

Abstract
Post-translational modifications (PTM) are key events in signal transduction since they affect protein function by regulating their abundance and/or activity. PTMs involve the covalent attachment of functional groups to specific amino acids. Since they tend to be generally reversible, PTMs serve as regulators of signal transduction pathways. GPCRs are major signaling proteins that undergo multiple types of PTMs. In this Review, we focus on the opioid receptors, members of family A GPCRs, and highlight recent advances in the field that have underscored the importance of PTMs in the functional regulation of these receptors. Since opioid receptor activity plays a central role in the development of tolerance and addiction to morphine and other drugs of abuse, understanding the molecular mechanisms regulating receptor activity is of fundamental importance.
Keywords: Receptor desensitization, phosphorylation, glycosylation, palmitoylation, ubiquitination, morphine, drug addiction
Relevance of Opioid Receptor post-translational modifications
The opioid receptor family:
Opioids were being used as pain relievers long before we began to understand the biochemical properties of opioid receptors. Opioids have been used for more than 4,000 years, and the analgesic effects of morphine have been explored since it was isolated in 1805. Although the chronic usage of opioids leads to the development of tolerance (see Glossary), physical dependence, and addiction, morphine remains the drug of choice for acute pain.
The opioid receptors, members of the family A G protein-coupled receptors (GPCRs), consist of µ-Opioid Receptor (MOR), δ-Opioid Receptor (DOR), κ-Opioid Receptor (KOR), and the Nociceptin/orphanin FQ receptor (NOP) [1]. Homology analysis shows that these receptors exhibit 49%−58% primary sequence identity with each other [1]. The ORs also exhibit high structural similarity with each other, which reinforces the notion that they all belong to the same family [2]. ORs appear to share a common evolutionary history, dating to ~450 millions of years ago [3].
Even though all ORs play a role in the modulation of pain, it is only in the past few decades that we have been able to clarify additional roles in behaviors and processes such as food intake, anxiety, depression, and immunomodulation [4,5]. MOR agonists are used in the treatment of pain but they are accompanied by undesirable side-effects such as respiratory depression and constipation [6,7]. KOR agonists produce dysphoric, hallucinogenic and psychotomimetic effects [8]. Interestingly, although DOR agonists have been shown to produce seizures [9], they have also been shown to have antidepressant and anxiolytic effects [10,11]. Finally, NOP agonists have been reported to cause somnolence and decrease blood pressure [12], although small molecule agonists have shown potential as anxiolytics, substance abuse medications, and antitussives [5]. Understanding the differences between side-effects and beneficial effects produced by OR agonists will be crucial to the development of safe analgesic drugs. The molecular mechanisms of ORs signaling are described in Box 1.
BOX 1: Steps of agonist-mediated opioid receptor signaling.
For the sake of simplicity, a linear cascade of events following receptor activation is described below. Opioid receptors are GPCRs that couple to inhibitory G proteins (Gi proteins). G proteins form a heterotrimeric complex comprised of α, β and γ subunits. GPCR activation induces the exchange of GDP for GTP at the α-subunit of the associated heterotrimeric G protein, and this in turn leads to the dissociation of the α-subunit from the βγ-subunit complex (reviewed in [79]). This results in the activation of downstream signaling cascades, including the modulation of calcium and potassium channels, the activation of phospholipase C, inhibition of adenylyl cyclase activity, enhancement of phosphorylation of protein kinases such as mitogen-activated protein kinase (MAPK), and activation of kinases such as PKC and GRKs [147]. Opioid receptor-mediated activation of inwardly rectifying potassium channels and inhibition of voltage-gated calcium channels leads to a decrease in neurotransmitter release (reviewed in [107]).
One of the mechanisms responsible for termination of this intracellular signaling is restoration of the heterotrimeric G-protein complex. This involves the hydrolysis of GTP bound to the Gα subunit that is facilitated by the regulator of G-protein signaling (RGS) [148,149]. Another mechanism involves receptor phosphorylation by GRKs followed by β-arrestin recruitment, and receptor internalization [70,87,132]. Interestingly, β-arrestin has also been shown to function as a multifunctional adaptor protein that in addition to mediating endocytosis, induces a second wave of signaling [141]. Following internalization, the receptor is either recycled back to the cell surface to initiate another wave of signaling, or targeted for degradation [77,101,131].
It is increasingly becoming apparent that GPCR signaling events are not hierarchical, in that multiple signaling pathways can be activated from the receptor at the cell surface as well as from different subcellular compartments (reviewed in [150]). Furthermore, GPCR activation by different agonists can lead to differential and at times ‘biased’ signaling (reviewed in [150]).
In summary, the classical steps of opioid receptor signaling are 1) agonist binding, 2) a G protein mediated signaling response, 3) receptor phosphorylation mediated by GRKs and internalization mediated by β-arrestin recruitment, followed by 4) recycling or degradation of the receptor (Fig. 2). In this review we discuss the relevance of different post translational modifications (PTM) in the regulation of these steps.
Post-translational modification (PTM) of GPCRs:
Receptor PTMs play a critical and important role in modulating signal transduction. Indeed, they represent the fine-tuning of receptor signaling. Among the PTMs that play key roles in GPCR signaling, glycosylation (sugar-linkage) [13,14], palmitoylation (palmitoyl-linkage) [15–18], phosphorylation (phosphate-linkage) [19–21] and ubiquitination (ubiquitin-linkage) [22–25], are the most frequently studied ones.
Glycosylation, which mainly takes place in the endoplasmic reticulum (ER) and Golgi apparatus, has been described as a quality control mechanism for GPCR synthesis, and has been shown to serve as a tag that directs the receptor to the plasma membrane [13,14]. Palmitoylation has been shown to play roles in receptor localization to lipid rafts on the plasma membrane, and in modifying protein-protein interactions including receptor dimerization [15–18]. Phosphorylation of GPCRs has been intensely studied, and results show that it plays a key role in regulating receptor activity including receptor desensitization and internalization [19,20]. Ubiquitination of GPCRs has been shown to affect receptor degradation, and in some cases this PTM serves as a regulator of the magnitude and the duration of GPCR signaling [22–25].
In this review, we focus on PTMs of opioid receptors. A thorough investigation of opioid receptor PTMs could be critical to understand the molecular mechanisms leading to the development of tolerance, as well as opioid-mediated side effects. This review is divided into three sections that are based on the location of the PTM on the receptor: extracellular (glycosylation), transmembrane (palmitoylation), and cytoplasmic (phosphorylation and ubiquitination) (Fig. 1). We mention the enzymes responsible for these PTMs in opioid receptors (ORs) and describe their biological relevance for opioid signaling (Fig. 2).
Figure 1: Post-translational modification of µ-Opioid Receptor.
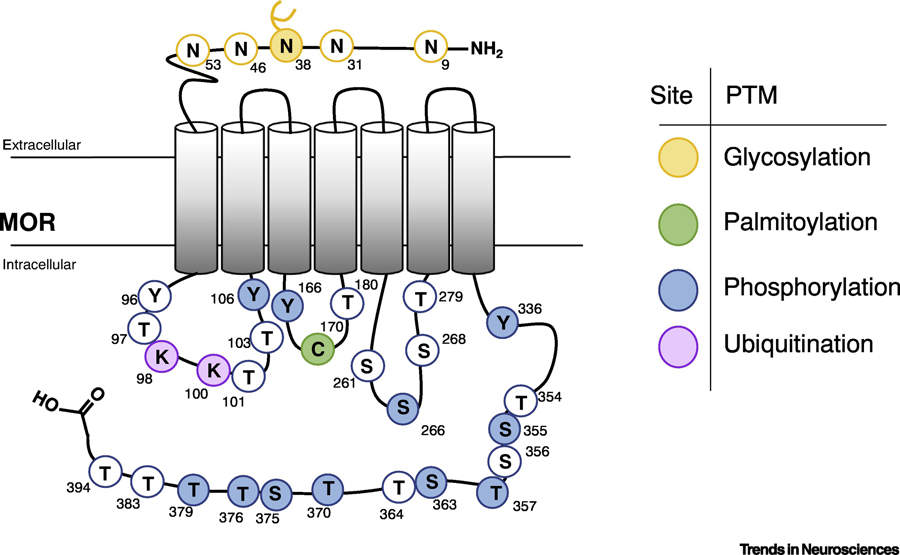
A schematic representation of rat µ-Opioid Receptor showing putative residues that could be post-translationally modified. Each post-translational modification category is represented by a different color. Filled circles represent known PTMs and white circles represent putative PTMs. The residue numbers in rat MOR are indicated above each reside. Since the N40 has been shown to be glycosylated, it is indicated with a branched chain.
Figure 2: Schematic summary of the major PTM events in µ-Opioid Receptor signaling.
