

Abstract
Cancer cell motility plays a central role in metastasis and tumor invasion but can be difficult to study accurately in vitro. A simple approach to address this challenge through the production of monolithic, photopatterned 3D tumor constructs in situ in a microfluidic device is described here. Through stepwise fabrication of adjoining hydrogel regions with and without incorporated cells, multidomain structures with defined boundaries are produced. By imaging cells over time, cellular activity with arbitrary control over medium conditions, including drug concentration and flow rate, is studied. First, malignant human colon carcinoma cells (HCT116) are studied for 10 days, comparing invasion dynamics and viability of cells in normal media to those exposed to two independent chemotherapeutic drugs: anti-proliferative 5-fluorouracil and anti-migratory Marimastat. Cytotoxicity is measured and significant differences are observed in cellular dynamics (migrating cell count, distance traveled, and rate) that correlate with the mechanism of each drug. Then, the platform is applied to the selective isolation of infiltrated cells through the photopatterning and subsequent dissolution of cleavable hydrogel domains. As a demonstration, the preferential collection of highly migratory cells (HCT116) over a comparable cell line with low malignancy and migratory potential (Caco-2) is shown.
Keywords: 3D cell cultures, biofabrication, cancer, microfluidics, migration
1. Introduction
Metastases are thought to be responsible for more than 90% of cancer-related deaths.[1] It is generally agreed that the process of metastasis involves multiple steps,[2] including movement of tumor cells into adjacent tissues, migration through the endothelium into blood vessels or lymphatics (intravasation), passage via circulation, and extravasation to and proliferation in distant tissue. However, the factors that contribute to this complex cascade of events are poorly understood; thus, there is a need for improved model systems to investigate them. Even the initial stage of metastasis, in which cancer cells infiltrate locally into adjacent tissue, is difficult to study in vitro because available technologies represent the in vivo environment poorly and have characteristics that may be practically or experimentally restrictive. For example, established techniques[3] like the classical transwell migration assay[4] and scratch assay[5] have proven to be valuable for probing chemotaxis and wound healing potential, respectively, however, they feature 2D cell cultures that do not reflect the 3D nature of tissues in which cancer cells reside and require large amounts of cells. Additionally, many conventional assays typically do not allow for the real-time assessment of cell viability; a capacity that is especially critical for studying the effects of therapeutic agents that may target metastasis.
Microfluidic systems can be used to offset several of the limitations encountered with conventional assays by requiring a low number of cells and enabling direct cell imaging and can also facilitate dynamic fluid delivery. However, most microfluidic-based approaches that have been reported to date probe single-cell[6] motility, often through artificial channels, or else involve 2D culture[7,8] motility. As a result, their relationship to physiological systems is uncertain. A limited number of strategies have been developed to integrate 3D cell cultures into the platform, including invasion from spheroids into surrounding gels.[9] However, most such efforts have employed either laminar flow profiles or physical barriers[10–15] to produce adjoining domains, thus restricting construct geometry, limiting parallelization and rapid fluid exchange, and increasing overall device complexity.
Here, we describe a simple and versatile approach for the in situ formation of 3D constructs in a pre-formed microfluidic architecture (Figure 1a) that can be used for concurrent cell migration and viability assessment under media exposure (Figure 1b). We utilize a serial in situ photopatterning technique (Figure 1c) to define multi-domain constructs composed of both cell-laden and cell-free regions. The total construct is composed of a single, monolithic hyaluronic acid (HA)/gelatin hydrogel, but features a defined boundary between regions (Figure 1c, right) for quantitative analysis of cell infiltration. As a demonstration, we integrated highly invasive HCT-116 human colon carcinoma cells that express mCherry fluorescent protein into our constructs to enable direct imaging of cells over time. We first showed that the interstitial flow-induced bias of cell movement can be mitigated through intermittent delivery of fresh buffer to the construct to maintain high viability and yield purely cell-driven motility. We then investigated the effects of two chemotherapeutic drugs: the thymidylate synthase inhibitor 5-fluorouracil (5FU), and the matrix metalloproteinases (MMP) inhibitor Marimastat. We demonstrated that the total number of cells infiltrating into the adjacent regions of the construct were reduced with increasing 5FU concentration, even while both the distance traveled by and the mean velocity of infiltrating cells were not significantly affected. In contrast, we found that increasing Marimastat exposure significantly reduced the total number of infiltrating cells, their distance migrated, and their mean velocity. Furthermore, we demonstrated our ability to biofabricate a cell-free region of the construct with a cleavable hydrogel, enabling the domain to be chemically dissolved to isolate and recover the infiltrating cells. Our results illustrate the efficacy of our platform in delivering quantitative cell migration and viability data with dynamic control of buffer and drug conditions using a simple and rapid system.
Figure 1.
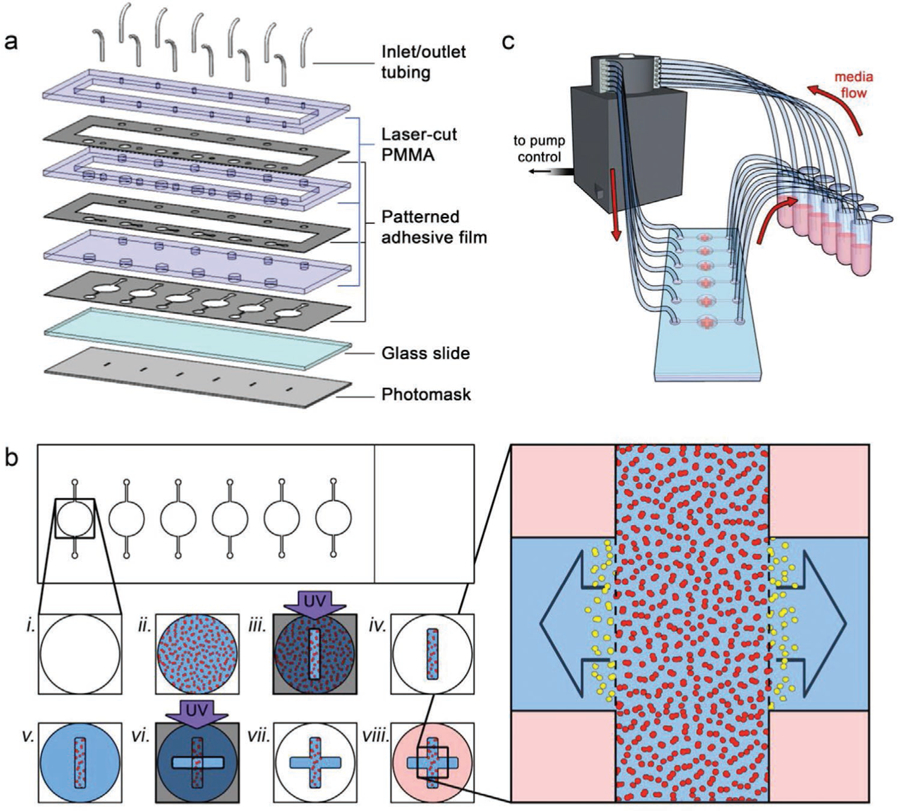
Device and construct fabrication. a) Exploded view of the components used to form the microfluidic system. See Section 4 for details. b) Process workflow for in situ biofabrication of monolithic, multi-region 3D cell culture constructs. A microfluidic chamber (i) is filled with cells (red) in a photopolymerizable HA hydrogel precursor (ii). Ultraviolet light exposure through a slot photomask is used to cross-link the precursor (iii) and the channel is flushed with clean buffer to leave a cell-laden oblong cuboid (iv). New photopolymerizable HA hydrogel precursor (no cells) is then added to the same chamber (v) and an additional UV exposure is performed through a second slot photomask, perpendicular to the first (vi), adding contiguous cell-free regions to the construct that remain after flushing with clean buffer (vii). Media is added to the final construct (viii) to support embedded cells. Right: expanded view of the construct; the inter-region border (dashed lines) defined by the construct geometry can be used to quantify distance and speed of invading cells (yellow) moving into the cell-free regions. c) Schematic view of experimental setup, showing flow from independent media reservoirs controlled via peristaltic pump.
