

Abstract
Influenza is one of the best examples of highly mutable viruses that are able to escape immune surveillance. Indeed, in response to influenza seasonal infection or vaccination, the majority of the induced antibodies are strain‐specific. Current vaccine against the seasonal strains with the strategy of surveillance‐prediction‐vaccine does not cover an unmet virus strain leading to pandemic. Recently, antibodies targeting conserved epitopes on the hemagglutinin (HA) protein have been identified, albeit rarely, and they often showed broad protection. These antibody discoveries have brought the feasibility to develop a universal vaccine. Most of these antibodies bind the HA stem domain and accumulate in the memory B cell compartment. Broadly reactive stem‐biased memory responses were induced by infection with antigenically divergent influenza strains and were able to eradicate these viruses, together indicating the importance of generating memory B cells expressing high‐quality anti‐stem antibodies. Here, we emphasize recent progress in our understanding of how such memory B cells can be generated and discuss how these advances may be relevant to the quest for a universal influenza vaccine.
Keywords: germinal center B cells, HA stem, immunodominant, TFH cell, universal
1. INTRODUCTION
Each year approximately one billion cases of influenza virus infection are reported, of which 3‐5 million are severe cases and 290‐650 thousand lead to deaths. 1 Influenza virus infection is also a global socioeconomic issue. 2
Influenza vaccine development was launched immediately after Smith et al 3 isolated influenza A virus in 1933, and the vaccines were tested 4 , 5 during the 1930s and 1940s.
A unique feature of this virus is its diversity. Influenza viruses are mainly divided into two types, A and B. The more virulent type A can be further classified into 18 different hemagglutinin (HA) subtypes and 11 different neuraminidase subtypes, theoretically 198 combinations (in fact 131 subtypes have been detected in nature). Type A influenza HA proteins are divided into two groups, group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18) and group 2 (H3, H4, H7, H10, H14, and H15), based on phylogenetic similarity (Figure 1). Not only that, what also makes it difficult to generate an effective vaccine is the fact that influenza viruses are constantly changing by “drift and shift.” As a result, the Global Influenza Surveillance and Response System (GISRS) was launched in 1952 and now includes over 150 institutions in more than 110 countries. The GISRS monitors the circulating influenza viruses year around and recommends the virus types, subtypes, lineages, and strains for the seasonal flu vaccine once a year. However, for making an annual vaccine, determining and targeting a specific virus strain, which is continuously changing, are not so easy. Although the vaccine effectiveness can differ from one subtype to the other and year to year, overall it ranges from 10% to 60% with an average of 40% between 2004 and 2019 in the United States (Past Seasons Vaccine Effectiveness Estimates, https://www.cdc.gov/flu/vaccines‐work/past‐seasons‐estimates.html). Note that these seasonal vaccines will provide little or no protection against unpredictable pandemic influenza viruses such as H5N1 and H7N9.
FIGURE 1.
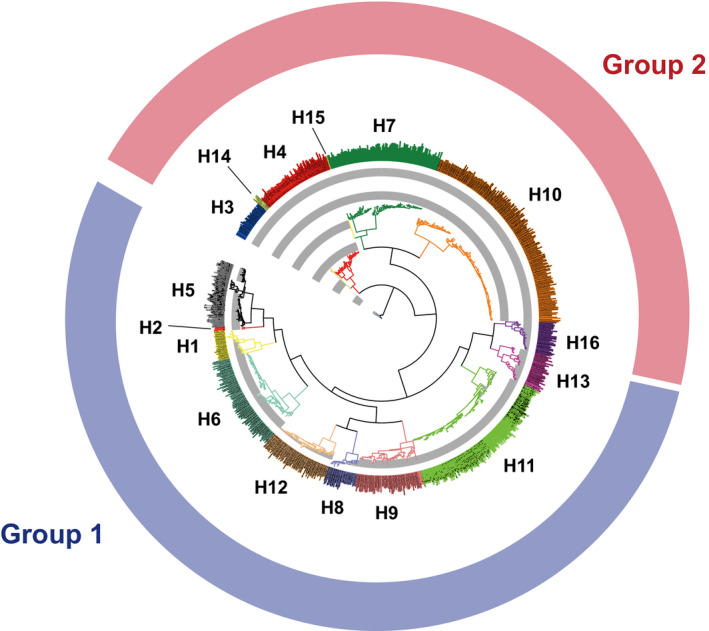
Phylogenetic tree of major influenza A virus HA subtypes. Sixteen HA subtypes are separated into group 1 (blue) and group 2 (red) based on phylogenetic relatedness. Nine hundred seventy‐five segments of the HA sequences were aligned, and the phylogenetic tree was generated using Influenza Research Database website 80 followed by iTOL. 81 H17N10 and H18N11 are not listed here
Multiple immune reactions contribute to protection in influenza infection. Among them, antibodies alone can adequately protect from infection, while T cell–mediated reactions seem to play an important role in recovery. 6 , 7 Influenza vaccines induce serum antibodies against two major glycoproteins on the surface of the virus, HA and NA. Antibodies against the globular head of HA are detected by the HI (HA inhibition) assay, and their titers boosted by vaccination are generally correlated with protection. 8 Interestingly, Clements et al 9 reported that serum titers detected by the NA inhibition assay are correlated with protection against virus challenge or illness after use of the inactivated vaccine. To date, the influenza vaccine is designed to target the HA protein.
The ultimate goal is to develop a universal vaccine against both a broad range of seasonal influenza virus strains and pandemic strains. In this regard, in 1993, Okuno et al 10 first isolated a broadly neutralizing antibody (bnAb) against group 1 influenza viruses. This finding demonstrated the feasibility of developing a universal vaccine. This mAb, called C179, recognizes a unique conformation formed by the HA1 and HA2 domains of the stem region of HA, which is evolutionally well‐conserved among many strains 11 (Figure 2). Since 2008, many groups have succeeded in isolating human monoclonal antibodies able to neutralize a broad spectrum of influenza virus strains. 12 Among them, some bnAbs even recognize both group 1 and group 2 viruses.
FIGURE 2.
