

Abstract
Coronavirus disease 2019 (COVID‐19) is a pandemic infectious disease caused by novel severe acute respiratory syndrome coronavirus‐2 (SARS CoV‐2). The SARS CoV‐2 is transmitted more rapidly and readily than SARS CoV. Both, SARS CoV and SARS CoV‐2 via their glycosylated spike proteins recognize the human angiotensin converting enzyme‐2 (ACE‐2) receptor. We generated multiple sequence alignments and phylogenetic trees for representative spike proteins of SARS CoV and SARS CoV‐2 from various host sources in order to analyze the specificity in SARS CoV‐2 spike proteins required for causing infection in humans. Our results show that among the genomes analyzed, two sequence regions in the N‐terminal domain “MESEFR” and “SYLTPG” are specific to human SARS CoV‐2. In the receptor‐binding domain, two sequence regions “VGGNY“ and ”EIYQAGSTPCNGV” and a disulfide bridge connecting 480C and 488C in the extended loop are structural determinants for the recognition of human ACE‐2 receptor. The complete genome analysis of representative SARS CoVs from bat, civet, human host sources, and human SARS CoV‐2 identified the bat genome (GenBank code: MN996532.1) as closest to the recent novel human SARS CoV‐2 genomes. The bat SARS CoV genomes (GenBank codes: MG772933 and MG772934) are evolutionary intermediates in the mutagenesis progression toward becoming human SARS CoV‐2.
Keywords: complete genomes, multiple sequence alignment, phylogenetic tree, receptor‐binding domain, severe acute respiratory syndrome coronavirus‐2, spike proteins
1. INTRODUCTION
In the last two decades, zoonotic coronaviruses, severe acute respiratory syndrome coronavirus (SARS CoV) (2002), 1 and Middle East Respiratory Syndrome coronavirus (MERS CoV) (2012) 2 have caused acute respiratory diseases in humans that have resulted in several deaths. The present coronavirus disease 2019 (COVID‐19) is a pandemic respiratory disease caused by the novel SARS CoV‐2. The initial infection started in Wuhan, Hubei province, China in December 2019 and very soon became a global outbreak, infecting populations in almost every country in the world causing a total of 184 248 deaths and 2 638 852 infections as of 23rd April 2020 (https://www.worldometers.info/coronavirus/). Within a short span of time, this pandemic has caused major social and economic disruptions. Compared to other coronaviruses, the novel SARS CoV‐2 appears to be spreading more rapidly and readily, posing a challenging task before the administrative and scientific communities. The SARS CoV‐2 is transmitted from person to person contact via respiratory secretions during coughing and sneezing. Infection of this highly pathogenic virus can cause acute respiratory distress syndrome which impacts the lung and heart functions. The prominent symptoms of this viral infection are flu, severe respiratory, enteric, and neurological disorders, resulting in increased white blood cells and kidney failure. There are no vaccines or drugs available to combat this deadly infectious disease and there is no strategic plan to treat the infected patients. Hence, there is a need to develop specific anti‐SARS CoV‐2 vaccines and drugs to treat infected patients, in order to reduce viral shedding and further transmission in populations.
