

Abstract
Infections caused by multidrug-resistant (MDR) Pseudomonas aeruginosa and Klebsiella pneumoniae are a serious worldwide public health concern due to the ineffectiveness of empirical antibiotic therapy. Therefore, research and the development of new antibiotic alternatives are urgently needed to control these bacteria. The use of cationic antimicrobial peptides (CAMPs) is a promising candidate alternative therapeutic strategy to antibiotics because they exhibit antibacterial activity against both antibiotic susceptible and MDR strains. In this study, we aimed to investigate the in vitro antibacterial effect of a short synthetic CAMP derived from the ΔM2 analog of Cec D-like (CAMP-CecD) against clinical isolates of K pneumoniae (n = 30) and P aeruginosa (n = 30), as well as its hemolytic activity. Minimal inhibitory concentrations (MICs) and minimal bactericidal concentrations (MBCs) of CAMP-CecD against wild-type and MDR strains were determined by the broth microdilution test. In addition, an in silico molecular dynamic simulation was performed to predict the interaction between CAMP-CecD and membrane models of K pneumoniae and P aeruginosa. The results revealed a bactericidal effect of CAMP-CecD against both wild-type and resistant strains, but MDR P aeruginosa showed higher susceptibility to this peptide with MIC values between 32 and >256 μg/mL. CAMP-CecD showed higher stability in the P aeruginosa membrane model compared with the K pneumoniae model due to the greater number of noncovalent interactions with phospholipid 1-Palmitoyl-2-oleyl-sn-glycero-3-(phospho-rac-(1-glycerol)) (POPG). This may be related to the boosted effectiveness of the peptide against P aeruginosa clinical isolates. Given the antibacterial activity of CAMP-CecD against wild-type and MDR clinical isolates of P aeruginosa and K pneumoniae and its nonhemolytic effects on human erythrocytes, CAMP-CecD may be a promising alternative to conventional antibiotics.
Keywords: Multidrug-resistant P aeruginosa strains, multidrug-resistant K pneumoniae strains, cationic antimicrobial peptides, cecropin D-derived peptide
Introduction
Antimicrobial resistance is currently a major public health concern worldwide.1 Resistant bacteria (RB) can cause serious infections that are becoming difficult to treat due to limited therapeutic options.2,3 The most critical group of RB includes multidrug-resistant (MDR) bacteria, which are defined as strains that are resistant to at least one agent in 3 or more antibiotic categories.3,4 These MDR strains represent a serious threat to human health, especially in clinics where they can cause nosocomial infections.3,5,6 Nonfermentative bacteria strains, such as Pseudomonas aeruginosa and Enterobacteriaceae (including Klebsiella pneumoniae), show resistance to multiple antibiotics and cause severe infections, such as bloodstream infections and pneumonia.3 Recently, resistant P aeruginosa and K pneumoniae were included in the World Health Organization (WHO) list of antibiotic-resistant bacteria that pose the greatest risk to human health.3 In this respect, both species were included in the critical category (priority 1) of the WHO priority pathogens list according to the urgency of the need for research and development of new antibiotics to fight against the growing global resistance to antimicrobial medicines.3
Within recent years, the resistance of P aeruginosa has increased, and they now show resistance to several antibiotics, such as β-lactams, aminoglycosides, fluoroquinolones, and polymyxins.7-9 To counter antibiotic attacks, P aeruginosa uses several mechanisms that can be classified as intrinsic, acquired, and adaptive resistance.7 Some intrinsic resistance mechanisms of P aeruginosa include the production of antibiotic-inactivating enzymes, low outer membrane permeability, and expression of efflux systems that pump antibiotics out of the cell.7,10,11 In addition to intrinsic resistance, their acquired resistance through mutational changes or acquisition of resistance genes via horizontal gene transfer greatly contributes to the development of MDR P aeruginosa (MDRPA) strains.7,12 Several causes, including the overprescription and excessive use of antibiotics, self-medication, and incomplete courses of treatment, can accelerate the development of MDRPA strains, leading to the ineffectiveness of empirical antibiotic therapy against P aeruginosa.7,13 As a result, an increase in the number of MDR P aeruginosa strains leads to more cases of persistent infections and increased mortality.12,13 Similarly, multidrug resistance of the Enterobacteriaceae family is an increasing global public health concern.14 Much of the resistance in Enterobacteriaceae species is acquired through the transfer of different mobile genetic elements to plasmids, which move between cells of different species, and chromosomal gene mutations.14 MDR K pneumoniae (MDRKP) isolates carry various resistance genes and display high resistance to a broad spectrum of antibiotics, including β-lactams, aminoglycosides, quinolones, tigecycline, and polymyxins.6,14-17 These MDRKP strains are known to cause hospital-acquired infections.6,16,17 The incidence of infections caused by MDRPA and KPMDR strains has increased patient morbidity and mortality in healthcare settings in Colombia and worldwide because they cause infections in hospitalized or immunocompromised individuals.5,9,16-19
Infections caused by these MDR bacteria are becoming more difficult to treat because of their limited susceptibility to antimicrobial agents, resulting in a growing problem regarding the selection of effective antibiotic treatments.6,7 Therefore, the development of new antibiotics or alternative therapeutic strategies for the treatment of infections caused by MDR bacteria is urgently needed.7 Antimicrobial peptides (AMP) are promising candidates as alternatives to antibiotics because they exhibit antimicrobial activity against both antibiotic susceptible and MDR strains of Gram-negative and Gram-positive bacteria.20-28 AMPs are a large group of naturally occurring low-molecular-weight peptides that protect their hosts against diverse microorganisms.20,22,29-31 These peptides have been identified in several species from plants to humans and play a fundamental role in the innate immunity of these organisms.21,22,29-31 Specifically, the use of cationic AMPs (CAMP) is emerging as a promising nonantibiotic therapeutic strategy to overcome resistance as they have shown to be highly effective in killing bacterial strains resistant to conventional antibiotics.7,20,25,32-34 The mechanisms of action of CAMPs vary widely, and they cause bacterial cell death very quickly in comparison with conventional antibiotics because they induce killing by acting on one or more bacterial targets.24,25,35 CAMPs can display direct activity by disrupting the integrity of bacterial membranes and cell wall synthesis or inhibiting intracellular functions, such as enzymatic activities or the synthesis of proteins and nucleic acids.24,36,37 In this respect, diverse groups of AMPs have exhibited activity against bacteria and other pathogens.31,32,34 In particular, cecropins (Cec) are a type of antimicrobial peptide with potential therapeutic applications.38 They belong to a group of naturally occurring AMPs in insects and have exhibited in vitro activity against Gram-positive and Gram-negative bacteria.38
In this study, we investigated the in vitro antibacterial activity of a novel short synthetic CAMP derived from the ΔM2 analog of Cec D-like (CAMP-CecD)27 against clinical strains of K pneumoniae and P aeruginosa. The antibacterial activity of ΔM2 against laboratory strains of Gram-positive and Gram-negative bacteria has been previously reported.27 Here, the effects of CAMP-CecD were explored against both susceptible wild-type K pneumoniae (WTKP) and MDRKP strains and susceptible wild-type P aeruginosa (WTPA) and MDRPA strains. In addition, an in silico molecular dynamic simulation was performed to predict the interaction between CAMP-CecD and membrane models of K pneumoniae and P aeruginosa.