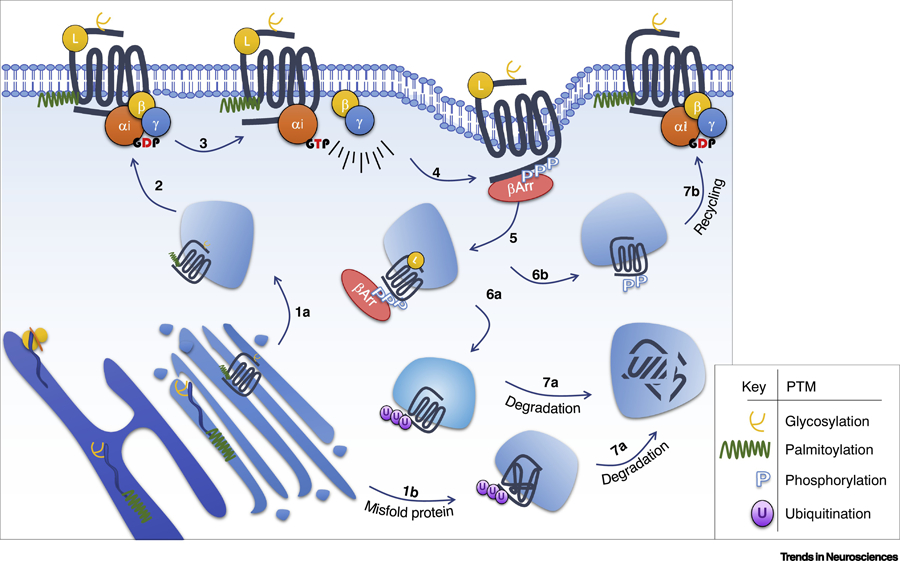
The opioid receptor undergoes glycosylation and palmitoylation in the endoplasmic reticulum and Golgi apparatus during maturation, which affect receptor’s cell surface expression (1a, 2). At the cell surface, agonist exposure leads to activation of G heterotrimeric proteins (3), followed by phosphorylation of the receptor (4), leading to β-arrestin recruitment (5) and receptor endocytosis. In the endosomal compartment the receptor can be ubiquitinated (6a) and degraded (7a), or it can be recycled (6b; 7b). Misfolded proteins are ubiquitinated (1b) and subsequently degraded (7a). Please note that the numbers do not reflect hierarchical events.
Glycosylation
Glycosylation consists of the attachment of sugar molecules to proteins. There are mainly two types of Glycosylation: N-linked and O-linked glycosylation. N-linked glycosylation consists of linking a sugar molecule to the nitrogen of an Asparagine (N) residue in a protein. The putative N-glycosylation motif is characterized by Asparagine-Xaa-Threonine/Serine (N-X-S/T) where Xaa can be any of the 20 natural amino acids except Proline, whereas O-linked glycosylation refers to linking a sugar molecule to the oxygen of Serine (S) or Threonine (T) residues in a protein [26,27]. During glycosylation, monosaccharide units such as galactose (Gal), mannose (Man), fucose (Fuc), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc) and sialic acids are covalently attached in linear or branching chains [28]. The initial step of glycosylation occurs co-translationally in the ER and this is followed by the formation of complex glycans in the Golgi apparatus. There are innumerable variations in monosaccharide composition, glycosidic linkages, and glycan branches, which leads to an incredibly diverse glycan repertoire [28]. Glycosylation has been described as a post-translational event that affects the maturation process of all secreted and transmembrane proteins. In the case of GPCRs, the correlation between receptor glycosylation, maturation and trafficking have been extensively studied (reviewed in [29–31]).
Functional significance of MOR glycosylation:
The initial evidence that ORs could be glycosylated came from studies with purified receptors [32–34]. Treatment with glycosidases such as Endo H, PNGase F, and O-glycosidase, or inhibition of GlcNac phosphotransferase with tunicamycin, led to changes in the molecular weight of the protein indicating N-linked and/or O-linked glycosylation of MOR [35–37]. The N-glycosylation motif is repeated 5 times in the amino-terminal extracellular portion of human MOR (hMOR) [38]. Among these, N-glycosylation at residue N40 in hMOR has been extensively studied [39–42] (Fig. 1; Table 1).
Table 1:
Summary of the PTM events in Opioid Receptors.
| PTM | Receptor | Enzyme | Residue | Occurrence | Role | Ref |
|---|---|---|---|---|---|---|
| Glycosylation | hMOR | Ribophorin I | N40 | Constitutive | Regulates levels of MOR at the cell surface; affects agonist-binding. N40D exhibits increased dependence to alcohol and opiates | [39,40,48,50,57] |
| hDOR | N.D. | N18; N33 | Constitutive | Regulates receptor trafficking. | [56] | |
| GalNac transferase | S6; S25; S29 | Constitutive | Enhances ligand-binding, and modulates cAMP signaling. | [59] | ||
| hKOR | N.D. | N25; N39 | Constitutive | Regulates receptor trafficking. Affects agonist-induced phosphorylation. | [37] | |
| Palmitoylation | rMOR | PATs | C170 | Constitutive | Facilitates membrane association and modulates subcellular localization. | [15,18,66] |
| rDOR | N.D. | C328; C333 | Constitutive/Induced | Modulates protein trafficking and subcellular localization. Palmitoylation is increased by agonist treatment. | [67,68] | |
| Phosphorylation | m/rMOR | Src | Y336 | Induced | Chronic morphine or etorphine treatment leads to phosphorylation by Src; this switches MOR signaling response from inhibitory to stimulatory. | [80] |
| Tyrosine Kinase | Y106; Y166 | Induced | DAMGO-mediated tyrosine kinase phosphorylation regulates receptor-G protein coupling efficacy. | [81] | ||
| GRKs | S355; T357; T370; S375; T376; T379 | Induced | DAMGO, fentanyl, Met-Enk and etonitazene induce hierarchical phosphorylation of these residues via GRK2/3, facilitating β-arrestin recruitment, MOR internalization, desensitization and tolerance. Morphine induces phosphorylation of S375 and weak phosphorylation of T370, T376 and T379, via GRK5/6. | [69,76,78,85,87–90,100] | ||
| PKC | S363; T370 | Constitutive/Induced | Modulates morphine-induced desensitization, and tolerance. PKC inhibition impairs DAMGO-mediated MOR recycling. | [69,77,91,92,101] | ||
| CAMKII | T370; S26 | Induced | S266 phosphorylation modulates DAMGO-induced desensitization. | [69,93] | ||
| mDOR | GRK2 | T358; S363 | Induced | Hierarchical phosphorylation required for clathrin-mediated receptor internalization. | [117,118] | |
| PKC | S344 | Constitutive | Modulates heterologous regulation via β-Arrestin- and clathrin-mediated receptor internalization. | [123] | ||
| Cdk5 | T161 | Induced | Regulates cell surface expression of DOR and facilitates the formation of DOR-MOR heterodimers. | [151] | ||
| mKOR | GRKs | S356; T357; T363; S369 | Induced | U50,488H treatment leads to GRK-mediated phosphorylation, β-arrestin recruitment and internalization. | [119] | |
| PKC | S356; T357; T363; S369 | Constitutive | Modulates agonist independent KOR internalization. | [124] | ||
| mNOP | GRK2/3 | S346; S351; T362; S363 | Induced | Affects receptor desensitization and internalization. | [116,128] | |
| Ubiquitination | mMOR | N.D. | K98; K100 | Induced | Regulates internalization and degradation induced by DAMGO and DADLE treatment. | [98,140,141] |
| hDOR | N.D. | N.D. | Induced | Induces receptor internalization and degradation. | [139,142] | |
| hKOR | N.D. | K63 | Induced | Modulates receptor expression. | [144,145]. |
The list includes (i) the PTM event, (ii) the enzyme responsible for the PTM (iii) the residues involved, (iv) whether the PTM event is agonist induced or constitutive and, (v) the role of the PTM in opioid signaling. N.D., non-described.
A major point of the clinical relevance of N40 glycosylation in MOR is related to the A118G polymorphism (SNP database [dsSNP] Accession No rs1799971). The frequency of this polymorphism is ~22% in all the 2,504 individuals sequenced in the 1000 Genome Project [43,44]. When translated, this polymorphism leads to an exchange of asparagine (N) for aspartic acid (D) at position 40 resulting in D40 and, consequently, the loss of this glycosylation site [39]. Patients carrying the A118G allele (N40D amino acid substitution) are reported to have lower pain thresholds [45], and to require higher opioid doses to get an analgesic response [46,47]. A118G has also been reported to be associated with increased dependence on alcohol [48] and opiates such as heroin [49,50].
A number of studies have explored the molecular consequences of N40D substitution on receptor activity. A recent study investigated the impact of N40D variant using induced inhibitory neuronal cells (iNs) generated from induced pluripotent stem (iPS) cell lines from human subjects carrying the homozygous D40 polymorphism [40]. The D40 iNs exhibit stronger suppression of spontaneous inhibitory postsynaptic currents in comparison to N40. Electrophysiological analyses of cultured neurons indicated that D40 iNs cells also exhibit altered sensitivity to the MOR agonist, DAMGO [40].
Studies examining the overexpression of D40 have reported an increase in the potency of MOR agonists [49,51,52]. Interestingly, the extent of glycosylation at N40 has also been reported to affect the affinity of some MOR agonists for the receptor in a cell- and agonist-dependent manner. In AV-12 and HEK293 cells, overexpression of the D40 hMOR isoform increased the binding affinity of β-endorphin in comparison to wild-type (N40) receptors, but not of other opioid peptides and alkaloids tested [49,53]. However, these results were not reproducible in COS cells [42]; this could be due to variation of the glycan profile between different cell lines [54]. The extent of N-glycosylation of MOR also differs among mouse brain regions; for example, the level of glycosylation is higher in the thalamus in comparison to the striatum [55]. Taken together, as in the case of other GPCRs, OR glycosylation has been described to modulates the dynamics of the steady-state levels of the receptor at the cell surface and consequently, of protein abundance [37,39,56,57]. The mechanisms of how variation in glycosylation affects MOR function in a brain region-specific manner remains to be investigated.
