

Abstract
Mitochondria are metabolic organelles essential not only for energy transduction, but also a range of other functions such as biosynthesis, ion and metal homeostasis, maintenance of redox balance, and cell signaling. A hallmark example of how mitochondria can rebalance these processes to adjust cell function is observed in macrophages. These innate immune cells are responsible for a remarkable breadth of processes including pathogen elimination, antigen presentation, debris clearance, and wound healing. These diverse, polarized functions often include similarly disparate alterations in the metabolic phenotype associated with their execution. In this chapter, mitochondrial bioenergetics and signaling are viewed through the lens of macrophage polarization: both classical, pro-inflammatory activation and alternative, anti-inflammatory activation are associated with substantive changes to mitochondrial metabolism. Emphasis is placed on recent evidence that aims to clarify the essential – rather than associative – mitochondrial alterations, as well as accumulating data suggesting a degree of plasticity within the metabolic phenotypes that can support pro- and anti-inflammatory functions.
1. Introduction
1.1. Mitochondria in cell physiology
Mitochondria play essential roles in cell physiology including, and extending far beyond, serving as the sites of oxidative phosphorylation. In addition to synthesizing the majority of ATP in most cell types (Nicholls and Ferguson, 2013; Pagliarini and Rutter, 2013), mitochondria are also hubs for biosynthesis, Ca2+ handling, iron homeostasis, redox balance, and signal transduction (Chandel, 2014; Murphy and Hartley, 2018; Shadel, 2012). Mitochondrially derived signals can be as varied as protein release from the intermembrane space (Jiang and Wang, 2004), metabolite efflux (Ryan et al., 2019), redox signals [e.g. reactive oxygen species (ROS) production] (Finkel, 2011; Murphy, 2009; Reczek and Chandel, 2015), and release of damage-associated molecular patterns (DAMPs) (Grazioli and Pugin, 2018; Picca et al., 2017).
It is increasingly appreciated that the roles of mitochondria in energy metabolism and as a signaling organelle are not necessarily discrete. In fact, one purpose of mitochondria as a signaling organelle is as a communicator of metabolic health status, serving a checkpoint or feedback function to instruct nuclear gene expression and cellular function (Chandel, 2015). Perhaps reflective of this, some proteins with essential roles in energy transduction have dual functions in pathways associated with cell death and disease. For example, cytochrome c is an essential component of the electron transport chain, but also triggers caspase-mediated apoptosis when released to the cytoplasm upon mitochondrial outer membrane permeabilization (Jiang and Wang, 2004; Tait and Green, 2010). Similarly, the FOF1-ATP synthase that generates ATP during oxidative phosphorylation also dimerizes to form the mitochondrial permeability transition pore, a channel causing inner membrane permeability and causative of pathologies such as ischemic heart injury and muscular dystrophy (Bernardi, 2013). This mitochondrial plasticity in shifting from energy powerhouse to signaling platform often involves a classical definition of reduced or impaired mitochondrial function (lowered oxidative phosphorylation, redox imbalance, etc.) (Jazwinski, 2013). Remarkably, however, emerging evidence in murine bone marrow-derived macrophages (BMDMs) suggests this does not necessarily translate to reduced or impaired cellular function.
1.2. Macrophage function and activation
Macrophages are cells of the innate immune system responsible for a remarkable breadth of functions essential to human health including killing pathogens, clearing subcellular debris, and repairing tissue (Adams and Hamilton, 1984). In addition to their role in healthy physiology, targeting macrophage function may lead to improved therapies for a variety of pathologies including tissue fibrosis, cancer, and metabolic disease (Wynn and Vannella, 2016).
Macrophages were first described over 130 years ago by Metchnikoff, who suggested that the observed phagocytosis by ameboid cells could be a means of host defense rather than a deleterious consequence of tissue damage or infection (Gordon, 2016). Specificity for the functional role and activation state of a macrophage at any given time is set by coordinating intrinsic, extrinsic, and environmental cues (Murray, 2017). This allows for the broad and sometimes opposing range of functions (i.e. “kill or repair”) these cells must execute. This so-called polarization state is often simplified through the classification of pro-inflammatory “M1” and anti-inflammatory “M2” macrophages (Gordon and Taylor, 2005). The nomenclature is drawn from the response to cytokines secreted by subsets of CD4+ helper T (Th) cells. A Th1 response that is associated with immunity to bacteria and infections and secretion of IFN-γ and tumor necrosis factor-α (TNF-α), whereas a Th2 response counteracts the microbicidal Th1 response and is associated with allergy and helminth immunity along with production of interleukin-4 (IL-4), IL-10, and IL-13.
Pro-inflammatory activation can occur via detection of pathogens by pattern recognition receptors (PRRs), proteins present at both the plasma and endosomal membranes (O’Neill et al., 2013). PRRs bind subsets of molecular scaffolds typically associated with microbes [pathogen associated molecular patterns (PAMPs)] or tissue damage [damage associated molecular patterns (DAMPS)]. Innate PRRs are broadly classified in two classes: membrane-bound, including Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic, such as NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs) (Akira and Takeda, 2004). Regardless of their localization, PRRs function by receptor-mediated activation of broad transcriptional programs, resulting in an orchestrated inflammatory response to eliminate pathogens or damaged cells (Barton and Medzhitov, 2003).
In addition to activation by PAMPs and DAMPs, macrophages can also be polarized to a range of activation states by secreted cytokines. This can occur in both paracrine [e.g. pro-inflammatory interferon-γ (IFN-γ) or anti-inflammatory IL-4 secreted by lymphocytes] and autocrine [e.g. pro-inflammatory interleukin-1β (IL-1β) or anti-inflammatory transforming growth factor-β (TGF-β)] manners upon receptor binding (Mosser and Edwards, 2008). In some cases, activation of PRR- and cytokine receptor-mediated activation integrate to amplify the inflammatory response. In the hallmark example, activation of TLR4, which recognizes the gram-negative bacterial membrane component lipopolysaccharide (LPS), synergizes with IFN-γ to create a powerful, composite pro-inflammatory signal (Held et al., 1999).
It is often accepted that the M1/M2 framework is based on rigidly prescribed conditions that rarely, if ever, occur physiologically, and do not necessarily reflect the spectral nature of macrophage activation (Martinez and Gordon, 2014). Nonetheless, the use of tightly controlled, in vitro polarization assays coupled with modern metabolic techniques has revealed profound and unexpected roles for several aspects of mitochondrial function in the control of the innate immune response.
An essential role for metabolism in macrophage function may be somewhat obvious in the traditional definition, as activation-induced phagocytosis, motility, and cytokine synthesis are all ATP-consuming processes that require commensurate changes in ATP production. Indeed, early biochemical studies showed that glucose consumption in naïve, unstimulated macrophages is far below its enzymatic capacity, suggesting a capacity to quickly upregulate metabolic pathways in response to external stimuli (Newsholme et al., 1986). Abundant genetic and pharmacologic evidence now exists to demonstrate several essential roles for metabolism in shaping macrophage function, and these are surveyed in multiple comprehensive reviews (Arts et al., 2016a; Benmoussa et al., 2018; Caputa et al., 2019; Geeraerts et al., 2017; Langston et al., 2017; O’Neill and Pearce, 2016; Odegaard and Chawla, 2011; Russell et al., 2019; Van den Bossche et al., 2017; Viola et al., 2019; Weinberg et al., 2015). Such roles include increased and/or rerouted metabolic flux through specific pathways, post-translational modifications by metabolic intermediates, and signaling initiated by metabolites, redox triggers, or nucleic acids.