Materials and Methods
Peptide design, sequence characteristics, and synthesis
CAMP-CecD is a short cationic peptide derived from the NH2-terminal region of ΔM2 peptide, a previously reported cecropin D Galleria mellonella analog.27 CAMP-CecD is composed of the first 18 residues of the ΔM2 peptide. The net charge and wheel projections of CAMP-CecD were analyzed using Heliquest software (https://heliquest.ipmc.cnrs.fr/). CAMP-CecD was provided by GenScript Corporation (Piscataway, NJ, USA) with a 98% purity. The lyophilized peptide was dissolved in phosphate-buffered saline (PBS, 138 mM NaCl, 3 mM KCl, 1.5 mM NaH2PO4, 8.1 mM Na2HPO4, and pH 7.4) at an initial concentration of 5000 μg/mL and then aliquoted to obtain the required concentration. Diluted solutions were prepared on the day of use.
Bacterial strains
All laboratory strains, including Escherichia coli ATCC 25922, K pneumoniae ATCC 2146, and P aeruginosa ATCC 27853, were obtained from the American Type Culture Collection (ATCC). A total of 60 clinical bacterial strains were tested, including 30 K pneumoniae and 30 P aeruginosa. All clinical isolates were recovered from two tertiary care hospitals in Cali, Colombia. The isolates were from intensive care units, and the sample sources were respiratory secretions, urine, and blood. The cultures of isolates were sent to a Microbiology Laboratory at Laboratorio de Salud Publica Departamental del Valle del Cauca (LSPD-Valle), in the context of laboratory surveillance, where the bacterial species were confirmed, and the antibiotic susceptibility characterization was performed. The strains were identified with the VITEK 2 Gram-Negative identification (VITEK 2 GN ID) card based on established substrates and biochemical methods measuring carbon source utilization, enzymatic activities, and resistance (Ref. 21341, Biomérieux). The VITEK 2 GN ID card was used with the VITEK 2 system for the automated identification of Enterobacteriaceae and non-Enterobacteriaceae Gram-negative bacilli.
Characterization of clinical isolates
Antibiotic susceptibility was initially assessed by disk diffusion according to the recommendations of the Clinical and Laboratory Standards Institute (CLSI).39 The antibiotic susceptibility of clinical isolates and the minimal inhibitory concentration (MIC) for each antibiotic were confirmed with the VITEK 2 Antimicrobial Susceptibility Testing (VITEK 2 AST) card in the VITEK 2 system (Biomérieux) according to the clinical breakpoints defined by the CLSI and European Committee on Antimicrobial Susceptibility Testing (EUCAST).39,40 The susceptibility of all K pneumoniae clinical isolates to antimicrobial agents, such as amikacin (AMK), ampicillin/sulbactam (SAM), cefepime (FEP), cefoxitin (FOX), ceftazidime (CAZ), ceftriaxone (CRO), ciprofloxacin (CIP), doripenem (DOR), ertapenem (ETP), gentamicin (GEN), imipenem (IPM), meropenem (MEM), piperacillin/tazobactam (TZP), and tigecycline (TGC), was determined. In addition, all isolates of P aeruginosa were tested against AMK, FEP, CAZ, CIP, Colistin (CST), DOR, GEN, IPM, MEM, TZP, and TGC. E coli ATCC 25922 and K pneumoniae ATCC 2146 strains were used as references for WTKP and MDRKP, respectively, while P aeruginosa ATCC 27853 was used as the reference for WTPA. The susceptibility and resistance of ATCC strains were also confirmed with the VITEK 2 AST card.
Antimicrobial assay of CAMP-CecD
The MIC values of CAMP-CecD were determined by the broth microdilution test, according to the protocol of CLSI.39,41 Briefly, pure bacterial cultures from specimens were obtained in brain heart infusion (BHI) agar and incubated at 37°C for 18 to 20 hours. A colony from the pure culture was initially resuspended in sterile water to reach the turbidity of 0.5 McFarland, and the resulting suspension contained approximately 1-4 × 108 colony forming units (CFU)/mL. Using this suspension, a final 1:1000 dilution was performed directly into cation-adjusted Mueller-Hinton broth to obtain a final concentration of 2-7 × 105 CFU/mL. These bacterial inoculums were incubated with different concentrations of CAMP-CecD. The highest tested concentration was 256 μg/mL, from which serial 1:2 dilutions were made. Mixtures of the peptide and inoculums in a final volume of 100 μL were incubated in sterile 96-well polypropylene microplates (Sigma-Aldrich) at 37°C. A peptide-free control was used for every isolate evaluated. In addition, the reference ATCC strains were used in each assay as controls to ensure reproducibility. The MIC of CAMP-CecD for each strain was defined as the lowest concentration that inhibited the visible growth of bacteria after incubation for 18 to 20 hours.39,41 The minimal bactericidal concentrations (MBC), defined as the lowest concentration of an antibacterial agent required to kill 99.9% of a particular bacterium, were determined by plating out the contents of the first 3 wells showing no visible bacterial growth onto Muller-Hinton agar plates incubated at 37°C for 18 to 20 hours.32,42
Hemolytic activity
Human erythrocytes were used to evaluate the hemolytic activity of CAMP-CecD at different concentrations (256, 128, 64, 32, 16, 8, 4, 2, and 1 μg/mL). Erythrocytes were isolated from fresh human peripheral blood, washed 3 times with PBS by centrifugation for 5 minutes at 3000 g, and then resuspended in PBS. The different peptide concentrations were added to 4% human erythrocytes in PBS, incubated at 37°C for 1 hour, and then centrifuged at 4000 g for 5 minutes. Aliquots of the supernatant were transferred to 96-well plates, and hemolysis was assessed by measuring the absorbance at 540 nm with a Multiskan Go spectrophotometer (Thermo). Erythrocytes in the absence and presence of Triton X100 (1%) were used as a negative (blank) and positive control, respectively. The equation [(Abs in the peptide solution − Abs in PBS)/(Abs in Triton X10 − Abs in PBS)] × 100 was used to calculate the percentage of hemolysis.27
In silico structural modeling of CAMP-CecD
First, structural models of the peptide were obtained using the I-TASSER platform (https://zhanglab.ccmb.med.umich.edu/I-TASSER/).43 Here, 3-dimensional (3D) atomic models of the peptide were generated using multiple threading alignments against the protein structure database RCSB Protein Data Bank (https://www.rcsb.org/), and the structural prediction with higher confidence according to its C-score was selected.43 From this model, a total of 100 molecular models were built for the peptide with MODELLER 9.14 using default parameters, and the best probable structures were selected based on the discrete optimized protein energy score (DOPE score).44 In addition, for the best models, their stereochemical quality was verified using Ramachandran plots in PROSA II (https://prosa.services.came.sbg.ac.at/prosa.php) and RAMPAGE (http://mordred.bioc.cam.ac.uk/). All selected models had more than 90% amino acid residues in the favored and additional regions allowed. All the structures were visualized with PyMOL (https://pymol.org/2/).
