Ustekinumab to INGAP Peptide for 12 Weeks in Adult Patients with T1D Mellitus | 5 | Ustekinumab + INGAP Peptide | Safety at 6 months | Nov-17; COMPLETED* |
| Grifols Therapeutics Inc. | NCT02093221 | Study of Human Plasma-Derived Alpha1-Proteinase Inhibitor in Subjects with New-Onset Type 1 Diabetes Mellitus | 76 | Alpha1-Proteinase Inhibitor | MMTT C-peptide at 12 months | Dec-17; TERMINATED (Follow-up data would not affect trial week 52 primary endpoint results) |
| Janssen Research & Development, LLC | NCT02641522 | Modulation of STAT3 Signalling With Siltuximab in Type 1 Diabetes | 10 | Siltuximab | Immunomodulation at 12 weeks | Dec-17; COMPLETED* |
| GlaxoSmithKline | NCT02000817 | Investigation of Otelixizumab in New-Onset, Autoimmune Type 1 Diabetes Mellitus Patients | 40 | Otelixizumab | Safety for 24 months | Sep-18 |
| GeNeuro Australia PTY Ltd | NCT03179423 | Randomised, Double-Blind, Placebo-Controlled Study to Investigate GNbAC1 in Patients With Onset of Type 1 Diabetes Within 4 Years | 60 | GNbAC1 | Safety at 24 weeks | Sep-18 |
| TrialNet | NCT02215200 | Antithymocyte Globulin (ATG) and Pegylated Granulocyte Colony Stimulating Factor (GCSF) in New Onset Type 1 Diabetes | 84 | ATG + pG-CSF | MMTT C-peptide at 12 months | Oct-18 |
| Novo Nordisk A/S | NCT02443155 | A Clinical Proof-of-principle Trial in Adult Subjects with Newly Diagnosed Type 1 Diabetes Mellitus Investigating the Effect of NNC0114–0006 and Liraglutide on Preservation of Β-cell Function | 304 | NNC0114–0006 /+ Liraglutide | MMTT C-peptide at 54 weeks | Apr-19 |
| University of Alabama at Birmingham | NCT02372253 | Repurposing of Verapamil as a Beta Cell Survival Therapy in Type 1 Diabetes | 52 | Verapamil | MMTT C-peptide at 12 months | Jul-19 |
| University of California, San Francisco | NCT01781975 | Imatinib Treatment in Recent Onset Type 1 Diabetes Mellitus | 67 | Imatinib | MMTT C-peptide at 12 months | Sep-19 |
| Caladrius Biosciences, Inc. | NCT02691247 | Safety and Efficacy of CLBS03 in Adolescents with Recent Onset Type 1 Diabetes (The Sanford Project T-Rex Study) | 111 | CLBS03 | MMTT C-peptide at 12 months | Mar-20 |
| Uppsala University Hospital | NCT02057211 | Mesenchymal Stem Cells to Intervene in the Development of Type 1 Diabetes: A Blinded Randomized Study | 50 | aMSCs | MMTT C-peptide at 24 months | May-20; SUSPENDED (Updated regulations for culture procedure) |
| TrialNet | NCT01030861 | Teplizumab for Prevention of Type 1 Diabetes in Relatives “At-Risk” | 170 | Teplizumab | T1D Diagnosis | Jan-22 |
denotes change in status of trial to ‘completed’ over the course of producing this review, however results are not yet reported in the literature.
AAT=Alpha-1 Antitrypsin, aMSC=autologous Mesenchymal Stem Cell, ATG=Antithymocyte Globulin, CLBS03= Caladrius Biosciences 03, DFIL2-Child=Dose Finding Study of IL-2 at Ultra-low Dose in Children with recently Diagnosed Type 1 Diabetes, DIL frequency=Adaptive Study of IL-2 Dose Frequency on Regulatory T Cells in Type 1 Diabetes, DMT1-MSC=MSC Administration for the Management of Type 1 Diabetic Patients, GNbAC1=Humanised monoclonal antibody against Multiple Sclerosis-Associated Retrovirus gene encoding an Envelope protein, IL-2=Interleukin-2, INGAP=Islet Neogenesis Associated Protein, ITN=Immune Tolerance Network, LLC=Limited Liability Company, MMTT=Mixed Meal Tolerance Test, MSC=Mesenchymal Stem Cell, NHS=National Health Service, NNC0114–0006=Novo Nordisk C0114–0006, pG-CSF=pegylated Granulocyte Colony Stimulating Factor, STAT3=Signal Transducer and Activator of Transcription 3, T1D=Type 1 Diabetes, Treg=Regulatory T cell, UST1D=Pilot Clinical Trial of Ustekinumab in Patients with New-Onset T1D.
Figure 1. Shifts in T1D clinical research emphasis over the past 10 years.