The SARS CoV‐2 comprises positive‐sense single‐stranded RNA genome of size 29 to 30 kb and belongs to the coronaviridae family and betacoronavirus subfamily. Mammals such as bats are the main reservoir of betacoronaviruses, but due to the zoonotic contacts and viral genomic mutations, SARS CoV‐2 has recently crossed species and caused infections in humans. 3 Research findings have pointed that previous zoonotic CoV infections such as SARS CoV that first infected humans in the Guangdong province of southern China in 2002 was transmitted from bats and civets, 4 , 5 , 6 , 7 , 8 the MERS CoV that originated in bats was first identified from camel to human transmission in Saudi Arabia in 2012. 2 , 9 , 10 , 11 , 12 These coronaviruses have crossed species and resulted in causing human infections leading to mortality. It is reported that civet SARS CoV can also infect humans. 13 , 14 The SARS‐like CoVs from some bats and civets are predicted to result in human infections 15 , 16 due to their changing genomic RNA sequences, importantly in the spike protein regions. 17 , 18 Since January 2020, several complete genome sequences of viral SARS CoV‐2 isolated from infected patients belonging to various geographical locations, such as Australia, China, Denmark, Finland, Hungary, India, Italy, Japan, South Korea, United States, and Vietnam, have been deposited in the GenBank (https://www.ncbi.nlm.nih.gov/genbank/). At the genomic level, the nucleotide sequences of SARS CoV and SARS CoV‐2 share 79.6% sequence identity. 19 The viral RNA stores the genetic information and also serves to translate into structural and nonstructural proteins of SARS CoV‐2. The SARS CoV uses angiotensin converting enzyme‐2 (ACE‐2) as receptor for entry into human epithelial cells 20 to cause the infection. Virus infectivity studies on HeLa cells have shown that SARS CoV‐2 also uses ACE‐2 as receptor for cellular entry. 19
It has been reported that the first SARS CoV‐2 infection in Wuhan, China is caused from the original host, bats, 19 and in less than 4 months transmitted among the human populations in the entire world. The initial contact between the SARS CoV/CoV‐2 and human host is via recognition between the heavily glycosylated cell envelope spike protein of the virus and the ACE‐2 receptor of the human host resulting in infection. In order to understand the specificity, estimate the extent of similarities and variations in the SARS CoV‐2 spike proteins required for binding the host receptor, we analyzed the representative spike protein sequences. Furthermore, in order to estimate the evolutionary progression of the bat SARS CoV genome, such that it is able to adapt to a human host as a novel coronavirus causing COVID‐19, we have analyzed the complete genomes of the bat, civet, human SARS CoV, and human SARS CoV‐2. We have carried out computational analyses on the nucleotide and protein sequences by generating multiple sequence alignments (MSAs), constructing phylogenetic trees, and analyzing the three‐dimensional structures of the spike proteins to address the above.
2. MATERIALS AND METHODS