In silico construction of Gram-negative bacteria membrane models
P aeruginosa and K pneumoniae membrane models were developed in silico using CHARMM-GUI.45 For the construction of the P aeruginosa membrane model, 3 different types of phospholipids were used, including 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylethanolamine (POPE), cardiolipin (PMCL1), and 1-Palmitoyl-2-oleyl-sn-glycero-3-(phospho-rac-(1-glycerol)) (POPG).46 The number of molecules in both the outer and inner monolayers of the in silico P aeruginosa membrane model was 21 POPE, 11 PMCL1, and 60 POPG.46 The K pneumoniae membrane model was constructed using 82 molecules of POPE, 6 molecules of PMCL1, and 5 of POPG in both the outer and inner monolayers.46 The peptide was localized in the center of each membrane model.
Molecular dynamic simulation: prediction of the interaction between CAMP-CecD and bacteria membrane models
Gromacs software version 2019.3 was used for the molecular dynamics simulation.47 The ion placement method was used with 0.15 M KCl, a water thickness of 22.5 Å, and CHARMM36 m as a force field, which is suitable for describing the distribution of molecules within a large system like membranes.48 The systems were adjusted by slowly heating to a temperature of 310°K at 1 femtosecond (fs)/step for 75 picoseconds (ps) to ensure the system did not present steric clashes or inappropriate geometry and at 2 fs/step for 300 ps in the equilibration step.48 Energy minimization of the system was obtained using the steepest descent algorithm with a tolerance value of 1000 kJ mol-1 nm-1 in 5000 steps with the Verlet cutoff scheme.47 The Berendsen algorithm to 125000 n-steps was used to equilibrate the temperature and pressure of the system. When the system was equilibrated, the production MD for data collection was run for 5 nanoseconds (ns) using the Nose–Hoover and Parrinello–Rahman algorithms to adjust the temperature and pressure. Particle Mesh Ewald (PME) summation was applied to correct for long-range electrostatic interactions.49
To understand the mechanism of action of CAMP-CecD, the interaction between CAMP-CecD and phospholipids was analyzed using GROMACS47 and visual molecular dynamics (VMD) programs.50 To this end, the root mean square deviation (RMSD), the radius of gyration, and the hydrogen bonds of the molecule were calculated. The partial density distribution of the molecule and phospholipids in the z-axis direction was counted.47,51 Finally, the interaction between the peptide and phospholipids was analyzed using the academic versions of PYMOL and Discovery Studio Visualizer (http://accelrys.com). POPE and POPG phospholipids were used for the membrane interaction analysis of K pneumoniae and P aeruginosa, respectively. The interactions between the corresponding phospholipids of each membrane model and the peptide residues were observed using the 2D graphic tool of Discovery Studio Visualizer (http://accelrys.com).
Statistical analysis
MICs and MBCs were determined in duplicate, and at least 3 independent assays were performed for each isolate. The results were analyzed using descriptive statistical tools with the median. Statistically significant differences in MICs between species and between wild-type and MDR strains were analyzed and compared using the nonparametric Kruskal–Wallis test with R-Project software Version 1.1.463. P values ⩽ .05 were considered statistically significant.
Results and Discussion
Peptide properties
CAMP-CecD was composed of 18 amino acid residues and exhibited a cationic net charge of +9 and a hydrophobicity index value of −0.002 (Figure 1). Helical wheel projections showed a strong amphipathic configuration as the hydrophilic face was clearly differentiated from the hydrophobic one (Figure 1). The hydrophilic face was composed of 10 residues, including R1,6,8,9,13,15; K5,12,16; and N2, whereas the hydrophobic core consisted of 8 residues, including A10,17; F3,4; I7,14,18; and G11.
Figure 1.
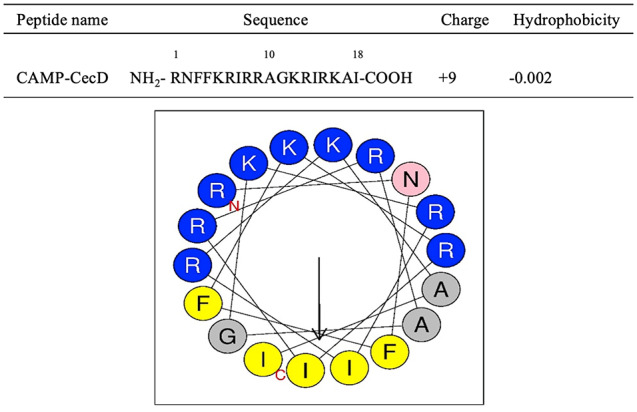
Helical wheel projection of CAMP-CecD. The residues are colored according to the classification: charged polar (blue) and neutral polar (pink). The hydrophobicity of residues is shown from yellow (the most hydrophobic residues) to gray (the least hydrophobic residues). The arrow points to the hydrophobic face. CAMP indicate cationic antimicrobial peptides.
Antibiotic susceptibility and resistance profiles of clinical isolates
The antibiotic susceptibility profiles of K pneumoniae and P aeruginosa clinical isolates and ATCC strains are shown in Figure 2. The susceptibility and resistance profiles were constructed using 7 and 6 antimicrobial categories for K pneumoniae and P aeruginosa, respectively (Figure 2). Fourteen K pneumoniae isolates were classified as WTKP, while the remaining 16 clinical isolates were considered MDRKP due to their resistance to at least 3 different antimicrobial categories, including penicillin + β-lactamase inhibitors, cephamycins, extended-spectrum cephalosporins, carbapenems, aminoglycosides, fluoroquinolones, and glycylcyclines (Figure 2A).4 Similarly, 14 P aeruginosa strains were considered wild-type strains with intrinsic resistance to tigecycline,52 and 16 were classified as MDRPA due to their nonsusceptibility to antimicrobial categories, such as penicillin + β-lactamase inhibitors, extended-spectrum cephalosporins, carbapenems, aminoglycosides, fluoroquinolones, and polymyxins (Figure 2B).4 The presence of MDRKP and MDRPA isolates is consistent with the real-world bacterial resistance situation in Colombia, where a high frequency of K pneumoniae and P aeruginosa MDR strains have been reported in addition to their successful spread during recent years around the country, representing a serious threat in Colombian hospitals.17,19
Figure 2.

Antimicrobial susceptibility profiles of selected K pneumoniae (A) and P aeruginosa (B) clinical isolates. The phenotypic profiles were constructed using several antimicrobial categories: penicillin + β-lactamase inhibitors (ampicillin/sulbactam and penicillin/tazobactam), cephamycins (cefoxitin), extended-spectrum cephalosporins (ceftazidime, ceftriaxone, and cefepime), carbapenems (doripenem, ertapenem, imipenem, and meropenem), aminoglycosides (amikacin and gentamicin), fluoroquinolones (ciprofloxacin), glycylcyclines (tigecycline), and polymyxins (colistin). The interpretative categories for antibiotic susceptibility were susceptible (green square), intermediate (yellow square), and resistant (red square) according to clinical breakpoints defined by the CLSI guidelines, except for tigecycline, which was interpreted according to EUCAST guidelines. CLSI indicates Clinical and Laboratory Standards Institute; Ec ATCC 25922, Escherichia coli ATCC 25922; EUCAST, European Committee on Antimicrobial Susceptibility Testing; Kp, K pneumoniae; Kp ATCC 2146, K pneumoniae ATCC 2146; Pa, P aeruginosa; Pa ATCC 27853, P aeruginosa ATCC 27853.