Glycosylation of other opioid receptor members:
There is evidence that N18, N33, S6, S25 and S29 at the N-terminal of DOR, and N25 and N29 of KOR are also glycosylated, [37,56,58,59] (Table 1). The glycosylation of DOR and KOR has been reported to affect receptor function by enabling receptor folding and cell surface localization [37,56]. O-glycosylation of hDOR was found to enhance ligand-binding and agonist-mediated inhibition of cAMP accumulation [59]. However, N-glycosylation of hDOR (and hKOR) do not affect diprenorphine (OR antagonist) binding [37,58]. However, studies with receptor mutants that could not be glycosylated (hDOR-N18Q/N33Q and hKOR-N25Q/N39Q) have reported increased rate of receptor internalization compared to wild-type receptors [37,56]. Furthermore, the N25Q/N39Q mutants in hKOR have been reported to exhibit increased agonist-induced receptor phosphorylation, internalization and desensitization [37]. Together, these studies suggest that glycosylation is able to affect receptor signaling in addition to facilitating receptor maturation.
Lipidation – Palmitoylation:
Lipid modification occurs through the covalent binding of lipids such as glycosylphosphatidylinositol (GPI) anchors, and fatty acids to distinct regions on the protein. There are three types of fatty acid modifications: N-myristoylation, palmitoylation, and isoprenylation (attachment of farnesyl or geranylgeranyl) [60]. Among these, palmitoylation has been most extensively studied in the case of GPCRs [15].
Palmitoylation is classically understood to involve the attachment of palmitate to one or more cysteine residues via a thioester bond (S-palmitate). Note that the attachment of palmitate is not exclusive to thioester bonds; recent studies have described palmitate linkage to an amide group (N-palmitate) in Gαs proteins [61]. The enzymes responsible for this PTM are the palmitoyl acyltransferases, members of the DHHC-CRD (Asp-His-His-Cys-cysteine-rich domain) family, and Rasp, a member of the membrane-bound O-acyltransferase (MBOAT) family [60]. Palmitoylation is a highly dynamic event and the balancing activity of palmitoyl acyltransferases (which add palmitate), and palmitoyl thioesterases (which remove palmitate) determine the stoichiometry of protein palmitoylation at steady state. There are three classes of depalmitoylating thioesterase enzymes: acyl protein thioesterases, α/β hydrolase domain-containing 17 proteins (ABHD17s), and palmitoyl-protein thioesterases (PPTs) [62].
Palmitoylation of GPCRs has been shown to play a major role in membrane anchoring. Furthermore, crystal structure analysis has shown cholesterol-palmitoyl interaction in the case of the β2-adrenergic receptor [63]. The role of GPCR palmitoylation and its functional implication has been reviewed elsewhere [23,64,65].
Functional significance of MOR palmitoylation:
The first evidence that MOR could be palmitoylated came from studies using a recombinant system where CHO cells overexpressing rMOR were labeled with [3H]palmitate [66]. Unlike other GPCRs that are palmitoylated on the C-terminal residues, the residue described to be palmitoylated, cysteine 170 (C170) of rMOR was shown to be in the intracellular loop of MOR [18,66] (Fig. 1, Table 1).
Studies have reported that palmitate at the residue C170 of MOR interacts with the cholesterol-enriched lipid raft microdomains at the plasma membrane and this is thought to facilitate receptor homodimerization and G protein coupling/activation [18] (Box 2). Furthermore, expression of the rMOR mutant (C170A) with impaired palmitoylation was reported to affect signaling (decrease ERK phosphorylation and modulate levels of cAMP) while this mutant was not found to affect the binding of morphine, naloxone or CTOP [18].
BOX 2: Relevance of PTM events for opioid dimerization.
Post-translational modifications have been thought to affect receptor dimerization [18,146,151,152]. For instance, µ-opioid receptor (MOR) palmitoylation at the second intracellular loop (C170) has been described to facilitate receptor homodimerization, via cholesterol-palmitoyl interactions [18]. In addition, δ-opioid receptor (DOR) phosphorylation at the second intracellular loop (T161) has been described to facilitate MOR-DOR heterodimerization [151]. This phosphorylation process is mediated by cyclin-dependent kinase 5 (Cdk5) [151]. Finally, studies have shown that the expression and cell trafficking of MOR-DOR heteromers are regulated by ubiquitination and by receptor transporter protein 4 (RTP4), a Golgi chaperone that protects the heteromer from ubiquitination and degradation leading to enhanced cell surface expression [146,152]. Taken together, these observations suggest that a combination of PTM events contribute to the regulation of levels and function of MOR-DOR heteromers.
Taken together, the primary role of palmitoylation appears to be facilitation of protein anchoring to the cell membrane and protein-protein interactions. Thus, palmitoylation of OR leads to pleiotropic effects involving plasma membrane distribution, subcellular localization, endocytosis, and recycling [15].
Palmitoylation of other opioid receptors:
Similar to rMOR, palmitoylation in hDOR was initially described after labeling with [3H]palmitate [67]. The C328 and C333 residues on the C-terminal region of mDOR have been described as potential palmitoylation sites [68] (Fig. 1; Table 1). In hDOR, palmitoylation plays a role in promoting protein trafficking from the ER to the membrane since lack of palmitoylation was found to decrease the expression of DOR at the cell surface [67]. The palmitoylation process occurs both in the ER and at the plasma membrane [67]. Receptor activation appears to modulate the extent of palmitoylation at the plasma membrane. For instance, leucine-enkephalin (LE) treatment was found to increase the incorporation of [3H]palmitate in hDOR. Interestingly, DOR palmitoylation mediated by the agonist does not appear to require G protein coupling or receptor internalization or recycling [67].
Phosphorylation
Protein phosphorylation is one of the most frequent PTM events. It has been estimated that 30% of all cellular proteins are phosphorylated on at least one residue [21]. During phosphorylation, the γ-phosphate from ATP is transferred to the hydroxyl oxygen of serine (S), threonine (T) or tyrosine (Y) residues of the target protein [21]. In GPCRs, phosphorylation affects the temporal dynamics of receptor signaling. Kinases, the enzymes that perform the phosphorylation reaction, represent a full 2% of the genome [21]. For the past few decades the kinases, G protein-coupled receptor kinases (GRKs), Ca2+/calmodulin-dependent protein kinase II (CAMKII), proto-oncogene tyrosine-protein kinase (Src) and protein kinase C (PKC), have been among the most intensely investigated kinases that modulate GPCR phosphorylation [69–72]. Given the importance of phosphorylation in affecting the dynamics of receptor signaling and trafficking, GPCR phosphorylation has been extensively studied (reviewed in [73,74]).
Functional significance of MOR phosphorylation:
Initial studies of MOR phosphorylation were directly motivated by the classical concept that GPCR phosphorylation by GRKs leads to β-arrestin recruitment and receptor internalization (Box 1) [75]. These early discoveries highlighted the complexity of opioid signaling by connecting phosphorylation of MOR to agonist-induced receptor internalization, desensitization, recycling, analgesia and tolerance [76–79].
There are ~20 predicted phosphorylation sites in MOR (Fig. 3) that are conserved between mouse and rat (reviewed in [79]). These residues are located at the intracellular loop, as well as at the C-terminal region of MOR (Fig. 1, and Fig. 3). Mass spectrometry analysis and/or studies with phospho-specific antibodies have identified Y106, Y166 and S266 in the intracellular loop [80,81]; and S363 and eight S/T residues within two cassettes (T354S355S356T357 and T370REHPS375T376ANT379) in the C-terminal tail to be phosphorylated [69,72,79,82–87]. These are seen in both cultured cells and in mouse brain tissue. [69,85,86].
Figure 3: Biological relevance of µ-Opioid Receptor phosphorylation.
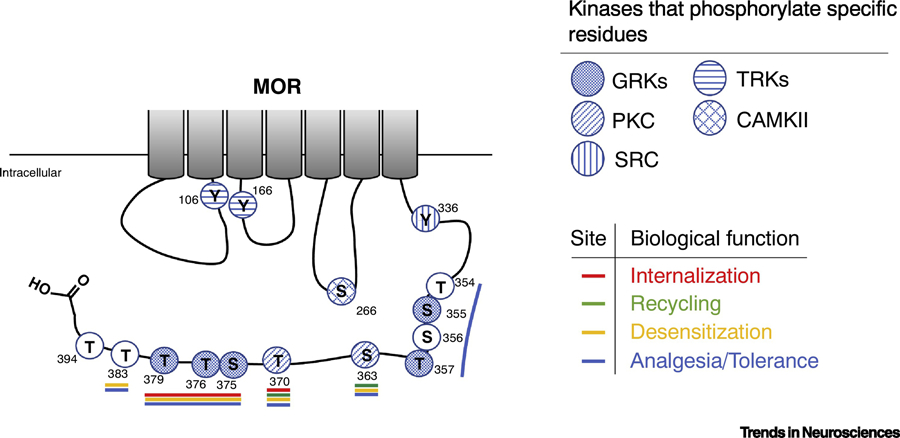
A schematic representation of the intracellular regions of rat µ-Opioid Receptor showing phosphorylatable residues and their biological relevance. Kinases that modify individual residues are indicated with filled hatches. The residue number is indicated next to each residue. Filled circles represent known phosphorylation sites and open circles represent putative phosphorylation residues. Residues that affect distinct biological activities are indicated with a colored line; in red are the residues described to play a role in MOR internalization; in green are the residues described to play a role in MOR recycling; in yellow are the residues described to play a role in MOR desensitization; and in blue are the residues described to play a role in opioid analgesia and tolerance. GRKs, G protein-coupled receptor kinases; RTKs, receptor tyrosine kinases; PKC, protein kinase C; CAMKII, Ca++/calmodulin kinase II; Src, Src kinase.