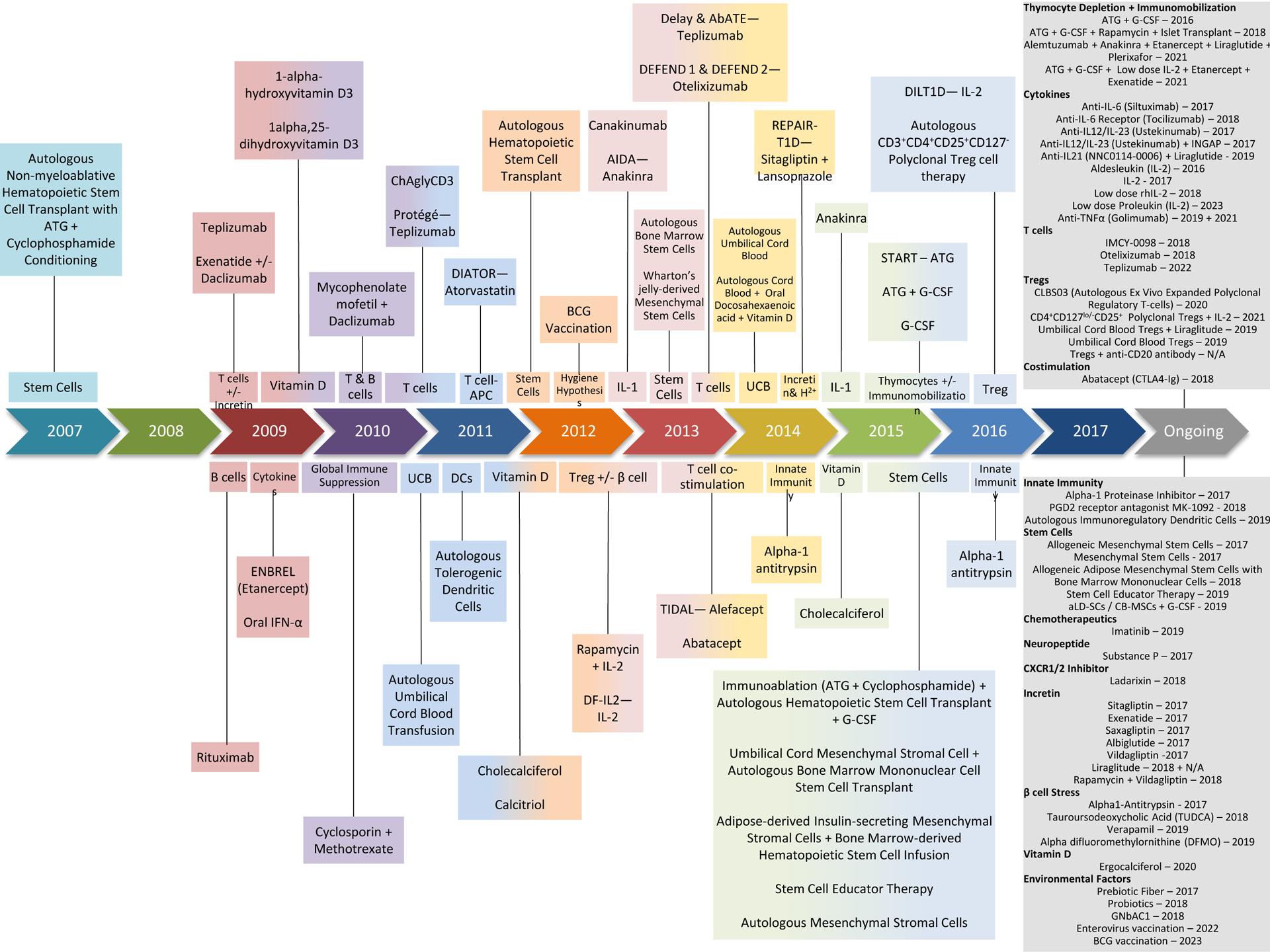
Therapeutic agents and target mechanisms/objectives are noted for T1D clinical trials over the past decade, organized by their year of first publication. Current ongoing studies are noted with their expected year of completion.
What have we learned from these studies, and did a positive change occur?
Providing a comprehensive summation of lessons learned from such an impressive level of clinical trial activity in this field represents a challenge. Against that background, we believe that the most important conceptual advance is the ever growing body of evidence that immunotherapy can be effective in altering the course of β-cell functional decline when commenced at symptomatic diagnosis of T1D.
Building on classical works demonstrating therapeutic efficacy with immune modulating/non-depleting monoclonal antibodies against CD3 (Otelixizumab, Teplizumab) on T-cells6,7, it is now evident from phase II/efficacy studies that B cell depletion (Rituximab, anti-CD20)8, reduction in circulating central and effector memory CD8+ T-cells (Alefacept [LFA3-Fc])9, as well as blockade of co-stimulation (Abatacept [CTLA4-Fc])10 each achieved similar levels of β-cell preservation for six to twelve months following disease onset in a subset of patients for each trial (Appendix Page 9–11). However, not every major therapeutic initiative proved efficacious: a Thymoglobulin-based effort and a combination trial utilizing anti-CD25/mycophenolate mofetil both failed to preserve C-peptide11,12. Moreover, Rapamycin/low-dose Proleukin (IL-2) demonstrated a temporal but reversible trend of deterioration of β-cell function13. In comparison, low-dose IL-2 monotherapy14–18 achieved immunomodulatory benchmarks (i.e., increased circulating Treg frequency), but it is not yet known whether this approach preserves C-peptide. Taken collectively, mechanistic findings from these studies support a view that agents (alone or in combination) acting upon both the effector and regulatory immune networks to an equal degree are likely to achieve a “zero sum game” in contrast with protocols that show greater selectivity for effector pathways (e.g., Alefacept [LFA3-Fc], Tocilizumab [IL-6 receptor antagonist]). These efforts afforded insights into potential mechanisms of beneficial therapeutic action, including the possibility that “exhaustion” of the effector arm and enhancement of the regulatory arm of the immune response may be associated with a positive outcome19. Such trials also paved the way for so-called “responder/non-responder” analysis (described in greater detail below and Appendix Page 9–11). Finally, seeing multiple therapeutic successes has also allowed for the design of combination or sequential strategies, with several currently at an early stage of planning. For now, their actual implementation unfortunately appears to be somewhat hindered for a number of reasons ranging from preference for conservative approaches to disputes related to trial design (i.e., drug selection, mechanistic and/or clinical endpoints, patient populations, stage of disease), and more.
These initial therapeutic successes/failures could increasingly guide the next generation of trials in patients with recent-onset disease and importantly, provide key information for efforts targeting disease prevention. But one example for the latter situation involves the performance of fully powered efficacy studies utilizing immune modulators to prevent diabetes development in high-risk subjects (i.e., multiple islet cell autoantibody-positive first-degree relatives, Stage 1–2 disease20). Indeed, prevention studies using Abatacept, a particularly well-tolerated immune modulatory fusion protein of CTLA-4 and the human immunoglobulin Fc region (TN18, NIH TrialNet), as well as anti-CD3 (Teplizumab; TN10, NIH TrialNet) are now under way. Longer-term data demonstrating improvements in safety with the latest generation of biologics (reviewed in21–24) support new trials in children with recent-onset or early-stage T1D.
Challenges for immune based interventions in T1D.
Knowledge voids, organizational or technological shortcomings, and the pathogenic puzzle that underlies T1D itself have each contributed to the current limitations of realizing immune based therapies capable of preventing T1D and/or preserving β-cell function. It is now recognized that complex and redundant mechanisms of β-cell destruction exist, involving various cell subsets and soluble factors25–29 (Table 1; described in Roep et al.). Hence, previous interventional efforts may have been too simplistic/biased in their adopted mode of action in order to overcome the redundancy afforded by the immune system in terms of contributing toward β-cell destruction.
Misguided utilization of preclinical data to direct clinical trials.
The availability of rodent models of T1D, most notably the non-obese diabetic (NOD) mouse, should theoretically accelerate efforts to prevent the disease or preserve C-peptide. However, despite the discovery of numerous immune-based therapeutic agents that initially demonstrated positive outcomes in NOD mice30 (for both prevention and reversal), such efforts have not, to a large extent, achieved their translational promise in human T1D31. Limited translational success32 can be partially ascribed to inappropriate standards for evaluating therapeutic efficacy in rodents (e.g., group sizes, definition of diabetes, use of controls), study designs that have no potential for translation due to pragmatic or equipoise issues, failure to recognize key physiological differences in the immune and metabolic systems of mice and humans in general and more specifically, in relation to T1D. Without question, insufficient attention has been paid towards scientific validation of experimental outcomes in preclinical studies of NOD mice. Indeed, recent highly organized efforts to replicate therapeutic events in NOD mice across laboratories have proven quite problematic33,34.