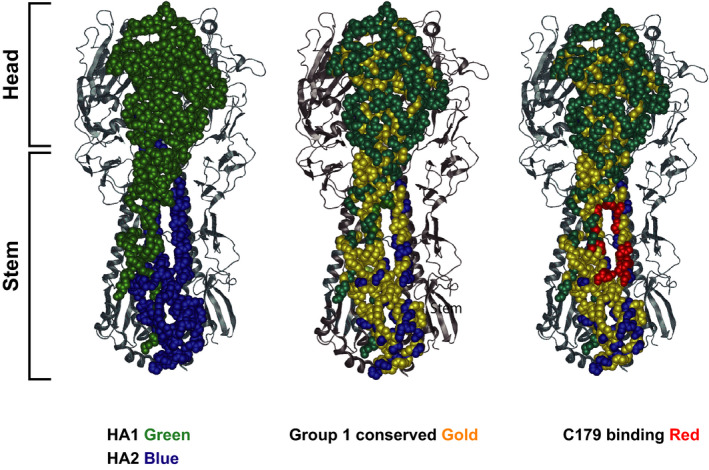
Hemagglutinin head and stem and their conserved regions. Structure view of trimerized hemagglutinin proteins of influenza virus. A homotrimer shown as a sphere model. HA1 and HA2 are colored in green and blue, respectively. The other polypeptides of HAs are depicted as a ribbon model. Conserved amino acids among group 1 influenza viruses are colored in gold overlaid on the model on the left. Amino acids of the C179 binding region are colored in red. These views were generated by Protean 3D using the structure PDB 4HLZ
Despite molecular identification for broadly reactive antibodies as mentioned above, it has not been clear which type of cells are majorly responsible for protection against re‐infection of variant influenza viruses in vivo. In this regard, we summarize recent progress to show the importance of memory B cells. Then, we discuss the potential ways to induce memory B cells carrying broadly reactive antibodies in breadth and affinity.
2. ROLES OF MEMORY B CELLS
2.1. Viral strategies for evading immunity
Influenza viruses acquire seasonal mutations, which results in the evolution of viral envelope proteins (primary targets of antibody‐mediated protection) comprising variable regions (head region) and conserved regions (stem region) (Figure 2); the stem region is essential for virus survival.
In order to protect the conserved domains from antibody surveillance, viruses are thought to employ at least two strategies. First, the conserved domains are weakly immunogenic or immunologically subdominant in contrast to the immunologically dominant head domains, 13 for instance, because the precursor frequency of B cells with germline ancestor antibodies recognizing the conserved regions is inherently low in humans 14 , 15 , 16 and mice (unpublished data). Even so, there are still significant numbers of such B cells; however, the conserved regions also provide steric hindrance to prevent antibody access. 17 Second, the structural mimicry of host self‐antigens by epitopes within the conserved domains moderates immunogenicity through immunological tolerance, thereby mitigating the generation of broadly reactive antibodies toward the conserved epitopes. Indeed, previous studies revealed that the majority of unmutated germline ancestors of broadly reactive antibodies against HIV 18 or influenza viruses 17 , 19 are autoreactive or polyreactive. Hence, as discussed later, targeting to block these virus survival strategies would be one possible approach when considering vaccine development.
2.2. Evidence for the importance of memory B cells in re‐infection by variant viruses
Among the two humoral memory compartments derived from GC, long‐lived plasma cells (LLPCs) and memory B cells, LLPCs secrete antibodies with the highest affinities for the primary infection virus; therefore, they are mostly directed toward the dominant HA head epitopes with the strain bias. On the other hand, memory B cells have been speculated to express protective antibodies recognizing subdominant stem epitopes of drifted virus (Figure 3A). This idea was initially based on studies of the antibody response in humans to vaccination with the pandemic 2009 H1N1 influenza vaccine, which showed that individuals who had lower levels of pre‐existing antibodies to this pandemic type virus generated broadly reactive anti‐stem memory responses. Moreover, these antibodies of plasmablasts derived from several branches of the clonotypes’ phylogeny over two consecutive years had undergone somatic hypermutation (SHM). 17 These observations suggested that those plasmablasts were activated from memory B cells.
FIGURE 3.
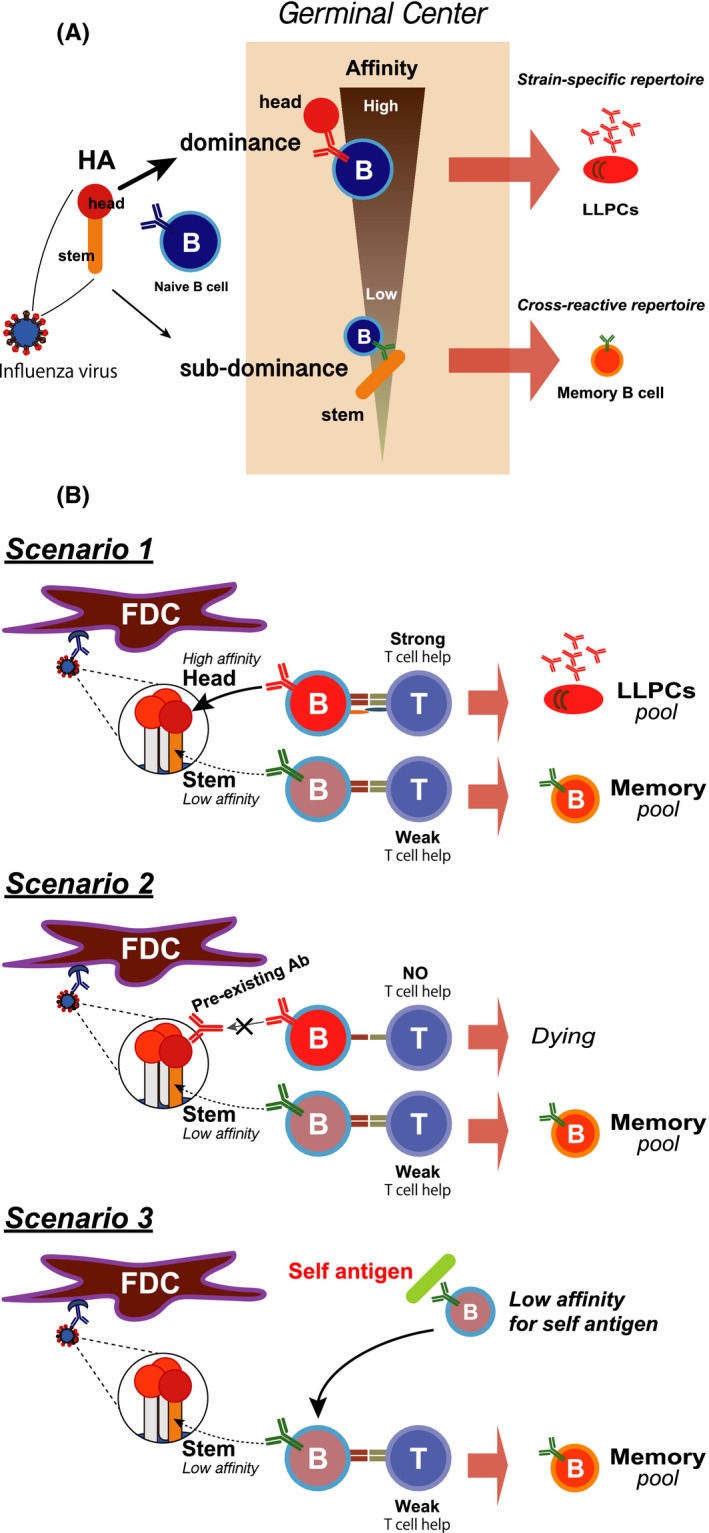
Two humoral memory compartments, LLPCs and memory B cells. A, Immunodominant head‐specific LLPCs and sub‐immunodominant stem‐specific memory B cells. The head region of HA contains epitopes that are immunodominant during infection or vaccination, while the stem region is subdominant. Antibodies against the head region have relatively higher affinity than those against the stem region and are usually strain‐specific. The immunodominant head region‐specific B cells differentiate into LLPCs. In contrast, the subdominant stem region‐specific B cells become memory B cells that are potentially cross‐reactive with other virus strains. B, Three mechanisms of generating stem‐specific memory B cells. As described in Figure 3A, stem‐specific B cells enter the memory B cell pool (Scenario 1). Once individuals are infected or vaccinated, antibodies against the head region are dominantly induced and continue to be produced. Those pre‐existing head‐specific antibodies mask the HA head epitope in a subsequent challenge so that unmasked stem‐specific B cells are able to respond and become memory cells (Scenario 2). Since several HIV and influenza bnAbs likely bind to self‐antigens, those Abs are believed to be derived from a B cell repertoire with low affinity for self‐antigens. Eventually, those B cells undergo SHM that triggers an override to recognize the stem region of HA. Those activated B cells can enter the memory pool (Scenario 3)