Antibacterial and hemolytic activity of CAMP-CecD
Because of their high potency and rapidity in causing bacterial cell death, several studies have evaluated the antibacterial activity of CAMPs.20,22,32 In this study, we investigated the in vitro antibacterial activity of CAMP-CecD against WTKP and MDRKP isolates and WTPA and MDRPA strains, which is summarized in Table 1. CAMP-CecD showed antibacterial activity against both K pneumoniae and P aeruginosa with MIC values between 32 and >256 μg/mL and against laboratory strains (Table 1). The MICs found in this study were comparable to those reported for other cationic peptides but slightly higher in comparison with those reported for the Cecropin A-melittin hybrid peptide.32 Overall, this activity against Gram-negative bacteria was consistent with previous reports in which different natural Cecs members and/or synthetic Cec-analogs were effective against laboratory strains of P aeruginosa and Enterobacteriaceae spp. (including K pneumoniae and E coli).27,32,38,53-56 Following the comparison of all tested isolates, statistical differences were found between both species, but there were no significant differences between wild-type strains of K pneumoniae and P aeruginosa or between MDR isolates of K pneumoniae and P aeruginosa (Table 1). In addition, the peptide was more efficient against MDRPA than MDRKP, with the lowest MIC value of 32 μg/mL for P aeruginosa MDR isolates (Table 1). The efficacy against MDR isolates found here was comparable to that reported previously for natural Cec members and Cec-like peptides, which killed MDR strains of Gram-negative bacteria, including Salmonella typhimurium, Acinetobacter baumannii, E coli, and P aeruginosa.57-60
Table 1.
In vitro antibacterial activity of CAMP-CecD against susceptible wild-type and MDR strains of K pneumoniae and P aeruginosa.
| K pneumoniae (number) | MIC (μg/mL) | MBC (μg/mL) | P aeruginosa (number) | MIC (μg/mL) | MBC (μg/mL) | P valuea |
|---|---|---|---|---|---|---|
| WTKP (14) | 32–>256 | 32–>256 | WTPA (14) | 64–256> | 64–256> | 0.14 |
| MDRKP (16) | 256–>256 | 256–>256 | MDRPA (16) | 32–256> | 32–256> | 0.12 |
| Total strains (30) | 32–>256 | 32–>256 | Total strains (30) | 32–256> | 32–256> | 0.03 |
| Ec ATCC 25922 | 128–256 | 128–256 | Pa ATCC 27853 | 32–64 | 32–64 | |
| Kp ATCC 2146 | 256–>256 |
Abbreviations: CAMP, cationic antimicrobial peptides; Ec ATCC 25922, Escherichia coli ATCC 25922; Kp ATCC 2146, K pneumoniae ATCC 2146; MBC, minimal bactericidal concentration; MDR, multidrug-resistant; MDRKP, multidrug-resistant K pneumoniae; MDRPA, multidrug-resistant P aeruginosa; MIC, minimal inhibitory concentration; Pa ATCC 27853, P aeruginosa ATCC 27853; WTKP, wild-type K pneumoniae; WTPA, wild-type P aeruginosa.
Significance level between the minimal inhibitory concentrations (MICs).
When intraspecific comparisons were performed, no significant differences were found between wild-type and MDR isolates of K pneumoniae (Figure 3), but WTKP strains were more susceptible to CAMP-CecD with the lowest MIC value of 32 μg/mL, whereas the lowest MIC value for MDRKP was 256 μg/mL (Figure 3). Similarly, when susceptible and MDR strains of P aeruginosa were compared, no statistical differences were found (Figure 4). Although no statistical differences were found, MDRPA strains showed a higher susceptibility to CAMP-CecD with MIC values from 32 to > 256 μg/mL, whereas the MIC values of the peptide for WTPA ranged between 64 to > 256 μg/mL (Figure 4). Interestingly, these findings suggest that the activity of CAMP-CecD against K pneumoniae and P aeruginosa is independent of their antibiotic resistance patterns (Figures 3 and 4). In addition, no statistical differences were found between the MICs and MBCs of CAMP-CecD against all bacteria tested independently of their resistance profiles (Table 1). Because the MBC values were equal to the MICs in all cases, CAMP-CecD may be considered a bactericidal agent (Table 1). These findings suggest that the antibiotic resistance mechanisms of K pneumoniae and P aeruginosa isolates tested in this study (Figure 2) do not alter the efficacy of CAMP-CecD. These results showed that CAMP-CecD was an active peptide against the clinical isolates of K pneumoniae and P aeruginosa, including wild-type and MDR strains. Previous studies have also reported the bactericidal effect of other CAMPs, including cecropin A hybrid peptides, cathelicidins, magainin, and nisin, against MDR strains. Similar to this study, no differences were detected between MICs and MBCs independently of the antibiotic resistance patterns.25,32,34,61
Figure 3.
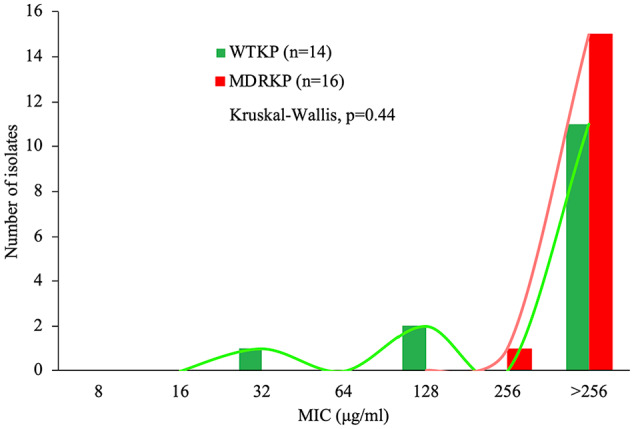
Minimal inhibitory concentration (MIC) distributions of CAMP-CecD for susceptible wild-type isolates (WTKP) and multidrug-resistant isolates of K pneumoniae (MDRKP). No significant differences were found between WTKP and MDRKP isolates, according to the Kruskal–Wallis test (P > .05). CAMP indicate cationic antimicrobial peptides.
Figure 4.
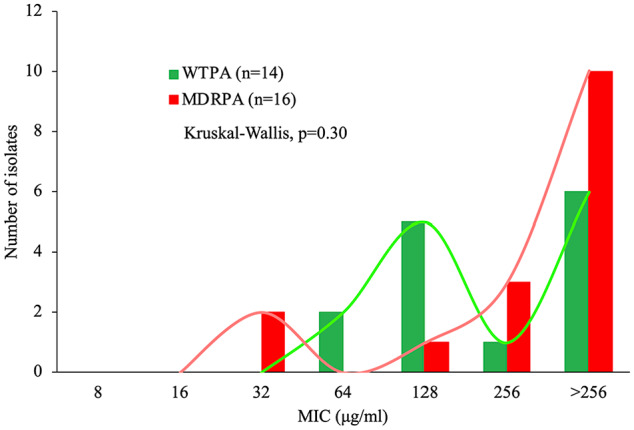
Minimal inhibitory concentration (MIC) distributions of CAMP-CecD for susceptible wild-type isolates (WTPA) and multidrug-resistant isolates of P aeruginosa (MDRPA). No significant differences were found between WTPA and MDRPA isolates, according to the Kruskal–Wallis test (P > .05). CAMP indicate cationic antimicrobial peptides.