2. Results and Discussion
2.1. Interstitial Flow
Interstitial hypertension is a hallmark of tumors and is thought to be an important driver of metastasis by promoting increased cell movement out of the primary tumor and into the surrounding tissue.[16] This role has been studied previously using microfluidic systems[12] through which interstitial flow, or the movement of fluid through the extracellular matrix (ECM), was shown to have a strong effect on cell migration through a combination of factors that includes shear forces. Our system is particularly well-suited to assess these directional effects, since the geometry of each construct enables analysis of cell motility in two opposing directions simultaneously (see Figure 1c, right). As an initial test, we prepared constructs with HCT-116 cell-laden regions that were oriented perpendicularly to the microfluidic channel such that cell migration into the two cell-free regions necessitated movement with and against the direction of flow, respectively. We then allowed the device to incubate under the constant flow of Dulbecco’s Modified Eagle’s Medium (DMEM, 4 µL min−1) for 10 days. Confocal micrographs of the constructs revealed an asymmetrical infiltration pattern (Figure 2a) with analyses of migration distance distributions (Figure 2b) showing that the mean distance traveled in the direction of flow was 44.3 µm and only 25.1 µm in the direction opposite to the flow. Comparison of the distributions using two-sample Kolmogorov–Smirnov (KS) test confirmed their statistical dissimilarity. Note that while we did not measure the pressure or shear stress generated in our system directly, we do not expect that these values impacted cell behavior. For example, in our past work using a variety of cell types,[17–20] we have utilized higher flow rates (≈10 uL min−1) in comparable microfluidic devices and observed no significant effects.
Figure 2.
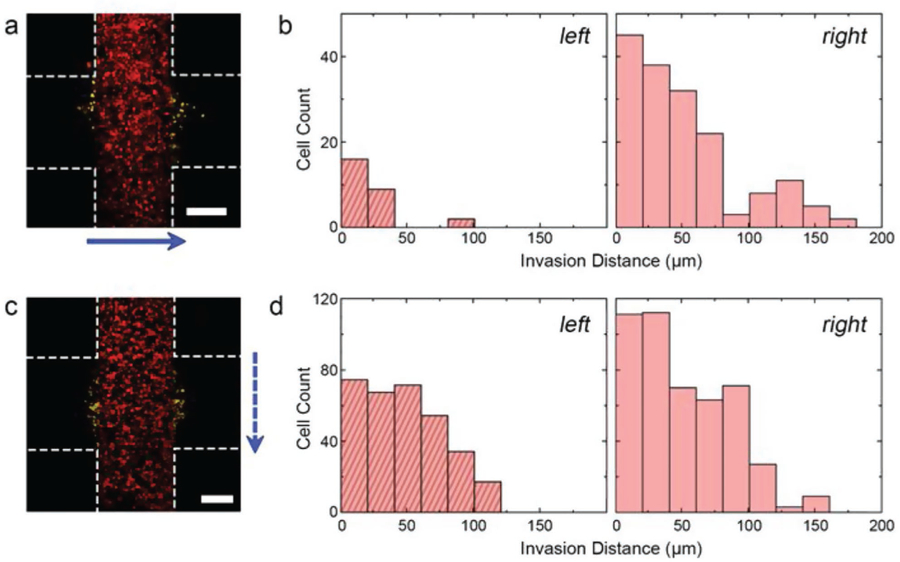
Interstitial flow effects a) maximum projection confocal micrograph of HCT-116 cells after 10 days of continuous flow (4 µL min−1). Blue arrow indicates direction of flow. Construct borders are roughly indicated by white dashed lines (inter-region border not indicated for clarity) and positions of invading cells (i.e., cells entering the cell-free region) are shaded yellow. b) Histograms of distances traveled into the left and right cell-free regions of (a), relative to the respective inter-region borders. c) Maximum projection confocal micrograph of HCT-116 cells after 10 days of intermittent flow (4 µL min−1 for 10 min, no flow for 120 min). Blue arrow shows direction of flow and other indicators match those found in (a). d) Histograms of distances traveled into the left and right cell-free regions of (c), relative to the respective inter-region borders. Scale bars are 300 µm. See also Figure S1, Supporting Information.
Given that the goals of this study also required probing the intrinsic, unbiased cell motility, we sought to establish conditions under which the effects of interstitial flow were mitigated while maintaining sufficient buffer exchange for high cell viability. To accomplish this, we made two changes to the system. First, we reoriented the constructs such that the cell-laden regions were parallel to the direction of flow so that fluid movement would neither oppose nor promote motion into the cell-free regions. Second, we reduced the overall exposure of the constructs to shear forces by incorporating intermittent flow, through which the same 4 µL min−1 flow rate of DMEM was applied for only 10 min followed by a 120 min flow interruption. Under these alternative conditions, we found that cell viability after 10 days remained high (mean, 77%) and also observed that cells moved equivalently into both the cell-free regions of our constructs (Figure 2c). The mean infiltration distance distributions (Figure 2d) on each of the two sides of the construct were 48.8 and 54.5 µm, respectively, with a two-sample KS test showing no statistical difference between populations. Consequently, we used the system under these conditions for all subsequent investigations and considered motility into both cell-free regions jointly.
2.2. Influence of the Chemotherapeutics Agents 5FU and Marimastat on HCT-116 Infiltration
5FU is an antimetabolite fluoropyrimidine analog that inhibits nucleotide synthase[21] and causes apoptosis in cells with high metabolic activity. Owing to this mechanism, it is an effective antiproliferative drug that is employed widely in the treatment of solid tumors, including as the principal chemotherapeutic agent used for the systematic treatment of colorectal cancer. We first used our system to study the effects of 5FU on the migration and viability of HCT-116 cells.
Fabricating four identical sets of cross-shaped migration constructs (three constructs per set), each individual structure was placed into intermittent circulation with DMEM spiked with 5FU at one of four concentrations: 0 (control), 1, 10, or 100 mm. Using maximum projection confocal images acquired across a 10-day incubation period, the number of migrating cells and their dynamics were then determined. Using infiltration histograms (Figure 3a), we observed that the migration of HCT-116 cells under control conditions evolved predictably, with more cells moving across larger distances throughout the experiment. Under 5FU insult, however, the number of cells crossing the boundary into the cell-free region was markedly reduced compared to the control; this reduction did not appear to be drug-concentration-dependent. The mean infiltration distances of cells progressed non-monotonically with time under all conditions, showing a quasi-asymptotic relationship (Figure 4a). We interpreted this shape to be the result of new cells crossing the boundary and thus reducing the mean. By considering the total number of infiltrating cells under all conditions, we found that their counts increased linearly with time (Figure 4c). We could therefore determine the infiltration rate for each condition (Figure 4e) and quantitatively confirm that exposure to 5FU reduced the number of migrating cells in a dose-independent manner. Critically, we found that infiltration dynamics were not sensitive to 5FU, with no significant differences observed between the mean distances traveled with versus without 5FU.