In the present manuscript, we focus on mitochondria as a hub for both energy transduction as well as metabolite and signal generation, and discuss how these different modes are used to shape macrophage function. It is unquestioned that, in murine macrophages, oxidative metabolism is associated with anti-inflammatory activation (stimulation with IL-4 ± IL-13), and classical inflammatory activation (LPS ± IFN-γ) involves the collapse of oxidative phosphorylation and repurposing of mitochondria towards accumulation of metabolites and other pro-inflammatory triggers. However, the essential, targetable metabolic requirements to adjust macrophage function may not be as black-or-white as initially thought. In fact, a crude analogy could be drawn to macrophage polarization itself. Similarly to how the M1/M2 paradigm is perhaps too bifurcated and discrete to reflect the graded nature of macrophage function (Martinez and Gordon, 2014; Murray, 2017), so too may be a one-size-fits-all approach that suggests ‘polarized’ metabolic phenotypes are indispensable for pro- or anti-inflammatory macrophage activation. Rather, accumulating evidence suggests bioenergetic and signaling roles for mitochondria in both pro- and anti-inflammatory macrophage activation, although these manifest in different ways.
2. Mitochondrial energy metabolism in macrophage activation
2.1. Overview of mitochondrial energy metabolism.
Of course, the best described function of mitochondria is energy metabolism and the production of ATP through oxidative phosphorylation (Nicholls and Ferguson, 2013). Mitochondria generate ATP through a series of energy transducing processes. The chemical energy in nutrients such as sugars, amino acids, and fatty acids is first harvested through the tricarboxylic acid (TCA) cycle (Figure 1). The TCA cycle (or Krebs cycle) is a series of eight consecutive enzymes that oxidize energy-rich substrates in order to generate the reduced electron carriers NADH and FADH2. During oxidative TCA cycle metabolism (conventionally drawn clockwise as in Figure 1), three dehydrogenases – isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (α-KGDH), and malate dehydrogenase (MDH) – generate NADH. Additionally, succinate dehydrogenase (SDH) acitivity generates FADH2, and ATP is generated through substrate-level phosphorylation by the succinyl-CoA synthetase reaction.
Figure 1 – Overview of TCA cycle bioenergetics.
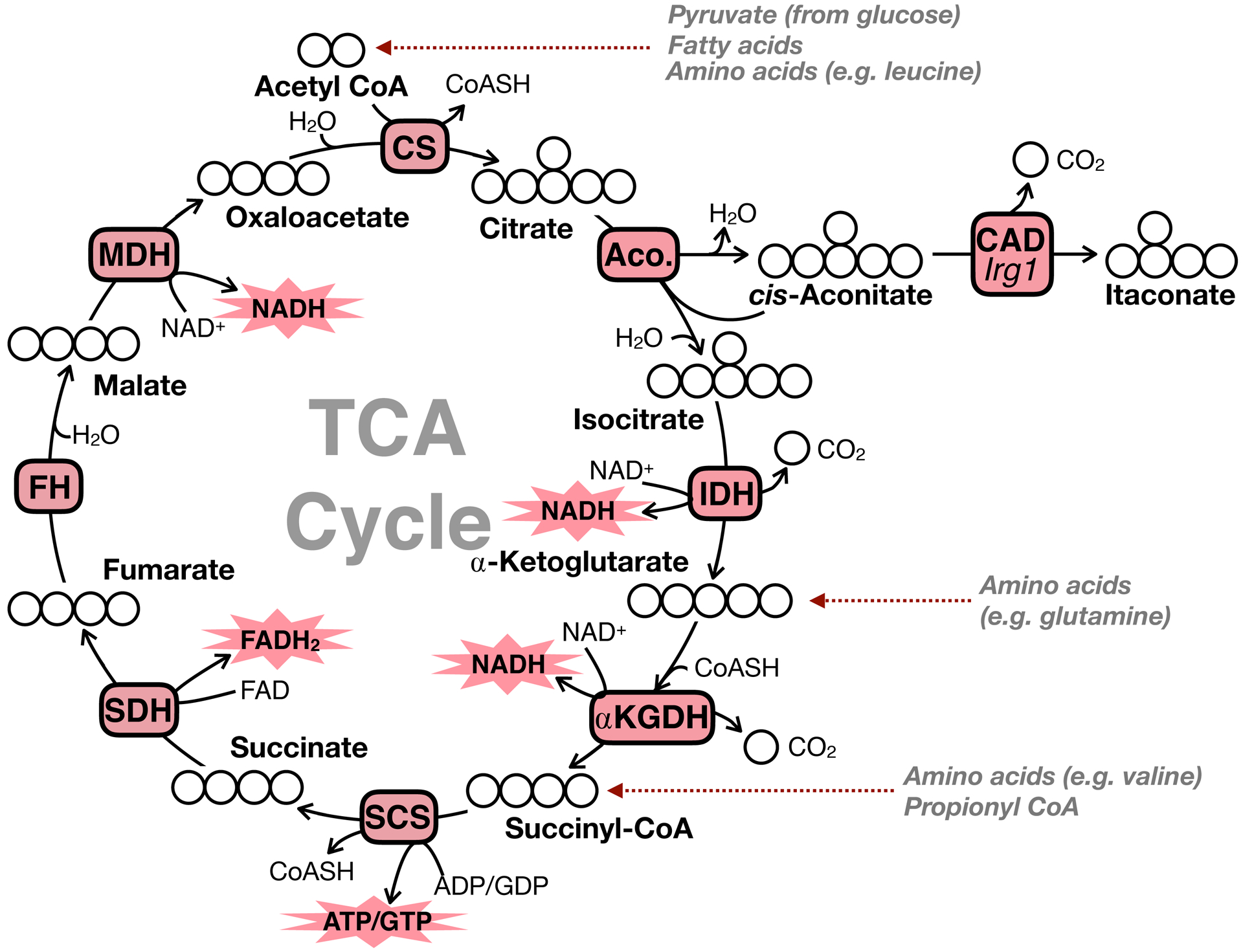
The TCA cycle is a series of 8 reactions which, from a bioenergetic perspective, provide electrons to the respiratory chain in the form of the reduced electron carriers NADH and FADH2. Isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase generate NADH when driven in the oxidative direction. Succinate dehydrogenase (respiratory complex II) oxidizes succinate to generate FADH2. CS, citrate synthase; Aco., aconitase; CAD, cis-aconitate decarboxylase; Irg1, immunoresponsive gene 1; IDH, isocitrate dehydrogenase; α-KGDH, α-ketoglutarate dehydrogenase; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase.
The reduced NADH and FADH2 generated by the TCA cycle and other mitochondrial dehydrogenases [e.g. pyruvate dehydrogenase (PDH), β-hydroxybutyrate dehydrogenase, glycerol-3-phosphate dehydrogenase] are then passed through the mitochondrial respiratory chain. The respiratory chain (or electron transport chain) is a series of energetically favorable electron transfer reactions that are coupled to pumping protons out of the matrix and against their concentration gradient (Rich and Maréchal, 2010). Respiratory complex I oxidizes NADH and transfers the electrons through a series of iron-sulfur clusters to ultimately reduce ubiquinone to ubiquinol (Q to QH2). This energy harvesting reaction is used to drive the translocation of protons out of the matrix (Hirst, 2013). Complex II (SDH) also generates QH2 using the electrons stripped from succinate during its oxidation. Importantly, SDH activity is not directly linked to complex I activity and, unlike complexes I, III, and IV, does not result in net proton translocation (Iverson, 2013). The reduced ubiquinol generated from activity of complexes I and II activity is re-oxidized by respiratory complex III: the electrons from QH2 are transferred to cytochrome c (Crofts, 2004). Finally, electron transfer from cytochrome c to reduce molecular oxygen (½O2 to H2O) at complex IV is in the last step of the respiratory chain.
The resulting potential energy harnessed from respiratory chain activity (10 H+ pumped for every electron pair passed from NADH to oxygen) is used to drive ATP synthesis during oxidative phosphorylation. Activity of the ATP synthase, often called respiratory complex V, relieves the proton gradient (i.e. H+ re-entry into the matrix) to catalyze phosphorylation of ADP (Walker, 2013). Best estimates currently show that per mole of glucose, oxidative phosphorylation generates a maximum of ~31.5 moles of ATP (Mookerjee et al., 2017).