This is not to say preclinical studies have not been useful, the development of anti-CD335 being perhaps the most noteworthy finding despite variant mechanisms of action between mice (induction of regulatory T-cells)36 and humans (T-cell depletion)37. But perhaps, we have relied too heavily on the NOD with translational outcomes being influenced by a limited understanding of murine diabetes pathogenesis, the model’s apparent fidelity to the human condition being misleading, or preclinical success simply representing too low a bar. Moving forward, with attention towards each of these facets, we must redefine the appropriate role for animal models in guiding human T1D trials. For example, the development of TNF-blocking biologic therapies for rheumatoid arthritis (RA) involved 1) identification of abnormal TNF production within joints of human RA patients, 2) in vitro investigation of TNF blocking agents, and 3) in vivo studies of two different murine RA models (reviewed in38). While an identical approach may not be possible in T1D, perhaps greater emphasis should be placed on observations in samples from human subjects (organ donor pancreas and serum from living patients) with in vitro assays, isogenic cellular systems39, NOD and humanized mouse models40 facilitating proof of concept/mechanism studies. While a certain degree of preclinical data must lay the groundwork for human T1D trials, it may be time to revise our position on conflicting NOD data as an uncompromising roadblock. Ultimately, human mechanistic evidence should trump mouse outcomes, particularly in situations where pathogenesis/drug action clearly differ between mouse and man.
Challenge of subject selection.
In settings of RA, multiple sclerosis (MS), psoriasis, Crohn’s disease, or ulcerative colitis, the therapeutic window extends well beyond diagnosis, and trials are often conducted in patients with established disease. In contrast, T1D trials are focussed on preserving residual β-cell mass/function in new-onset T1D, which significantly limits subject availability and eliminates the possibility of repeat or crossover study design. Further limitations are imposed by patient demographics wherein at least half of new-onset T1D cases occur in children and adolescents41–43.
Industry support for immune-mediated therapies.
For decades, pharmaceutical companies expressed limited interest in this field, in part, due to relatively rare occurrence of T1D in the general population44,45, limited knowledge of human T1D pathogenesis, availability of a treatment deemed adequate by many (i.e., insulin), the need to develop safe/effective therapies for use in children, and a lack of biomarkers that reflect therapeutic success. In addition, competing/complex priorities in industry sometimes limit dialogue between entities exclusively concerned with metabolic disease (all forms of diabetes) and immunotherapy (autoimmune/inflammatory conditions, allergy, asthma, and certain cancers), with T1D straddling the two arms. Navigating this divide has frustrated many clinician scientists seeking to prevent T1D or preserve β-cell function.
Despite these historical challenges, two major phase III programs were attempted, both utilizing anti-CD3 monoclonal antibodies (i.e., Otelixizumab, Teplizumab). Sadly, both failed at the Phase III stage, for reasons that appear related to complexity and inconsistency of endpoints selection, entry criteria, as well as finding efficacious doses with minimal toxicity. For example, the DEFEND-1 and DEFEND-2 phase III trials46,47 involved a 3.1mg dose of otelixizumab which was well tolerated but failed to preserve C-peptide compared to the phase II BDR study48 (48mg otelixizumab) in which patients experienced prolonged C-peptide secretion and reduced insulin dose but also, cytokine release syndrome and Epstein-barr virus reactivation (further reviewed in49). Hence, efforts at dose optimization to minimize side effects resulted in catastrophic failure of a drug with potential for efficacy. Indeed, we must re-evaluate the acceptable risk/benefit ratio given that certain side effects, once considered non-starters, may now be considered acceptable with sufficient therapeutic efficacy.
Access to agents of potential interest is frequently hampered by the well-known vicissitudes of pharmaceutical company-led drug development. Although the pharmaceutical industry appears to maintain a watching interest, it is unusual for T1D immunotherapy to be a high priority area, with progress largely relying on the re-purposing of immune modulators (e.g., Rituximab [anti-CD20], Alefacept [LFA3-Fc] and Alemtuzumab [anti-CD52]) effective in other autoimmune and inflammatory diseases. Furthermore, business needs drove the decision to discontinue Alefacept production in November of 2011 (https://www.psoriasis.org/media/press-releases/amevive-alefacept-voluntarily-discontinued-us), despite its approval for the treatment of Psoriasis and early indications of efficacy for T1D50,51.
Paucity of informative biomarkers.
Apart from the obvious benefits provided by genetic markers of disease (i.e., HLA and risk alleles identified by genome-wide association studies (GWAS)52), autoantibodies and C-peptide represent hallmark indicators of autoimmunity and β-cell function, respectively, and remain vital tools for determining T1D risk, diagnosis, and most recently, disease staging20. However, aside from the accelerated decline in C-peptide in the months surrounding diagnosis53,54, changes in C-peptide occur slowly within a given individual; hence, trials aimed at preserving C-peptide are lengthy and involve large cohorts, often requiring at least a full year of follow-up. As a result, future therapeutic developments will increasingly depend upon new/additional surrogates of response enabling rapid, more affordable screening to identify lead candidates for larger clinical trials.
The list of needs in terms of an idealized panel of biomarkers for T1D is extensive and includes those capable of guiding attempts to: 1) improve and/or maintain optimal glycaemic control (following diagnosis); 2) decrease the effector mechanisms underlying β-cell death and dysfunction; 3) correct inherent defects in immune regulation; 4) quantitate existing β-cell mass, and 5) directly indicate the degree of β-cell death. With regard to this latter notion, two recent efforts are especially noteworthy: “β-cell death” assays (i.e., circulating demethylated insulin transcripts)55–57 and assessment of circulating proinsulin levels in association with C-peptide and insulin determinations58. The ability to separate treatment failures (i.e., an indicator of failed pathway engagement) from treatment efficacy, in the face of which a trial participant failed to respond (i.e., clinical non-responder) would be transformative. Any such biomarkers, if analysed both at baseline and following treatment, may prove useful for 1) eventual stratification of subjects who may be more likely to respond to treatment versus those who should be excluded from the study and 2) for identifying those “non-responders” who need additional treatments, as will be discuss later.