Figure 3.
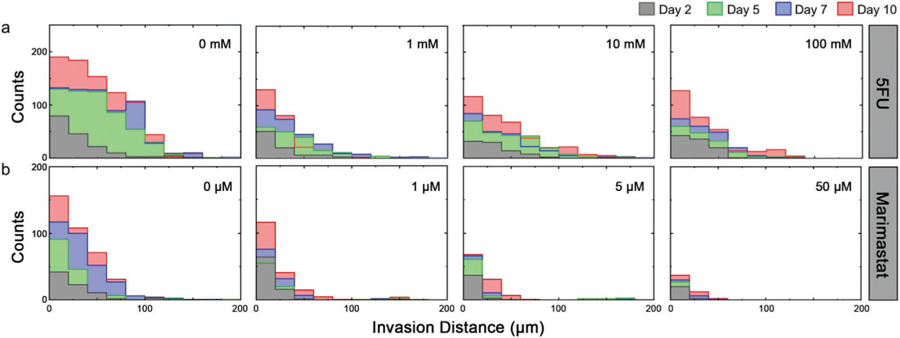
Effects of drug insult on in vitro HCT-116 invasion. Time evolution of histograms for HCT-116 invasion distances (total count across all constructs) under conditions a) 0, 1, 10, and 100 mm 5FU and b) 0, 1, 5, and 50 µm Marimastat. Plots show data for day 2 (grey), 5 (green), 7 (blue), and 10 (red). Source images shown in Figures S2 and S3, Supporting Information.
Figure 4.
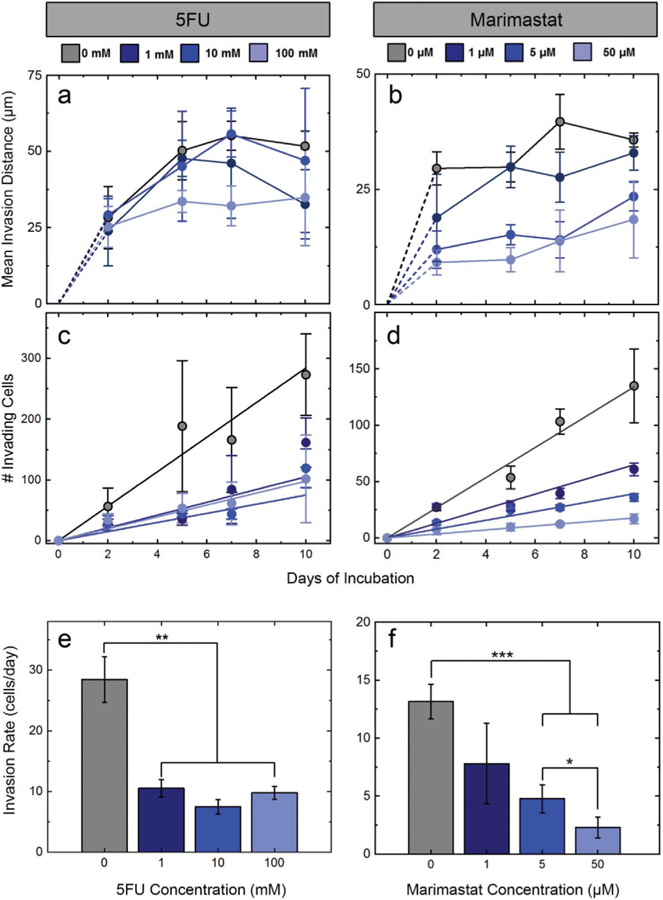
HCT-116 invasion quantification for all conditions and all time points. Colors represent different concentrations of 5FU (left) and Marimastat (right) (indicated at top). a,b) Mean invasion distances and c,d) number of cells crossing into the cell-free region for increasing drug concentrations. Rates of invasion for all concentrations of e) 5FU and f) Marimastat. Drug insult reduces invasion rate for each, but a concentration dependence is observed only for the antimigratory Marimastat. Significance: *p < 0.05, **p < 0.01, ***p < 0.001.
As a final metric, we also analyzed HCT-116 viability in response to 5FU exposure (Figure 5a). Because of the long (10 day) duration of our incubation, decomposition of early-dying cells following apoptosis could significantly influence cell quantification, resulting in an overestimation of the total viability at the conclusion of the experiment. This possibility is supported by the reduction in total cell count (LIVE plus DEAD) observed as a function of 5FU (Figure 5b) despite the use of a uniform initial cell density. To account for this, we calculated L/D ratios by comparing live cell count under each condition to the total average number of cells in the control (0 mm 5FU) constructs on the same day (Figure 5c). Under this alternative scaling, we observed a strong decrease in relative cell viability, reaching as low as 37 ± 5.0% under 100 mm 5FU. Note that a similar but less severe decrease was also observed in direct (non-relative) viability quantification (Figure S4, Supporting Information).
Figure 5.
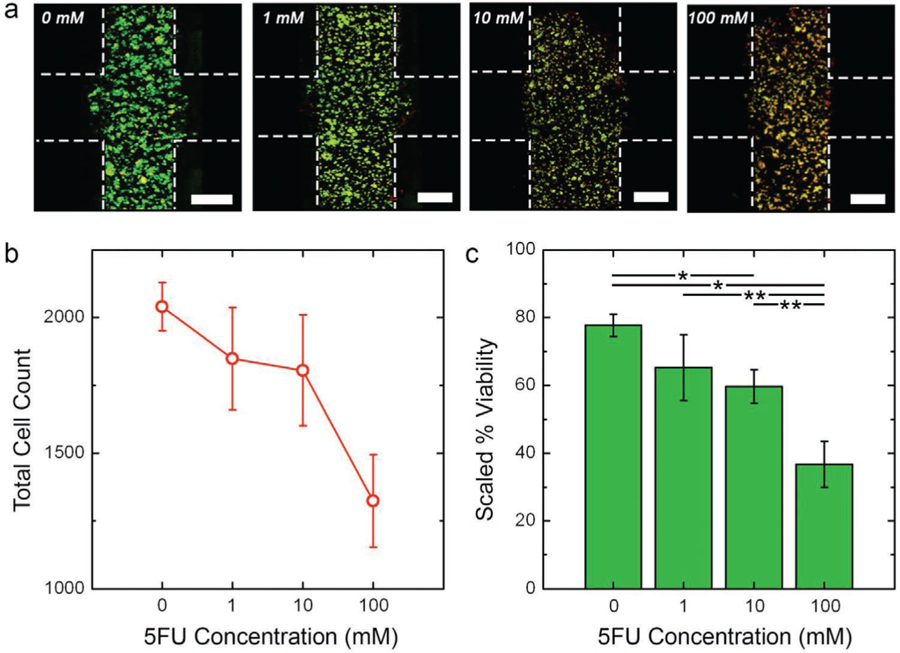
HCT-116 viability under 5FU insult. a) Maximum projection L/D confocal micrographs of HCT-116 cells after 10 days intermittent flow of indicated 5FU concentration. Green cells are live and red cells are dead. Construct borders are roughly indicated by white dashed lines (inter-region border not indicated for clarity) and scale bars are 300 µm. b) Total cell count (live plus dead) on day ten, indicating the net loss of cells as a function of 5FU concentration. c) Scaled viability on day 10 calculated as the ratio of live cells under a given condition to total number of cells in the control (0 mm) construct. This value accounts for dead cell decomposition during the long-term measurement. Significance: *p < 0.01, **p < 0.05.