Broadly, mitochondrial energetics can be considered as a balance between process that generate this potential energy and those that consume it (Figure 2) (Divakaruni and Brand, 2011). Under most conditions, the mitochondrial electron transport chain is the dominant processes that generates the proton motive force (pmf). As described earlier, the respiratory chain links exergonic (energetically “downhill”) electron transfer reactions, beginning with the oxidation of NADH by complex I or FADH2 by complex II, to endergonic (energetically “uphill”) proton translocation. Importantly, though, many enzymes in mitochondria are reversible and respond to thermodynamic cues (Murphy, 2015). For example, when the membrane potential drops below threshold levels, the ATP synthase and NAD(P) transhydrogenase can operate in the ‘reverse’ direction to help maintain the pmf (Nicholls and Ferguson, 2013; Nickel et al., 2015).
Figure 2 – Overview of mitochondrial bioenergetics and membrane potential.
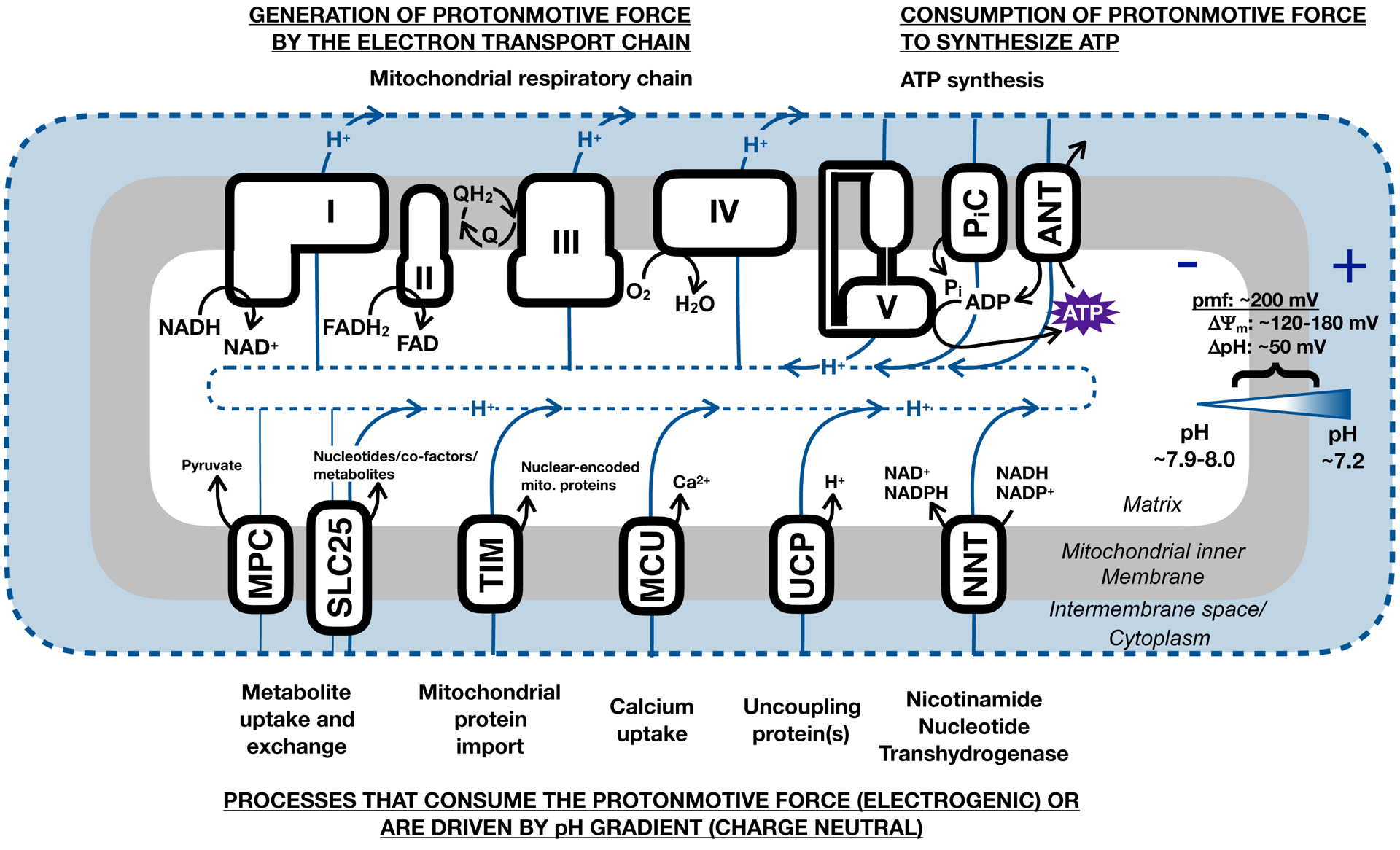
A reservoir of potential energy exists across the mitochondrial inner membrane due to the H+ gradient across the inner membrane. This consists of both an electrical component (Δψm), due to disparate charge across the membrane, and a chemical component, due to the differential pH (ΔpH). The resulting protonmotive force (pmf) can be used to drive a range of energy-demanding reactions central to mitochondrial activity. I-V, mitochondrial respiratory complexes I-IV and the ATP synthase (complex V); PiC, inorganic phosphate carrier; ANT, adenine nucleotide translocase; NNT, nicotinamide nucleotide transhydrogenase [also NAD(P) transhydrogenase]; UCP, uncoupling protein; MCU, mitochondrial calcium uniporter; TIM, translocase of the inner membrane; SLC25, solute carrier family 25; MPC, mitochondrial pyruvate carrier.
The pmf is used to drive a range of mitochondrial processes (Brand and Nicholls, 2011), some of which are presented here. During oxidative phosphorylation, the membrane potential is used to drive not just the rotary catalysis of the ATP synthase, but also the prerequisite import of inorganic phosphate and ADP into the matrix against the charge gradient (both are anionic) (LaNoue and Schoolwerth, 1979). The NAD(P) transhydrogenase, or nicotinamide nucleotide transhydrogenase, helps maintain reducing power in the matrix by oxidizing NADH to generate NADPH, an energetically unfavorable process that consumes the potential energy across the inner membrane to proceed (Hoek and Rydstrom, 1988; Murphy, 2015). Mitochondrial import of nuclear encoded proteins also consumes the membrane potential (Wiedemann and Pfanner, 2017), as does the electrogenic exchange of glutamate and aspartate by SLC25A12 in the malate-aspartate shuttle (LaNoue and Tischler, 1974). Mostly, uptake of metabolites into the matrix is charge neutral due to exchange with other metabolites, although uniport of substrates such as pyruvate is aided by the pH gradient to bring anions into the matrix along with H+ co-transport (or OH− antiport) (Palmieri, 2013; Taylor, 2017). Ion channels, notably the mitochondrial calcium uniporter (MCU), will rapidly consume the membrane potential to drive Ca2+ uptake (De Stefani et al., 2014; Duchen, 2000). Additionally, uncoupling proteins are H+ channels that dissipate the membrane potential to unlink (or ‘uncouple’) the activity of the respiratory chain to ATP synthesis (Divakaruni and Brand, 2011). UCP1 activity drives non-shivering thermogenesis in brown adipose tissue (Cannon and Nedergaard, 2004), and it remains contested as to whether unannotated UCPs catalyze so-called proton leak (Vozza et al., 2014). Clearly, a role for the mitochondrial membrane potential includes, but importantly extends far beyond, mitochondrial ATP synthesis.
2.2. Energy metabolism and inflammatory macrophage activation
Although perhaps the best studied feature of classical inflammatory macrophage activation is a striking increase in glycolytic flux (Cramer et al., 2003; Haschemi et al., 2012; Tannahill et al., 2013), profound mitochondrial remodeling occurs as well. Experiments since the 1970s have sought to link inflammatory triggers such as LPS to altered mitochondrial function (Kato, 1972), though recent appreciation of the integration of metabolism and signaling (Chandel, 2015) coupled to breakthrough technologies that increased the scope of functional mitochondrial measurements in their cellular context (Buescher et al., 2015; Divakaruni et al., 2014) have brought exponential progress. A landmark study in 2010 noticed similarities between the signaling pathways activated upon growth factor ligation to their cognate receptors and those activated during TLR ligation (Krawczyk et al., 2010). Given that activation of signaling pathways downstream of growth factor receptors leads to altered cellular metabolism (Christofk et al., 2008; Shaw, 2006), it was hypothesized that similar metabolic changes would also be observed upon activation of dendritic cells (DCs, antigen-presenting innate immune cells). Upon activation with LPS, DCs increase glycolysis and decrease respiration (Krawczyk et al., 2010). This hallmark metabolic shift is also observed upon classic inflammatory activation with LPS ± IFN-γ in BMDMs. This was initially demonstrated at the level of gene expression (Rodríguez-Prados et al., 2010) and later reinforced with functional assays that showed that metabolic changes can present as early as 4 hours (Haschemi et al., 2012; Tannahill et al., 2013).