The spike proteins were retrieved from the NCBI database using the sequence similarity search BLAST program, 21 with human SARS CoV‐2 spike protein as the query sequence (NCBI code: QHD43416) from the genome (GenBank code: MN908947.3). 3 The complete genome nucleotide sequences of SARS CoV from bats, civets, and SARS CoV and CoV‐2 from human host were obtained from NCBI virus database (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide) in FASTA format. Only complete genome sequences without any ambiguity in nucleotide composition were considered for analyses. The redundancy in each data set was removed using the CD‐HIT program. 22
The nucleotide and protein sequence homology analyses based on MSA reveal the substitutions, deletions, and insertions at each position. To understand the evolutionary relationships between the members, the MSAs were further processed to generate phylogenetic trees—a pictorial representation of the evolutionary relationships between related members of various sequences analyzed. The MSAs and phylogenetic trees of the spike proteins and complete genomes were generated using the Next Generation Phylogeny.fr web service available at https://NGPhylogeny.fr. 23 The protocol takes all sequences (nucleotide or protein) as input in FASTA file format and generates the MSA and phylogenetic tree. In the NGPhylogeny server, we have selected FastME 2.0 program that infers phylogenies using a distance approach since it is capable of handling large data sets. 24 Based on the input FASTA sequences, an MSA is generated that adopts Multiple Alignment using Fast Fourier Transform (MAFFT) 25 with gap extension penalty 0.123 and gap opening penalty 1.53. The MSA generated is parsed through Block Mapping and Gathering with Entropy (BGME) software for selecting regions suitable for phylogenetic inference. 26 This method uses a sliding window size 3, maximum entropy threshold 0.5, gap rate cutoff 0.5, minimum block size 5, matrix: PAM250 for DNA, and BLOSUM62 for proteins. FastME estimates phylogenies that employ distance‐based methods from MSAs using TN93 and LG as substitution models for DNA and proteins, respectively. In the distance‐based methods, pairwise distances between all pairs of sequences are generated as a square matrix. The sequence pairs with shorter pairwise distances are clustered together more closely in the phylogenetic tree. Tree refinement was performed using Subtree Pruning and Regrafting (SPR) with Balanced version of Minimum Evolution (BalME), with a decimal precision for branch length set to 6. Finally, the phylogenetic trees were generated using Interactive Tree Of Life program (iTOL) v4. 27
3. RESULTS AND DISCUSSION
3.1. Analyses of the spike proteins of SARS CoV and SARS CoV‐2
The SARS CoV and SARS CoV‐2 spike proteins retrieved from various host sources have a sequence length ranging between 1240 to 1273 amino acids. Structurally, a spike protein is characterized by three regions: (a) the N‐terminal extracellular domain, (b) a transmembrane anchor domain, and (c) an intracellular segment. The N‐terminal extracellular domain comprises a receptor binding subunit (S1) and a membrane‐fusion subunit (S2). The S1 subunit comprises two domains: N‐terminal domain (NTD) and receptor‐binding domain (RBD). The sequence analyses of spike proteins from the various host sources are shown in the MSA in Figure S1 and the phylogenetic tree in Figure 1. From Figure 1, it is observed that the paralogous proteins from individual host sources are associated with a distinct clade, the spike proteins from human SARS CoV‐2 share highest sequence similarity according to the least pairwise distances. Also, the orthologous spike proteins from other host species are highly similar according to the low pairwise distances. Therefore, it is intriguing to see that despite high sequence identity between the spike proteins from various host sources, only some SARS CoV and SARS CoV‐2 are able to bind the human host ACE‐2 receptor. In order to understand this, we have analyzed the MSA of the spike proteins.