Supporting the collateral roles of memory B cells against drifted viruses, Purtha et al showed that mouse memory B cell responses to West Nile virus retain the ability to engage variant viruses, whereas antibody derived from LLPCs is poorly able to inhibit the variant viruses. More recently, mouse and human studies have further strengthened this concept. In a mouse model infection (Narita influenza infection followed by re‐infection with Narita or PR8 virus), pre‐existing antibodies secreted by LLPCs protected from the homologous challenge (Narita re‐infection), whereas protection from heterologous challenge (PR8 re‐infection) required memory B cell activation. These memory B cells were mostly GC‐experienced and primarily directed toward the conserved HA stem regions. 20 In addition, human longitudinal studies of Dengue and Zika virus responses showed that memory B cell responses are more diverse (cross‐reactive with both Dengue and Zika) than those of LLPC‐derived antibodies. 21
Further question will be whether influenza HA stem‐specific memory B cells can differentiate into LLPCs and secrete anti‐stem antibodies for long time? Miller et al 22 performed a longitudinal study of serum antibody titers against HA stem. The titer increases over 20‐year period. Further Nachbagauer also reported that the elderly had the highest antibody titers against the stem. 23 However, immunodominance of specific region of the HA such as head globular domain might interfere the anti‐stem humoral responses upon conventional vaccination 17 as we discuss later.
2.3. Potential mechanisms by which broadly reactive GC B cells enter the memory B cell compartment
Then, in the case of influenza, how do broadly reactive GC B cells enter the memory B cell pool, rather than the LLPC pool? While many previous GC studies have focused on understanding how the B cells undergo maturation toward the production of higher‐affinity antibodies, 24 , 25 there has been an increased realization that the GC responses simultaneously support the development of a diverse population of antigen‐specific B cells. Excellent reviews have been published describing recent progress in GC biology, 26 , 27 which are highly recommended to the reader.
Considering these recent advances in GC biology, at least three non‐mutually exclusive possible mechanisms for the generation of anti‐stem antibodies can be envisaged (Figure 3B). First, since almost 100 clones are known to be able to enter the GC response in an individual GC, 28 anti‐stem B cell clones can have a chance to get into a GC. Because inter‐clonal selection seems to be not so stringent during the early GC period, anti‐stem GC cells could be maturated to some extent during the early time point. During GC reactions, memory B cells with not so high‐affinity antibodies are preferentially generated at early time points, 29 , 30 and these anti‐stem GC cells are allowed to enter the memory pool during this time frame. According to this temporal model, at a later time point these anti‐stem clones are outcompeted by immunodominant anti‐head clones. Second, as another type of regulation, GC diversity may be promoted by antibody‐mediated feedback, as dominant GC clones bearing antibodies specific for particular epitopes, for instance in the head region, give rise to plasma cells that secrete antibody that masks these epitopes, thereby enhancing the selection of clones that bind other epitopes, for instance in an originally subdominant stem region. In this case, GC clones with anti‐stem antibodies would not necessarily come up only at the early phase. Finally, as mentioned above, many of the unmutated germline ancestors of anti‐stem antibodies are autoreactive. In this regard, studies of Sabouri et al have important implications. 31 They showed that autoreactive GC B cells first acquire mutations that decrease their affinity for both the self‐antigen and foreign antigen. This state may be positively selected during GC reactions, because the large decrease in receptor occupancy by self‐antigen reciprocally permits increased uptake of the foreign antigen, thereby obtaining increased T cell help. Then, these GC cells might have a better chance to be recruited into the memory B cell pool.
2.4. Re‐diversification of pre‐existing memory B cells
In the case of HIV vaccination, iterative entry of memory B cells into GC reactions is thought to be required for the generation of more broadly neutralizing antibodies to various HIV variants. Indeed, the known antibodies with this capability have a remarkably large number of mutations generated by SHM. Hence, it will be important to know whether conventional influenza vaccination induces reentry of pre‐existing, particularly broadly reactive, memory B cells into GCs and, if not, to understand why this does not take place.