Taken together, our results were indicative of the anti-proliferative mechanism of 5FU: the drug kills cells efficiently, but resistant phenotypes[22,23] retain the same migratory activity as observed under control conditions. Because cellular motility thways are not known to be directly impacted by 5FU, it is perhaps unsurprising that surviving cells retain native motility. However, decoupling viability from infiltration adds a valuable perspective; for example, previous studies using conventional transwell migration and scratch assays concluded that 5FU produces an apparent decrease in HCT-116 invasiveness.[24,25] However, without accompanying viability data to account for cell death, it is unclear that the observed decrease in the number of migrated cells is a result of direct drug activity or simply a reduction in the total number of viable cells owing to the increasing 5FU exposure. Our results demonstrate that active proliferation and high metabolism (i.e., the cellular states that are prone to 5FU sensitivity) do not necessarily predict invasiveness. This suggests that the effectiveness of 5FU that drives its clinical use in colorectal cancer may only prevent metastasis[26] insomuch as it kills cells that could otherwise metastasize; this concept is also supported by previous work.[27]
As a counter-test to the 5FU measurements, we next investigated an alternate chemotherapeutic drug known to operate through a different mechanism. Marimastat is a synthetic anti-migratory drug that inhibits broad spectrum of MMPs, which are secreted by cancer cells to degrade type IV collagens present in the surrounding ECM, thereby promoting migration and ultimately metastasis.[28] Denatured collagen is a major component of the HA hydrogel scaffold surrounding our cells, suggesting a pathway for Marimastat may significantly impact cell migration in our system. Consequently, we followed the precedent of the 5FU measurements and fabricated four sets of cross-shaped migration constructs to determine the effect of Marimastat on HCT-116 cell migration and viability. The four drug concentrations used were 0 (control), 1, 5, and 50 µm and constructs were probed for the same 10 day incubation period as described above.
Like 5FU, infiltration histograms (see Figure 3b) demonstrated that Marimastat insult reduced the total number of HCT-116 cells migrating into the cell-free region. However, in contrast to prior results, we also observed a strong dose-dependence to this quality. Indeed, direct quantification of the infiltrating cell counts for all conditions (Figure 4d) showed linear increases with time, similar to 5FU above, but their rises were strongly impacted by drug concentration. As a result, significant decreases in infiltration rates (i.e., slopes of cell count data) were found with increasing Marimastat exposure (Figure 4f). Mean infiltration distances were again found to follow a quasi-asymptotic trend, with distances traveled increasing quickly at early time points but then moderating later. However, under Marimastat exposure, we observed a considerable dose-dependent reduction in the distances traveled by the cells (Figure 4b). Considering cell viability (Figure 6a), because the total cell count across drug concentrations was considerably more stable than those treated with 5FU (Figure 6b), the non-scaled viability analysis at the highest concentration (50 µm) of Marimastat on day 10 yielded a value (54 ± 1.5%, Figure S5, Supporting Information) that was comparable to what was found relative to control, as described above (47 ± 2.7%) (Figure 6c). These results again corresponded with mechanism: as an antimigratory drug, Marimastat caused fewer cells to infiltrate the cell-free regions of the constructs and those that did had decreased migratory capacity.
Figure 6.
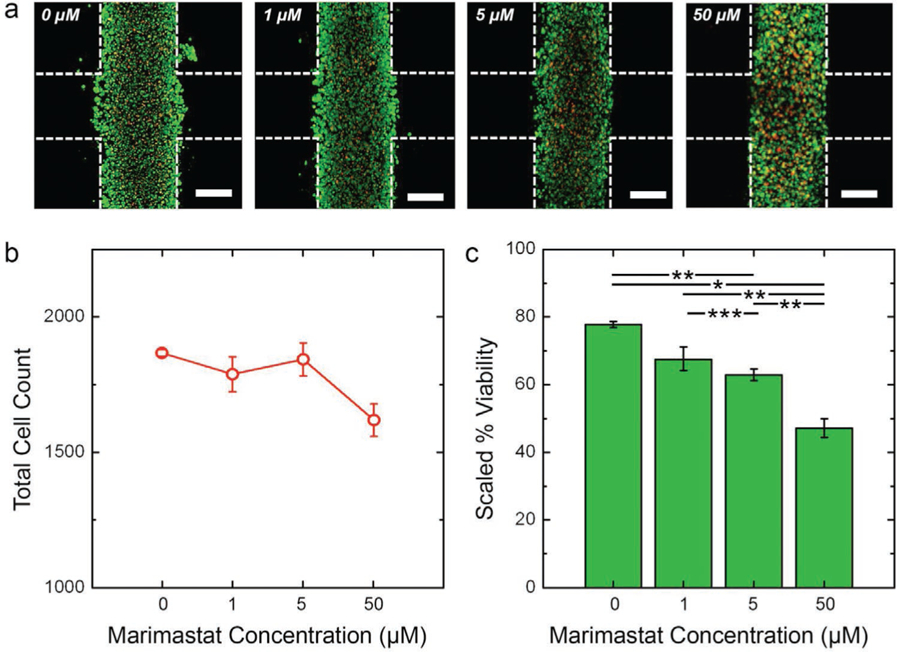
HCT-116 viability under Marimastat insult. a) Maximum projection L/D confocal micrographs of HCT-116 cells after 10 days intermittent flow of indicated 5FU concentration. Green cells are live and red cells are dead. Construct borders are roughly indicated by white dashed lines (inter-region border not indicated for clarity) and scale bars are 300 µm. b) Total cell count (live plus dead) on day ten, indicating the net loss of cells as a function of 5FU concentration. c) Scaled viability on day 10 calculated as the ratio of live cells under a given condition to total number of cells in the control (0 mm) construct. This value accounts for dead cell decomposition during the long-term measurement. Significance: *p < 0.001, **p < 0.01, ***p < 0.05.
Previous studies[29–31] have shown that Marimastat is an anti-metastatic agent that is not effective in inhibiting proliferation of tumor cells.[32,33] It is not a cytotoxic agent, as the pharmacokinetics of the drugs specifically chelates the active site of the MMP and only potently inhibits cell invasion by preventing ECM degradation. It was clear from our experimental results that Marimastat was able to significantly inhibit migration in a dose-dependent manner as seen in preclinical studies.[29,34] Although Marimastat was shown to be a promising chemotherapeutic agent in cell lines and animal models, it has shown no efficacy in clinical trials, and requires combination with anti-proliferative drugs to increase selective cancer cell cytotoxicity.[35] Nevertheless, it serves as an effective mechanistic tool for in vitro studies such as those presented here.