We also evaluated the hemolytic activity of CAMP-CecD. Unlike the pattern peptide ΔM2 that showed slight hemolytic activity, CAMP-CecD did not show hemolytic activity at the highest evaluated concentration (256 µg/mL).27 Therefore, in this case, the hemolytic activity did not vary depending on the cationic charge as both peptides had a net charge of +9. In contrast to our results, a previous study reported a positive association between charge and hemolytic activity of the peptide V13K.62 However, variations in other structural properties could influence the hemolytic activity of each peptide. For example, the reported hydrophobicity of the peptide ΔM2 is substantially higher (0.178)27 than that of CAMP-CecD (−0.002) (Figure 1). According to previous reports, increased hydrophobicity can enhance hemolytic activity.63,64 Furthermore, increasing the length in an RW peptide has been shown to increase hemolytic activity.65
In silico interaction between CAMP-CecD and membrane models of K pneumoniae and P aeruginosa
The behavior of the peptide in the two membrane models of Gram-negative bacteria is shown in Figure 5. In the K pneumoniae membrane model, a greater variation in peptide structure was observed in comparison with its structure in the P aeruginosa membrane model (Figure 5). Specifically, at the end of the 5 ns in K pneumoniae membranes, the peptide folded in half, losing its initial structure (Figure 5). This change in peptide structure in K pneumoniae increased the radius of gyration, which reached up to 0.90 nm at 1 ns (Figure 6). This flexibility in the central region of the peptide reflects a hinge that facilitates contact with the nonpolar region of phospholipids, allowing the peptide to be inserted into the K pneumoniae membrane.66 In contrast, the peptide remained stable most of the time in the P aeruginosa membrane, except for a slight variation in the amino-terminal end at 3 ns (Figure 5). Stability is also an important structural property for antibacterial activity.67 These results could explain the statistical differences in the activity of CAMP-CecD between both species when all tested isolates were compared (Table 1).
Figure 5.
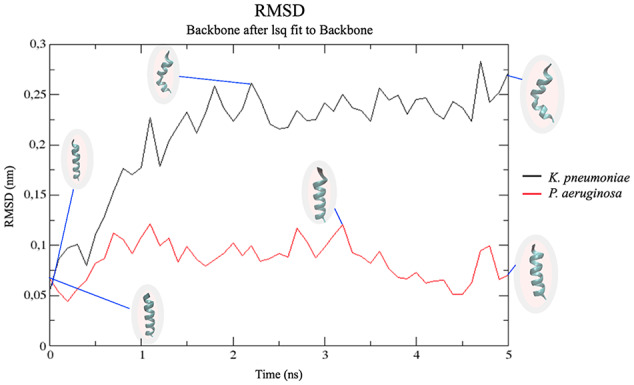
RMSD analysis of the backbone atoms of CAMP-CecD in K pneumoniae and P aeruginosa membrane models. A higher variation in peptide structure was observed in the membrane model of K pneumoniae in comparison with the P aeruginosa membrane model. CAMP indicates cationic antimicrobial peptides; RMSD, root-mean-square deviation.
Figure 6.
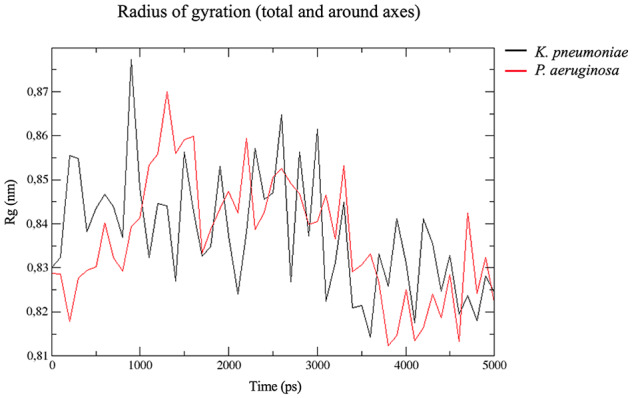
Radius of gyration analysis of CAMP-CecD in K pneumoniae and P aeruginosa membrane models. A higher variation in the radius of gyration was detected in the membrane model of K pneumoniae compared with the P aeruginosa membrane model. CAMP indicates cationic antimicrobial peptides.
Furthermore, the density profile of phospholipids and water (TIP3) from the bilayers of K pneumoniae and P aeruginosa in the presence of the ligand along the z axis was obtained (Figure 7). A greater asymmetry in the density of POPE compared with the rest of the phospholipids in K pneumoniae membranes was observed (Figure 7A), whereas in the P aeruginosa membrane, a larger asymmetry in POPG compared with the other phospholipids was detected (Figure 7B). These asymmetries in the density of membrane phospholipids can be explained by the interactions with the peptide. Interactions, such as hydrogen bonds, were observed between these phospholipids and CAMP-CecD (Figure 8). Specifically, more hydrogen bonds between the peptide and POPE in the K pneumoniae membrane were detected, reaching up to 12 hydrogen bonds between 4 and 5 ns, while fewer interactions were found with POPG (Figure 8A). In the case of P aeruginosa membranes, a greater interaction of the peptide with POPG was observed, leading to the formation of up to 20 hydrogen bonds throughout the simulation (Figure 8B).
Figure 7.

Partial density profile (PMCL, POPE, POPG, TIP3-water) in the Z direction in K pneumoniae (A) and P aeruginosa (B) membrane systems. CAMP indicates cationic antimicrobial peptides; PMCL, cardiolipin; POPE, 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylethanolamine; POPG, 1-Palmitoyl-2-oleyl-sn-glycero-3-(phospho-rac-(1-glycerol)); TIP3, density profile of phospholipids and water.
Figure 8.

Hydrogen bonds between CAMP-CecD and the phospholipids of K pneumoniae (A) and P aeruginosa (B) membranes. CAMP indicates cationic antimicrobial peptides; PMCL, cardiolipin; POPE, 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylethanolamine; POPG, 1-Palmitoyl-2-oleyl-sn-glycero-3-(phospho-rac-(1-glycerol)).
The detailed interactions between CAMP-CecD and both phospholipids, including POPE in K pneumoniae membranes and POPG in P aeruginosa membranes, are shown in Figure 9. The peptide folded in half at 5 ns and interacted with POPE in K pneumoniae mainly through hydrogen bonds (Figure 9A). However, some residues of CAMP-CecD, such as the arginine at positions 6, 9, and 13, interacted with POPE through Van der Waals interactions, salt bridges, hydrogen bridges, and hydrophobic interactions (Figure 9B). Meanwhile, the arginine at positions 1 and 8 interacted with POPG through Van der Waals interactions, salt bridges, and hydrogen bridges in P aeruginosa membranes (Figure 8C and D).