To this end, two human studies analyzed B cell repertoires upon conventional influenza vaccination. Since they could not directly analyze GC reactions, these studies had to employ blood samples. Lau et al 32 sorted CD20loCD38hiCD27+ plasmablasts at day 7 and CD27+CD21lo B cells and classical CD27+CD21hi memory B cells at day 14. (CD21lo cells are thought to be another subset of memory B cells, being more prone to differentiate into plasmablasts upon antigen stimulation.) Ellebedy et al 33 sorted HA‐specific plasmablasts at day 7 and HA‐specific CD20+ memory B cells at day 14 (a mixture of CD21lo and CD21hi memory B cells). Both studies demonstrated some clonal overlap (~30%) between plasmablasts and memory B cells, indicating that an individual B cell clone can generate both plasmablasts and memory B cells. The clones found in day 7 HA‐specific plasmablasts and day 14 HA‐specific CD20+ memory B cells were present in the pre‐existing memory B cell population, indicating that pre‐existing memory B cells were indeed activated. Importantly, both studies showed no overall increase in SHM over time in these de novo generated plasmablasts and memory B cells after conventional vaccination.
Differing from the above two human studies analyzing whole anti‐HA B cells, Andrews et al traced specifically anti‐stem human memory B cells using H7N9 monovalent inactivated vaccine for H7N9‐naive adults. 34 Upon vaccination, pre‐existing memory B cells to the stem region expanded, but these expanded B cells underwent little additional SHM. In contrast, naive or freshly generated memory B cells specific for H7N9 head epitopes evolved and maturated over several months. Collectively, these three human studies indicate that, upon vaccination with the current influenza vaccine, pre‐existing memory B cells, particularly anti‐stem memory B cells, can be activated, but it is difficult for them to enter the GC and undergo further diversification.
A mouse study has reached the same conclusion and provided more mechanistic insights into why pre‐existing memory B cells cannot be recruited into GCs. Their experiments used a prime‐boost mouse model (influenza PR8 infection followed by vaccination with homologous PR8 HA proteins). Upon secondary vaccination, recall GCs were mostly derived from new naive B cells, but not from pre‐existing memory B cells. 35
So, why then do pre‐existing memory B cells, upon conventional vaccination, behave in a seemingly non‐beneficial way for us (Figure 4)? First, previous transfer experiments demonstrate that memory B cells defined by surface IgG expression or by markers of more mature memory phenotype are prone to differentiate into plasmablasts, 36 rather than entering into GCs. Such types of memory B cells prone to plasmablasts might be preferentially activated, probably because of their higher affinity for the secondary antigen. Second, it is generally thought that memory B cells have higher affinity and are present at higher precursor frequencies than naive B cells specific for the same antigen. However, given that several different memory subsets coexist, only one of which has the propensity to enter GCs, the actual number of generated memory B cells capable of entering the GC might be small. In contrast, the number of naive B cells with sufficient affinity to enter a primary GC reaction is large. Hence, the precursor frequency of memory B cells capable of reentering GCs might be very small. Finally, in contrast to transfer experiments into naive mice, in non‐transfer conditions, negative feedback by pre‐existing and/or de novo generated antibodies might operate, which could limit GC entry of clones specific for epitopes targeted by these antibodies. It will be important to elucidate which mechanisms are predominantly responsible for the inability of memory B cells to reenter the GC.
FIGURE 4.
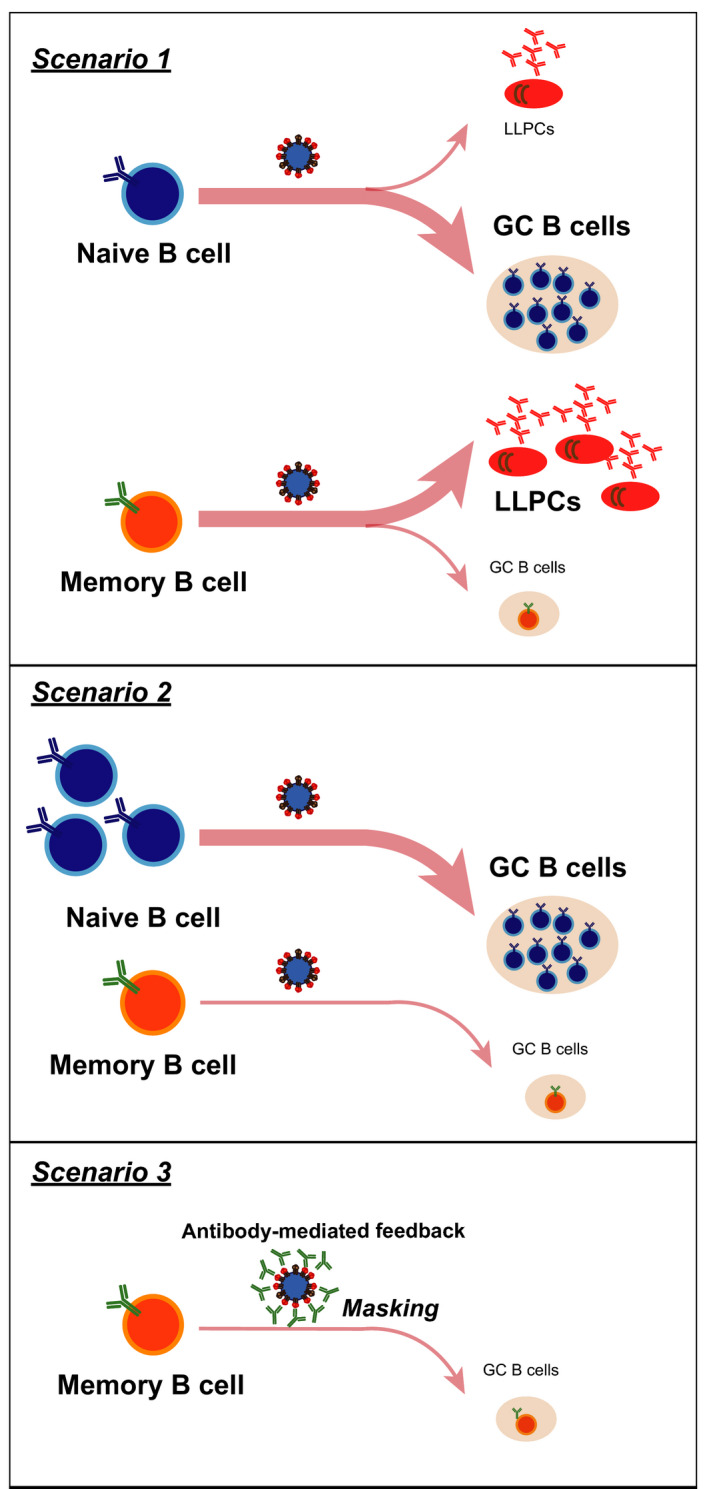
Mechanisms by which pre‐existing memory B cells are at a disadvantage for GC entry. (Scenario 1) In transfer experiments, memory B cells are prone to become plasmablasts without entering the GC. (Scenario 2) Naive B cells have sufficient affinity, and the majority of them are licensed to enter the GC. Although memory B cells are believed to have high affinity and should be licensed to enter the GC, in fact, only a small subset of memory B cells has the capacity to enter the GC. (Scenario 3) Pre‐existing antibodies can mask epitopes that are supposed to guide memory B cells to enter the GC so that the number of memory B cells able to enter the GC is limited
3. DESIGN OF UNIVERSAL VACCINES INDUCING ANTI‐STEM ANTIBODIES
It is likely that an effective vaccination approach for influenza will depend on several rounds of vaccination and the generation of memory B cells with good quality of anti‐stem antibodies at each round that can further reenter the GC. So, the important factors are as follows: (a) Which kind of antibodies these memory B cells should have; (b) which immunogen design should be used; and (c) how we can recruit memory B cells into GCs.