In sum, while the ability of immune-mediated therapies to modulate anti-β-cell autoimmunity has clearly been established, major voids exist in terms of our having a cadre of biomarkers capable of providing vital information regarding mechanisms of treatment efficacy (prevention and C-peptide preservation), effectiveness of a drug in achieving its desired mechanism of action (particularly in a given individual), as well as information beneficial to the design of future efforts. We also support biomarker evaluation with an eye to their use in other immune-mediated/autoimmune diseases, consideration of best practices, and in consultation with regulatory agencies. From the latter, advice for phase I or II trials that include analyses of immune biomarker identification and mechanism of action should provide useful information towards trial designs where issues of safety, treatment efficacy, and meeting desired clinical outcomes are paramount, forming what has been referred to as a “response signature”.
Remarkable disease heterogeneity.
Within each of the successful intervention trials conducted to date, it appears that reaching a primary endpoint (typically measured as preservation of C-peptide secretion) with study-wide statistical significance usually results from achieving marked efficacy in subgroups of patients (i.e., responders and non-responders) rather than a consistent, but modest change across all treated subjects (Appendix Page 9–11). Thus, as with many complex diseases, this could reflect patient heterogeneity in terms of the disease itself or, in terms of response to the therapeutic agent. A stark example of this observation would be the repeated practice of enrolling subjects across a broad age range, including both young children and adults. Children developing T1D have higher genetic risk predisposition, a more aggressive disease presentation, faster clinical progression and a broader autoimmune response, compared to adults59. Thus, age of onset is a surrogate of disease heterogeneity, and remarkably, several immunotherapies appear more effective in the younger subgroup (e.g., Rituximab60, Diamyd GAD-alum61; reviewed in62,63). However, the notion that particular clinical benefit may arise in a certain subgroup of patients is not new or restricted to T1D. Indeed, approximately 40% of patients with RA fail to respond to TNF inhibitors64 (Appendix Page 6–8), but the drugs have regulatory approval and significantly improve quality of life for those patients who respond to treatment.
In the absence of prior knowledge of specific therapeutic actions in individuals carrying certain disease variants, selection of a wide range of patients is preferable as long as power is sufficient to detect subset effects. As discussed above, a greater emphasis on enrolment of children could reap benefits, but on the whole, given our current understanding, we do not yet consider subject stratification to be favourable since selection bias may limit the opportunities for unexpected findings or identification of responder subgroups. However, perspectives are changing given the improved ability to diagnose disease prior to symptom onset20. Our focus as a field is turning toward trial efficiency, the goal being to successfully predict therapeutic response and enrol subjects who are most likely to see benefit from a given intervention.
Contribution of β-cells themselves to T1D.
The concept that β-cells contribute to the autoimmune attack (Table 1) was first heralded in the classical “Homicide versus Suicide” lecture by the late Dr. Franco Bottazzo in the mid-1980s65 and was recently reconsidered66. Since, we have learned more about the β-cell, and at least four ways have been put forward, by which they might participate in their own demise. First, because of their high biosynthetic rate, β-cells may be especially vulnerable to stress-induced changes during local infections or other inflammatory events, resulting in production of specific autoantigenic peptides recognized by pathogenic T-cells. Second, increased β-cell sensitivity to cytokine-mediated killing has been noted. Third, persistent changes in β-cell physiology once autoimmunity has been initiated may enhance autoimmune destruction. Fourth and finally, β-cells appear extremely prone to self-directed cellular destruction, as they are sensitive to multiple forms of endoplasmic reticulum (ER) stress. Each of these facets finds support from observations made in human T1D donor tissues (discussed in Roep et al.). Although the inimitable metabolic wiring of the β-cell works extraordinarily well to control their function under normal conditions, these processes may contribute to the pathologic circumstances underlying T1D.
β-cell function does not equal β-cell mass.
Recent immune-histopathological studies of the human T1D pancreas point to a disconnection between β-cell function and mass. Even when circulating C-peptide levels are very low, significant numbers of insulin-positive islet cells can be detected in a large number of patients, albeit in declining numbers as time passes after diagnosis25,67,68. This insight challenges the dogma that T1D diagnosis reflects the end-stage of disease with little to play for in terms of β-cell mass. Rather, many remaining β-cells are likely dysfunctional69,70, perhaps in a fashion that hides them from immune attack. If this is a non-specific, reversible consequence of inflammation, there may be more benefit to be gained from a prompt intervention than previously appreciated. A further complicating factor may be that the balance between β-cell loss and dysfunction likely differs from patient to patient67, yet we currently have no way to measure β-cell mass in living individuals. Thus, the success of a particular immunotherapy approach may vary, and our limited ability to monitor the islet response to autoimmunity/immunotherapy deserves priority attention.
Moving Forward – New Directions
Compared to efforts in other autoimmune disease fields where appreciable headway has been made bringing immunomodulatory treatments to the clinical space (Appendix Page 6–8), critical factors have prevented therapeutic progress for T1D: 1) high bar of safety/efficacy, 2) short intervention window (within 3 months of diagnosis), 3) lack of short-term surrogate markers of efficacy, 4) insufficient understanding of the complex etiology (i.e., β-cell and immunological contributions), and 4) paucity of biomarkers (beyond autoantibodies) to predict time to diagnosis and select tailored therapies. To improve on past efforts seeking to prevent T1D and preserve C-peptide, a series of modifications to immune-based intervention practices (Figure 2) would potentially prove of high value going forward.
Figure 2. Considerations to improve T1D immune therapy trials.

Implementing these eight notions may accelerate identification of effective immune therapies for T1D and application in clinical settings.
Interventional staging and intervention change after disease progression.
The field has recently seen movements toward reconsidering the definition of T1D as well as evaluation of therapies at specific, pre-specified stages of the pathological processes. For example, a recent revision to T1D nosology allows for the presence of multiple (≥ 2) autoantibodies in an individual to be considered either Stage 1 disease20 or a manifestation of an Autoimmune Beta Cell Disorder (“ABCD”)71. This framework offers the potential to meet the goal of introducing a therapy having the highest capacity for efficacy at the appropriate stage of disease.
Introducing this concept of staged interventions will not occur without challenges. First and foremost, will T1D care providers, patients and their families accept a disease diagnosis in the absence of symptoms? Depending on the particular health care or educational system, such an action might impart obstacles related to insurance, education, and more. As at least 85% of new T1D cases emanate from the general population (i.e., non-relatives), effective identification of large numbers of staged individuals will require extensive population-based screening programmes; an effort that has, until recently, seen limited enthusiasm due to logistical, technical, and financial challenges. Hence, careful attention must be extended to increase enthusiasm and application of this staging model, with hope that recent programs directed at general screening [e.g., Fr1da (https://www.typ1diabetes-frueherkennung.de/), ASK (https://www.askhealth.org/)] will provide a pathway for this in the future.