FIGURE 1.
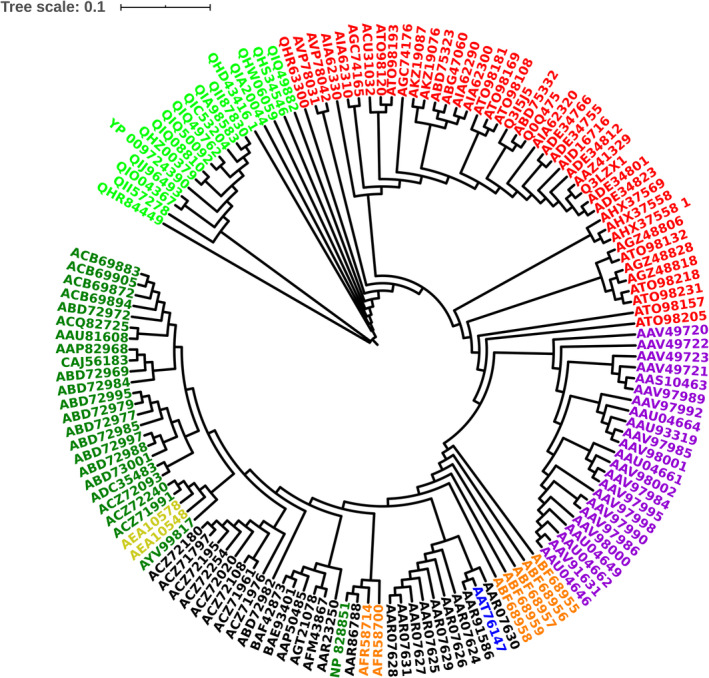
Phylogenetic tree of SARS CoV and SARS CoV‐2 spike proteins. Human CoV‐2 (green), human CoV (dark green), bat CoV (red), civet CoV (violet), cat CoV (orange), swine CoV (blue), mouse CoV (bright yellow), SARS CoV recombinant spike proteins and from lab adapted cells (black). SARS CoV‐2, severe acute respiratory syndrome coronavirus‐2
From Figure S1, it is observed that the N‐terminal ~500 amino acids comprising S1 subunit vary to a moderate extent among all host sources relative to the later region that share high sequence identity. The region approximately between 300 and 500 amino acid residues is crucial in spike proteins as it forms the RBD that recognizes the ACE‐2 receptor which allows entry of the virus into host cells. A sequence motif “PRRA” from 681P to 684A (amino acid numbering is according to NCBI code: QHD43416) that is gained only in the human SARS CoV‐2 spike proteins is referred to as furin cleavage site. 28 , 29 In this work, we identify the sequence regions in human SARS CoV‐2; a six residues insertion sequence “MESEFR” from 153M to 158R and another six residues insertion sequence “SYLTPG” from 247S to 252G. The bat SARS CoV spike protein QHR63300 (GenBank code: MN996532.1) also comprises the identical sequence regions as above. In two bat spike proteins, AVP78042 (GenBank code: MG772934) and AVP78031 (MG772933), a six residues sequence “SIREFA” and a three residues sequence “GDP” are present at equivalent positions, respectively. The insertion sequence regions present in human SARS CoV‐2, 153MESEFR158, and 247SYLTPG252 are associated with the NTD and are located distant from the ACE‐2 binding site. From the three‐dimensional structure of human SARS CoV‐2, we infer that these regions are likely to be exposed toward the surface of the spike protein in NTD. In human SARS CoV‐2, there are two insertion regions: a five residues insertion “VGGNY” from 445V to 449Y, and a 13 residues insertion “EIYQAGSTPCNGV” from 471E to 483V. The bat SARS CoV spike protein QHR63300 has also gained equivalent insertions with the sequences; “EGGNF” and “EIYQAGSKPCNGQ.” It is interesting to note that the bat SARS CoV QHR63300 has already acquired a 13 residues sequence region, whereas all other spike proteins that recognize the ACE‐2 receptor in SARS CoV comprise a 12 residues sequence region. The Figure 2A was generated by editing the MSA in Figure S1 to depict the sequence regions discussed above in representative spike proteins from the four host sources (bat, civet, human SARS CoV, and human SARS CoV‐2).
FIGURE 2.
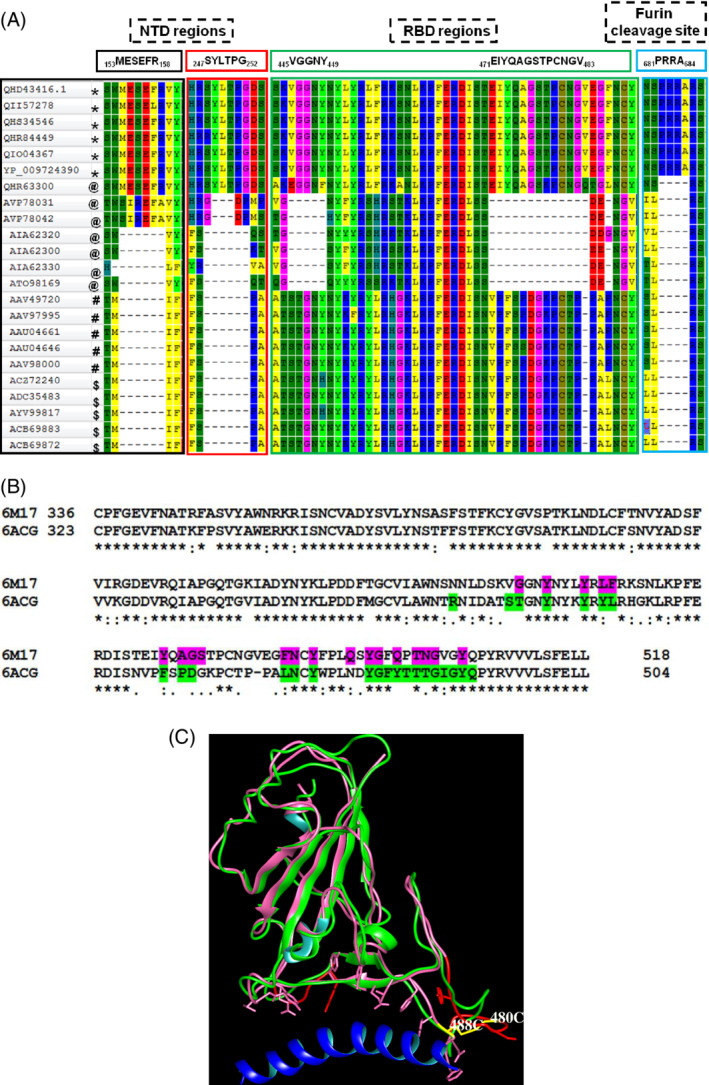