The kinases responsible for phosphorylating specific MOR residues and their phosphorylation sites are shown in Table 1 and Fig. 3. The kinases are: Src for residue Y336 [80]; tyrosine kinases for residues Y106 and Y166 [81]; GRKs for residues S355, T357, T370, S375, T376 and T379 [69,85,87–90], PKC for residues S363 and T370 [69,91,92], and CAMKII for the residues S366 and T370 [69,93]. The exact molecular mechanism of how agonist-binding triggers the activation of these kinases and consequent receptor phosphorylation remains unclear. Studies have described that the only residues that are found to be phosphorylated under basal conditions are S363 and T370, all the others are thought to be phosphorylated following receptor activation [69,82,86].
Most studies that aim at understanding the role of MOR phosphorylation focus on the S/T residues T354, S355, S356, T357, S363, T370, S375, T376, T379, T383 and T394 at the C-terminal tail (Fig. 3, Table 1) [76,82,94,95]. These residues have been described to play an important role in the modulation of receptor desensitization, recycling, opioid analgesia and tolerance [78,94–96]. Recent studies in animals harboring mutations of phosphorylation residues described above (phosphorylation deficient mice) reported an increase in agonist-mediated analgesic effects with no changes (or exacerbation) of side effects such as respiratory depression and constipation [76]. These recent findings have raised a renewed interest in exploring the implications of MOR phosphorylation to the biological effects of opioids (Box 3).
BOX 3: The role of MOR phosphorylation and β-arrestin recruitment in regulating opioid effects.
Recent findings highlight the importance of receptor phosphorylation and β-arrestin recruitment in regulating opioid receptor function. These include studies that show that, (i) mutations of C-terminal MOR phosphorylation residues (to alanine) impair interactions with β-arrestin, enhance analgesia, diminish analgesic tolerance, but do not suppress respiratory depression, constipation and opioid withdrawal symptoms [76,87]; (ii) phosphorylation of MOR upon morphine or DAMGO treatment is not impaired in β-arrestin 1/2 KO cells [98]; (iii) the lack of β-arrestin-2 suppresses the lethal side effects of opioid treatment without affecting the analgesic response [98,114,153,154]. Therefore, the role of β-arrestin needs to be systematically explored considering its relevance to different aspects of MOR PTM events.
The role of different agonists in mediating MOR phosphorylation, the kinases responsible for phosphorylation, and the biological relevance of this PTM are described in detail below (Fig. 3; Table 1). Since these phosphorylation events have been described to be involved in different processes of MOR signaling, we have structured the section of MOR phosphorylation by focusing on the correlation between this PTM and the distinct cell biological/pharmacological effects, including internalization, recycling, desensitization, antinociception and tolerance (Fig. 3; Table 1).
Effect of phosphorylation on MOR internalization:
Early studies reported that different OR agonists differentially phosphorylate MOR and the extent of phosphorylation could be correlated to the extent of receptor internalization [79]. The mechanisms underlying the differences in agonist activation of MOR and its internalization were found to be directly related to the phosphorylation of specific residues by distinct GRKs isoforms [70,72,87,97]. Furthermore these studies reported that treatment with high-efficacy opioids such DAMGO ([D-Ala2-MePhe4-Gly-ol]), fentanyl, and etonitazene facilitated robust internalization [72,87,97,98] whereas, a partial agonist such as morphine did not [72,83,99]. Correlating this with MOR phosphorylation, studies have reported that high-efficacy opioids initially induce phosphorylation of S375, followed by phosphorylation of T370, T376 and T379 [72,83]. This process is mediated by GRK2/3, and serves to facilitate MOR diffusion in the plasma membrane [100] and subsequent β-arrestin recruitment and receptor internalization [72,87,97,98]. Supporting these findings, mutation of residues S375T376ANT379 to A375A376ANA379 decreased β-arrestin recruitment and internalization after treatment with Met-Enk [78,85]. Notably, mutation of residues T354S355S356T357 (which are not involved in the aforementioned hierarchical phosphorylation of MOR by GRK2/3) to A354A355A356A357 did not affect receptor internalization [85,94–96] (Fig. 3).
In contrast, morphine treatment was found to induce robust phosphorylation of S375, but only a weak phosphorylation of T370, T376, and T379 [87]. Recent studies indicate that MOR phosphorylation at S375 is primarily by the kinase GRK5 [90,97] and to a lesser extent by GRK2/3 [87,90]. Increasing the expression of GRK2/3 led to enhanced mMOR internalization in response to morphine treatment supporting the idea that MOR phosphorylation at T370, T376, and T379 contribute to MOR internalization. [83,97].
Effect of phosphorylation on MOR recycling:
The field has only recently begun to investigate the role of MOR phosphorylation on receptor recycling [77,101]. One of the strategies to explore MOR recycling is via the overexpression of MOR N-terminally tagged with a pH-sensitive GFP, which produces a fluorescence signal during vesicle fusion with the plasma membrane [77,101]. Studies have suggested a role for PKC in receptor recycling since treatment with a PKC inhibitor was found to reduce DAMGO-mediated MOR recycling [77]. Furthermore, mutation of S363 and T370 residues of mMOR (known PKC phosphorylated residues) to alanine led to impaired MOR recycling [101] (Fig. 3).
The exact role of PKC in MOR recycling is still unknown. Preliminary studies suggest that PKC phosphorylation of MOR occurs at multiple cellular compartments [77,101]. Although additional studies are necessary, the following evidence corroborate this hypothesis: (i) S363 and T370 residues of mMOR appear to be phosphorylated by PKC in the basal state [69,82,86,92] and the phosphorylation level is not increased after agonist treatment [85,86], (ii) DAMGO treatment does not lead to PKC activation [102]; however, PKC inhibition affects MOR recycling following receptor internalization by DAMGO [77]. These findings support a role for PKC-mediated receptor phosphorylation in multiple compartments (at cell membrane in the basal state and in an intracellular compartment following agonist treatment-mediated receptor internalization).
Effect of phosphorylation on MOR desensitization:
A hallmark of MOR is the rapid desensitization following acute treatment with an opioid Opioid-induced acute MOR desensitization has been studied electrophysiologically in neurons of the thalamus or locus coeruleus of mice or rats expressing different MOR mutants. This method has allowed the investigation of receptor desensitization at different time points and opioid concentrations [79]. It has been hypothesized that acute desensitization requires receptor phosphorylation, followed by β-arrestin recruitment and/or receptor internalization. Therefore, similar to the receptor internalization, the mechanism that drives the desensitization process is probe specific [70,72,76,78,85].
Electrophysiological studies showed that mutation of 10 S/T C-terminal residues (T354, S355, S356, T357, S363, T370, S375, T376, T379, and T383) significantly reduced the acute desensitization mediated by Met-Enk in locus coeruleus neurons [76,78,94]. Attenuation of Met-Enk-mediated acute desensitization was also observed in mice with mutations of both phosphorylation casettes i.e. S375T376ANT379 and T354S355S356T357 [96]. However, this attenuation was not seen when the S375T376ANT379 cassette alone was mutated [94,96]. Together, these data point towards the contribution of phosphorylation of specific C-terminal residues in the acute desensitization to Met-Enk.
Studies comparing acute desensitization by Met-Enk to that by morphine showed that mutation of the 11 S/T C-terminal residues to alanine (T393 in addition to the 10 S/T residues described earlier) did not abolish morphine-mediated receptor desensitization [95]. One potential explanation is that a different set of kinases is involved in morphine-mediated desensitization. For example, morphine-mediated PKC activation has been described to play a role in MOR desensitization [71,95,103–106]. Also, morphine-induced desensitization (but not Met-Enk) was reduced by PKC inhibition [95]. The molecular mechanism of how PKC modulates desensitization in a probe specific manner remains unclear. Recent proteomic analyses have reported that PKC, activated by acute treatment with morphine, can subsequently interact with regulators of MOR signaling [107]. Among these regulators, Raf kinase inhibitory protein (RKIP) is an interesting substrate since it has been found to inhibit GRK, and RKIP/GRK interactions have been found to enable Gαβγ-binding and signaling [107–109].
Effect of phosphorylation on opioid-mediated antinociception and tolerance:
Studies have used transgenic animals to correlate MOR phosphorylation with antinociception and development of tolerance to opioids [76,78,94,110,111]. Studies using mice expressing MOR with alanine mutations in 10 S/T C-terminal phosphorylation sites (T354, S355, S356, T357, S363, T370, S375, T376, T379 and T383) show that these animals exhibit enhanced antinociception compared to wild-type mice in response to fentanyl or morphine [76]; this suggests that these phosphorylatable residues play a role in modulating opioid-induced analgesia [76].
Chronic opioid administration induces antinociceptive tolerance [76,78,94]. The related changes in MOR activation can be evaluated using classical tests of pain response in animals such as the hot-plate test [76,110,111]. Such studies show that tolerance to fentanyl and morphine is abrogated in mice with alanine mutations of 11 S/T C-terminal phosphorylation sites (or in 10 S/T C-terminal phosphorylation sites except for T394) [76].
The role of specific residues in the C-terminal of MOR (listed above) that mediate opioid tolerance has also been investigated [76,110,111]. Interestingly, phosphorylation of S375 has been described to regulate tolerance to high-efficacy opioid agonists such as fentanyl, DAMGO, Met-Enk, or etonitazene, but not morphine [76,110]. In addition, mice expressing MOR with a T394A mutation did not exhibit acute tolerance to either morphine or etorphine [111].
Studies examining specific enzymes responsible for receptor phosphorylation have implicated a number of kinases and phosphatases in mediating opioid tolerance [78,104,112,113]. For instance, PKC inhibition [104,114,115] and GRK3 knock-down was found to reverse tolerance induced by chronic morphine [113]. In contrast, inhibition of protein phosphatase PP2A, was found to enhance morphine antinociceptive tolerance [104,112].