Beyond this, the model for staged interventions should be extended beyond “pre-T1D”. The vast majority of available agents are first tried as interventions within 100 days after diagnosis as the setting in which they can show their potential and their safety. As noted, a limited number of therapies have shown some ability to extend the ability to produce C-peptide for months following classical diagnosis (Appendix Page 9–11), but this benefit ultimately wanes for all but a few. Could there be a rationale to test for efficacy at other stages post-diagnosis such as the “honeymoon” 3–6 months after diagnosis when there is temporary post-diagnosis recovery of C-peptide secretion (and immunohistochemistry suggests that as few as 10% of islets are immune infiltrated)? Or, perhaps in a “rolling trials model” where, if an individual…throughout the natural history of T1D fails to respond to a given therapy, they would be afforded the opportunity to receive additional treatments. At present, there is a paucity of appropriate biomarkers directly related to drug action, but cautious optimism underlies current efforts to standardise new surrogate markers, such as immune subset frequency measured by multicolour flow cytometry.
Biomarker improvements – setting the stage for tailored therapies.
Collective observations have essentially formed a metanarrative where metabolic (rate of decline of C-peptide/loss of β-cell mass), immunologic (inflammatory versus regulatory signatures), and pathologic (islet infiltration) features of the disease are causally inter-related. However, neither β-cell mass nor insulitis can currently be visualized in living patients. Regardless, it is appealing to apply the ever-growing list of confirmed and increasingly accepted number of immune/metabolic biomarkers combined with anthropometric features (e.g., age, BMI) to categorise patients into a relatively limited number of phenotypic bins whereby their response to an intervention strategy will be most optimal.
New approaches to stratification, including immune and metabolic correlates that signify heterogeneity and thereby, likelihood of response to treatment, will be required to optimize therapeutic strategies. To make this first concept a reality will require the ability to detect distinct immune signatures72. Studies of pancreatic tissues from T1D patients show that T-cell autoimmunity to islet antigens, detectable in circulation, might also be found in the disease lesion; pointing to their potential relevance as (surrogate) measures of insulitis, disease activity and immunotherapeutic intervention25,73–75. Next-generation immunosequencing of the T-cell receptor (TCR) repertoire has also demonstrated that CD8+ T-cell clones are less tissue restricted than their CD4+ counterparts76 and prone to have structural features consistent with autoimmune potential77. Techniques to detect antigen-specific CD8+ T-cells in peripheral blood are well developed and could likely be further improved by newer high-throughput single cell applications78. Current T-cell biomarker assays either require fresh cells (e.g., T-cell proliferation) or are HLA-restricted (e.g., MHC multimer staining), but efforts are underway to develop immunosequencing-based biomarkers that can be applied using fresh or frozen samples regardless of HLA (recently reviewed in79).
Early evidence emphasizes the value of baseline immune phenotypes to guide treatment strategies. In islet transplantation, the degree of CD4+ T-cell reactivity to GAD65 or IA-2 predicts clinical outcome80,81. Similar baseline signatures have been established around CD8+ T-cell autoreactivity82: autoimmune signatures can guide selection of T1D patients for autologous hematopoietic stem cell therapy (aHSCT) with a superior chance of clinical benefit (i.e., low frequencies of circulating islet autoreactive CD8+ T-cells)83. Finally, since β-cell function rather than β-cell mass is the typical primary endpoint for intervention trials in T1D, clinical successes in subgroups of patients may have been missed62; thus, the T1D research and care communities must develop better measures of β-cell mass (i.e., imaging in living patients), homeostasis, resilience and function, so that end-points are better able to reflect efficacy.
Team science approach to clinical trials.
These early steps toward tailored therapies (i.e., precision or personalized medicine) for those with T1D appear encouraging but will require both validation and expansion to other groups of patients and treatments. This calls for a bolder approach to “Team Science” in which all clinical trials are embellished with a similar if not cross-referencing suite of mechanistic assays. Such efforts will require standardization, ideally, in the form of centralized IRBs and perhaps even the establishment of core laboratories where mechanistic assays are performed.
Additional consideration must be given to how we define clinical response for consistency across studies (Appendix Page 9–11), to ensure appropriate categorization of subjects. A meta-analysis of GAD-alum vaccination studies highlights one negative study as having more than twice as many placebo “responders” compared to the two studies with positive outcomes84. Hence moving forward, there is a need to apply advanced statistical analyses (e.g., Bayesian Network Analysis) to detect potentially clinically relevant changes in autoimmunity/immunoregulation as well as metabolic effects of therapy and when possible, to compare clinical trial data against historical controls.
Trials of other autoimmune diseases grade clinical response by the degree of symptomatic improvement (e.g., ACR20/50/70 for RA64; Appendix Page 6–8). We propose the use of similar metrics for T1D patients (e.g., preservation of baseline 2hr C-peptide AUC to 6, 12, and 24 months [Cpep6/12/24]). Along with the elucidation of responder and non-responder phenotypes at the level of C-peptide retention85, the same principles may be applied in the detection of adverse events, such as disease acceleration. Immune and metabolic monitoring can give early warnings and insights into these mechanisms86.
Complex mechanisms of T1D raise the need to treat β-cells in combination therapy.
Combination therapies of two immunomodulatory agents may beget excessive immunosuppressive risk; hence, combinations with agents directed at β-cell function or regeneration may be preferable. Recent knowledge gains noted above (i.e., heterogeneity in the extent and composition of the insulitic lesions, metabolic decline25,67,87,88, circulating T-cell and autoantibody immune signatures82,87, and detectable β-cell function at diagnosis and in long-standing diabetes) underscore the potential applications for incretin treatments, which improve capacity for insulin production by residual β-cells and suppress glucagon secretion, as well as the need for therapeutics to reduce β-cell stress co-administered with immunomodulatory therapy to reverse autoimmunity in symptomatic T1D. Hence, therapies once considered only applicable to those with T2D may be of potential benefit for those with T1D, and their use in appropriate research studies should be encouraged. Indeed, combinations of these agents, along with those traditionally thought of for treatment of T1D, are likely warranted for consideration. Finally and realistically, in order to achieve the first licenced T1D therapy, we must consider that drug development in T1D remains a profitable enterprise.
Concluding Thoughts
The suggestions presented herein represent a “roadmap” for the next decade of clinical trials in T1D, starting with patient selection through to end-points and embedded mechanistic studies, and as a field, we must shift gears in three key areas. First, rather than classifying trials as failures or otherwise, they should be viewed as contributing to the canon of knowledge. Second, dissemination and use of the new Stage 1–320/ABCD71 approach will yield greater patient engagement and in turn, exert a greater pressure on research funders (government, charitable and private) and the pharmaceutical industry to lead this effort. Third and perhaps the greatest “win” will be the opportunity for more coordination of study templates across networks, investigators and industry. This alignment of exclusion/inclusion criteria, stratification tools, embedded biomarker studies, outcomes, mechanistic platforms, sample archiving and long-term follow-up with public availability of data is required to perform studies as inexpensively and efficiently as possible whilst still acquiring maximum information. Several networks are already in place to underpin this effort, including Type 1 Diabetes TrialNet and Immune Tolerance Network in the U.S. and the Innovative Medicines Initiative programme INNODIA in Europe, each of which has strong pharma engagement. Population screening initiatives for Stages 1–2 disease are paving the way for translation into public health settings.