3.1. Which kind of antibodies
Do antibodies induced by vaccination require neutralization activity? Antibodies raised by the seasonal vaccine are directed to the globular head of HA and block virus entry into host cells. The level of HA inhibition activity reflecting the blocking level of virus entry is well‐correlated with neutralization activity and in vivo protection. In the mouse model, Fc receptor–mediated antibody‐dependent cellular cytotoxicity (ADCC) plays an important role in in vivo protection through 37 (Figure 5). Adding the neutralizing activity to the ADCC activity will certainly enhance protection in vivo. However, to what degree ADCC contributes to in vivo protection over neutralization activity in humans has not yet been determined. Another interesting question is whether the way an antibody recognizes antigen is also important to its ADCC function. Ravetch's group reported that bnAb targeting to the HA stem also carry out ADCC. 38 , 39 Inducing these dual functioned antibodies by vaccination ideally would maximize the efficacy of a universal vaccine.
FIGURE 5.
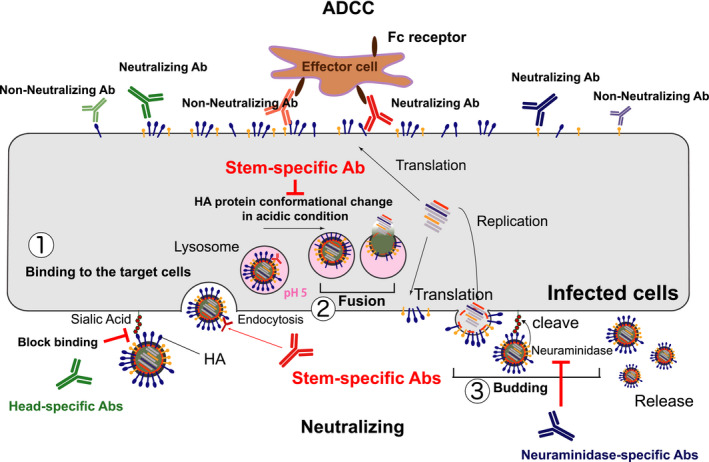
Two major functions of antibodies for protection. Antibodies directly target viruses and prevent their entry into the host cell by blocking the virus‐host cell interaction (1) or membrane fusion (2). Recent studies also demonstrated that some antibodies can inhibit the virus neuraminidase activity (3) critical for virus budding. 82 In addition, after virus infection, viral envelope proteins expressed on the infected cell surface are opsonized by antibodies. Effector cells, for example, NK cells, recognize these antibody‐opsonized infected cells via their Fc receptors and then eliminate the infected cells by antibody‐dependent cellular cytotoxicity
Several structural studies of the protein complex of HA and bnAbs have been performed. Surprisingly, these antibodies, including FI6v3, 40 CR9114, 41 CR6261, 42 CR8020, 43 and S9‐3‐37 44 from humans and C179 10 from mouse, recognize a common structural epitope of the hydrophobic groove formed by the HA1 and HA2 domains. These findings clearly demonstrate that the area of the epitope with broadly neutralizing activity is very narrow.
3.2. Which immunogen should be used?
How can we induce activation of B cells harboring stem‐specific antibodies? In the past, several attempts were made to develop a universal vaccine. In 1996, Sagawa et al 45 first expressed a headless HA (A/Okuda/57 (H2N2)) on CV‐1 cells and used them to immunize mice. They could show limited cross‐protection against influenza viruses A/FM/1/47 (H1N1). In 2010, Bommakanti et al 46 designed an HA2‐based immunogen and showed cross‐strain protection but not the cross‐subtypic protection. In 2013, Palese's group generated a series of recombinant chimeric HAs with variable globular head domains and stem domains. 47 Sequential immunization with these chimeric HAs predominantly induced production of stem‐specific antibodies. The strategies described above did demonstrate cross‐protection from virus challenges but not cross‐neutralization. In 2015, protein structure–based design and screenings provided two headless HA recombinant proteins that could induce cross‐protection. 48 , 49 One of them even showed the induction of broadly neutralizing antibodies using the pre‐clinical adjuvant matrix M. 49 These structure studies clearly showed the importance of conformation of the HA to fully induce broadly neutralizing and protective antibodies. Surprisingly, many of these stem‐targeted bnAbs shared a common conformational epitope with C179. 11 Targeting this small region of the conformational epitope is a challenge for designing a stable vaccine antigen.
For immunogen design, two concerns exist. First, choosing an antigen epitope enabling induction of autoantibody had better be avoided. There exists a trade‐off between autoreactivity and breadth of the cross‐reactive antibody. Various BCR transgenic mouse studies of bnAbs against HIV demonstrated that bnAbs are autoreactive and designated to be inactivated by central tolerance. 50 , 51 , 52 , 53 , 54 , 55 In fact, Bajic et al 19 showed biochemically that 6 influenza bnAbs are poly‐ and self‐reactive. Our recent investigation of one bnAb revealed that the bnAb‐bearing B cell is self‐reactive and targeted for receptor editing in vivo (unpublished results). Given these observations, we need to design an antigen able to induce antibodies that broadly neutralize virus entry and at the same time cannot be a target for tolerance. In the case of HIV, the b12 antibody seems to be in this category. 54
Second, as mentioned above in the mouse prime‐boost model, only a minority of memory‐derived clones reenter the GC, demonstrating that the constriction of the relatively divergent primary memory B cell repertoire takes place by boosting. 35 Therefore, in order to be efficiently targeted by sequential vaccination, an epitope should be rendered dominant from the beginning. Thus, efforts to target epitopes of interest by engineering high‐affinity germline antibody‐targeting immunogens 56 or occluding normally immunodominant regions 57 are needed.