Taken together, several studies point to the relevance of MOR C-terminal tail phosphorylation on the control of opioid analgesia and tolerance. The mechanisms responsible for opioid tolerance are not completely understood and require additional studies.
Phosphorylation of other opioids receptor: Phosphorylation of other ORs has been shown to play roles in receptor desensitization and internalization [109,116–121]. For instance, agonist-induced mDOR desensitization and internalization is regulated by residues T358 and S363 [117], in a mechanism potentially mediated by GRK2/3 and β-arrestin recruitment [120,122]. Interestingly, DOR phosphorylation at S344 is mediated by PKC in an agonist-independent manner [123].
Studies with mKOR reported agonist-mediated phosphorylation at S356, T357, T363 and S369. Notably, the extent of phosphorylation at these residues is higher after U50,488H treatment as compared to etorphine treatment [119,124]. Agonist-induced KOR phosphorylation involves a mechanism mediated by GRK2/3 and GRK5/6 activation [121,124]. Similarly to other opioid receptors, PKC has also been implicated in agonist-independent KOR phosphorylation [124].
The activation of NOP by its endogenous ligand, nociceptin/orphanin FQ, has been reported to lead to the activation of kinases such as PKC, extracellular signal–regulated kinase 1 (ERK1) and ERK2, p38 mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK) [125–127]. Agonist-induced mNOP activation recruits GRK2/3 [116], leading to activation of the latter and consequently receptor phosphorylation at S346, followed by S351, T362 and S363. These events suggest that NOP phosphorylation facilitates receptor desensitization and internalization [116,128].
Ubiquitination
Ubiquitination is the formation of a covalent bond between ubiquitin, a 76-amino acid protein, and the protein substrate [25]. This bond is usually formed between the C-terminal glycine (G76) of ubiquitin and the epsilon amino group of the lysine of the target substrate, but in some cases, the ubiquitin can be attached to the amino group at the N-terminus of the substrate [129]. Ubiquitin has eight potential attachment points, allowing for the formation of polyubiquitin chains with a high structural diversity (reviewed in [130]). The ubiquitination process is mediated by three enzymatic reactions. In the first reaction, ubiquitin is activated at its C-terminus in an ATP-dependent manner by an E1-activating enzyme. Next, the activated ubiquitin is transferred to a cysteine residue on an E2-conjugating enzyme. Finally, the E2-ubiquitin intermediate interacts with an E3-ubiquitin ligase, which transfers the ubiquitin to the lysine residue on the substrate [23]. The E3-ubiquitin ligases are categorized into two families: the E6AP C terminus (HECT), which possess inherent catalytic activity, and the Really Interesting New Gene (RING), which facilitate the interaction between the substrate and the E2 enzymes (reviewed in [131,132]). Recent studies indicate that β-arrestin can function as an adaptor protein that facilitates the ubiquitination process by interacting with E3-ubiquitin ligases [22,98].
Ubiquitination is a transient PTM that is reversed by deubiquitinating enzymes (reviewed in [131,132]). Deubiquitinating enzymes are divided into five families: the ubiquitin carboxy-terminal hydrolases, ubiquitin–specific proteases, ovarian tumor-related proteases, Machado-Joseph disease protein domain proteases, and jab1/MPN domain-associated metalloisopeptidases (JAMM) (reviewed in [131,132]). The role of ubiquitination in regulating receptor levels and trafficking has been extensively studied in the case of GPCRs such as β2-adrenergic and vasopressin V2 receptors (reviewed in [132,133]).
Initially, ubiquitin was characterized as a degradation-tag that directs proteins towards the proteasome pathway. Indeed, in the context of GPCRs, the ubiquitination process is generally related to degradation of internalized receptors, in a mechanism linked to long-term desensitization of transmembrane signaling (reviewed in [22,130]). In addition, GPCRs can undergo agonist-mediated ubiquitination, and a few exhibit constitutive ubiquitination that modulates correct receptor trafficking to and from the plasma membrane [131,134]. While agonist-mediated ubiquitination occurs at the plasma membrane, requires receptor phosphorylation and promotes GPCR internalization and down-regulation, constitutively ubiquitinated receptors undergo reversible agonist-mediated deubiquitination at the plasma membrane [131,134]. In addition, some newly synthetized proteins require deubiquitination to translocate to the cell surface [135]. Depending of the ubiquitin lysine-linkage on the modified substrate different pathways can be activated [130]. For instance, poly-ubiquitin chains with lysine-48 (K48) linkage have been implicated in substrate degradation [136], while ubiquitin-chains with lysine-63 (K63) linkage have been associated with vesicular trafficking or kinase activation [137]. More comprehensive studies are needed to fully elucidate how ubiquitination regulates GPCR turnover and activity.
Functional significance of MOR ubiquitination:
Ubiquitination of ORs regulates their endocytosis and degradation [98,138,139]. In addition, misfolding of ORs in the ER can also activate the ubiquitination process and induce OR degradation [138,139] (Fig. 2). Interestingly, MOR ubiquitination is ligand specific [98,140], suggesting a possible role for this PTM in biased signaling (Box 3). Different MOR agonists can differentially activate ubiquitin attachment, in a process mediated by β-arrestin [132]. For instance, treatment with DAMGO, but not morphine, leads to increased receptor ubiquitination; β-arrestin-1 appears to play a role in this process since the increase in ubiquitination was abrogated in β-arrestin KO cells treated with DAMGO [98]. An interesting study reported that DADLE ([D-Ala2, D-Leu5]-Enkephalin) triggers ubiquitination of the first intracellular loop of MOR in a process mediated by β-arrrestin-2, which results in receptor down-regulation by lysosomal proteolysis [140,141] (Fig. 1; Table 1). Inhibition of MOR ubiquitination leads to a delay of the endocytic process via scission of clathrin-coated vesicles [141].
Ubiquitination of other opioids receptors: The ubiquitination of DOR has been described in different subcellular compartments. During biosynthesis, DOR is ubiquitinated in a process that works as a quality control that targets the misfolded receptor and labels it for degradation by the proteasome [139,142]. Interestingly, treatment with DOR agonists (Deltorphin I and II) leads to DOR endocytosis, followed by ubiquitination and lysosomal degradation [143].
KOR ubiquitination is enhanced by treatment with the agonists U50,488H and Dyn A [144]. The residue K63 of hKOR is polyubiquitinated in a process that takes place after receptor phosphorylation. Polyubiquitination contributes to changes in KOR expression, and this is an agonist-dependent process [144,145] (Table 1).
Finally, studies examining the maturation of MOR-DOR interacting complexes have found that the expression of DOR protects MOR from ubiquitination and degradation [146] and this, in turn, leads to increased cell surface expression of the MOR-DOR heteromer.
Concluding remarks and future directions
The binding of different agonists to ORs leads to different sets of molecular changes, which contributes to the complex pharmacological profile for opioids. The regulation of this complex process relies on the combination of all the PTMs described in this review: (i) the glycosylation of OR modulates the steady-state levels of the receptor at the cell surface, (ii) the hierarchical phosphorylation process affects receptor internalization/recycling as well as desensitization/tolerance, (iii) the palmitoylation process influences receptor distribution at the plasma membrane, and (iv) the ubiquitination process regulates receptor abundance (Fig. 2). In future studies it will be critical to investigate the crosstalk between PTM events, agonist-specificity and spatio-temporal dynamics of receptor signaling regulated by these PTMs (see Outstanding Questions).
Outstanding question box:
What roles do PTMs play in the spatio-temporal dynamics of OR signaling?
What are the effects of the glycan profile of the host cells in OR glycosylation and binding of opioid ligands?
What is the contribution of receptor palmitoylation to the extent of phosphorylation and how does it affect agonist-mediated receptor distribution and internalization?
What is the role of phosphorylation at non-C-terminal sites (i.e. intracellular loops) in modulating receptor activity?
What is the role of β-arrestin recruitment in receptor ubiquitination, and how is this connected to differences in agonist-mediated receptor activation?
Highlights.
Post-translational modifications in G-protein coupled receptors are responsible for fine-tuning receptor signaling. Receptor subcellular localization, membrane distribution dynamics, protein-protein interactions, and receptor signaling are all events mediated by post translational modifications on specific amino acid residues.
Glycosylation of the opioid receptor plays a role in modulating the steady-state levels at the cell surface. In human µ-Opioid Receptor, asparagine 40 (N40) is one residue that has been described to be glycosylated, and N40D polymorphism has been implicated in pain sensitivity and dependence on alcohol and heroin.
Palmitoylation and phosphorylation modulate the dynamics of movement of µ-Opioid Receptor at the plasma membrane and specifically, diffusion into lipid rafts. The crosstalk between these two post-translational modification events regulates agonist-mediated receptor distribution and internalization.
Phosphorylation of the C-terminus of µ-Opioid Receptor is associated with agonist-induced receptor internalization, recycling, desensitization, as well as analgesia and development of tolerance.
Ubiquitination of the µ-Opioid Receptor affects receptor degradation in a process dependent on β-arrestin recruitment.
Acknowledgements:
We would like to thank Dr. Ivone Gomes, Dr. Deborah Schechtman and Seshat Mack for critical reading and constructive comments. The work was supported in part by NIH grants (DA008863 and NS026880 to LAD).