2.3. Selective Hydrogel Dissolution to Isolate Migrated Cells
An advantage of the conventional transwell assay is the ability to retrieve migrated cells for subsequent analyses. The cells from the top chamber pass through the porous membrane of the transwell insert to a lower chamber[36,37] where they can be collected, enabling the examination of the phenotypes that promote invasion. Typically, infiltrating cells are fixed and stained using cytological dyes for quantification using a fluorescent reader; the non-migrated cells in the top chamber are removed prior to staining, commonly with a cotton swab. This is considered tedious and inconsistent; moreover, information about non-migrated cells is lost.[3]
To address this limitation, we employed a strategy of dissolving the hydrogel only in the cell-free region, thus releasing infiltrated cells selectively for downstream analysis. This was achieved by replacing polyethylene glycol diacrylate (PEGDA) in the hydrogel with disulfide-containing PEGDA (PEGSSDA) crosslinkers in the hydrogel precursor, through which disulfide bonds could be formed between thiol-modified hyaluronan and thiol-modified gelatin to form a hydrogel with properties that are comparable to the gel used above.[38] However, the disulfide bonds enabled cleaving through chemical reduction to rapidly dissociate regions formed with this gel in a convenient, non-enzymatic way.[39] Consequently, by using PEGSSDA to form the cell-free regions of our cross-shaped migration construct, the reducing agent N-Acetyl-l-Cysteine (NAC) could subsequently be used to release infiltrated cells selectively into the microfluidic channel for retrieval (Figure 7a).
Figure 7.
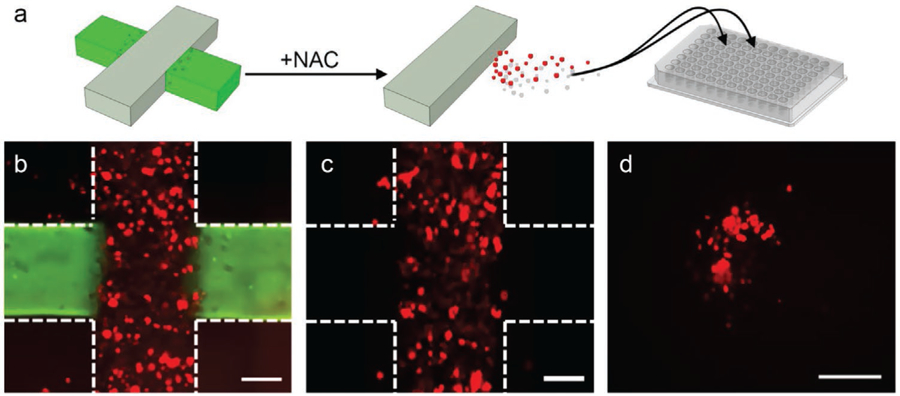
Dissolving of PEGSSDA hydrogel domains. a) Schematic representation of selective recovery of infiltrated cells (red) from a cross structure (left). Non-infiltrating cells in grey region not shown for clarity. NAC is used to dissolve the gel in the cell-free domain (green), releasing only invaded cells into circulation (center) where they can then be recovered and loaded onto a 96-well plate (right) for analysis. Overlaid 2D images of the multi-domain construct on day 10 before (b) and after (c) dissolving the gel with NAC. The vertical cell-laden domain encapsulates mCherry cells (red) and the horizontal domain is stained with Alexafluor 488 for structure visualization. Construct borders are roughly indicated by white dashed lines and scale bars are 300 µm. Following NAC (c), the green fluorescent region (hydrogel with PEGSSDA crosslinker) is selectively removed along with any infiltrated cells it contained. d) 2D image of cells recovered from the construct in a 96-well plate and incubated for 4 h. Scale bar is 300 µm.
As an initial experiment, we incorporated mCherry HCT 116 cells into the PEGDA/PEGSSDA cross structures. After a 10-day incubation period as described above, constructs were imaged to ensure the presence of both the cell-laden and cell-free domains and to confirm cell movement (Figure 7b). We then introduced media containing 50 mm NAC to the device and incubated. Subsequent imaging after treatment (Figure 7c) showed selective dissolution of the cell-free regions only, leaving the cell-laden zone intact and releasing infiltrated cells into the surrounding fluid. Indeed, by recovering the media after dissolution and transferring it into a cell culture plate, we observed cell growth (Figure 7d) that confirmed the liberation of infiltrated cells.
For heterogeneous cell mixtures, selective retrieval could enable the identification and differentiation of invasive and non-invasive phenotypes. To demonstrate this concept, we implemented a co-culture approach in which the cell-laden region was populated with both the highly invasive HCT-116 cells and minimally invasive[40] Caco-2 cells (Figure 8a). After a 7-day incubation, the PEGSSDA matrix with infiltrated cells was again dissociated using NAC, and the media was collected and imaged (Figure 8b). Based on these analyses, we found that the invasive HCT-116 cells accounted for 92.4 ± 2.1% of retrieved cells compared to 7.6 ± 2.1% for minimally invasive Caco-2 cells (Figure 8c). This result demonstrates that highly invasive phenotypes can be isolated selectively from cell mixtures, and shows the utility of the assay in investigating patient-derived samples.
Figure 8.
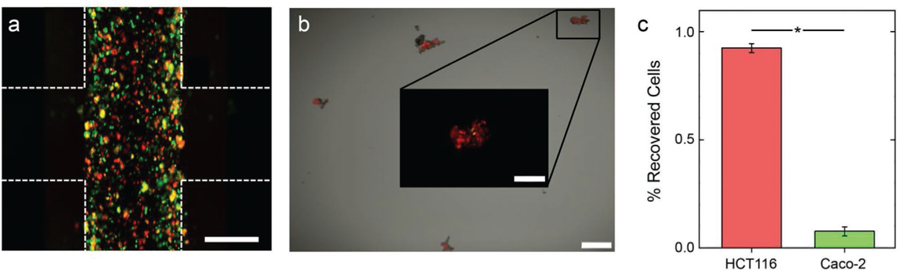
Isolating migrated cells in co-culture of mCherry HCT116 and Caco-2. a) Overlaid 2D image of the multi-domain construct with horizontal cell-laden domain encapsulating mCherry HCT116 cells (red) and Qtracker 525-labeled Caco-2 cells (green) on day 10 of the experiment before dissolving the PEGSSDA gel. Construct borders are roughly indicated by white dashed lines and scale bar is 300 µm. b) 2D image of cells recovered from the construct, loaded on a 96-well plate, and incubated for 4 h. Scale bar is 150 µm. Inset shows red and green signals for a zoom of a typical region. c) Percentage of all recovered cells analyzed in the red (HCT116) and green (Caco-2), showing the invasive phenotype is dominant in the infiltrated population. Significance: *p < 0.005.
3. Conclusions
In this study, we have presented a simple and powerful new assay for the direct co-assessment of cellular migration and viability. Using serial photopatterning, we biofabricated monolithic 3D hydrogel constructs featuring localized cell-laden and cell-free regions with well-defined borders inside a pre-formed microfluidic device. The approach enabled straightforward, parallel control over buffer conditions and supported the direct imaging and assessment of cell invasion over extended timeframes and of viability via conventional double-stain fluorescence assays.
As a demonstration, we used the platform to probe highly invasive human colon carcinoma cells (HCT-116) over 10 days, showing that extrinsic effects of shear can be negated. We exploited the parallel nature of our microfluidic device to perform a drug study by investigating the concentration-dependent effects of two chemotherapeutic agents in common use: 5FU, a thymidylate synthase inhibitor, and Marimastat, an MMP inhibitor. We showed that increasing chronic exposure to 5FU decreased cell viability and consequently the number of invading cells but did not significantly impact the invasive activity of surviving cells. In contrast, we found that Marimastat reduced both the number and extent of infiltration. Our observations are in line with the antiproliferative and antimigratory mechanisms of the drugs, respectively, and highlight that 5FU alone prevents migration only by decreasing the probability of metastatic invasion through reducing the total cell population. Cells capable of escaping 5FU cytotoxicity through low metabolism, cell cycle arrest, or another means of chemoresistance can retain an invasive phenotype.