There are likely multiple mechanisms that cause the reduction in mitochondrial ATP production. A contributing factor is undoubtedly the production of high concentrations of nitric oxide upon activation. Induction of inducible nitric oxide synthase (iNOS, the Nos2 gene), which catalyzes NO production via metabolism of arginine to citrulline (Leone et al., 1991), is dramatically upregulated upon activation with LPS ± IFN-γ (Han et al., 1999). Nitric oxide production is important for the bactericidal and tumoricidal function or macrophages (Adams et al., 1990; MacMicking et al., 1997; Stuehr and Nathan, 1989). Moreover, it also inhibits oxidative phosphorylation in BMDMs and DCs, as rates of ATP-linked mitochondrial respiration are rescued (to varying degrees) upon both genetic ablation or pharmacologic inhibition of iNOS (Bailey et al., 2019; Everts et al., 2012; Garedew and Moncada, 2008; Van den Bossche et al., 2017). Inhibitory effects of NO on oxidative mitochondrial metabolism can manifest via direct inhibition of respiratory complexes, such as competitive inhibition of respiratory complex IV by NO or S-nitrosation of respiratory complex I (Chouchani et al., 2013; Cleeter et al., 1994; Clementi et al., 1998). Additionally, nitric oxide may affect expression of mitochondrial proteins, as expression of the NADH-binding domain of complex I Ndufv2 is reduced upon activation with LPS + IFN-γ in wild-type BMDMs but unchanged on a Nos−/− background (Bailey et al., 2019). However, in many cases the respiratory inhibition upon inflammatory activation is only partially rescued with genetic or pharmacologic inhibition of iNOS (Bailey et al., 2019; Van den Bossche et al., 2016), suggesting nitric oxide-independent means of reprogramming mitochondrial metabolism during inflammatory activation.
Additional mechanisms likely include disruption of oxidative TCA cycle metabolism. Of course, an inability to generate NADH or FADH2 due to decreased expression/activity of TCA cycle dehydrogenases necessarily slows respiratory chain activity and, as a result, oxidative phosphorylation. LPS stimulation results in a decrease in gene expression of both IDH and MDH in BMDMs (Jha et al., 2015; Tannahill et al., 2013), and changes observed with metabolomics and stable isotope tracing confirm reduced IDH activity and a bonafide break in the TCA cycle (De Souza et al., 2019; Jha et al., 2015). Multiple mechanisms likely contribute to the reduced IDH activity observed during pro-inflammatory activation. IDH expression is reportedly repressed by the orphan nuclear receptor Nur77 (Koenis et al., 2018) as well as type I interferon signaling (De Souza et al., 2019). Although genetic downregulation does not always manifest in altered IDH protein abundance (Bailey et al., 2019), it is well established that enzyme activity is decreased. Other TCA cycle enzymes and mitochondrial dehydrogenases show reduced activity upon pro-inflammatory activation as well (Cordes et al., 2016; Palmieri et al., 2018), and the extent to which control over respiratory inhibition is distributed among these various enzymes is not completely understood.
Additionally, mitochondrial reactive oxygen species (ROS) production may be another mechanism by which mitochondrial ATP production is reduced. In addition to microbicidal superoxide produced by the plasma membrane-bound NADPH oxidase (Cathcart, 2004), mitochondrial ROS production is thought to be an important signal during inflammatory macrophage activation (West et al., 2011). Mitochondrial adaptations that support ROS production may necessarily involve reductions in oxidative phosphorylation. Of course, mitochondrial ROS production is directly affected by the bioenergetic status of mitochondria, though it is important to draw a distinction between ROS production associated with mitochondrial dysfunction (Adam-Vizi and Starkov, 2010) versus broader programs that reappropriate mitochondria away from oxidative phosphorylation and towards a generation of ROS as a signal. For example, mitochondria alter respiratory chain supercomplex assembly upon bacterial recognition in a way that may support mitochondrial ROS production but reduce oxidative capacity through specific pathways over time (Garaude et al., 2016). Moreover, during LPS activation, BMDM mitochondria are repurposed away from oxidative phosphorylation and towards production of reactive oxygen species. In fact, mitochondria remarkably maintain an elevated membrane potential despite a profound collapse of oxidative phosphorylation, suggesting a reset balance between pmf production and consumption and perhaps hydrolysis of glycolytically-derived ATP to help maintain the membrane potential (Mills et al., 2016).
Although it is well established that profound respiratory inhibition occurs in murine BMDMs upon activation with LPS (± IFN-γ) treatment, the functional reason for this remains somewhat unclear. For example, a reduced respiratory rate during inflammatory activation could be necessary for (i) enhancing glycolytic flux to fuel cytoplasmic NADPH production and anabolism, (ii) generating redox signals such as ROS from reverse electron transport, or (iii) increasing abundance of TCA cycle-associated signaling metabolites by slowing Krebs cycle turnover. However, each of these processes can occur, in principle, without the near-total collapse of respiratory chain activity observed during LPS + IFN-γ treatment.
Moreover, it is likely that our current models for mitochondrial repurposing during classical pro-inflammatory macrophage activation may not be one-size-fits-all. For example, inflammatory stimuli such as IFN-γ treatment (without LPS) (Wang et al., 2018a), the TLR4 agonist monophospholipid A (Fensterheim et al., 2018), or low concentrations of LPS (10 ng/mL) (Cordes et al., 2016) do not elicit similarly dramatic reductions in mitochondrial ATP production in BMDMs. Moreover, maximal respiratory capacity is actually increased early upon bacterial recognition (Garaude et al., 2016) or LPS + IFN-γ stimulation (Cameron et al., 2019), and genetic models that blunt NO production (thereby enhancing respiration) increase production of succinate and itaconate in response to LPS + IFN-γ stimulation. The mitochondrial bioenergetic changes observed in murine BMDMs upon LPS activation are also largely absent in human peripheral blood monocyte (PBMC)-derived macrophages (Vijayan et al., 2019). As such, the precise functional role of mitochondria during inflammatory activation in murine BMDMs, and whether the same mechanisms are present in human macrophages, are not fully understood.
2.3. Energy metabolism and anti-inflammatory activation
Given the array of different macrophage functions coupled with the requisite role of metabolism in macrophage activation, it follows that there is tremendous diversity in the metabolic phenotypes as well. Of course, the most straightforward examples of this are the almost entirely disparate phenotypes observed between LPS- and IL-4-polarized macrophages (Van den Bossche et al., 2017). While LPS ± IFN-γ-activated macrophages depress oxidative phosphorylation, anti-inflammatory activation with IL-4 is associated with increased respiratory capacity and mitochondrial metabolism.
The first indication that mitochondrial metabolism was associated with IL-4-driven (or “alternative”) activation was increased expression of enzymes associated with the TCA cycle, fatty acid oxidation genes, and drivers of mitochondrial biogenesis such as Pgc1b (Haschemi et al., 2012; Vats et al., 2006). Mitochondrial mass increases, as does maximal respiratory capacity upon IL-4 activation (Huang et al., 2014; Vats et al., 2006). Additionally, biochemical and functional evidence is supported by genetic evidence for an essential role for mitochondria and/or lipid metabolism, as the nuclear receptor PPAR-γ, a master transcription factor regulating lipid homeostasis, is essential for IL-4-driven polarization (Odegaard et al., 2007).