Glossary
- Post-translational modification
any modification that a protein undergoes after its translation, such as phosphorylation, glycosylation, palmitoylation or ubiquitination
- Glycosylation
the covalent linkage of a sugar to an amino acid residue on secreted or membrane-bound proteins. Amino acid residues that are primarily glycosylated are asparagine, serine, and threonine
- Phosphorylation
the covalent attachment of a phosphate group to a serine, tyrosine or threonine residue on a substrate in eukaryotes
- Palmitoylation
lipid modification characterized by the addition of a palmitate (16-carbon saturated fatty acid) to a cysteine residue via a thioester linkage
- Ubiquitination
attachment of one or more ubiquitin proteins (8.6 kDa) to a lysine residue on the substrate
- Efficacy
ability of an agonist to effectively activate the receptor once it is bound to it; determines how efficient an agonist is at producing a desired effect
- Opioid-induced desensitization
the decrease in signaling response produced by the receptor after acute exposure to opioid agonists. This effect takes place in seconds to minutes after exposure to the opioid agonist and is reversible upon removal of opioid agonist from the system
- Tolerance
the reduced effect of a drug after chronic use. In contrast to opioid-induced desensitization, tolerance develops over hours in cellular contexts, and days to weeks in animal models. Tolerance is characterized by the need to increase the dose of the drug in order to maintain the desired effect, leading to a rightward shift in the dose-response curve. One of the hallmarks of tolerance is a prolonged recovery period after desensitization
- Opioid addiction
a chronic disease characterized by a compulsive and continuous need to use opioid drugs
- Withdrawal
symptoms that occur upon discontinuing usage of a drug. In mouse models, the opioid withdrawal symptoms are jumping, “wet-dog” shakes, excessive grooming, weight loss and diarrhea
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer section: We declare that none of the authors have competing financial or non-financial interests as defined by Cell Press.
References:
- 1.Cox BM et al. (2015) Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br. J. Pharmacol 172, 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filizola M and Devi LA (2013) Grand opening of structure-guided design for novel opioids. Trends Pharmacol. Sci 34, 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dreborg S et al. (2008) Evolution of vertebrate opioid receptors. Proc. Natl. Acad. Sci. U. S. A 105, 15487–15492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lutz PE and Kieffer BL (2013) Opioid receptors: Distinct roles in mood disorders. Trends Neurosci 36, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darcq E and Kieffer BL (2018) Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci 19, 499–514 [DOI] [PubMed] [Google Scholar]
- 6.Imam MZ et al. (2018) Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 131, 238–255 [DOI] [PubMed] [Google Scholar]
- 7.Paronis CA and Woods JH (1997) Ventilation in morphine-maintained rhesus monkeys. II: Tolerance to the antinociceptive but not the ventilatory effects of morphine. J. Pharmacol. Exp. Ther 282, 355–362 [PubMed] [Google Scholar]
- 8.Pfeiffer A et al. (1986) Psychotomimesis mediated by κ opiate receptors. Science (80-.). 233, 774–776 [DOI] [PubMed] [Google Scholar]
- 9.Snead OC (1986) Opiate-induced seizures: A study of μ and δ specific mechanisms. Exp. Neurol 93, 348–358 [DOI] [PubMed] [Google Scholar]
- 10.Nieto MM et al. (2005) Physiological control of emotion-related behaviors by endogenous enkephalins involves essentially the delta opioid receptors. Neuroscience 135, 305–313 [DOI] [PubMed] [Google Scholar]
- 11.Javelot H et al. (2010) Human opiorphin is a naturally occurring antidepressant acting selectively on enkephalin-dependent δ-opioid pathways. J. Physiol. Pharmacol 61, 355–362 [PubMed] [Google Scholar]
- 12.Zaveri NT (2016) Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J. Med. Chem 59, 7011–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiro RG (2002) Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, [DOI] [PubMed] [Google Scholar]
- 14.Goth CK et al. (2018) Fine-Tuning Limited Proteolysis: A Major Role for Regulated Site-Specific O-Glycosylation. Trends Biochem. Sci 43, 269–284 [DOI] [PubMed] [Google Scholar]
- 15.Neve KA et al. (2003) Role of palmitoylation/depalmitoylation reactions in G-protein-coupled receptor function. Pharmacol. Ther 97, 1–33 [DOI] [PubMed] [Google Scholar]
- 16.Resh S, M.D. (2013) Covalent Lipid Modifications of Proteins. Curr. Biol 23, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang P et al. (2007) Cholesterol reduction by methyl-β-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem. Pharmacol 73, 534–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng H et al. (2012) Palmitoylation and membrane cholesterol stabilize μ-opioid receptor homodimerization and G protein coupling. BMC Cell Biol 13, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z et al. (2017) Phosphorylation of g protein-coupled receptors: From the barcode hypothesis to the flute model. Mol. Pharmacol 92, 201–210 [DOI] [PubMed] [Google Scholar]
- 20.Shen A et al. (2018) Functionally distinct and selectively phosphorylated GPCR subpopulations co-exist in a single cell. Nat. Commun 9, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ubersax JA and Ferrell JE (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 22.Shenoy SK (2007) Seven-transmembrane receptors and ubiquitination. Circ. Res 100, 1142–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alonso V and Friedman PA (2013) Minireview: Ubiquitination-regulated G Protein-Coupled Receptor Signaling and Trafficking. Mol. Endocrinol 27, 558–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jean-Charles PY et al. (2016) Ubiquitin-Related Roles of β-Arrestins in Endocytic Trafficking and Signal Transduction. J. Cell. Physiol 231, 2071–2080 [DOI] [PubMed] [Google Scholar]
- 25.Hislop JN and Von Zastrow M (2011) Role of Ubiquitination in Endocytic Trafficking of G-Protein-Coupled Receptors. Traffic 12, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohtsubo K and Marth JD (2006) Glycosylation in Cellular Mechanisms of Health and Disease. Cell 126, 855–867 [DOI] [PubMed] [Google Scholar]
- 27.Mrázek H et al. (2013) Carbohydrate synthesis and biosynthesis technologies for cracking of the glycan code: Recent advances. Biotechnol. Adv 31, 17–37 [DOI] [PubMed] [Google Scholar]
- 28.Brooks SA (2004) Appropriate glycosylation of recombinant proteins for human use. Mol. Biotechnol 28, 241–255 [DOI] [PubMed] [Google Scholar]
- 29.Duvernay MT et al. (2005) The regulatory mechanisms of export trafficking of G protein-coupled receptors. Cell. Signal 17, 1457–1465 [DOI] [PubMed] [Google Scholar]
- 30.Achour L et al. (2008) An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol. Sci 29, 528–535 [DOI] [PubMed] [Google Scholar]
- 31.Zhang M and Wu G (2019) Mechanisms of the anterograde trafficking of GPCRs: Regulation of AT1R transport by interacting proteins and motifs. Traffic 20, 110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eppler CM et al. (1993) Purification and partial amino acid sequence of a μ opioid receptor from rat brain. J. Biol. Chem 268, 26447–26451 [PubMed] [Google Scholar]
- 33.Liu-Chen LY et al. (1993) Beta-[3H]funaltrexamine-labeled mu-opioid receptors: species variations in molecular mass and glycosylation by complex-type, N-linked oligosaccharides. Mol. Pharmacol 44, 749–56 [PubMed] [Google Scholar]
- 34.Kieffer BL et al. (1994) The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci 91, 1193–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garzon J et al. (1995) Antibodies to the cloned mu-opioid receptor detect various molecular weight forms in areas of mouse brain. Mol. Pharmacol 47, 738–44 [PubMed] [Google Scholar]
- 36.Law PY et al. (1985) Effects of cycloheximide and tunicamycin on opiate receptor activities in neuroblastoma X glioma NG108–15 hybrid cells. Biochem. Pharmacol 34, 9–17 [DOI] [PubMed] [Google Scholar]
- 37.Li JG et al. (2007) N-glycosylation of the human κ opioid receptor enhances its stability but slows its trafficking along the biosynthesis pathway. Biochemistry 46, 10960–10970 [DOI] [PubMed] [Google Scholar]
- 38.Li-Na (2012) Post-Transcriptional Regulation of Opioid Receptors in the Nervous System. Fron Biosci [DOI] [PMC free article] [PubMed]
- 39.Huang P et al. (2012) A common single nucleotide polymorphism A118G of the μ opioid receptor alters its N-glycosylation and protein stability. Biochem. J 441, 379–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halikere A et al. (2019) Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Mol. Psychiatry 1, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y et al. (2005) Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J. Biol. Chem 280, 32618–32624 [DOI] [PubMed] [Google Scholar]
- 42.Befort K et al. (2001) A Single Nucleotide Polymorphic Mutation in the Human μ-Opioid Receptor Severely Impairs Receptor Signaling. J. Biol. Chem 276, 3130–3137 [DOI] [PubMed] [Google Scholar]
- 43.Zerbino DR et al. (2018) Ensembl 2018. Nucleic Acids Res 46, D754–D761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auton A et al. (2015) A global reference for human genetic variation. Nature 526, 68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fillingim RB et al. (2005) The A118G single nucleotide polymorphism of the μ-opioid receptor gene (OPRM1) is associated with pressure pain sensitivity in humans. J. Pain 6, 159–167 [DOI] [PubMed] [Google Scholar]
- 46.Chou WY et al. (2006) Association of μ-opioid receptor gene polymorphism (A118G) with variations in morphine consumption for analgesia after total knee arthroplasty. Acta Anaesthesiol. Scand 50, 787–792 [DOI] [PubMed] [Google Scholar]