These aspirations will need to be tempered with a more realistic handling of expectations across scientists, clinicians and the lay population. The management of T1D in terms of treating to glycaemic targets is challenging, with as few as 17% of teenaged patients achieving target HbA1c levels of <7.5% (58 mmol/mol) and significant proportions experiencing recent severe hypoglycaemic or diabetic ketoacidotic events89. Against this backdrop, achieving modest improvements in C-peptide secretion (known to facilitate blood sugar control by injected insulin) for a period of months/years in a subset of patients could be transformative. Indeed, “breakthroughs” in the treatment of other autoimmune diseases (i.e., MS, RA, Crohn’s disease, ulcerative colitis) have enhanced disease management but do not represent curative interventions (Appendix Page 6–8). To this end, more emphasis on T1D patient-proximal outcomes and less on “cure” and “remission” are needed, albeit with the added challenge of capturing these benefits as robust endpoints that also fit the bill for drug regulators. Though perhaps less sensational, we believe these objectives to be more immediately achievable with potential to provide clinically meaningful improvement for patients living with T1D.
Supplementary Material
Key Messages.
To date, trials of non-antigen-specific therapies for type 1 diabetes (T1D) have targeted immunomodulation, cytokines and anti-inflammation, vitamin D, and cell-based treatment, and 51 trials of non-antigen-specific immunotherapy trials for T1D are currently awaiting, actively recruiting, or active/ongoing but not recruiting participants.
Failures to meet trial endpoints and advance therapies into clinical settings can be ascribed to a series of flawed guiding hypotheses and/or current unknowns related to possible subcategories of T1D etiologies, potential impact of environmental, temporal, and demographic/geographic factors, patterns of loss of β-cell mass/function, our ability to quantify functional and dysfunctional β-cell mass in living patients; ability of residual β-cells to compensate for declining β-cell mass/function prior to clinical onset of the disease, the need for earlier intervention to prevent progression from multi-autoantibody positivity (Stage 1 disease) to dysglycemia (Stage 2).
Key concepts to improve trials of immune based therapy include mechanistic data to guide trials and agent selection, treat to mechanistic outcomes, combination therapies, treat to disease stage based on genetic, immune, and metabolic data, globally coordinated efforts with standardized SOPs, enrolment of subjects from the general population in addition to relatives of T1D patients, develop protocols that address disease heterogeneity, rolling or adaptive trial designs.
Emphasis should be placed on patient-proximal outcomes, such as modest improvements in C-peptide secretion for a period of months/years in a subset of patients, rather than on sensational outcomes such as “cure” and “remission.
Search Strategy and Selection criteria.
To identify references for this Review, we searched PubMed for articles published between January 1, 2007 and February 15, 2018. The search terms used were “immune” OR “immunomodulat*” OR “immunosuppress*” OR “immuni*” OR “immunologic*” OR “Immunotherap*” OR “autoimmun*” OR “antibodies” OR “autologous” OR “stem cell” OR “mesenchymal stromal cell” AND “type 1 diabetes” OR “insulin-dependent diabetes” OR “IDDM” OR “autoimmune diabetes”. We filtered for studies published in English, carried out in Humans, and for the article type that included Clinical Study, Clinical Trial, Clinical Trial Phase I, Clinical Trial Phase II, Clinical Trial Phase III, Clinical Trial Phase IV, Multicenter Study, and Randomized Controlled Trial. To identify relevant articles, we reviewed the titles and abstracts of the resultant references for exclusion, and additionally we included relevant articles cited in the subsequent publications, and widely cited older publications that we deemed of importance. We identified relevant recruiting and active clinical trials by searching ClinicalTrials.gov, most recently on February 15th, 2018. Search results for “Type 1 Diabetes” (Condition/Disease) results were reviewed and trials of immunotherapies selected; trials of devices, insulin analogues, and lifestyle adjustments (e.g., exercise) were removed.
Funding
Atkinson efforts were supported by grants from the NIH (P01 AI42288), JDRF, the Jeffrey Keene Family Professorship, the Leona M. and Harry B. Helmsley Charitable Trust, and the Brehm Coalition for Diabetes Research.
Related work in the Peakman laboratory receives funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115797 INNODIA. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and “EFPIA”, ‘JDRF International” and “The Leona M. and Harry B. Helmsley Charitable Trust”. The laboratory is also supported via the National Institute of Health Research Biomedical Research Centre Award to Guy’s and St Thomas National Health Service Foundation Trust and King’s College London.
The Roep studies were supported by the Dutch Diabetes Research Foundation, Stichting DON, the European Commission (INNODIA-115797, EE-ASI-305305, NAIMIT-241447), the Dutch Arthritis Foundation (LLP-16), The Juvenile Diabetes Research Foudation and the Wanek Family Project for Type 1 Diabetes.
No funding sources influenced the writing of the manuscript or decision to submit for publication.
Footnotes
Conflict of Interest Statement
Dr. Atkinson has a patent US 8758761 B2 issued for combination therapies including ATG plus G-CSF for the treatment of type 1 diabetes. The authors declare that no other conflicts of interest exist pertaining to the contents of this manuscript.