It is unquestionable that enhanced mitochondrial metabolism is associated with anti-inflammatory activation, and multiple metabolic pathways likely fuel this phenotype. For example, glucose-driven oxidative phosphorylation increases in response to IL-4 (Covarrubias et al., 2016; Huang et al., 2016; Vats et al., 2006). The inhibitory analog 2-deoxyglucose (2-DG), which blocks glucose oxidation after its cellular uptake, also decreases many markers of IL-4 polarization in vitro and blunts the in vivo response to infection with the gastrointestinal helminth H. polygyrus (Covarrubias et al., 2016; Huang et al., 2016; Tan et al., 2015). Indeed, data suggests that some process dependent on mitochondrial glucose oxidation, rather than glycolysis, may explain the dependency. 2-DG blocks the IL-4-response, but cells grown in medium supplemented with galactose can polarize normally (Wang et al., 2018b). Moreover, knockdown of the pyruvate dehydrogenase kinase-1 (PDK1), which relieves kinase-mediated repression of PDH activity (Harris et al., 1997), also can reportedly boost expression of some anti-inflammatory markers (Tan et al., 2015). Likewise, inhibition of the mitochondrial pyruvate carrier, which blocks uptake of glucose-derived pyruvate into mitochondria, lowers expression of the IL-4 associated markers resistin-like molecule alpha (RELMα) and programmed cell death 1 ligand 2 (PD-L2) (Huang et al., 2016).
In addition to increases in glucose metabolism, increased oxidation of glutamine and fatty acids are also enhanced in BMDMs upon alternative activation. IL-4 enhances expression of the plasma membrane glutamine transporter ASCT2 and steady-state glutamine levels (Palmieri et al., 2017; Tavakoli et al., 2017). Glutamine withdrawal from the experimental medium or inhibition of glutaminase also blunt the IL-4 response (Jha et al., 2015; Liu et al., 2017). Increased fatty acid oxidation (FAO) is also universally recognized as being strongly associated with anti-inflammatory macrophage activation (Gonzalez-Hurtado et al., 2017; Namgaladze and Brüne, 2014; Odegaard et al., 2007; Vats et al., 2006). A suite of genes related to lipid metabolism and oxidation increase in response to IL-4, and functional assays with radiolabeled long chain fatty acids also show a substantial increase in fatty acid uptake and oxidation (Gonzalez-Hurtado et al., 2017; Vats et al., 2006).
Whether this fatty acid oxidation in an essential and targetable feature of the IL-4 response, however, has been the subject of debate and may be context-dependent (Van den Bossche and van der Windt, 2018). Indeed, the carnitine palmitoyltransferase-1 (CPT-1) inhibitor etomoxir can block IL-4-driven polarization at 200 μM (Covarrubias et al., 2016; Huang et al., 2016, 2014). However, accumulating pharmacologic and genetic evidence suggests this is likely due to an off-target effect of the drug, a reactive epoxide with several non-specific effects (Ceccarelli et al., 2011; Nomura et al., 2016; Raud et al., 2018). Lower concentrations of the drug that demonstrably block fatty acid oxidation do not inhibit the IL-4 response (Divakaruni et al., 2018; Namgaladze and Brüne, 2014; Tan et al., 2015), and high concentrations of etomoxir block IL-4-driven activation even in models where CPT-1a or CPT-2 have been genetically ablated (Divakaruni et al., 2018; Nomura et al., 2016).
The association between enhanced activities of multiple metabolic pathways with IL-4-associated differentiation suggests a degree of metabolic plasticity with IL-4-polarized macrophages (Wang et al., 2018b), and raises the question of whether the role of mitochondria is primarily bioenergetic or more closely aligned with biosynthesis and signaling. Indeed, IL-4 polarization can proceed when BMDMs are cultured in galactose (Wang et al., 2018b), which will increase reliance on oxidative phosphorylation for ATP, and in the presence of a range of respiratory chain inhibitors (which will result in glycolysis needing to meet almost all of the cellular ATP demand) (Divakaruni et al., 2018). These results suggest a threshold requirement of ATP, irrespective of the pathway used for its generation. However, this is not without contradictory data. Multiple, independent reports have shown that chemical inhibition of oxidative phosphorylation or blocking the expression of mitochondrial proteins via reduced hypusination of the translation factor eukaryotic initiation factor 5A (eIF5A) reduces markers of the IL-4 response (Puleston et al., 2019; Van den Bossche et al., 2016; Vats et al., 2006).
Multiple, non-exclusive reasons might underlie the discrepancy. Importantly, it is well accepted that metabolic regulation of alternative activation is mosaic, as metabolic modulators frequently only regulate a subset of IL-4-associated genes (Covarrubias et al., 2015; Divakaruni et al., 2018; Sanin et al., 2018). As such, varying results may be due to differential readouts of genes and/or markers which are used to classify the IL-4 response. For example, it is possible that respiratory chain inhibition could affect the abundance of individual cell surface markers, but perhaps change neither gene expression nor the population of cells positive for multiple markers. Results may also be less reflective of the role of oxidative phosphorylation but rather attributable to different macrophage preparations and their glycolytic capacity to meet a threshold ATP demand. The broader use of functional assays or larger-scale -OMICS data beyond expression of select genes and proteins could therefore prove powerful in linking essential metabolic processes to alternative macrophage activation. Additional clarity could also come from using standardized, minimal concentrations of tool compounds to avoid potential off-target effects of respiratory chain inhibitors, which are often used at concentrations far exceeding the concentrations required for maximal effects. Nonetheless, our current understanding remains that mitochondrial metabolism is crucial to the IL-4 response, though this dependence may not be solely attributable to bioenergetic function.
3. Mitochondrial signaling and macrophage activation
3.1. Mitochondrial signaling in cell physiology
One of the more exciting and remarkable developments in mitochondrial biology over the past decade has been appreciation of the myriad of ways in which the organelle links metabolism to cell physiology via TCA cycle metabolites (Dang and Su, 2017; Lu and Thompson, 2012; Mills et al., 2017; Ryan et al., 2019; Sciacovelli and Frezza, 2017; Sivanand et al., 2018). Much like bioenergetics, early inspiration likely came from the field of cancer metabolism following the discovery that cancers linked to TCA cycle mutations accumulate metabolites that can alter transcription factor activation and epigenetic remodeling (Lu et al., 2012; Selak et al., 2005; Xiao et al., 2012; Xu et al., 2011). Indeed, some of these same principles underlie how TCA cycle metabolites can promote genetic and epigenetic changes during macrophage activation (Covarrubias et al., 2016; Liu et al., 2017; Tannahill et al., 2013).
However, the unique induction of the immunometabolite itaconate upon inflammatory macrophage activation stands out as an archetypal example of repurposing mitochondrial metabolism towards metabolite production to execute cell-specific functions (Cordes et al., 2015; O’Neill and Artyomov, 2019). In addition to TCA cycle metabolites, ROS (Garaude et al., 2016; West et al., 2011) and release of nucleic acids (Dhir et al., 2018; Shimada et al., 2012; Zhong et al., 2018) are also emerging as mitochondrially derived signals that can shape the immune response.
3.2. Mitochondrial signaling in inflammatory macrophage activation
Roles for many TCA cycle metabolites have been identified as important in shaping macrophage phenotypes (Figure 3) (Arts et al., 2016b; Infantino et al., 2011; Liu et al., 2017), and production of the immunomodulatory metabolites itaconate and succinate are hallmark examples of how mitochondria can be repurposed to support macrophage function (Cordes et al., 2015; Mills and O’Neill, 2014; O’Neill and Artyomov, 2019). The anti-microbial properties of itaconic acid have well known for decades as an inhibitor of the isocitrate lyase, an enzyme in the glyoxylate shunt (McFadden and Purohit, 1977). This pathway, non-functional or absent in mammals but necessary for long-term persistence of some bacterial infections (Muñoz-Elías and McKinney, 2005), allows biosynthesis and anaplerotic reactions to occur from acetyl-CoA-producing nutrients such fatty acids, thereby allowing bacterial survival under glucose-limiting conditions (Dolan and Welch, 2018). Additionally, the coenzyme A ester of itaconate (itaconyl-CoA) can inhibit bacterial methylmalonyl-CoA mutase, an integral enzyme in bacterial strains which catabolize propionate as an energy source (Ruetz et al., 2019).