- 47.Klepstad P et al. (2004) The 118 A > G polymorphism in the human μ-opioid receptor gene may increase morphine requirements in patients with pain caused by malignant disease. Acta Anaesthesiol. Scand 48, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 48.Sloan ME et al. (2018) The OPRM1 A118G polymorphism: Converging evidence against associations with alcohol sensitivity and consumption. Neuropsychopharmacology 43, 1530–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bond C et al. (1998) Single-nucleotide polymorphism in the human mu opioid receptor gene alters β-endorphin binding and activity: Possible implications for opiate addiction. Proc. Natl. Acad. Sci 95, 9608–9613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y et al. (2015) Mouse Model of the OPRM1 (A118G) Polymorphism: Differential Heroin Self-Administration Behavior Compared with Wild-Type Mice. Neuropsychopharmacology 40, 1091–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahmoud S et al. (2011) Pharmacological consequence of the A118G μ opioid receptor polymorphism on morphine-and fentanyl-mediated modulation of Ca2+ channels in humanized mouse sensory neurons. Anesthesiology 115, 1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Margas W et al. (2007) Modulation of Ca2+ channels by heterologously expressed wild-type and mutant human μ-opioid receptors (hMORs) containing the A118G single-nucleotide polymorphism. J. Neurophysiol 97, 1058–1067 [DOI] [PubMed] [Google Scholar]
- 53.Miller GM et al. (2004) A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: Association with stress response and aggression. Mol. Psychiatry 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 54.Goh JB and Ng SK (2018) Impact of host cell line choice on glycan profile. Crit. Rev. Biotechnol 38, 851–867 [DOI] [PubMed] [Google Scholar]
- 55.Huang AM et al. (2008) Brain region-specific N-glycosylation and lipid rafts association of the rat mu opioid receptor. Biochem. Biophys. Res. Commun 365, 82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markkanen PMH and Petäjä-Repo UE (2008) N-glycan-mediated quality control in the endoplasmic reticulum is required for the expression of correctly folded δ-opioid receptors at the cell surface. J. Biol. Chem 283, 29086–29098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge X et al. (2009) m-Opioid Receptor Cell Surface Expression Is Regulated by Its Direct Interaction with Ribophorin I. Mol. Pharmacol 75, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lackman JJ et al. (2014) N-glycan-dependent and -independent quality control of human δ opioid receptor N-terminal variants. J. Biol. Chem 289, 17830–17842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lackman JJ et al. (2018) Site-specific O-glycosylation of N-terminal serine residues by polypeptide GalNAc-transferase 2 modulates human δ-opioid receptor turnover at the plasma membrane. Cell. Signal 42, 184–193 [DOI] [PubMed] [Google Scholar]
- 60.Resh MD (2006) Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol 2, 584–590 [DOI] [PubMed] [Google Scholar]
- 61.Kleuss C and Krause E (2003) Gαs is palmitoylated at the N-terminal glycine. EMBO J 22, 826–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koster KP and Yoshii A (2019) Depalmitoylation by palmitoyl-protein thioesterase 1 in neuronal health and degeneration. Front. Synaptic Neurosci 11, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cherezov V et al. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science (80-.). 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goddard AD and Watts A (2012) Regulation of G protein-coupled receptors by palmitoylation and cholesterol. BMC Biol 10, 2–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greaves J and Chamberlain LH (2007) Palmitoylation-dependent protein sorting. J. Cell Biol 176, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen C et al. (1998) Palmitoylation of the rat μ opioid receptor. FEBS Lett 441, 148–152 [DOI] [PubMed] [Google Scholar]
- 67.Petäjä-Repo UE et al. (2006) Distinct subcellular localization for constitutive and agonist-modulated palmitoylation of the human δ opioid receptor. J. Biol. Chem 281, 15780–15789 [DOI] [PubMed] [Google Scholar]
- 68.Merkouris M et al. (1996) Identification of the critical domains of the δ-opioid receptor involved in G protein coupling using site-specific synthetic peptides. Mol. Pharmacol 50, 985–993 [PubMed] [Google Scholar]
- 69.Chen YJ et al. (2013) Identification of phosphorylation sites in the COOH-terminal tail of the μ-opioid receptor. J. Neurochem 124, 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lowe JD et al. (2015) Role of G Protein-Coupled Receptor Kinases 2 and 3 in μ-Opioid Receptor Desensitization and Internalization. Mol. Pharmacol 88, 347–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ingram SL and Traynor JR (2009) Role of protein kinase C in functional selectivity for desensitization at the μ-opioid receptor: From pharmacological curiosity to therapeutic potential. Br. J. Pharmacol 158, 154–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Just S et al. (2013) Differentiation of Opioid Drug Effects by Hierarchical Multi-Site Phosphorylation. Mol. Pharmacol 83, 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magalhaes AC et al. (2012) Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol 165, 1717–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gurevich VV and Gurevich EV (2019) GPCR signaling regulation: The role of GRKs and arrestins. Front. Pharmacol 10, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang J et al. (1998) Role for G protein-coupled receptor kinase in agonist-specific regulation of μ-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U. S. A 95, 7157–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kliewer A et al. (2019) Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kunselman JM et al. (2019) Homologous regulation of mu opioid receptor recycling by Gβγ, Protein Kinase C, and receptor phosphorylation. Mol. Pharmacol 48109, mol.119.117267 [DOI] [PMC free article] [PubMed]
- 78.Arttamangkul S et al. (2018) Cellular tolerance at the µ-opioid receptor is phosphorylation dependent. Elife 7, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Williams JT et al. (2013) Regulation of m -Opioid Receptors : Desensitization, Phosphorylation, Internalization, and Tolerance. 10.1124/pr.112.005942 [DOI] [PMC free article] [PubMed]
- 80.Zhang L et al. (2009) Src phosphorylation of μ-receptor is responsible for the receptor switching from an inhibitory to a stimulatory signal. J. Biol. Chem 284, 1990–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McLaughlin JP and Chavkin C (2001) Tyrosine phosphorylation of the μ-opioid receptor regulates agonist intrinsic efficacy. Mol. Pharmacol 59, 1360–1368 [DOI] [PubMed] [Google Scholar]
- 82.El Kouhen R et al. (2001) Phosphorylation of Ser 363, Thr 370, and Ser 375 Residues within the Carboxyl Tail Differentially Regulates μ-Opioid Receptor Internalization. J. Biol. Chem 276, 12774–12780 [DOI] [PubMed] [Google Scholar]
- 83.Schulz S et al. (2004) Morphine induces terminal μ-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J 23, 3282–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doll C et al. (2011) Agonist-selective patterns of μ-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br. J. Pharmacol 164, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lau EK et al. (2011) Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci. Signal 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moulédous L et al. (2015) Phosphoproteomic analysis of the mouse brain mu-opioid (MOP) receptor. FEBS Lett 589, 2401–2408 [DOI] [PubMed] [Google Scholar]
- 87.Miess E et al. (2018) Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid-opioid receptor desensitization. Sci. Signal 11, 1–16 [DOI] [PubMed] [Google Scholar]
- 88.Deng HB et al. (2000) Role for the C-terminus in agonist-induced μ opioid receptor phosphorylation and desensitization. Biochemistry 39, 5492–5499 [DOI] [PubMed] [Google Scholar]
- 89.Wang HL et al. (2002) Identification of two C-terminal amino acids, Ser355 and Thr357, required for short-term homologous desensitization of μ-opioid receptors. Biochem. Pharmacol 64, 257–266 [DOI] [PubMed] [Google Scholar]
- 90.Glück L et al. (2014) Loss of morphine reward and dependence in mice lacking G protein-coupled receptor kinase 5. Biol. Psychiatry 76, 767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feng B et al. (2011) Protein Kinase C-Mediated Phosphorylation of the μ-Opioid Receptor and Its Effects on Receptor Signaling. Mol. Pharmacol 79, 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Illing S et al. (2014) Heterologous regulation of agonist-independent μ-opioid receptor phosphorylation by protein kinase C. Br. J. Pharmacol 171, 1330–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koch T et al. (1997) Site Mutation in the Rat μ-Opioid Receptor Demonstrates the Involvement of Calcium/Calmodulin-Dependent Protein Kinase II in Agonist-Mediated Desensitization. J. Neurochem 69, 1767–1770 [DOI] [PubMed] [Google Scholar]
- 94.Arttamangkul S et al. (2019) Separation of Acute Desensitization and Long-Term Tolerance of µ-Opioid Receptors Is Determined by the Degree of C-Terminal Phosphorylation. Mol. Pharmacol 96, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yousuf A et al. (2015) Role of Phosphorylation Sites in Desensitization of m-Opioid Receptor. Mol. Pharmacol. Mol Pharmacol 88, 825–835 [DOI] [PubMed] [Google Scholar]
- 96.Birdsong WT et al. (2015) Agonist Binding and Desensitization of the -Opioid Receptor Is Modulated by Phosphorylation of the C-Terminal Tail Domain. Mol. Pharmacol 88, 816–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Doll C et al. (2012) Deciphering μ-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br. J. Pharmacol 167, 1259–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Groer CE et al. (2011) Agonist-directed interactions with specific β-arrestins determine μ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem 286, 31731–31741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McPherson J et al. (2010) μ-opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol. Pharmacol 78, 756–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gondin AB et al. (2019) GRK Mediates μ-Opioid Receptor Plasma Membrane Reorganization. Front. Mol. Neurosci 12, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bowman SL et al. (2015) Cell-autonomous regulation of mu-opioid receptor recycling by substance P. Cell Rep 10, 1925–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Halls ML et al. (2016) Plasma membrane localization of the mu-opioid receptor controls spatiotemporal signaling. Sci. Signal 9, ra16. [DOI] [PubMed] [Google Scholar]