Figure 9.

Interactions between CAMP-CecD and the phospholipids of K pneumoniae and aeruginosa membranes. (A) 3D model of hydrogen bonds between the peptide (in violet arginine residues) and POPE in K pneumoniae. (B) 2D image of interactions between the peptide and POPE in K pneumoniae. (C) 3D model of hydrogen bonds between the peptide (in violet arginine residues) and POPG in P aeruginosa. (D) 2D image of interactions between the peptide and POPG in P aeruginosa. CAMP indicates cationic antimicrobial peptides; POPE, 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylethanolamine; POPG, 1-Palmitoyl-2-oleyl-sn-glycero-3-(phospho-rac-(1-glycerol)).
According to the molecular dynamic simulation results, a loss of the peptide structure occurred when K pneumoniae was treated with CAMP-CecD due to the presence of greater interactions with POPE, mainly hydrogen bonds that affected the stability of the peptide. In contrast, the secondary structure of CAMP-CecD showed greater stability in P aeruginosa membranes due to the increased number of interactions with POPG, especially hydrogen bonds. This is consistent with previous studies that show a strong association between the arginine residues of CecB members and P aeruginosa membranes due to interactions with the anionic POPG lipid moieties.53 Although both K pneumoniae and P aeruginosa are Gram-negative bacteria, their membrane structures differ greatly, particularly in the proportion of POPE and POPG phospholipids.46 For example, K pneumoniae has a higher proportion of POPE, which increased the interaction of the peptide with this phospholipid. This destabilized the secondary structure of the peptide, which may be related to the decreased antimicrobial activity in K pneumoniae compared with P aeruginosa in which the peptide had higher stability and effectiveness, even in MDR isolates. The relationship between the structural stability of the peptide and its antimicrobial activity provided by the interaction of arginine residues and membrane phospholipids has been previously reported.68 Although a relationship between arginine residues and the antibacterial effect of the peptides is evident, it is not entirely clear what specific properties contribute to this activity.69 A probable relationship could be inferred for the interaction between arginine residues from helical CAMPs and anionic phospholipids in bacteria membranes, such as POPG.70-72 Hence, salt bridges and electrostatic interactions between the side chain (amino groups) of arginine residues at the ends of the alpha-helix of the peptide and phosphates in the headgroup of POPG can be formed.70-72 These interactions allow the entry of the peptide into the bacterial membrane and simultaneously contribute to the stability of its helical structure.68 In addition, these interactions disrupt the lipid bilayer structure, likely inducing the lateral separation of phospholipids because of interactions between their polar heads and the cationic residues of the peptide.25 This interaction facilitates the insertion of CAMP-CecD into the membrane by laterally displacing the phospholipids and rearranging the anionic lipids in a separate domain.46,73 The reorganization of the lipids around the domain induces defects in the bacterial membrane, such as the recruitment of anionic lipids from other locations on the membrane and the disturbance of the existing domains on the lipid bilayer.73 This causes membrane pore formation and likely results in a rapid killing effect of the bacteria.25
Regarding peptide structural characteristics, some parameters, such as helicity, hydrophobicity, and amphipathicity, may increase the antibacterial activity and improve the prokaryotic selectivity of CAMPs.74 It has been reported that a range of hydrophobicity between 40% and 60% facilitates the electrostatic interaction of the peptide with the membrane.75,76 CAMP-CecD exhibited a hydrophobicity of 39% (Figure 1), which indicates a high probability of its interaction with the bacterial membranes and subsequent insertion. Finally, the enhanced prokaryotic selectivity of CAMP-CecD may be explained by its high affinity for anionic phospholipids in P aeruginosa, such as POPG (Figure 8), as well as the net charge of +9 (Figure 1),77 which was confirmed by the antibacterial activity and low hemolytic activity of this peptide.
Conclusion
In conclusion, the results of this study demonstrated the activity of CAMP-CecD against Gram-negative bacteria with significant differences between K pneumoniae and P aeruginosa, possibly explained by the increased stability of the peptide in the P aeruginosa membrane model. This peptide exhibited antibacterial activity against both wild-type and MDR strains of K pneumoniae and P aeruginosa that show different antibiotic phenotypes. In this respect, and due to its minimal hemolytic effect, CAMP-CecD can be considered as a potential alternative drug to conventional antibiotics to control MDR bacteria associated with severe infectious diseases.
Acknowledgments
The authors thank Helen Agudelo for her technical help at the laboratory.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by Universidad Santiago de Cali and Laboratorio de Salud Publica Departamental del Valle del Cauca (grant number 440-621118-47).
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: Iván Darío Ocampo-Ibáñez: Conceptualization, methodology, supervision, project administration, funding acquisition, writing of the main manuscript text, and figure and table preparation. Yamil Liscano: Conceptualization, writing, and figure preparation. Sandra Patricia Rivera-Sanchez: Methodology, supervision, project administration, and funding acquisition. José Oñate-Garzón: Conceptualization, writing, and figure and table preparation. Ashley Dayan Lugo-Guevara: Methodology and investigation. Liliana Janeth Flórez-Elvira: Statistical analysis and table preparation. Maria Cristina Lesmes: Funding acquisition and supervision. All authors reviewed and approved the final manuscript.
Research Ethics and Patient Consent: The study did not use human subjects or tissues. Bacterial isolates used in this study were obtained from existing clinical collections routinely collected as part of the laboratory practices of hospitals, so no specimens were collected for the purpose of this study. The studied isolates were not accompanied by personal patient identification (names, IDs, addresses, phone numbers, and birth dates were unknown).
ORCID iDs: Iván Darío Ocampo-Ibáñez  https://orcid.org/0000-0002-9833-3537
https://orcid.org/0000-0002-9833-3537
Sandra Patricia Rivera-Sánchez
https://orcid.org/0000-0002-6564-3061
Liliana Janeth Flórez-Elvira
https://orcid.org/0000-0001-8836-9088
References
- 1. Voulgaris GL, Voulgari ML, Falagas ME. Developments on antibiotics for multidrug resistant bacterial gram-negative infections. Expert Rev Anti Infect Ther. 2019;17:387-401. doi: 10.1080/14787210.2019.1610392. [DOI] [PubMed] [Google Scholar]
- 2. Moradali MF, Ghods S, Rehm BHA. Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front Cell Infect Microbiol. 2017;7:39. doi: 10.3389/fcimb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization. WHO publishes list of bacteria for which new antibiotics are urgently needed; 2017. https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed. Published 2017.