Figure 3 – Non-bioenergetic roles of TCA cycle metabolites.
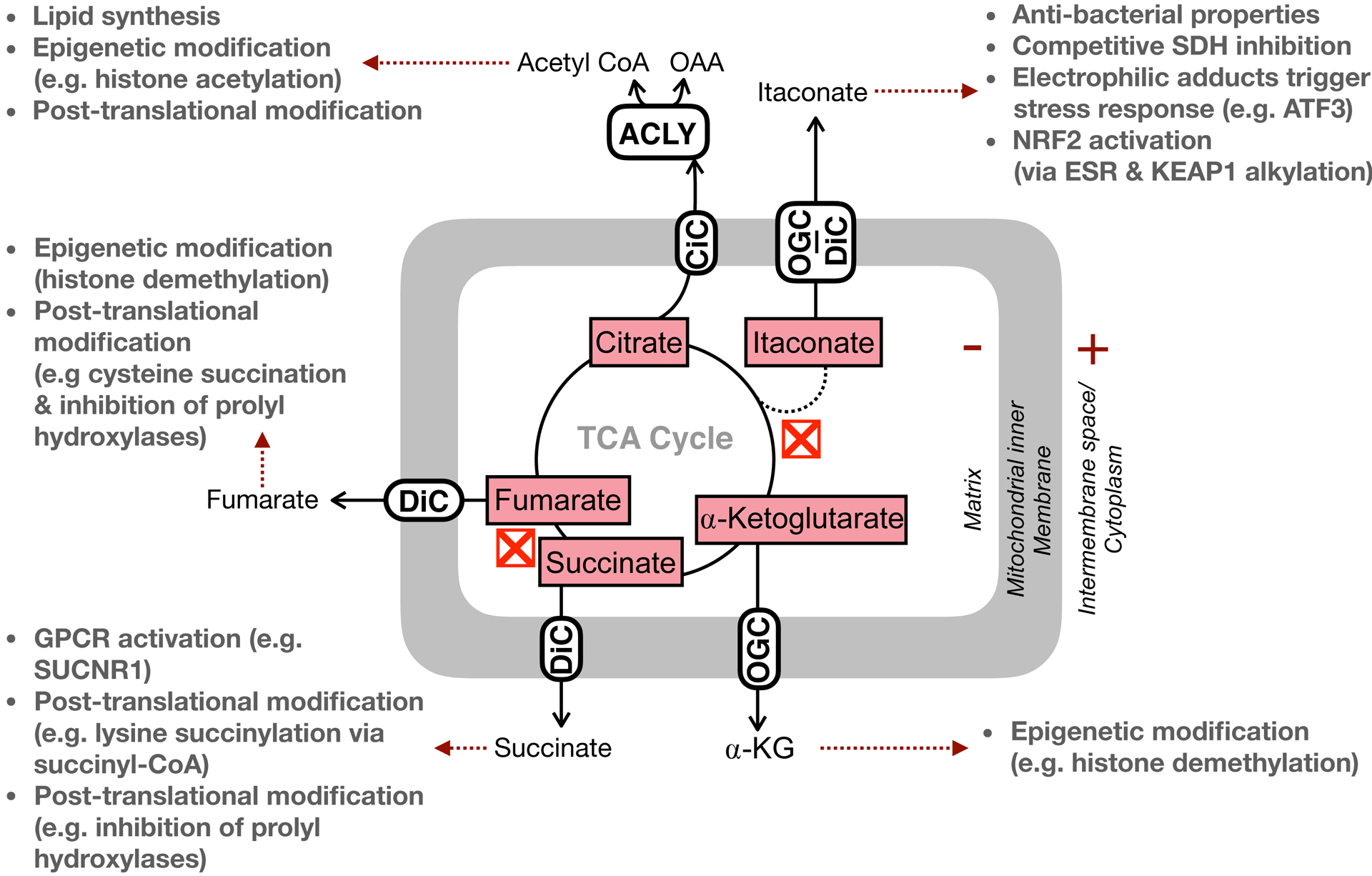
In addition to linking substrate oxidation to the respiratory chain, TCA cycle metabolites also serve critical roles is regulating cell function independently of direct effects on energy metabolism. These include post-translational modifications (e.g. hydroxylation, acetylation, alkylation, etc.), epigenetic modifications, ligand-mediated alterations of protein activity, and even direct functional effects. Red “X” symbols indicate ‘breaks’ in the TCA cycle at IDH and SDH as detailed in the manuscript text. ACLY, ATP citrate lyase; DiC, dicarboxylate carrier; OGC, oxoglutarate carrier; CiC, citrate carrier.
Multiple findings were integrated to reveal Irg1 as the gene encoding the enzyme generating itaconate as an offshoot of the mitochondrial TCA cycle (Michelucci et al., 2013). Previous work had identified Irg1 as heavily transcribed upon LPS activation (Lee et al., 1995), and later work showed macrophages produced itaconate upon infection (Shin et al., 2011; Strelko et al., 2011; Sugimoto et al., 2012). Finally, using sequence homology with Aspergillus cis-aconitase decarboxylase (CAD), Irg1 was identified and characterized as the mammalian CAD responsible for itaconate production (Michelucci et al., 2013).
In addition to a putative role as a bactericidal metabolite, itaconate may also have anti-inflammatory roles via posttranslational modification and direct effects on macrophage metabolism. The electrophilic metabolite, which is produced at millimolar concentrations upon activation (Cordes et al., 2016; Michelucci et al., 2013), can act as a Michael acceptor and affect the cellular oxidative stress response. Itaconate can alkylate a critical cysteine on Kelch-like ECH-associated protein 1 (KEAP1) (E. L. Mills et al., 2018), an endogenous repressor of the transcription factor NRF2 (Nuclear factor erythroid 2-related factor 2). This slows the degradation of NRF2, thereby increasing its activity and promoting an anti-inflammatory phenotype (Ahmed et al., 2017). Moreover, by reacting with glutathione as a Michael acceptor, itaconate can also decrease IκBζ pathway activity and, in turn, lower pro-inflammatory gene expression (Bambouskova et al., 2018).
Itaconate is also directly responsible for the increase in succinate observed during pro-inflammatory activation, as itaconate (methylene succinate) is a competitive inhibitor of succinate dehydrogenase (Cordes et al., 2016; Lampropoulou et al., 2016). The orders-of-magnitude increase in itaconate upon LPS activation is coupled to an increase in succinate, both of which are absent in Irg−/− BMDMs (Lampropoulou et al., 2016). Much like itaconate, succinate can also shape macrophage activation in multiple, distinct ways.
Increased levels of succinate observed during LPS activation can adjust pro-inflammatory gene expression via altered stability of hypoxia inducible factor-1α (HIF-1α) (Tannahill et al., 2013). Inhibition of prolyl hydroxylases by the altered succinate/α-ketoglutarate ratio allows continued transcription of the inflammatory cytokine interleukin-1β (IL-1β) due to a hypoxia response element on the IL-1β promoter (Tannahill et al., 2013; Zhang et al., 2006). Adjusting succinate levels with cell permeant succinate analogs can also support a pro-inflammatory function for the metabolite, as these decrease expression of the anti-inflammatory IL-1RA and IL-10 (Mills et al., 2016).
In addition to stabilization of prolyl hydroxylases, succinate can regulate cell signaling via activation of extracellular receptors. It is an endogenous ligand for the G-protein coupled receptor GPR91 (or SUCNR1) (He et al., 2004). First identified in the kidney as a mediator of hypertension (He et al., 2004), SUCNR1 is also expressed in DCs and BMDMs (Littlewood-Evans et al., 2016; Rubic et al., 2008). Its activation enhances the pro-inflammatory effect of LPS, and cells from Gpr91−/− mice have reduced HIF-1α and IL-1β production upon activation relative to wild-type controls (Littlewood-Evans et al., 2016).