In addition, we showed an ability to retrieve invading cells selectively via the targeted dissociation of hydrogel. By incorporating PEGSSDA as the matrix for the cell-free regions, infiltrated cells could be released and collected for downstream analysis. We demonstrated this concept by isolating invasive HCT-116 cells relative to the minimally invasive Caco-2 cell line.
Our overall assay is well-positioned to expand toward further applications and address additional aspects of tumor cell invasion. For example, an identical biofabrication approach could be used to incorporate a broad range of other cell types, including both cell lines and patient-derived cells.[18] To support this, the hydrogel can be engineered to match the properties of arbitrary tissue types,[41–43] and can be supplemented with additional factors to better represent physiological ECM.[41,44,45] In addition, the flexibility of photopatterning supports the ability to produce more complex constructs, potentially featuring multiple, discrete regions containing different cell types to investigate the effects of cell–cell interactions. The adaptability of the microfluidic-architecture itself allows for integration with more complex subsystems for complementary processing. It also supports the rapid temporal control of conditions, potentially allowing clinical treatment schedules to be reproduced in vitro. Finally, selective retrieval of infiltrated cells will allow the assessment of mutations or biomarkers associated with the invasive phenotype and enable independent drug studies on that subpopulation alone. Taken together, our simple system is a powerful addition to the tumor-on-a-chip[46] toolbox.
4. Experimental Section
Microfluidic Device Fabrication:
Microfluidic devices were produced using a low-cost, rapid-prototyping approach pioneered by Cooksey et al.[47] based on patterned adhesive films. Six discrete channels were formed in an adhesive film (140 µm thickness, part number 3M9495MPF, Strouse, Westminster, MD) using a computer-controlled razor plotter (CE6000–40, GraphTec, Irvine, CA) and layered on a clean glass microscope slide (VWR, Radnor, PA). Next, a polymethylmethacrylate (PMMA, McMaster-Carr, Elmhurst, IL) sheet cut to the same slide dimensions was furnished with inlet/outlet openings using a laser etcher (Full Spectrum Laser H-series, Las Vegas, NV) and subsequently layered on the patterned film to facilitate access to the enclosed channels. Even pressure was applied to ensure proper sealing of the device. The completed structure was further layered with four layers of patterned adhesive films and laser etched PMMA to form an on-device bubble trap based on the design of Zheng et al.[48] to aid in preventing bubble formation or deposition in the channel that could interfere with performance. The functioning of the bubble trap component is described in more detail in Figure S6, Supporting Information. Finally, polytetrafluoroethylene tubing was inserted into each port in the PMMA slide for fluid delivery and was secured using a UV-cure polyester resin (Solarez, Vista, CA). Each channel in the device was connected to an independent reservoir by Silastic tubing (Corning, Inc., Corning, NY) through a micro-peristaltic pump (MP2 Precision, Elemental Scientific, Inc., Omaha, NE) to facilitate flow through all channels simultaneously. Cell culture constructs were maintained under intermittent flow (4 µL min−1 for 10 min, followed by 120 min with no flow), as controlled by an in-house-designed LabView program (National Instruments, Austin, TX) for the duration of the experiments unless otherwise noted. The purpose of the intermittent flow was to introduce a fresh supply of nutrients and oxygen at regular intervals as well as remove any high local concentration of waste products. Because the system was closed, secreted signaling molecules were recirculated.
Culturing of Cells:
A human colon carcinoma cell line (HCT-116) engineered to express mCherry[49] was used in all experiments. For co-culture experiments, human epithelial colorectal adenocarcinoma cells (Caco-2) cells were also used. Both cell types were cultured independently in 15 cm round cell culture dishes using 15 mL of DMEM-10 supplemented with 10% v/v fetal bovine serum, 1% v/v l-glutamine, and 1% v/v penicillin–streptomycin solution. Cultures were maintained in an incubator at 37 °C with 5% CO2. Cells were passaged twice a week upon reaching 70–80% confluency using trypsin (0.05%, Difco Laboratories, Detroit, MI). Cells were collected using the same trypsinization approach prior to their incorporation into microfluidic devices.
Cell Labeling Using Qtracker:
Caco-2 cells with no intrinsic fluorophore expression were tracked using Qtracker 525 (ThermoFisher, Waltham, MA) according to the manufacturer’s instructions; this delivered green-fluorescent Qdot 525 nanocrystals into the cytoplasm of live cells. Briefly, a 20 nm labeling solution was prepared by mixing 2 µL each of Qtracker Components A and B in DMEM-10 media. The cell suspension (≈1.8 × 107 cells mL−1) was added to the labeling solution and incubated at 37 °C for 60–90 min. The cells were mixed thoroughly in the labeling solution by pipetting every 15 min during the incubation to improve cell labeling efficiency. The cells were then washed twice with DMEM-10 and the labeling was confirmed through fluorescence imaging of a small aliquot. The labeled cells were then centrifuged to form a pellet for use in experiments.
Hydrogel Preparation:
The HA/gelatin hydrogel (HyStem-HP, ESI-BIO, Alameda, CA) was prepared as described elsewhere.[17,43,50] Briefly, sterile water was mixed with 0.05% w/v 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone photoinitiator (Sigma Aldrich, St. Louis, MO) and then used to produce solutions of thiol-modified hyaluronan (Heparasil), thiol-modified gelatin (Gelin-S), and thiol-reactive PEGDA crosslinker (Extralink) at concentrations of 1% w/v each. The Heparasil, Gelin-S, and Extralink solutions were then mixed at a ratio of 2:2:1 v/v, respectively, to form the HA hydrogel precursor. For experiments requiring downstream dissolving of hydrogel, the PEGDA crosslinker was replaced with PEGSSDA crosslinker. Remaining components were prepared as described above. To accelerate cell migration in co-culture, 200 ng mL−1 of human stromal cell-derived factor 1-alpha (SDF-1α or CXCL12, Peprotech, Inc., Rocky Hill, NJ) were added to the PEGSSDA mixture.
In Situ Biofabrication of 3D Cell Culture Constructs:
Hydrogel constructs were produced in a pre-formed microfluidic device using in situ photopatterning.[51] To achieve this, photomasks were prepared from an aluminum foil/adhesive film bilayer using a razor plotter and attached directly to the bottom surface of the glass slide (see Figure 1a). The photomasks featured rectangular slots with dimensions 3.0 × 0.5 mm at the center of each microfluidic chamber. Unless otherwise noted, slots were oriented parallel to the direction of flow. The HA hydrogel precursor mixed with HCT-116 cells at a concentration of 3.6 × 107 cells mL−1 were introduced to all the channels in the device (Figure 1c,i and ii) through the inlet ports using a syringe or a pipette, and was subsequently exposed through the photomask (Figure 1c, iii) to initiate rapid thiol-ene crosslinking with UV light using a handheld source (BlueWave 75, Dymax, Torrington, CT; 365 nm wavelength, 18 W cm−2) held ≈12 cm from the device surface for 3 s. Given the distance from the UV source, we estimate a total dose delivered to the cells of <0.2 W cm−2. Note that this exposure level has not been found to induce any significant perturbation to cell behavior.[52] The uncrosslinked precursor was then flushed away using fresh phosphate buffered saline (PBS, Figure 1c, iv), leaving discrete 3D constructs of hydrogel-encapsulated cells spanning from the bottom surface of the channel to the top surface as defined by the photomask. For co-culturing HCT-116 and Caco-2 cells, the HA precursor was mixed with both cell types, each at a concentration of 1.8 × 107 cells mL−1 to maintain the same total cell density as above. Next, the photomask was replaced with another that featured identical slots rotated by 90°. The same HA hydrogel precursor without cells was then added to the device channels (Figure 1c, v) and exposed to UV light as above (Figure 1c, vi) to yield cross-shaped hydrogel constructs with cell-free regions in two arms (Figure 1c, vii). Precursor containing PEGSSDA (with or without SDF-1α) was used in experiments requiring select regions to be dissolved. Clean PBS was again used to flush any uncrosslinked precursor before the introduction of culture media (DMEM-10, Figure 1c, viii).