A, Portions of the alignment of spike proteins extracted from the multiple sequence alignment (Figure S1). Insertion sequence regions and their locations within the NTD, RBD, and furin cleavage sites for human SARS CoV‐2 (*), bat SARS CoV (@), civet SARS CoV (#), human SARS CoV ($). B, Pairwise sequence alignment corresponding to the RBD from human SARS CoV (6ACG) and human SARS CoV‐2 (6M17). The residues that lie within 5 Å in RBD from ACE‐2 are highlighted in 6ACG (green) and 6M17 (magenta). The start and end amino acid residues numbers in RBD are shown. “*” indicates identical residues; “:” indicates conservative amino acid residue substitutions; “.” indicates weakly conserved amino acid residue substitutions in the alignment. C, Structural superposition of human SARS CoV (6ACG, green) and human SARS CoV‐2 (6M17, magenta) and the long H1 helix in ACE‐2 (blue). The side chains of amino acid residues in RBD that lie within 5 Å from ACE‐2 are shown. The deletion region in RBD of bat SARS CoV is shown in the structure of 6M17 (red). The C480‐C488 disulfide bond (yellow) connects the extended loop in 6M17. ACE‐2, angiotensin converting enzyme‐2; NTD, N‐terminal domain; RBD, receptor‐binding domain; SARS CoV‐2, severe acute respiratory syndrome coronavirus‐2 [Color figure can be viewed at wileyonlinelibrary.com]
The two sequence regions, “VGGNY” and “EIYQAGSTPCNGV,” are part of the RBD and involved in recognition of the ACE‐2 receptor in human host. Their absence in the bat SARS CoV at equivalent positions may be responsible for their inability to bind human ACE‐2. To study this, we have analyzed the three‐dimensional structures of human SARS CoV (PDB code: 6ACG) 30 and the RBD domain of human SARS CoV‐2 (6M17) complexed with ACE‐2 receptor. 31 The structures were superimposed and amino acid residues within 5 Å distance from the ACE‐2 receptor were identified and highlighted in the pairwise sequence alignment of RBD (Figure 2B). Despite amino acid mutations in these regions in both proteins, the structures are highly superimposable. The location corresponding to the deletions of the two sequence regions in bat SARS CoV with respect to human SARS CoV‐2 matches with the ACE‐2 binding region (Figure 2C). The second insertion sequence region, that is, 471EIYQAGSTPCNGV483 in the RBD domain forms an extended loop in human SARS CoV‐2 and is stabilized by a disulfide bond between 480C and 488C (Figure 2C). This disulfide bond is conserved in all spike proteins that comprise the insertion sequence regions in RBD. In bat SARS CoV spike proteins, the position equivalent to 488C is replaced by a conserved glycine residue and therefore the above disulfide bond will be absent. Interestingly, the bat SARS CoV spike protein QHR63300 which has acquired the insertion sequences in RBD also possesses the disulfide bond. We believe that the presence of the above sequence regions in RBD and the disulfide bond that stabilizes the conformation of the extended loop are required for the recognition of human ACE‐2.
The sequence regions identified in this work serve as potential candidate epitopes for the design of antibodies specific for human SARS CoV‐2 recognition. Our analyses suggest that the bat spike protein QHR63300 has undergone significant evolutionary changes such that it resembles the human SARS CoV‐2 spike protein more than the bat SARS CoV which may have led to the transmission of SARS CoV from bat to human as the novel SARS CoV‐2. Our results also suggest that the bat SARS CoV spike proteins—AVP78042 and AVP78031—are in the progression of acquiring mutations toward becoming SARS CoV‐2‐like proteins. The phylogenetic tree in Figure 1 showing the proteins—QHR63300, AVP78042, and AVP78031 close to the human SARS CoV‐2—is in support of our hypothesis.