- 4. Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2011;18:268-281. [DOI] [PubMed] [Google Scholar]
- 5. Prakash V, Mishra P, Premi H, Walia A, Dhawan S, Kumar A. Increasing incidence of multidrug resistant Pseudomonas aeruginosa in inpatients of a tertiary care hospital. Int J Res Med Sci. 2014;2:1302. doi: 10.5455/2320-6012.ijrms20141111. [DOI] [Google Scholar]
- 6. Ferreira RL, Da Silva BCM, Rezende GS, et al. High prevalence of multidrug-resistant Klebsiella pneumoniae harboring several virulence and β-lactamase encoding genes in a brazilian intensive care unit. Front Microbiol. 2019;9:3198. doi: 10.3389/fmicb.2018.03198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv. 2019;37:177-192. doi: 10.1016/j.biotechadv.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 8. Dößelmann B, Willmann M, Steglich M, et al. Rapid and consistent evolution of colistin resistance in extensively drug-resistant Pseudomonas aeruginosa during morbidostat culture. Antimicrob Agents Chemother. 2017;61:e00043-17. doi: 10.1128/AAC.00043-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matos EC, Matos HJ, Conceição ML, Rodrigues YC, Carneiro IC, Lima KV. Clinical and microbiological features of infections caused by Pseudomonas aeruginosa in patients hospitalized in intensive care units. Rev Soc Bras Med Trop. 2016;49:305-311. doi: 10.1590/0037-8682-0446-2015. [DOI] [PubMed] [Google Scholar]
- 10. Breidenstein EBM, De La Fuente-Núñez C, Hancock RE. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19:419-426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 11. Drenkard E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003;5:1213-1219. doi: 10.1016/j.micinf.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 12. Henrichfreise B, Wiegand I, Pfister W, Wiedemann B. Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob Agents Chemother. 2007;51:4062-4070. doi: 10.1128/AAC.00148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirsch EB, Tam VH. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev Pharmacoecon Outcomes Res. 2010;10:441-451. doi: 10.1586/erp.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Partridge SR. Resistance mechanisms in Enterobacteriaceae. Pathology. 2015;47:276-284. doi: 10.1097/PAT.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 15. Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem. 2014;6:25-64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ocampo AM, Vargas CA, Sierra PM, Cienfuegos AV, Jiménez JN. Caracterización molecular de un brote de Klebsiella pneumoniae resistente a carbapenémicos en un hospital de alto nivel de complejidad de Medellín, Colombia. Biomédica. 2015;35:496-504. doi: 10.7705/biomedica.v35i4.2610. [DOI] [PubMed] [Google Scholar]
- 17. Ocampo A, Chen L, Cienfuegos A, et al. A two-year surveillance in five Colombian tertiary care hospitals reveals high frequency of non-CG258 clones of carbapenem-resistant Klebsiella pneumoniae with distinct clinical characteristics. Antimicrob Agents Chemother. 2016;60:332-342. doi: 10.1128/AAC.01775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. World Health Organization and Pan American Health Organization. Epidemiological Alert: Enterobacteriacea With Plasmid-Mediated Transferable Colistin Resistance, Public Health Implications in the Americas. Washington, DC: World Health Organization and Pan American Health Organization; 2016. [Google Scholar]
- 19. Correa A, Del Campo R, Perenguez M, et al. Dissemination of high-risk clones of extensively drug-resistant Pseudomonas aeruginosa in Colombia. Antimicrob Agents Chemother. 2015;59:2421-2425. doi: 10.1128/AAC.03926-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guaní-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Terán LM. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin Immunol. 2010;135:1-11. doi: 10.1016/j.clim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 21. Brown KL, Hancock REW. Cationic host defense (antimicrobial) peptides. Curr Opin Immunol. 2006;18:24-30. doi: 10.1016/j.coi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 22. Reddy KVR, Yedery RD, Aranha C. Antimicrobial peptides: premises and promises. Int J Antimicrob Agents. 2004;24:536-547. doi: 10.1016/j.ijantimicag.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 23. Mataraci E, Dosler S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob Agents Chemother. 2012;56:6366-6371. doi: 10.1128/AAC.01180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang L, Gallo RL. Antimicrobial peptides. Curr Biol. 2016;26:R14-R19. doi: 10.1016/J.CUB.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 25. Bahar AA, Ren D. Antimicrobial peptides. Pharmaceuticals (Basel). 2013;6:1543-1575. doi: 10.3390/ph6121543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oñate-Garzón JF, Manrique-Moreno M, Patiño Gonzalez E. Antimicrobial activity of cationic peptides designed from neutral peptide. Acta Biol Colomb. 2017;22:157-164. doi: 10.15446/abc.v22n2.56809. [DOI] [Google Scholar]
- 27. Oñate-Garzón J, Manrique-Moreno M, Trier S, Leidy C, Torres R, Patiño E. Antimicrobial activity and interactions of cationic peptides derived from Galleria mellonella cecropin D-like peptide with model membranes. J Antibiot (Tokyo). 2016;70:238-245. doi: 10.1038/ja.2016.134. [DOI] [PubMed] [Google Scholar]
- 28. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389-395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 29. Aslam B, Wang W, Arshad MI, et al. Antibiotic resistance: a rundown of a global crisis. Infect Drug Resist. 2018;11:1645-1658. doi: 10.2147/IDR.S173867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang S, Zeng X, Yang Q, Qiao S. Antimicrobial peptides as potential alternatives to antibiotics in food animal industry. Int J Mol Sci. 2016;17:603. doi: 10.3390/ijms17050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fry DE. Antimicrobial peptides. Surg Infect (Larchmt). 2018;19:804-811. doi: 10.1089/sur.2018.194. [DOI] [PubMed] [Google Scholar]
- 32. Geitani R, Ayoub Moubareck C, Touqui L, Karam Sarkis D. Cationic antimicrobial peptides: alternatives and/or adjuvants to antibiotics active against methicillin-resistant Staphylococcus aureus and multidrug-resistant Pseudomonas aeruginosa. BMC Microbiol. 2019;19:54. doi: 10.1186/s12866-019-1416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mandal SM, Roy A, Ghosh AK, Hazra TK, Basak A, Franco OL. Challenges and future prospects of antibiotic therapy: from peptides to phages utilization. Front Pharmacol. 2014;5:105. doi: 10.3389/fphar.2014.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lei J, Sun LC, Huang S, et al. The antimicrobial peptides and their potential clinical applications. Am J Transl Res. 2019;11:3919-3931. [PMC free article] [PubMed] [Google Scholar]
- 35. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238-250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 36. Sharma S, Sahoo N, Bhunia A. Antimicrobial peptides and their pore/ion channel properties in neutralization of pathogenic microbes. Curr Top Med Chem. 2015;16:46-53. doi: 10.2174/1568026615666150703115454. [DOI] [PubMed] [Google Scholar]
- 37. Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. 2016;6:194. doi: 10.3389/fcimb.2016.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brady D, Grapputo A, Romoli O, Sandrelli F. Insect cecropins, antimicrobial peptides with potential therapeutic applications. Int J Mol Sci. 2019;20:5862. doi: 10.3390/ijms20235862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing (CLSI Supplement M100). 29th ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2019. [Google Scholar]
- 40. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters (Version 10.0.). Basel, Switzerland: The European Committee on Antimicrobial Susceptibility Testing; 2020. [Google Scholar]