Cancer Cell Motility Assessment:
Direct imaging of constructs was performed using an Olympus FluoView FV1000 confocal microscope. For all time points except day 10, an image of the complete construct was formed by stitching together multiple z-stacks (5 µm steps) collected using a 559 nm laser excitation wavelength (mCherry; red) and performing a maximum intensity projection into a single 2D image. On day 10, a comparable image was acquired by combining the red and green channel (λ = 405 nm) signals following LIVE/DEAD (L/D) staining of the sample (see below). Note that HCT-116 mCherry fluorescence intensity decreased by 90–95% by day 10 due to photobleaching and other effects so that it was not a significant perturbation to the combined L/D signal. Microscope sensitivity was sufficient to resolve mCherry fluorescence easily at all other time points despite reduction in signal intensity. For all data sets, infiltration distances were quantified using the Imaris MeasurementPro software (Bitplane, Concord, MA) by determining the position of each cell (see Figures S2 and S3, Supporting Information) relative to the border defined by the adjacent arms of the cross-shaped structure (see the dashed line in Figure 1c, right, as an example).
Live/Dead Cell Viability Determination:
L/D solution comprised 2 µm calcein-AM and 2 µm ethidium homodimer-1 (LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells, ThermoFisher, Waltham, MA) in 1 mL of DMEM-10 and PBS mixed in a 1:1 ratio. After flushing each channel with clean PBS, the L/D solution was introduced and incubated for 1 h. Channels were flushed again with clean PBS prior to imaging. Cell viability for each construct was determined by calculating the ratio of the number of cells in the 405 nm (green) channel (LIVE) to the total number of cells in both the green (LIVE) and the 559 nm (red) channels (DEAD).
Selective Recovery of Migrated Cells:
The cell-free PEGSSDA regions were dissolved to release migrated cells by cleaving the disulfide bonds with NAC (Sigma-Aldrich, St. Louis, MO) using a modified version of the manufacturer’s protocol (ESI-BIO, Alameda, CA) as described elsewhere.[39,44] To aid with visualization, fluorescent dye (Alexa Fluor 488 maleimide) was mixed with the PEGSSDA hydrogel precursor to covalently bond with the thiols in the hydrogel network. For the co-culture experiments, the chemokine SDF-1α was added directly to the PEGSSDA precursor that was used for the cell-free domain to promote directed cell migration. The heparin component of the hydrogel provides a role similar to heparan sulfate proteoglycans in ECM,[53] forming an ionic bond with the SDF-1α and facilitating slow and localized release of the chemokine. This method promoted the cells to specifically migrate into the cell-free domain in contrast to mixing the chemoattractant into the media which would promote non-directional migration. After 7–10 days of cell migration in the construct (as indicated in the text), the channels were flushed with clean DMEM-10 to wash away any cells that were present outside of the construct. Next, 50 mm NAC was prepared in DMEM-10 and the pH adjusted to 7.4. Then 30 µL of the NAC solution was added to each channel using a pipette or syringe and the device was incubated for 2 h at 37 °C in 5% CO2. After incubation, the fluid in each channel was slowly removed using a syringe and introduced into the individual wells of a 96 well plate, and 100 µL of clean DMEM-10 media was added to each well containing any retrieved cells. Each construct in the microfluidic device was imaged after the experiment to confirm that the cell-free domain was completely dissolved and that the cell-laden region remained intact. The well plate was incubated at 37 °C with 5% CO2 for 4 h to promote cell adhesion. To remove residual dissolved hydrogel, the media was aspirated from the well plate and each well was rinsed with PBS before fresh DMEM-10 was added to the migrated cells recovered from the cell construct. The wells were imaged in fluorescence using Olympus IX83 Inverted Microscope. It was important to image prior to 12 h to avoid any cell proliferation effects on the isolated cells.
Statistical Analysis:
For each experimental condition, identical constructs were prepared in triplicate to enable statistical analyses. Student’s t-tests were used to determine differences between means with p < 0.05 considered statistically significant. The two-sample KS test was used for non-parametric comparison of distributions. Confidence intervals of 95% or better were considered to be significant.
Supplementary Material
Acknowledgements
A.R.H. acknowledges start-up funds from the Wake Forest School of Medicine. A.S. acknowledges funds from the Wake Forest Baptist Medical Center Clinical and Translational Science Institute Open Pilot Program. The authors gratefully acknowledge the Wake Forest Comprehensive Cancer Center’s Cellular Imaging Shared Resource (through NCI Support Grant P30CA012197) and Dr. Frank Marini for confocal microscopy support.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Shiny A. P. Rajan, Virginia Tech-Wake Forest School of Biomedical Engineering and Sciences, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA Wake Forest Institute for Regenerative Medicine, Wake Forest School of Medicine Medical Center, Winston-Salem, NC 27101, USA.
Aleksander Skardal, Virginia Tech-Wake Forest School of Biomedical Engineering and Sciences, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA; Wake Forest Institute for Regenerative Medicine, Wake Forest School of Medicine Medical Center, Winston-Salem, NC 27101, USA; Comprehensive Cancer Center at Wake Forest Baptist Medical, Medical Center Boulevard, Winston-Salem, NC 27157, USA; Department of Cancer Biology, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA; Department of Biomedical Engineering, The Ohio State University and The Ohio State University Comprehensive, Cancer Center, The Ohio State University Wexner Medical Center, USA.
Adam R. Hall, Virginia Tech-Wake Forest School of Biomedical Engineering and Sciences, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA Wake Forest Institute for Regenerative Medicine, Wake Forest School of Medicine Medical Center, Winston-Salem, NC 27101, USA; Comprehensive Cancer Center at Wake Forest Baptist Medical, Medical Center Boulevard, Winston-Salem, NC 27157, USA.