3.2. Complete genome analyses of SARS CoV and SARS CoV‐2
The representative complete genomes of nucleotide sequences from bat, civet, human CoV, and human CoV‐2 were analyzed. The MSA is shown in Figure S2 and the phylogenetic tree in Figure 3. From Figure 3, it is observed that the human SARS CoV‐2 genomes cluster into one clade (pairwise distance is lower than 0.002) revealing high identity that suggest their recent evolution. The bat SARS CoV genome (GenBank code: MN996532.1) is also member of this clade (pairwise distance between 0.042 and 0.043) indicating that it is the closest homolog to the human SARS CoV‐2 among the bat genomes. The two bat SARS CoV genomes (GenBank codes: MG772933.1 and MG772934.1) are also close to the human SARS CoV‐2 clade. The human and civet SARS CoV genomes cluster into another distinct clade. The members of bat SARS CoV clade have undergone maximum evolutionary changes as observed in Figure 3. Based on these results, we propose that the bat SARS CoV genomes have diverged the most during the last 18 years (since its detection in 2002) and have evolved closer to civet and human SARS CoV genomes. The bat SARS CoV genome (GenBank code: MN996532.1) has diverged significantly into the recent novel human SARS CoV‐2 genomes, whereas the bat SARS CoV genomes (GenBank codes: MG772933 and MG772934) are intermediates during the evolution of bat SARS CoV into human novel SARS CoV‐2. We believe that the bat SARS CoV genomes such as these are likely to undergo further evolutionary mutations and become adaptable to infecting human populations.
FIGURE 3.
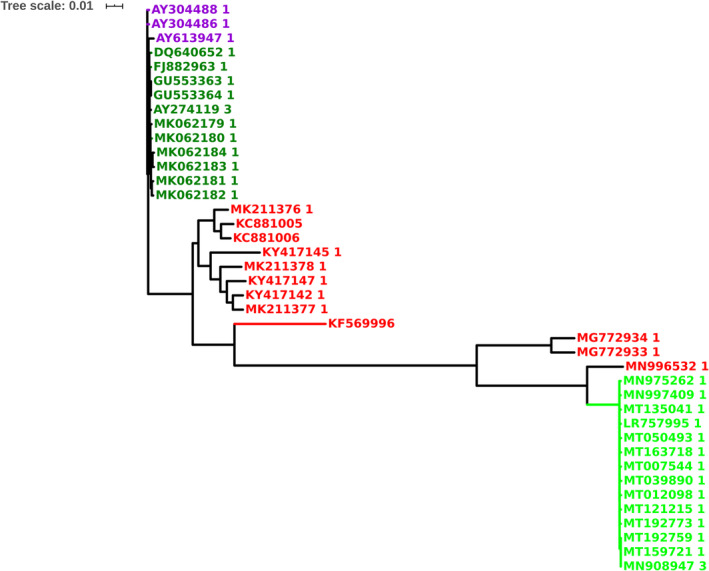
Phylogenetic tree of SARS CoV and SARS CoV‐2 complete genomes. Human SARS CoV‐2 (green), human SARS CoV (dark green), bat SARS CoV (red), and civet SARS CoV (violet). SARS CoV‐2, severe acute respiratory syndrome coronavirus‐2 [Color figure can be viewed at wileyonlinelibrary.com]
4. CONCLUSIONS
Two sequence regions “MESEFR” and “SYLTPG” in the NTD of spike protein in human SARS CoV‐2 and two sequence regions “VGGNY” and “EIYQAGSTPCNGV” in the RBD that interact with ACE‐2 may be exploited as potential candidates for antibody design. The phylogenetic analyses of the bat, civet, human SARS CoV, and human SARS CoV‐2 genomes show that the bat SARS CoV genome (GenBank code: MN996532.1) is closest homolog of human SARS CoV‐2. The two other bat SARS CoV genomes (GenBank codes: MG772933 and MG772934) are intermediates in the evolution of bat genomes into human SARS CoV‐2.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Figure S1 Multiple sequence alignment of SARS CoV and SARS CoV‐2 spike proteins from different host sources.
Figure S2 Multiple sequence alignment of representative complete genomes from human SARS CoV‐2, human SARS CoV, bat SARS CoV and civet SARS CoV.
ACKNOWLEDGMENT
L. G. thanks School of Chemistry and CAS, UGC for providing research facilities.
Guruprasad L. Evolutionary relationships and sequence‐structure determinants in human SARS coronavirus‐2 spike proteins for host receptor recognition. Proteins. 2020;88:1387–1393. 10.1002/prot.25967
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1002/prot.25967.