Efflux of mitochondrially-derived citrate by the mitochondrial citrate carrier (CiC) is also important for aspects of pro-inflammatory macrophage activation. Citrate released from mitochondria is converted to malonyl CoA by first generating cytoplasmic acetyl CoA via ATP citrate lyase (ACLY) and subsequent conversion of acetyl CoA to malonyl CoA by acetyl CoA carboxylase (ACC). Inhibition of both the mitochondrial citrate carrier (CiC) responsible for efflux as well as ACLY decrease markers of pro-inflammatory activation such as NO production (Infantino et al., 2013, 2011). Malonyl CoA is primarily considered as a building block for fatty acid synthesis, as well as an endogenous CPT-1 inhibitor that slows fatty acid oxidation upon increased mitochondrial pyruvate metabolism (Hue and Taegtmeyer, 2009; McGarry et al., 1978). However, recent work shows that the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) can be malonylated, and this post-translational modification relieves GAPDH-mediated repression of TNFα mRNA thereby enhancing translation (Galván-Peña et al., 2019). Regardless of the TCA cycle metabolite, it appears as if the pronounced changes in abundance are dynamic, as the increased levels of itaconate, succinate, and citrate observed during pro-inflammatory activation are restored to basal levels after 48 hours (Seim et al., 2019).
ROS is also a well-established signal for mitochondrial control of macrophage activation, and may be an additional, essential link between succinate levels and macrophage activation. Production of superoxide is, of course, tightly associated with inflammatory macrophage activation given the important role of the plasma membrane NADPH oxidase in pathogen clearance (Cathcart, 2004). Mitochondrial superoxide and ROS production was initially linked to macrophage activation through genetic studies with proteins indirectly associated with oxidative stress (Arsenijevic et al., 2000; Kizaki et al., 2002; Tal et al., 2009), and direct evidence came from studies linking cytokine signaling and TLR activation to mitochondrial ROS production and microbial clearance (Sonoda et al., 2007; West et al., 2011).
Succinate is strongly associated with mitochondrial ROS production through mitochondrial reverse electron transport (RET) (Murphy, 2009). Under conditions of a high membrane potential along with reduced NADH and quinone pools, succinate oxidation is associated with RET (Robb et al., 2018). These conditions can push electrons in the ‘backwards’ direction through complex I and generate substantial superoxide (Chouchani et al., 2016). This form of ROS production is thought to underlie much of the damage associated with cardiac ischemia reperfusion injury – where succinate also increases upon ischemia and is oxidized upon reperfusion (Chouchani et al., 2014) – and occurs at physiologically relevant membrane potentials and quinone reduction states (Robb et al., 2018).
Indeed, the mitochondrial membrane potential increases upon inflammatory activation in a subpopulation of macrophages (Mills et al., 2016), linking the bioenergetic phenotype observed during inflammatory activation with increased succinate oxidation and RET. Moreover, bacterial recognition causes a shift away from reliance on complex I and towards a phenotype with enhanced complex II (succinate dehydrogenase) activity and sensitive to blocking mitochondrial ROS production, further establishing the respiratory chain as a link between TLR activation and enhanced mitochondrial ROS (Garaude et al., 2016). A requirement for RET in the LPS-induced regulation of IL-1β production is given by blunting the response with rotenone (a complex I inhibitor that will block RET-driven superoxide from complex I), FCCP (an uncoupler that will dissipate the pmf), and heterologous expression of alternative oxidase [AltOx, an enzyme found in some plants that will re-oxide the quinone pool (El-Khoury et al., 2014)] (Mills et al., 2016). Remarkably, mice expressing AltOx are resistant to LPS-induced toxicity, demonstrating the in vivo relevance of mitochondrial redox status in mediating the inflammatory response.
Recent work also suggests that ROS production by respiratory complex III may play a role in pro-inflammatory activation: the complex III inhibitor myxothiazol can blunt increases in cell surface marker expression and cytokine release upon LPS + IFN-γ treatment (Cameron et al., 2019). However, the exact links between enhanced mitochondrial superoxide production and nuclear gene expression remain unclear. Release of oxidized mitochondrial nucleic acids, though, is a likely mechanism (Shimada et al., 2012; West and Shadel, 2017; Zhong et al., 2018).
3.3. Mitochondrial signaling in anti-inflammatory activation
Given the association between IL-4 polarization and increased oxidative metabolism, a role for mitochondrial signaling has been somewhat overshadowed by bioenergetic function. Nonetheless, mitochondrially derived metabolites can play important roles in regulating the anti-inflammatory response, particularly via epigenetics. Glutamine-derived α-ketoglutarate has been shown to be important as a co-factor for the histone demethylase Jumonji domain-containing protein D3 (JmjD3) (Liu et al., 2017), which regulates epigenetic modification during IL-4-driven activation. Similarly to pro-inflammatory macrophage activation, acetyl CoA derived from mitochondrial citrate is also important for the IL-4 response. In addition to its role in lipogenesis, genetic and pharmacologic evidence support a role for ACLY-derived acetyl CoA in histone acetylation during alternative activation (Covarrubias et al., 2016).
Unexpectedly, the metabolic co-factor coenzyme A (CoA) has also emerged as an essential mediator of IL-4-driven macrophage activation. CoA is a ubiquitous metabolite facilitating acyl transfer reactions and is essential to a range of physiological processes, most notably carbohydrate and lipid metabolism (Leonardi et al., 2005). Initial links between CoA and alternative activation were discovered while exploring which of the many off-target effects of etomoxir was responsible for its efficacy in blocking anti-inflammatory activation (Divakaruni et al., 2018). Exogenous delivery of CoA to macrophages restored the etomoxir-induced decrease in intracellular CoA levels, as well as the rescue the expression of IL-4-associated cell surface markers, even in models with genetic disruption of long-chain fatty acid oxidation. Importantly, exogenous supplementation of CoA to the assay medium enhanced the IL-4-associated cell surface markers above IL-4-treatment alone. The result suggests that intracellular CoA levels may be limiting, and therefore regulatory and targetable, in anti-inflammatory polarization. However, the precise mechanism by which CoA affects alternative activation, and whether this involves mitochondrial metabolism, remain unclear. A mechanism could be as straightforward as CoA abundance directly affecting acetyl CoA levels and histone acetylation (Covarrubias et al., 2016), or rather CoA levels could be limiting for a host of oxidative or biosynthetic metabolic reactions necessary for the IL-4 response.
Similarly unclear is whether mitochondrial reactive oxygen species, a pro-inflammatory trigger, plays any role in dampening the anti-inflammatory response. Supportive evidence at the level of gene expression comes from the use of different mitochondrial inhibitors. For example, complex I inhibition with rotenone, which blocks reverse electron transport and IL-1β production upon LPS activation (Mills et al., 2016), remarkably increases some markers of the IL-4 response (Divakaruni et al., 2018). Moreover, rotenone and the ATP synthase inhibitor oligomycin both inhibit oxidative phosphorylation but can have markedly different effects on anti-inflammatory activation, perhaps suggesting this may be due to differential effects on the mitochondrial membrane potential and downstream redox triggers (Divakaruni et al., 2018). Clearly, more work is needed to better understand the precise role of mitochondria in anti-inflammatory activation, particularly in the discovery of signals that reciprocally regulate pro- and anti-inflammatory markers and work within the graded, spectral nature of macrophage activation.