- 41. Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Tenth Edition (CLSI Document M07-A10). Wayne, PA: Clinical and Laboratory Standards Institute; 2015. [Google Scholar]
- 42. Giacometti A, Cirioni O, Barchiesi F, et al. In vitro susceptibility tests for cationic peptides: comparison of broth microdilution methods for bacteria that grow aerobically. Antimicrob Agents Chemother. 2000;44:1694-1696. doi: 10.1128/aac.44.6.1694-1696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725-738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinforma. 2016; 54:5.6.1-5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang J, Mackerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135-2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Epand RM, Epand RF. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim Biophys Acta. 2009;1788:289-294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 47. Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19-25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 48. Pink DA, Belaya M, Levadny V, Quinn B. A model of polar group statics in lipid bilayers and monolayers. Langmuir. 1997;13:1701-1711. doi: 10.1021/la950343o. [DOI] [Google Scholar]
- 49. Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys. 1995;103:3668-3679. doi: 10.1063/1.470043. [DOI] [Google Scholar]
- 50. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33-8, 27-8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 51. Shearer J, Khalid S. Communication between the leaflets of asymmetric membranes revealed from coarse-grain molecular dynamics simulations. Sci Rep. 2018;8:1805. doi: 10.1038/s41598-018-20227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanz-García F, Hernando-Amado S, Martínez JL. Mutational evolution of Pseudomonas aeruginosa resistance to ribosome-targeting antibiotics. Front Genet. 2018;9:451. doi: 10.3389/fgene.2018.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Romoli O, Mukherjee S, Mohid SA, et al. Enhanced silkworm cecropin B antimicrobial activity against Pseudomonas aeruginosa from single amino acid variation. ACS Infect Dis. 2019;5:1200-1213. doi: 10.1021/acsinfecdis.9b00042. [DOI] [PubMed] [Google Scholar]
- 54. Kaushal A, Gupta K, Shah R, Van Hoek ML. Antimicrobial activity of mosquito cecropin peptides against Francisella. Dev Comp Immunol. 2016;63:171-180. doi: 10.1016/j.dci.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 55. Lockey TD, Ourth DD. Formation of pores in Escherichia coli cell membranes by a cecropin isolated from hemolymph of Heliothis virescens larvae. Eur J Biochem. 1996;271:263-271. [DOI] [PubMed] [Google Scholar]
- 56. Wang J, Ma K, Ruan M, et al. A novel cecropin B-derived peptide with antibacterial and potential anti-inflammatory properties. PeerJ. 2018;6:e5369. doi: 10.7717/peerj.5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee E, Kim JK, Jeon D, Jeong KW, Shin A, Kim Y. Functional roles of aromatic residues and helices of papiliocin in its antimicrobial and anti-inflammatory activities. Sci Rep. 2015;5:12048. doi: 10.1038/srep12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee E, Shin A, Kim Y. Anti-inflammatory activities of cecropin A and its mechanism of action. Arch Insect Biochem Physiol. 2015;88:31-44. doi: 10.1002/arch.21193. [DOI] [PubMed] [Google Scholar]
- 59. Saugar JM, Rodríguez-Hernández MJ, De La Torre BG, et al. Activity of cecropin A-melittin hybrid peptides against colistin-resistant clinical strains of Acinetobacter baumannii: molecular basis for the differential mechanisms of action. Antimicrob Agents Chemother. 2006;50:1251-1256. doi: 10.1128/AAC.50.4.1251-1256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jayamani E, Rajamuthiah R, Larkins-Ford J, et al. Insect-derived cecropins display activity against Acinetobacter baumannii in a whole-animal high-throughput Caenorhabditis elegans model. Antimicrob Agents Chemother. 2015;59:1728-1737. doi: 10.1128/AAC.04198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brumfitt W, Salton MRJ, Hamilton-Miller JMT. Nisin, alone and combined with peptidoglycan-modulating antibiotics: activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci. J Antimicrob Chemother. 2002;50:731-734. doi: 10.1093/jac/dkf190. [DOI] [PubMed] [Google Scholar]
- 62. Jiang Z, Vasil AI, Hale JD, Hancock REW, Vasil ML, Hodges RS. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial pept. Biopolymers. 2008;90:369-383. doi: 10.1002/bip.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother. 2007;51:1398-1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chou HT, Kuo TY, Chiang JC, et al. Design and synthesis of cationic antimicrobial peptides with improved activity and selectivity against Vibrio spp. Int J Antimicrob Agents. 2008;32:130-138. doi: 10.1016/j.ijantimicag.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 65. Liu Z, Brady A, Young A, et al. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob Agents Chemother. 2007;51:597-603. doi: 10.1128/AAC.00828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liscano Y, Salamanca CH, Vargas L, Cantor S, Laverde-Rojas V, Oñate-Garzón J. Increases in hydrophilicity and charge on the polar face of alyteserin 1c helix change its selectivity towards gram-positive bacteria. Antibiotics. 2019;8:238. doi: 10.3390/antibiotics8040238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tossi A, Scocchi M, Skerlavaj B, Gennaro R. Identification and characterization of a primary antibacterial domain in CAP18, a lipopolysaccharide binding protein from rabbit leukocytes. FEBS Lett. 1994;339:108-112. doi: 10.1016/0014-5793(94)80395-1. [DOI] [PubMed] [Google Scholar]
- 68. Zompra AA, Galanis AS, Werbitzky O, Albericio F. Manufacturing peptides as active pharmaceutical ingredients. Future Med Chem. 2009;1:361-377. doi: 10.4155/fmc.09.23. [DOI] [PubMed] [Google Scholar]
- 69. Chan DI, Prenner EJ, Vogel HJ. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim Biophys Acta. 2006;1758:1184-1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 70. Forood B, Feliciano EJ, Nambiar KP. Stabilization of alpha-helical structures in short peptides via end capping. Proc Natl Acad Sci U S A. 1993;90:838-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Glukhov E, Stark M, Burrows LL, Deber CM. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J Biol Chem. 2005;280:33960-33967. doi: 10.1074/jbc.M507042200. [DOI] [PubMed] [Google Scholar]
- 72. Zhou HX, Lyu PC, Wemmer DE, Kallenbach NR. Structure of a C-terminal alpha-helix cap in a synthetic peptide. J Am Chem Soc. 1994;116:1139-1140. doi: 10.1021/ja00082a049. [DOI] [Google Scholar]
- 73. Polozov IV, Polozova AI, Molotkovsky JG, Epand RM. Amphipathic peptide affects the lateral domain organization of lipid bilayers. Biochim Biophys Acta. 1997;1328:125-139. doi: 10.1016/S0005-2736(97)00080-1. [DOI] [PubMed] [Google Scholar]
- 74. Dathe M, Wieprecht T. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim Biophys Acta. 1999;1462:71-87. doi: 10.1016/S0005-2736(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 75. Yeaman MR, Yount NY. The metamorphosis of the kappa. Pharmacol Rev. 2003;57:27-55. [DOI] [PubMed] [Google Scholar]
- 76. Ntwasa M, Goto A, Kurata S. Coleopteran antimicrobial peptides: prospects for clinical applications. Int J Microbiol. 2012;2012:101989. doi: 10.1155/2012/101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feder R, Dagan A, Mor A. Structure-activity relationship study of antimicrobial dermaseptin S4 showing the consequences of peptide oligomerization on selective cytotoxicity. J Biol Chem. 2000;275:4230-4238. doi: 10.1074/jbc.275.6.4230. [DOI] [PubMed] [Google Scholar]