3.3. How do memory B cells enter secondary GCs?
As discussed above, at least three factors can be considered to block reentry of pre‐existing memory B cells into GCs: (a) Among several coexisting distinct subsets of memory B cells, there is preferential activation of plasmablast‐poised memory B cell subsets; (b) the small frequency of memory B cells; and (c) negative feedback by antigen‐specific antibodies. In regard to the first issue, skewing to generating more GC‐poised memory B cells seems to be difficult at present, because of the factors that control the formation of each memory B cell subset which will be an important challenge in this field now. For the second point, it is better to use only the target epitope of interest (occluding normally immunodominant regions) from the outset, thereby increasing the frequency of memory B cells bearing antibodies to the epitope of interest. In regard to the third possibility, immunogens had better be specifically designed to escape antibody feedback by B cell clones already represented in the serum. For instance, we would design an antigen consisting of either a “no man(antibody)’s land” epitope in the HA protein or a de novo synthetic antigen proficient for vaccine efficacy.
Apart from manipulating memory B cells, fueling TFH (T follicular helper) cells in GC in vaccine development is also important. TFH cells control antigen‐specific GC responses quantitatively and qualitatively. Cognitive interaction between TFH cells and B cells through TCR and MHCII in GC facilitates proliferation of antigen‐specific B cells and introduction of SHMs in B cells for affinity maturation. 58 Several human studies showed that the numbers of peripheral memory TFH cells are correlated with the amount of HIV bnAbs. 59 , 60 Furthermore, an experiment using the macaque animal model demonstrated that GC TFH cells are associated with bnAbs. 61 Antigen‐specific TFH cells are required for GC expansion, and several adjuvants are reported to be able to expand TFH cells. These include squalene‐based adjuvants such as MF59, 62 , 63 Alum + TLR7 ligand, 63 and other TLR ligands, 64 , 65 , 66 , 67 , 68 , 69 , 70 and glucopyranosyl lipid adjuvant‐stable emulsion, GLA‐SE. 71 Hence, adjuvants that facilitate TFH formation could contribute to enhancing GC formation and maintenance, which in turn could facilitate recruiting memory B cells into recall GCs. In this context, recent observations that IL‐12 receptor signaling and T‐bet in T cells are required for TFH suppression might have important implications. 72 If mechanisms can be identified that suppress Th1 immunity, they might enhance GC reactions through facilitating TFH cell differentiation.
Recently, we demonstrated that active vitamin D3 also serves as an adjuvant by expanding TFH cells, leading to GC expansion and enhanced humoral immune responses. By being repurposed, application of the psoriasis drug Oxarol (active vitamin D3 analog) ointment on the skin after intradermal injection of influenza split vaccine antigen protected from influenza virus infection. 73
4. CURRENT STATUS AND PERSPECTIVES
A reasonable goal for the development of a universal influenza vaccine has been proposed: “A vaccine with ≥75% protection against symptomatic disease caused by group 1 and group 2 influenza A viruses lasting ≥12 months in all populations”. 74 Currently, several universal vaccines are in clinical trials. 75 , 76 The leading candidate is M‐001: a single polypeptide consisting of three repetitions of 9 linear, conserved peptides from influenza A and influenza B. These peptides are derived from the M1 matrix protein, the nucleoprotein, and the conserved regions of the HA. 77 The other vaccines in clinical trials are MVA‐NP + M1, an adenovirus vector expressing NP and M1 from influenza A, Flu‐V, four conserved peptides from NP, M1, and M2 to induce T cell responses, 78 and OXV83, a recombinant NP in a virus‐like particle. 79 These vaccines mainly induce cellular immunity by T cells or ADCC. However, they are not optimal for protecting from the infection; more likely, these vaccines would prevent death. Vaccines inducing humoral responses are ideal to protect from virus infection. H1ssF_3928, composed of an HA stem fused with ferritin nanoparticles, 48 is in this category and is under clinical trial. Other recent preclinical vaccines seem to target humoral responses.
Current progress in the research field of humoral memory responses based on cellular dynamics provides us with new insights. These insights will facilitate development of the ideal universal influenza vaccine. Different from the trial‐and‐error type of vaccine development in the past, these mechanistically based vaccine design strategies should also be easily applied to other vaccines against SARS‐CoV‐2 or other emerging viruses causing respiratory tract infections.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
The authors acknowledge P. Burrows for critical reading. This work was supported by grants to HF, RS, and TK. from the Ministry of Education, Culture, Sports, Science and Technology in Japan, grants to RS from the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and grants to TK and HF from the Secom Science and Technology Foundation.
Fukuyama H, Shinnakasu R, Kurosaki T. Influenza vaccination strategies targeting the hemagglutinin stem region. Immunol Rev. 2020;296:132–141. 10.1111/imr.12887
This article is part of a series of reviews covering B and Th cell response to Ag in vivo: implications for vaccine development and diseases appearing in Volume 296 of Immunological Reviews
Contributor Information
Hidehiro Fukuyama, Email: hidehiro.fukuyama@riken.jp.
Tomohiro Kurosaki, Email: kurosaki@ifrec.osaka-u.ac.jp.