REFERENCES
- 1. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967‐1976. [DOI] [PubMed] [Google Scholar]
- 2. Azhar EI, El‐Kafrawy SA, Farraj SA, et al. Evidence for camel‐to‐human transmission of MERS coronavirus. N Engl J Med. 2014;370(26):2499‐2505. [DOI] [PubMed] [Google Scholar]
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu RH, He JF, Evans MR, et al. Epidemiologic clues to SARS origin in China. Emerg Infect Dis. 2004;10(6):1030‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS‐associated coronavirus. Science. 2003;300(5624):1399‐1404. [DOI] [PubMed] [Google Scholar]
- 6. Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394‐1399. [DOI] [PubMed] [Google Scholar]
- 7. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953‐1966. [DOI] [PubMed] [Google Scholar]
- 8. Holmes KV, Enjuanes L. The SARS coronavirus: a postgenomic era. Science. 2003;300(5624):1377‐1378. [DOI] [PubMed] [Google Scholar]
- 9. Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS‐like disease. Clin Microbiol Rev. 2015;28(2):465‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sabir JS, Lam TT, Ahmed MM, et al. Co‐circulation of three camel coronavirus species and recombination of MERS‐CoVs in Saudi Arabia. Science. 2016;351(6268):81‐84. [DOI] [PubMed] [Google Scholar]
- 11. Azhar EI, Hashem AM, El‐Kafrawy SA, et al. Detection of the Middle East respiratory syndrome coronavirus genome in an air sample originating from a camel barn owned by an infected patient. MBio. 2014;5(4):e01450‐e01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Omrani AS, Al‐Tawfiq JA, Memish ZA. Middle East respiratory syndrome coronavirus (MERS‐CoV): animal to human interaction. Pathog Glob Health. 2015;109(8):354‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang M, Yan M, Xu H, et al. SARS‐CoV infection in a restaurant from palm civet. Emerg Infect Dis. 2005;11(12):1860‐1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Wong SK, Li F, et al. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2‐S‐protein interactions. J Virol. 2006;80(9):4211‐4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Menachery VD, Yount BL Jr, Debbink K, et al. A SARS‐like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. 2015;21(12):1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang N, Li SY, Yang XL, et al. Serological evidence of bat SARS‐related coronavirus infection in humans, China. Virol Sin. 2018;33(1):104‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song HD, Tu CC, Zhang GW, et al. Cross‐host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. 2005;102(7):2430‐2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menachery VD, Yount BL, Sims AC, et al. SARS‐like WIV1‐CoV poised for human emergence. Proc Natl Acad Sci USA. 2016;113(11):3048‐3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li W, Moore MJ, Vasilieva N, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schäffer AA, Aravind L, Madden TL, et al. Improving the accuracy of PSI‐BLAST protein database searches with composition‐based statistics and other refinements. Nucleic Acids Res. 2001;29(14):2994‐3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W, Godzik A. Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658‐1659. [DOI] [PubMed] [Google Scholar]
- 23. Lemoine F, Correia D, Lefort V, et al. NGPhylogeny.fr: new generation phylogenetic services for non‐specialists. Nucleic Acids Res. 2019;47(W1):W260‐W265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lefort V, Desper R, Gascuel O. FastME 2.0: a comprehensive, accurate, and fast distance‐based phylogeny inference program. Mol Biol Evol. 2015;32(10):2798‐2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10(1):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256‐W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Q, Qiu Y, Li JY, Zhou ZJ, Liao CH, Ge XY. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019‐nCoV) potentially related to viral transmissibility. Virol Sin. 2020;20:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun. 2020;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song W, Gui M, Wang X, Xiang Y. Cryo‐EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14(8):e1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan R, Zhang Y, Guo Y, Xia L, Zhou Q. Structural basis for the recognition of the 2019‐nCoV by human ACE2. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Multiple sequence alignment of SARS CoV and SARS CoV‐2 spike proteins from different host sources.
Figure S2 Multiple sequence alignment of representative complete genomes from human SARS CoV‐2, human SARS CoV, bat SARS CoV and civet SARS CoV.