References
- [1].Chaffer CL, Weinberg RA, Science 2011, 331, 1559. [DOI] [PubMed] [Google Scholar]
- [2].van Zijl F, Krupitza G, Mikulits W, Mutat. Res./Rev. Mutat. Res 2011, 728, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kramer N, Walzl A, Unger C, Rosner M, Krupitza G, Hengstschläger M, Dolznig H, Mutat. Res./Rev. Mutat. Res 2013, 752, 10. [DOI] [PubMed] [Google Scholar]
- [4].Boyden S, J. Exp. Med 1962, 115, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Todaro GJ, Lazar GK, Green H, J. Cell. Comp. Physiol 1965, 66, 325. [DOI] [PubMed] [Google Scholar]
- [6].Chen Y-C, Allen SG, Ingram PN, Buckanovich R, Merajver SD, Yoon E, Sci. Rep 2015, 5, 9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nie F-Q, Yamada M, Kobayashi J, Yamato M, Kikuchi A, Okano T, Biomaterials 2007, 28, 4017. [DOI] [PubMed] [Google Scholar]
- [8].Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE, Cell 2012, 148, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chan KT, Asokan SB, King SJ, Bo T, Dubose ES, Liu W, Berginski ME, Simon JM, Davis IJ, Gomez SM, Sharpless NE, Bear JE, J. Cell Biol 2014, 207, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chung S, Sudo R, Mack PJ, Wan C-R, Vickerman V, Kamm RD, Lab Chip 2009, 9, 269. [DOI] [PubMed] [Google Scholar]
- [11].Jeong GS, Kwon GH, Kang AR, Jung BY, Park Y, Chung S, Lee S-H, Biomed. Microdevices 2011, 13, 717. [DOI] [PubMed] [Google Scholar]
- [12].Polacheck WJ, Charest JL, Kamm RD, Proc. Natl. Acad. Sci. USA 2011, 108, 11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Businaro L, De Ninno A, Schiavoni G, Lucarini V, Ciasca G, Gerardino A, Belardelli F, Gabriele L, Mattei F, Lab Chip 2013, 13, 229. [DOI] [PubMed] [Google Scholar]
- [14].Mi S, Du Z, Xu Y, Wu Z, Qian X, Zhang M, Sun W, Sci. Rep 2016, 6, 35544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blaha L, Zhang C, Cabodi M, Wong JY, Biofabrication 2017, 9, 045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lunt SJ, Chaudary N, Hill RP, Clin. Exp. Metastasis 2009, 26, 19. [DOI] [PubMed] [Google Scholar]
- [17].Skardal A, Murphy SV, Devarasetty M, Mead I, Kang H-W, Seol Y-J, Shrike Zhang Y, Shin S-R, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Jin Lee S, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE, Atala A, Sci. Rep 2017, 7, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mazzocchi AR, Rajan SAP, Votanopoulos KI, Hall AR, Skardal A, Sci. Rep 2018, 8, 2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aleman J, George SK, Herberg S, Devarasetty M, Porada CD, Skardal A, Almeida-Porada G, Small 2019, 15, 1902971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aleman J, Skardal A, Biotechnol. Bioeng 2019, 116, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Longley DB, Harkin DP, Johnston PG, Nat. Rev. Cancer 2003, 3, 330. [DOI] [PubMed] [Google Scholar]
- [22].De Angelis PM, Svendsrud DH, Kravik KL, Stokke T, Mol. Cancer 2006, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bose D, Zimmerman LJ, Pierobon M, Petricoin E, Tozzi F, Parikh A, Fan F, Dallas N, Xia L, Gaur P, Samuel S, Liebler DC, Ellis LM, Br. J. Cancer 2011, 105, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhu P, Zhao N, Sheng D, Hou J, Hao C, Yang X, Zhu B, Zhang S, Han Z, Wei L, Zhang L, Sci. Rep 2016, 6, 20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Seo K, Ki SH, Park EY, Shin SM, Arch. Pharmacal Res 2017, 40, 231. [DOI] [PubMed] [Google Scholar]
- [26].Otsubo D, Yamashita K, Fujita M, Nishi M, Kimura Y, Hasegawa H, Suzuki S, Kakeji Y, Anticancer Res 2015, 35, 4425. [PubMed] [Google Scholar]
- [27].Warusavitarne J, Ramanathan P, Kaufman A, Robinson BG, Schnitzler M, Int. J. Colorectal Dis 2006, 21, 625. [DOI] [PubMed] [Google Scholar]
- [28].Deryugina EI, Luo GX, Reisfeld RA, Bourdon MA, Strongin A, Anticancer Res 1997, 17, 3201. [PubMed] [Google Scholar]
- [29].Rasmussen HS, McCann PP, Pharmacol. Ther 1997, 75, 69. [DOI] [PubMed] [Google Scholar]
- [30].Hidalgo M, Eckhardt SG, Natl J Cancer Inst 2001, 93, 178. [DOI] [PubMed] [Google Scholar]
- [31].Fraley SI, Wu P, He L, Feng Y, Krisnamurthy R, Longmore GD, Wirtz D, Sci. Rep 2015, 5, 14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Waleh NS, Murphy BJ, Zaveri NT, Cancer Lett 2010, 289, 111. [DOI] [PubMed] [Google Scholar]
- [33].Lv Y, Zhao X, Zhu L, Li S, Xiao Q, He W, Yin L, Theranostics 2018, 8, 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kimata M, Otani Y, Kubota T, Igarashi N, Yokoyama T, Wada N, Yoshimizu N, Fujii M, Kameyama K, Okada Y, Kumai K, Kitajima M, Jpn. J. Cancer Res 2002, 93, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brown PD, Expert Opin. Invest. Drugs 2000, 9, 2167. [DOI] [PubMed] [Google Scholar]
- [36].Albini A, Iwamoto Y, Kleinman HK, Martin GR, Aaronson SA, Kozlowski JM, McEwan RN, Cancer Res 1987, 47, 3239. [PubMed] [Google Scholar]
- [37].Albini A, Benelli R, Nat. Protoc 2007, 2, 504. [DOI] [PubMed] [Google Scholar]
- [38].Ogawa M, Jing H, Kitts DD, Nakai S, Nakamura S, Med J Food 2003, 6, 317. [DOI] [PubMed] [Google Scholar]
- [39].Zhang J, Skardal A, Prestwich GD, Biomaterials 2008, 29, 4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].de Both NJ, Vermey M, Dinjens WN, Bosman FT, Br. J. Cancer 1999, 81, 934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Skardal A, Zhang J, Prestwich GD, Biomaterials 2010, 31, 6173. [DOI] [PubMed] [Google Scholar]
- [42].Skardal A, Devarasetty M, Kang H-W, Mead I, Bishop C, Shupe T, Lee SJ, Jackson J, Yoo J, Soker S, Atala A, Acta Biomater 2015, 25, 24. [DOI] [PubMed] [Google Scholar]
- [43].Skardal A, Murphy SV, Crowell K, Mack D, Atala A, Soker S, J. Biomed. Mater. Res., Part B 2017, 105, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Skardal A, Sarker SF, Crabbe A, Nickerson CA, Prestwich GD, Biomaterials 2010, 31, 8426. [DOI] [PubMed] [Google Scholar]
- [45].Skardal A, Devarasetty M, Kang H-W, Seol Y-J, Forsythe SD, Bishop C, Shupe T, Soker S, Atala A, J. Vis. Exp 2016, 110, e53606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tsai H-F, Trubelja A, Shen AQ, Bao G, Soc JR, Interface 2017, 14, 20170137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cooksey GA, Atencia J, Lab Chip 2014, 14, 1665. [DOI] [PubMed] [Google Scholar]
- [48].Zheng W, Wang Z, Zhang W, Jiang X, Lab Chip 2010, 10, 2906. [DOI] [PubMed] [Google Scholar]
- [49].Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA, Cancer Res 2012, 72, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Skardal A, Devarasetty M, Forsythe S, Atala A, Soker S, Biotechnol. Bioeng 2016, 113, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Skardal A, Devarasetty M, Soker S, Hall AR, Biofabrication 2015, 7, 031001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ciesielska S, Bil P, Gajda K, Poterala-Hejmo A, Hudy D, Rzeszowska-Wolny J, PLoS One 2019, 14, e0205215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Amara A, Lorthioir O, Valenzuela A, Magerus A, Thelen M, Montes M, Virelizier JL, Delepierre M, Baleux F, Lortat-Jacob H, Arenzana-Seisdedos F, J. Biol. Chem 1999, 274, 23916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.