REFERENCES
- 1. Iuliano AD, Roguski KM, Chang HH, et al. Estimates of global seasonal influenza‐associated respiratory mortality: a modelling study. Lancet. 2018;391(10127):1285‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peasah SK, Azziz‐Baumgartner E, Breese J, Meltzer MI, Widdowson MA. Influenza cost and cost‐effectiveness studies globally–a review. Vaccine. 2013;31(46):5339‐5348. [DOI] [PubMed] [Google Scholar]
- 3. Smith W, Andrewes CH, Laidlaw PP. A virus obtained from influenza patients. Lancet. 1933;2:66‐68. [Google Scholar]
- 4. Stokes J, Chenoweth AD, Waltz AD, Gladen RG, Shaw D. Results of immunization by means of active virus of human influenza 1. J Clin Investig. 1937;16(2):237‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davenport FM. Current knowledge of influenza vaccine. JAMA. 1962;182(1):11‐13. [DOI] [PubMed] [Google Scholar]
- 6. Murphy BR, Clements ML. The systemic and mucosal immune response of humans to influenza A virus. Curr Top Microbiol Immunol. 1989;146:107‐116. [DOI] [PubMed] [Google Scholar]
- 7. Thomas PG, Keating R, Hulse‐Post DJ, Doherty PC. Cell‐mediated protection in influenza infection. Emerg Infect Dis. 2006;12(1):48‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hobson D, Curry RL, Beare AS, Ward‐Gardner A. The role of serum haemagglutination‐inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. Epidemiol Infect. 1972;70(4):767‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild‐type virus. J Clin Microbiol. 1986;24(1):157‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Okuno Y, Isegawa Y, Sasao F, Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol. 1993;67(5):2552‐2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dreyfus C, Ekiert DC, Wilson IA. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J Virol. 2013;87(12):7149‐7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705‐742. [DOI] [PubMed] [Google Scholar]
- 13. Angeletti D, Gibbs JS, Angel M, et al. Defining B cell immunodominance to viruses. Nat Immunol. 2017;18(4):456‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Avnir Y, Tallarico AS, Zhu Q, et al. Molecular signatures of hemagglutinin stem‐directed heterosubtypic human neutralizing antibodies against influenza A viruses. PLoS Pathog. 2014;10(5):e1004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaur K, Sullivan M, Wilson PC. Targeting B cell responses in universal influenza vaccine design. Trends Immunol. 2011;32(11):524‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lingwood D, McTamney PM, Yassine HM, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 2012;489(7417):566‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews SF, Huang Y, Kaur K, et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med. 2015;7(316):316ra192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mouquet H, Scheid JF, Zoller MJ, et al. Polyreactivity increases the apparent affinity of anti‐HIV antibodies by heteroligation. Nature. 2010;467(7315):591‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bajic G, van der Poel CE, Kuraoka M, et al. Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Sci Rep. 2019;9(1):3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leach S, Shinnakasu R, Adachi Y, et al. Requirement for memory B‐cell activation in protection from heterologous influenza virus reinfection. Int Immunol. 2019;31(12):771‐779. [DOI] [PubMed] [Google Scholar]
- 21. Pérez‐Guzmán EX, Pantoja P, Serrano‐Collazo C, et al. Time elapsed between Zika and dengue virus infections affects antibody and T cell responses. Nat Commun. 2019;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller MS, Gardner TJ, Krammer F, et al. Neutralizing antibodies against previously encountered influenza virus strains increase over time: a longitudinal analysis. Sci Transl Med. 2013;5(198):198ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nachbagauer R, Choi A, Izikson R, Cox MM, Palese P, Krammer F. Age Dependence and isotype specificity of influenza virus hemagglutinin stalk‐reactive antibodies in humans. MBio. 2016;7:e01996‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maclennan ICM. Germinal Centers. Ann Rev Immunol. 1994;12(1):117‐139. [DOI] [PubMed] [Google Scholar]
- 25. Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381(6585):751‐758. [DOI] [PubMed] [Google Scholar]
- 26. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity. 2016;45(3):471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol. 2017;45:21‐30. [DOI] [PubMed] [Google Scholar]
- 28. Tas JMJ, Mesin L, Pasqual G, et al. Visualizing antibody affinity maturation in germinal centers. Science. 2016;351(6277):1048‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shinnakasu R, Inoue T, Kometani K, et al. Regulated selection of germinal‐center cells into the memory B cell compartment. Nat Immunol. 2016;17(7):861‐869. [DOI] [PubMed] [Google Scholar]
- 30. Weisel FJ, Zuccarino‐Catania GV, Chikina M, Shlomchik MJ. A Temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44(1):116‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sabouri Z, Schofield P, Horikawa K, et al. Redemption of autoantibodies on anergic B cells by variable‐region glycosylation and mutation away from self‐reactivity. Proc Natl Aca Sci USA. 2014;111(25):E2567‐E2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lau D, Lan LY‐L, Andrews SF, et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci Immunol. 2017;2(7):eaai8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellebedy AH, Jackson KJL, Kissick HT, et al. Defining antigen‐specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol. 2016;17(10):1226‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews SF, Chambers MJ, Schramm CA, et al. Activation dynamics and immunoglobulin evolution of pre‐existing and newly generated human memory B cell responses to influenza hemagglutinin. Immunity. 2019;51(2):398‐410 e395. [DOI] [PubMed] [Google Scholar]
- 35. Mesin L, Schiepers A, Ersching J, et al. Restricted clonality and limited germinal center reentry characterize memory B cell reactivation by boosting. Cell. 2020;180(1):92‐106.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15(3):149‐159. [DOI] [PubMed] [Google Scholar]
- 37. Von Holle TA, Moody MA. Influenza and antibody‐dependent cellular cytotoxicity. Front Immunol. 2019;10:1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dilillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk–specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med. 2014;20(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dilillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti‐influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Investig. 2016;126(2):605‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Corti D, Voss J, Gamblin SJ, et al. A Neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850‐856. [DOI] [PubMed] [Google Scholar]
- 41. Dreyfus C, Laursen NS, Kwaks T, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337(6100):1343‐1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ekiert DC, Bhabha G, Elsliger M‐A, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324(5924):246‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ekiert DC, Friesen RHE, Bhabha G, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333(6044):843‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu NC, Yamayoshi S, Ito M, Uraki R, Kawaoka Y, Wilson IA. Recurring and adaptable binding motifs in broadly neutralizing antibodies to influenza virus are encoded on the D3–9 segment of the Ig gene. Cell Host Microbe. 2018;24(4):569‐578.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sagawa H, Ohshima A, Kato I, Okuno Y, Isegawa Y. The immunological activity of a deletion mutant of influenza virus haemagglutinin lacking the globular region. J Gen Virol. 1996;77:1483‐1487. [DOI] [PubMed] [Google Scholar]