References
- 1.Ziegler AG, Bonifacio E, Powers AC, Todd JA, Harrison LC, Atkinson MA. Type 1 Diabetes Prevention: A Goal Dependent on Accepting a Diagnosis of an Asymptomatic Disease. Diabetes 2016; 65(11): 3233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiller CR, Dupre J, Gent M, et al. Effects of cyclosporine in recent-onset juvenile type 1 diabetes: impact of age and duration of disease. J Pediatr 1987; 111(6 Pt 2): 1069–72. [DOI] [PubMed] [Google Scholar]
- 3.Chase HP, Butler-Simon N, Garg SK, et al. Cyclosporine A for the treatment of new-onset insulin-dependent diabetes mellitus. Pediatrics 1990; 85(3): 241–5. [PubMed] [Google Scholar]
- 4.Cook JJ, Hudson I, Harrison LC, et al. Double-blind controlled trial of azathioprine in children with newly diagnosed type I diabetes. Diabetes 1989; 38(6): 779–83. [DOI] [PubMed] [Google Scholar]
- 5.Sobel DO, Henzke A, Abbassi V. Cyclosporin and methotrexate therapy induces remission in type 1 diabetes mellitus. Acta Diabetol 2010; 47(3): 243–50. [DOI] [PubMed] [Google Scholar]
- 6.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes. N Engl J Med 2002; 346: 1692–8. [DOI] [PubMed] [Google Scholar]
- 7.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. NEnglJMed 2005; 352(25): 2598–608. [DOI] [PubMed] [Google Scholar]
- 8.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009; 361(22): 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest 2015; 125(8): 3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orban T, Beam CA, Xu P, et al. Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower C-peptide decline. Diabetes 2014; 63(10): 3449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gitelman SE, Gottlieb PA, Felner EI, et al. Antithymocyte globulin therapy for patients with recent-onset type 1 diabetes: 2 year results of a randomised trial. Diabetologia 2016; 59(6): 1153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb PA, Quinlan S, Krause-Steinrauf H, et al. Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new- onset type 1 diabetes. Diabetes Care 2010; 33(4): 826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long SA, Rieck M, Sanda S, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes 2012; 61(9): 2340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartemann A, Bensimon G, Payan CA, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2013; 1(4): 295–305. [DOI] [PubMed] [Google Scholar]
- 15.Rosenzwajg M, Churlaud G, Mallone R, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 2015; 58: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu A, Snowhite I, Vendrame F, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 2015; 64(6): 2172–83. [DOI] [PubMed] [Google Scholar]
- 17.Truman LA, Pekalski ML, Kareclas P, et al. Protocol of the adaptive study of IL-2 dose frequency on regulatory T cells in type 1 diabetes (DILfrequency): a mechanistic, non-randomised, repeat dose, open-label, response-adaptive study. Bmj Open 2015; 5(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Todd JA, Evangelou M, Cutler AJ, et al. Regulatory T Cell Responses in Participants with Type 1 Diabetes after a Single Dose of Interleukin-2: A Non-Randomised, Open Label, Adaptive Dose-Finding Trial. PLoS Med 2016; 13(10): e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long SA, Thorpe J, DeBerg HA, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol 2016; 1(5): eaai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Insel RA, Dunne JL, Atkinson MA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015; 38(10): 1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.AlMutairi N, Abouzaid HA. Safety of biologic agents for psoriasis in patients with viral hepatitis. J Dermatolog Treat 2018: 1–4. [DOI] [PubMed] [Google Scholar]
- 22.McKinnon RA, Cook M, Liauw W, et al. Biosimilarity and Interchangeability: Principles and Evidence: A Systematic Review. BioDrugs 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saldanha M, Dandekar P, Jain R. A Regulatory Perspective on Testing of Biological Activity of Complex Biologics. Trends Biotechnol 2018. [DOI] [PubMed] [Google Scholar]
- 24.Lockwood SJ, Prens LM, Kimball AB. Adverse Reactions to Biologics in Psoriasis. Curr Probl Dermatol 2018; 53: 1–14. [DOI] [PubMed] [Google Scholar]
- 25.Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. The Journal of experimental medicine 2012; 209(1): 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leete P, Willcox A, Krogvold L, et al. Differential Insulitic Profiles Determine the Extent of beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016; 65(5): 1362–9. [DOI] [PubMed] [Google Scholar]
- 27.Willcox A, Gillespie KM. Histology of Type 1 Diabetes Pancreas. Methods Mol Biol 2016; 1433: 105–17. [DOI] [PubMed] [Google Scholar]
- 28.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia 2010; 53(9): 2020–8. [DOI] [PubMed] [Google Scholar]
- 29.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155(2): 173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999; 5(6): 601–4. [DOI] [PubMed] [Google Scholar]
- 31.Roep BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nat Rev Immunol 2004; 4(12): 989–97. [DOI] [PubMed] [Google Scholar]
- 32.Atkinson MA. Evaluating preclinical efficacy. Sci Transl Med 2011; 3(96): 96cm22. [DOI] [PubMed] [Google Scholar]
- 33.Sarikonda G, Sachithanantham S, Manenkova Y, et al. Transient B-cell depletion with anti-CD20 in combination with proinsulin DNA vaccine or oral insulin: immunologic effects and efficacy in NOD mice. PLoS One 2013; 8(2): e54712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gill RG, Pagni PP, Kupfer T, et al. A Preclinical Consortium Approach for Assessing the Efficacy of Combined Anti-CD3 Plus IL-1 Blockade in Reversing New-Onset Autoimmune Diabetes in NOD Mice. Diabetes 2016; 65(5): 1310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A 1994; 91(1): 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 1997; 158(6): 2947–54. [PubMed] [Google Scholar]
- 37.Alkemade GM, Hilbrands R, Vandemeulebroucke E, et al. Preservation of recall immunity in anti-CD3-treated recent onset type 1 diabetes patients. Diabetes Metab Res Rev 2011; 27(8): 925–7. [DOI] [PubMed] [Google Scholar]
- 38.McNamee K, Williams R, Seed M. Animal models of rheumatoid arthritis: How informative are they? Eur J Pharmacol 2015; 759: 278–86. [DOI] [PubMed] [Google Scholar]
- 39.Wallet MA, Santostefano KE, Terada N, Brusko TM. Isogenic Cellular Systems Model the Impact of Genetic Risk Variants in the Pathogenesis of Type 1 Diabetes. Front Endocrinol (Lausanne) 2017; 8: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan S, Li Y, Xia J, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci U S A 2017; 114(41): 10954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer-Davis EJ, Lawrence JM, Dabelea D, et al. Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002–2012. N Engl J Med 2017; 376(15): 1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross-sectional, genetically stratified survival analysis from UK Biobank. Lancet Diabetes Endocrinol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oram RA, Thomas NM, Jones SE, Weedon MN, Hattersley AT. Analysis of Diabetes Incidence Against Type 1 Diabetes Genetic Risk Score in 150,000 Shows Autoimmune Diabetes Presents as Often Between 30–60 Years as Before 30 Years. Diabetes 2016; 65(Supplement 1): A413. [Google Scholar]
- 44.Raab J, Haupt F, Scholz M, et al. Capillary blood islet autoantibody screening for identifying pre-type 1 diabetes in the general population: design and initial results of the Fr1da study. BMJ Open 2016; 6(5): e011144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ehehalt S, Dietz K, Willasch AM, Neu A. Prediction model for the incidence and prevalence of type 1 diabetes in childhood and adolescence: evidence for a cohort-dependent increase within the next two decades in Germany. Pediatr Diabetes 2012; 13(1): 15–20. [DOI] [PubMed] [Google Scholar]