- 103.Mousa SA et al. (2016) Protein kinase C-mediated mu-opioid receptor phosphorylation and desensitization in rats, and its prevention during early diabetes. Pain 157, 910–921 [DOI] [PubMed] [Google Scholar]
- 104.Levitt ES and Williams JT (2012) Morphine Desensitization and Cellular Tolerance Are Distinguished in Rat Locus Ceruleus Neurons. Mol. Pharmacol 82, 983–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arttamangkul S et al. (2015) Does PKC activation increase the homologous desensitization of μ opioid receptors? Br. J. Pharmacol 172, 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chu J et al. (2010) Agonist-dependent μ-opioid receptor signaling can lead to heterologous desensitization. Cell. Signal 22, 684–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pena DA et al. (2018) Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA. Proteomes 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deiss K et al. (2012) Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein-coupled receptor kinase (GRK) 2. J. Biol. Chem 287, 23407–23417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brackley AD et al. (2016) GRK2 Constitutively Governs Peripheral Delta Opioid Receptor Activity. Cell Rep 16, 2686–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grecksch G et al. (2011) Analgesic tolerance to high-efficacy agonists but not to morphine is diminished in phosphorylation-deficient S375A μ-opioid receptor knock-in mice. J. Neurosci 31, 13890–13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang XF et al. (2016) T394A mutation at the µ opioid receptor blocks opioid tolerance and increases vulnerability to heroin self-administration in mice. J. Neurosci 36, 10392–10403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gabra BH et al. (2007) Evidence for an important role of protein phosphatases in the mechanism of morphine tolerance. Brain Res 1159, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Terman GW et al. (2004) G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br. J. Pharmacol 141, 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bohn LM et al. (2002) Differential mechanisms of morphine antinociceptive tolerance revealed in βarrestin-2 knock-out mice. J. Neurosci 22, 10494–10500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hull LC et al. (2010) The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by μ-opioid agonists of different efficacy. J. Pharmacol. Exp. Ther 332, 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mann A et al. (2019) Agonist-selective NOP receptor phosphorylation correlates in vitro and in vivo and reveals differential post-activation signaling by chemically diverse agonists. Sci. Signal 12, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kouhen OM El et al. (2000) Hierarchical phosphorylation of δδ-opioid receptor regulates agonist-induced receptor desensitization and internalization. J. Biol. Chem 275, 36659–36664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.DiCello JJ et al. (2019) Clathrin and GRK2/3 inhibitors block delta opioid receptor internalization in myenteric neurons and inhibit neuromuscular transmission in the mouse colon. Am. J. Physiol. Liver Physiol 10.1152/ajpgi.00085.2019 [DOI] [PubMed]
- 119.Chen C et al. (2016) Determination of sites of U50,488H-promoted phosphorylation of the mouse κ opioid receptor (KOPR): disconnect between KOPR phosphorylation and internalization. Biochem. J 473, 497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qiu Y et al. (2007) Phosphorylation of the δ-opioid receptor regulates its β-arrestins selectivity and subsequent receptor internalization and adenylyl cyclase desensitization. J. Biol. Chem 282, 22315–22323 [DOI] [PubMed] [Google Scholar]
- 121.McLaughlin JP et al. (2004) Prolonged Kappa Opioid Receptor Phosphorylation Mediated by G-protein Receptor Kinase Underlies Sustained Analgesic Tolerance. J. Biol. Chem 279, 1810–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo J et al. (2000) Identification of G protein-coupled receptor kinase 2 phosphorylation sites responsible for agonist-stimulated δ-opioid receptor phosphorylation. Mol. Pharmacol 58, 1050–1056 [DOI] [PubMed] [Google Scholar]
- 123.Xiang B et al. (2001) Heterologous Activation of Protein Kinase C Stimulates Phosphorylation of δ-Opioid Receptor at Serine 344, Resulting in β-Arrestin- and Clathrin-mediated Receptor Internalization. J. Biol. Chem 276, 4709–4716 [DOI] [PubMed] [Google Scholar]
- 124.Chiu YT et al. (2017) Agonist-dependent and-independent k opioid receptor phosphorylation: Distinct phosphorylation patterns and different cellular outcomes. Mol. Pharmacol 92, 588–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chan ASL and Wong YH (2000) Regulation of c-Jun N-terminal Kinase by the ORL1 receptor through multiple G proteins. J. Pharmacol. Exp. Ther 295, 1094–1100 [PubMed] [Google Scholar]
- 126.Lou LG et al. (1997) Nociceptin/Orphanin FQ activates protein kinase C, and this effect is mediated through phospholipase C/Ca2+ pathway. Biochem. Biophys. Res. Commun 240, 304–308 [DOI] [PubMed] [Google Scholar]
- 127.Zhang Z et al. (1999) Endogenous δ-opioid and ORL1 receptors couple to phosphorylation and activation of p38 MAPK in NG108–15 cells and this is regulated by protein kinase A and protein kinase C. J. Neurochem 73, 1502–1509 [DOI] [PubMed] [Google Scholar]
- 128.Zhang NR et al. (2012) Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization, and arrestin signaling. J. Biol. Chem 287, 42019–42030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hershko A and Ciechanover A (1998) The ubiquitin system. Annu. Rev. Biochem 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 130.Jean-Charles PY et al. (2016) Chapter One - Ubiquitination and Deubiquitination of G Protein-Coupled Receptors, 141 Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 131.Kennedy JE and Marchese A (2015) Regulation of GPCR trafficking by ubiquitin. Prog. Mol. Biol. Transl. Sci 132, 15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Skieterska K et al. (2017) Regulation of G protein-coupled receptors by ubiquitination. Int. J. Mol. Sci 18, 10–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wojcikiewicz RJH (2004) Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol. Sci 25, 35–41 [DOI] [PubMed] [Google Scholar]
- 134.Canals M et al. (2012) Ubiquitination of CXCR7 controls receptor trafficking. PLoS One 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Milojević T et al. (2006) The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2a receptor. Mol. Pharmacol 69, 1083–1094 [DOI] [PubMed] [Google Scholar]
- 136.Ikeda F and Dikic I (2008) Atypical ubiquitin chains: New molecular signals. “Protein Modifications: Beyond the Usual Suspects” Review Series. EMBO Rep 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen ZJ and Sun LJ (2009) Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol. Cell 33, 275–286 [DOI] [PubMed] [Google Scholar]
- 138.Chaturvedi K et al. (2001) Proteasome Involvement in Agonist-induced Down-regulation of μ and δ Opioid Receptors. J. Biol. Chem 276, 12345–12355 [DOI] [PubMed] [Google Scholar]
- 139.Petäjä-Repo UE et al. (2001) Newly Synthesized Human δ Opioid Receptors Retained in the Endoplasmic Reticulum Are Retrotranslocated to the Cytosol, Deglycosylated, Ubiquitinated, and Degraded by the Proteasome. J. Biol. Chem 276, 4416–4423 [DOI] [PubMed] [Google Scholar]
- 140.Hislop JN et al. (2011) Ubiquitination in the first cytoplasmic loop of μ-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J. Biol. Chem 286, 40193–40204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Henry AG et al. (2012) Regulation of Endocytic Clathrin Dynamics by Cargo Ubiquitination. Dev. Cell 23, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Petäjä-Repo UE et al. (2000) Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human δ opioid receptor. J. Biol. Chem 275, 13727–13736 [DOI] [PubMed] [Google Scholar]
- 143.He SQ et al. (2011) Facilitation of μ-Opioid Receptor Activity by Preventing δ-Opioid Receptor-Mediated Codegradation. Neuron 69, 120–131 [DOI] [PubMed] [Google Scholar]
- 144.Li JG et al. (2008) Agonist-promoted Lys63-linked polyubiquitination of the human κ-opioid receptor is involved in receptor down-regulation. Mol. Pharmacol 73, 1319–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li JG et al. (2000) Mechanisms of agonist-induced down-regulation of the human κ-opioid receptor: Internalization is required for down-regulation. Mol. Pharmacol 58, 795–801 [PubMed] [Google Scholar]
- 146.Decaillot FM et al. (2008) Cell surface targeting of - opioid receptor heterodimers by RTP4. Proc. Natl. Acad. Sci 105, 16045–16050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Canals M (2015) The Complex Roles of -Opioid Receptor Phosphorylation: A Key Determinant in Receptor Signaling and Regulation. Mol. Pharmacol 88, 814–815 [DOI] [PubMed] [Google Scholar]
- 148.Wang Q et al. (2009) Differential modulation of μ-and δ-opioid receptor agonists by endogenous RGS4 protein in SH-SY5Y cells. J. Biol. Chem 284, 18357–18367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rodríguez-Muñoz M et al. (2007) RGS14 prevents morphine from internalizing Mu-opioid receptors in periaqueductal gray neurons. Cell. Signal 19, 2558–2571 [DOI] [PubMed] [Google Scholar]
- 150.Kenakin T (2019) Biased receptor signaling in drug discovery. Pharmacol. Rev 71, 267–315 [DOI] [PubMed] [Google Scholar]
- 151.Xie WY et al. (2009) Disruption of Cdk5-associated phosphorylation of residue threonine-161 of the δ-opioid receptor: Impaired receptor function and attenuated morphine antinociceptive tolerance. J. Neurosci 29, 3551–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fujita W et al. (2019) Regulation of _an opioid receptor chaperone protein, RTP4, by morphine S. Mol. Pharmacol 95, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schmid CL et al. (2017) Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 171, 1165.e13–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bohn LM et al. (2000) Μ-Opioid Receptor Desensitization By Β-Arrestin-2 Determines Morphine Tolerance But Not Dependence. Nature 408, 720–723 [DOI] [PubMed] [Google Scholar]