- 46. Bommakanti G, Citron MP, Hepler RW, et al. Design of an HA2‐based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc Natl Acad Sci USA. 2010;107(31):13701‐13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krammer F, Pica N, Hai R, Margine I, Palese P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk‐specific antibodies. J Virol. 2013;87(12):6542‐6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Impagliazzo A, Milder F, Kuipers H, et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science. 2015;349(6254):1301‐1306. [DOI] [PubMed] [Google Scholar]
- 49. Yassine HM, Boyington JC, McTamney PM, et al. Hemagglutinin‐stem nanoparticles generate heterosubtypic influenza protection. Nat Med. 2015;21(9):1065‐1070. [DOI] [PubMed] [Google Scholar]
- 50. Verkoczy L, Diaz M, Holl TM, et al. Autoreactivity in an HIV‐1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc Natl Acad Sci USA. 2010;107(1):181‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verkoczy L, Chen Y, Bouton‐Verville H, et al. Rescue of HIV‐1 broad neutralizing antibody‐expressing B cells in 2F5 VH x VL knockin mice reveals multiple tolerance controls. J Immunol. 2011;187(7):3785‐3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen Y, Zhang J, Hwang KK, et al. Common tolerance mechanisms, but distinct cross‐reactivities associated with gp41 and lipids, limit production of HIV‐1 broad neutralizing antibodies 2F5 and 4E10. J Immunol. 2013;191(3):1260‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doyle‐Cooper C, Hudson KE, Cooper AB, et al. Immune tolerance negatively regulates B cells in knock‐in mice expressing broadly neutralizing HIV antibody 4E10. J Immunol. 2013;191(6):3186‐3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ota T, Doyle‐Cooper C, Cooper AB, et al. B cells from knock‐in mice expressing broadly neutralizing HIV antibody b12 carry an innocuous B cell receptor responsive to HIV vaccine candidates. J Immunol. 2013;191(6):3179‐3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dosenovic P, von Boehmer L, Escolano A, et al. Immunization for HIV‐1 broadly neutralizing antibodies in human Ig knockin mice. Cell. 2015;161(7):1505‐1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sangesland M, Ronsard L, Kazer SW, et al. Germline‐encoded affinity for cognate antigen enables vaccine amplification of a human broadly neutralizing response against influenza virus. Immunity. 2019;51(4):735‐749.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Adachi Y, Tonouchi K, Nithichanon A, et al. Exposure of an occluded hemagglutinin epitope drives selection of a class of cross‐protective influenza antibodies. Nat Commun. 2019;10(1):3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Crotty S. T Follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Locci M, Havenar‐Daughton C, Landais E, et al. Human circulating PD‐1(+)CXCR3(‐)CXCR5(+) memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39(4):758‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moody MA, Pedroza‐Pacheco I, Vandergrift NA, et al. Immune perturbations in HIV‐1‐infected individuals who make broadly neutralizing antibodies. Sci Immunol. 2016;1(1):aag0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamamoto T, Lynch RM, Gautam R, et al. Quality and quantity of T‐FH cells are critical for broad antibody development in SHIVAD8 infection. Sci Transl Med. 2015;7(298):298ra120. [DOI] [PubMed] [Google Scholar]
- 62. Mastelic Gavillet B, Eberhardt CS, Auderset F, et al. MF59 mediates Its B cell adjuvanticity by promoting T follicular helper cells and thus germinal center responses in adult and early life. J Immunol. 2015;194(10):4836‐4845. [DOI] [PubMed] [Google Scholar]
- 63. Liang F, Lindgren G, Sandgren KJ, et al. Vaccine priming is restricted to draining lymph nodes and controlled by adjuvant‐mediated antigen uptake. Sci Transl Med. 2017;9(393):eaal2094. [DOI] [PubMed] [Google Scholar]
- 64. Desbien AL, Dubois Cauwelaert N, Reed SJ, et al. IL‐18 and subcapsular lymph node macrophages are essential for enhanced B cell responses with TLR4 agonist. Adjuvants. 2016;197(11):4351‐4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ugolini M, Gerhard J, Burkert S, et al. Recognition of microbial viability via TLR8 drives TFH cell differentiation and vaccine responses. Nat Immunol. 2018;19(4):386‐396. [DOI] [PubMed] [Google Scholar]
- 66. Rookhuizen DC, Defranco AL. Toll‐like receptor 9 signaling acts on multiple elements of the germinal center to enhance antibody responses. Proc Natl Acad Sci USA. 2014;111(31):E3224‐E3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Madan‐Lala R, Pradhan P, Roy K. Combinatorial delivery of dual and triple TLR agonists via polymeric pathogen‐like particles synergistically enhances innate and adaptive immune responses. Sci Rep. 2017;7(1):2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dowling JK, Mansell A. Toll‐like receptors: the Swiss army knife of immunity and vaccine development. Clin Transl Immunol. 2016;5(5):e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brahmakshatriya V, Kuang Y, Devarajan P, et al. IL‐6 production by TLR‐activated APC broadly enhances aged cognate CD4 helper and B cell antibody responses in vivo. J Immunol. 2017;198(7):2819‐2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Radtke AJ, Anderson CF, Riteau N, et al. Adjuvant and carrier protein‐dependent T‐cell priming promotes a robust antibody response against the Plasmodium falciparum Pfs25 vaccine candidate. Sci Rep. 2017;7:40312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hill DL, Pierson W, Bolland DJ, et al. The adjuvant GLA‐SE promotes human Tfh cell expansion and emergence of public TCRbeta clonotypes. J Exp Med. 2019;216(8):1857‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Elsner RA, Shlomchik MJ. IL‐12 blocks Tfh cell differentiation during salmonella infection, thereby contributing to germinal center suppression. Cell Rep. 2019;29(9): 2796‐2809. e2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sato R, Makino‐Okamura C, Lin Q, et al. Repurposing the Psoriasis Drug Oxarol to an ointment adjuvant for the influenza vaccine. Int Immunol. 2019;31(12):771–779. [DOI] [PubMed] [Google Scholar]
- 74. Paules CI, Marston HD, Eisinger RW, Baltimore D, Fauci AS. The Pathway to a universal influenza vaccine. Immunity. 2017;47(4):599‐603. [DOI] [PubMed] [Google Scholar]
- 75. Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14(3):167‐182. [DOI] [PubMed] [Google Scholar]
- 76. Phillipson JE, Babecoff R, Ben‐Yedidia T. Is a universal influenza vaccine feasible? Ther Adv Vaccines Immunother. 2019;7:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Levi R, Arnon R. Synthetic recombinant influenza vaccine induces efficient long‐term immunity and cross‐strain protection. Vaccine. 1996;14(1):85‐92. [DOI] [PubMed] [Google Scholar]
- 78. Pleguezuelos O, Robinson S, Stoloff GA, Caparros‐Wanderley W. Synthetic Influenza vaccine (FLU‐v) stimulates cell mediated immunity in a double‐blind, randomised, placebo‐controlled Phase I trial. Vaccine. 2012;30(31):4655‐4660. [DOI] [PubMed] [Google Scholar]
- 79. Del Campo J, Pizzorno A, Djebali S, et al. OVX836 a recombinant nucleoprotein vaccine inducing cellular responses and protective efficacy against multiple influenza A subtypes. NPJ Vaccines. 2019;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696‐704. [DOI] [PubMed] [Google Scholar]
- 81. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256‐W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen YQ, Wohlbold TJ, Zheng NY, et al. Influenza infection in humans induces broadly cross‐reactive and protective neuraminidase‐reactive antibodies. Cell. 2018;173(2):417‐429 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]