- 46.Aronson R, Gottlieb PA, Christiansen JS, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 2014; 37(10): 2746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ambery P, Donner TW, Biswas N, Donaldson J, Parkin J, Dayan CM. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med 2014; 31(4): 399–402. [DOI] [PubMed] [Google Scholar]
- 48.Keymeulen B, Walter M, Mathieu C, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010; 53(4): 614–23. [DOI] [PubMed] [Google Scholar]
- 49.Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol 2013; 149(3): 268–78. [DOI] [PubMed] [Google Scholar]
- 50.Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest 2015; 125(8): 3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rigby MR, DiMeglio LA, Rendell MS, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol 2013; 1(4): 284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baxter AG, Jordan MA. From markers to molecular mechanisms: type 1 diabetes in the post-GWAS era. Rev Diabet Stud 2012; 9(4): 201–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sosenko JM, Skyler JS, Beam CA, et al. Acceleration of the loss of the first-phase insulin response during the progression to type 1 diabetes in diabetes prevention trial-type 1 participants. Diabetes 2013; 62(12): 4179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenbaum CJ, Beam CA, Boulware D, et al. Fall in C-Peptide During First 2 Years From Diagnosis: Evidence of at Least Two Distinct Phases From Composite Type 1 Diabetes TrialNet Data. Diabetes 2012; 61(8): 2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herold KC, Usmani-Brown S, Ghazi T, et al. beta cell death and dysfunction during type 1 diabetes development in at-risk individuals. J Clin Invest 2015; 125(3): 1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirmira RG, Sims EK, Syed F, Evans-Molina C. Biomarkers of beta-Cell Stress and Death in Type 1 Diabetes. Curr Diab Rep 2016; 16(10): 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lehmann-Werman R, Neiman D, Zemmour H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A 2016; 113(13): E1826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ludvigsson J, Heding L. Abnormal proinsulin/C-peptide ratio in juvenile diabetes. Acta Diabetol Lat 1982; 19(4): 351–8. [DOI] [PubMed] [Google Scholar]
- 59.Arif S, Gibson VB, Nguyen V, et al. beta-cell specific T-lymphocyte response has a distinct inflammatory phenotype in children with Type 1 diabetes compared with adults. Diabet Med 2017; 34(3): 419–25. [DOI] [PubMed] [Google Scholar]
- 60.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009; 361(22): 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med 2012; 366(5): 433–42. [DOI] [PubMed] [Google Scholar]
- 62.Woittiez NJ, Roep BO. Impact of disease heterogeneity on treatment efficacy of immunotherapy in Type 1 diabetes: different shades of gray. Immunotherapy 2015; 7(2): 163–74. [DOI] [PubMed] [Google Scholar]
- 63.Wherrett DK, Chiang JL, Delamater AM, et al. Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care 2015; 38(10): 1975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma X, Xu S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep. England; 2013: 177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bottazzo GF. Lawrence lecture. Death of a beta cell: homicide or suicide? DiabetMed 1986; 3: 119–30. [DOI] [PubMed] [Google Scholar]
- 66.Atkinson MA, Bluestone JA, Eisenbarth GS, et al. How does type 1 diabetes develop?: the notion of homicide or beta-cell suicide revisited. Diabetes 2011; 60(5): 1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campbell-Thompson M, Fu A, Kaddis JS, et al. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016; 65(3): 719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oram RA, Jones AG, Besser RE, et al. The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 2014; 57(1): 187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wasserfall C, Nick HS, Campbell-Thompson M, et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab 2017; 26(3): 568–75.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. beta Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab 2017; 25(3): 727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonifacio E, Mathieu C, Nepom GT, et al. Rebranding asymptomatic type 1 diabetes: the case for autoimmune beta cell disorder as a pathological and diagnostic entity. Diabetologia 2017; 60(1): 35–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roep BO, Atkinson MA, van Endert PM, Gottlieb PA, Wilson SB, Sachs JA. Autoreactive T cell responses in insulin-dependent (Type 1) diabetes mellitus. Report of the first international workshop for standardization of T cell assays. J Autoimmun 1999; 13(2): 267–82. [DOI] [PubMed] [Google Scholar]
- 73.Pathiraja V, Kuehlich JP, Campbell PD, et al. Proinsulin-specific, HLA-DQ8, and HLA-DQ8-transdimer-restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes 2015; 64(1): 172–82. [DOI] [PubMed] [Google Scholar]
- 74.Babon JA, DeNicola ME, Blodgett DM, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016; 22(12): 1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Michels AW, Landry LG, McDaniel KA, et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 2017; 66(3): 722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seay HR, Yusko E, Rothweiler SJ, et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 2016; 1(20): e88242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomez-Tourino I, Kamra Y, Baptista R, Lorenc A, Peakman M. T cell receptor beta-chains display abnormal shortening and repertoire sharing in type 1 diabetes. Nat Commun 2017; 8(1): 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu H, Caen O, Vrignon J, et al. High throughput single cell counting in droplet-based microfluidics. Sci Rep 2017; 7(1): 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jacobsen LM, Posgai A, Seay HR, Haller MJ, Brusko TM. T Cell Receptor Profiling in Type 1 Diabetes. Curr Diab Rep 2017; 17(11): 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hilbrands R, Huurman VA, Gillard P, et al. Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes 2009; 58(10): 2267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huurman VA, Hilbrands R, Pinkse GG, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS One 2008; 3(6): e2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Velthuis JH, Unger WW, Abreu JR, et al. Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes 2010; 59(7): 1721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Malmegrim KC, de Azevedo JT, Arruda LC, et al. Immunological Balance Is Associated with Clinical Outcome after Autologous Hematopoietic Stem Cell Transplantation in Type 1 Diabetes. Front Immunol 2017; 8: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beam CA, MacCallum C, Herold KC, Wherrett DK, Palmer J, Ludvigsson J. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: findings from a Bayesian meta-analysis. Diabetologia 2017; 60(1): 43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beam CA, Gitelman SE, Palmer JP. Recommendations for the definition of clinical responder in insulin preservation studies. Diabetes 2014; 63(9): 3120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Long SA, Rieck M, Sanda S, et al. Rapamycin/IL-2 Combination Therapy in Patients with Type 1 Diabetes Augments Tregs yet Transiently Impairs beta-Cell Function. Diabetes 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arif S, Leete P, Nguyen V, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014; 63(11): 3835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leete P, Willcox A, Krogvold L, et al. Differential insulitic profiles determine the extent of beta cell destruction and the age at onset of type 1 diabetes. Diabetes 2016. [DOI] [PubMed] [Google Scholar]
- 89.Miller KM, Foster NC, Beck RW, et al. Current state of type 1 diabetes treatment in the U.S.: updated data from the T1D Exchange clinic registry. Diabetes Care 2015; 38(6): 971–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.