4. Concluding Remarks
In addition to autoimmune diseases that may be expected to have a dysregulated balance in the macrophage polarization state, it is now well accepted that an imbalance between pro- and anti-inflammatory function can underlie a range of diseases as varied as diet-induced obesity (Lumeng et al., 2008), insulin resistance (Olefsky and Glass, 2010), cardiovascular disease (Bolego et al., 2013; Zhou and Tian, 2018), non-alcoholic steatotic hepatitis (Kazankov et al., 2019), neurodegeneration (Baik et al., 2019), and cancer (Noy and Pollard, 2014). It therefore follows that ‘re-polarizing’ the macrophage population may be a therapeutic strategy (Sica and Mantovani, 2012), and pre-clinical studies have investigated pharmacologic and biologic means to repolarize macrophages to adjust disease progression (Chen et al., 2018; Georgoudaki et al., 2016). Given the requisite metabolic changes that occur during macrophage activation, and that mitochondrial changes may explain limitations in reprogramming macrophages after initial polarization (Van den Bossche et al., 2016), mitochondrial metabolism is an attractive therapeutic target (Murphy and Hartley, 2018). Strategies could include not only repurposing existing drugs that target metabolic enzymes and transcriptional programs, but also cell-permeant metabolite analogs (Martin et al., 2019; E. A. Mills et al., 2018) given the dramatic effects of TCA cycle metabolites on macrophage function. Indeed, application of these concepts to targeting macrophage metabolism is likely to only further enhance our appreciation for the breadth of mitochondrial repurposing upon macrophage activation.
ACKNOWLEDGEMENTS
Anthony E. Jones is supported by the UCLA Tumor Cell Biology Training Program (USHHS Ruth L. Kirschstein Institutional National Research Service Award # T32 CA009056).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adam-Vizi V, Starkov AA, 2010. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimer’s Dis. 10.3233/JAD-2010-100465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DO, Hamilton TA, 1984. The Cell Biology of Macrophage Activation. Annu. Rev. Immunol 10.1146/annurev.iy.02.040184.001435 [DOI] [PubMed] [Google Scholar]
- Adams LB, Hibbs JB, Taintor RR, Krahenbuhl JL, 1990. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from L-arginine. J. Immunol [PubMed] [Google Scholar]
- Ahmed SMU, Luo L, Namani A, Wang XJ, Tang X, 2017. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta - Mol. Basis Dis 10.1016/j.bbadis.2016.11.005 [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, 2004. Toll-like receptor signalling. Nat. Rev. Immunol 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D, 2000. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet 10.1038/82565 [DOI] [PubMed] [Google Scholar]
- Arts RJW, Joosten LAB, Netea MG, 2016a. Immunometabolic circuits in trained immunity. Semin. Immunol 10.1016/j.smim.2016.09.002 [DOI] [PubMed] [Google Scholar]
- Arts RJW, Novakovic B, ter Horst R, Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC, Wang SY, Habibi E, Gonçalves LG, Mesquita I, Cunha C, van Laarhoven A, van de Veerdonk FL, Williams DL, van der Meer JWM, Logie C, O’Neill LA, Dinarello CA, Riksen NP, van Crevel R, Clish C, Notebaart RA, Joosten LAB, Stunnenberg HG, Xavier RJ, Netea MG, 2016b. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 10.1016/j.cmet.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J-I, Mook-Jung I, 2019. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 10.1016/j.cmet.2019.06.005 [DOI] [PubMed] [Google Scholar]
- Bailey JD, Diotallevi M, Nicol T, McNeill E, Shaw A, Chuaiphichai S, Hale A, Starr A, Nandi M, Stylianou E, McShane H, Davis S, Fischer R, Kessler BM, McCullagh J, Channon KM, Crabtree MJ, 2019. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 10.1016/j.celrep.2019.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH, Duncan D, Bregman H, Keskin A, Santeford A, Apte RS, Sehgal R, Johnson B, Amarasinghe GK, Soares MP, Satoh T, Akira S, Hai T, De Guzman Strong C, Auclair K, Roddy TP, Biller SA, Jovanovic M, Klechevsky E, Stewart KM, Randolph GJ, Artyomov MN, 2018. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature. 10.1038/s41586-018-0052-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R, 2003. Toll-like receptor signaling pathways. Science (80-.). 10.1126/science.1085536 [DOI] [PubMed] [Google Scholar]
- Benmoussa K, Garaude J, Acín-Pérez R, 2018. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J. Mol. Biol 10.1016/j.jmb.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Bernardi P, 2013. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol 10.3389/fphys.2013.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolego C, Cignarella A, Staels B, Chinetti-Gbaguidi G, 2013. Macrophage function and polarization in cardiovascular disease a role of estrogen signaling? Arterioscler. Thromb. Vasc. Biol 10.1161/ATVBAHA.113.301328 [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG, 2011. Assessing mitochondrial dysfunction in cells. Biochem. J 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B, Gottlieb E, Hiller K, Jones RG, Kamphorst JJ, Kibbey RG, Kimmelman AC, Locasale JW, Lunt SY, Maddocks ODK, Malloy C, Metallo CM, Meuillet EJ, Munger J, Nöh K, Rabinowitz JD, Ralser M, Sauer U, Stephanopoulos G, St-Pierre J, Tennant DA, Wittmann C, Vander Heiden MG, Vazquez A, Vousden K, Young JD, Zamboni N, Fendt SM, 2015. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol 10.1016/j.copbio.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, Kyle RL, Patterson AE, Hässler F, Buescher JM, Kelly B, Pearce EL, Pearce EJ, 2019. Inflammatory macrophage dependence on NAD + salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat. Immunol 10.1038/s41590-019-0336-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J, 2004. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev 10.1152/physrev.00015.2003 [DOI] [PubMed] [Google Scholar]
- Caputa G, Castoldi A, Pearce EJ, 2019. Metabolic adatations of tissue-resident immune cells. Nat. Immunol 10.1038/s41590-019-0407-0 [DOI] [PubMed] [Google Scholar]
- Cathcart MK, 2004. Regulation of Superoxide Anion Production by NADPH Oxidase in Monocytes/Macrophages: Contributions to Atherosclerosis. Arterioscler. Thromb. Vasc. Biol 10.1161/01.ATV.0000097769.47306.12 [DOI] [PubMed] [Google Scholar]
- Ceccarelli SM, Chomienne O, Gubler M, Arduini A, 2011. Carnitine palmitoyltransferase (CPT) modulators: A medicinal chemistry perspective on 35 years of research. J. Med. Chem 10.1021/jm100809g [DOI] [PubMed] [Google Scholar]
- Chandel NS, 2015. Evolution of Mitochondria as Signaling Organelles . Cell Metab. 10.1016/j.cmet.2015.05.013 [DOI] [PubMed] [Google Scholar]
- Chandel NS, 2014. Mitochondria as signaling organelles. BMC Biol. 10.1186/1741-7007-12-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, Jin X, Liu J, Mo S, Zhang T, Cheng F, Zhou Y, Zhang H, Tang K, Ma J, Liu Y, Huang B, 2018. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun 10.1038/s41467-018-03225-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RAJ, Krieg T, Brookes PS, Murphy MP, 2013. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex i. Nat. Med 10.1038/nm.3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP, 2014. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP, 2016. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 10.1016/j.cmet.2015.12.009 [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC, 2008. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 10.1038/nature06734 [DOI] [PubMed] [Google Scholar]
- Cleeter MWJ, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AHV, 1994. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 10.1016/0014-5793(94)00424-2 [DOI] [PubMed] [Google Scholar]
- Clementi E, Brown GC, Feelisch M, Moncada S, 1998. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. U. S. A 10.1073/pnas.95.13.7631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes T, Michelucci A, Hiller K, 2015. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu. Rev. Nutr 10.1146/annurev-nutr-071714-034243 [DOI] [PubMed] [Google Scholar]
- Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K, Metallo CM, 2016. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem 10.1074/jbc.M115.685792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Horng T, 2015. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol 10.1016/j.smim.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, Zhang Y, Blair IA, Horng T, 2016. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 10.7554/eLife.11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS, 2003. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 10.1016/S0092-8674(03)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts AR, 2004. The Cytochrome bc 1 Complex: Function in the Context of Structure. Annu. Rev. Physiol 10.1146/annurev.physiol.66.032102.150251 [DOI] [PubMed] [Google Scholar]
- Dang L, Su S-SM, 2017. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu. Rev. Biochem 10.1146/annurev-biochem-061516-044732 [DOI] [PubMed] [Google Scholar]
- De Souza DP, Achuthan A, Lee MKS, Binger KJ, Lee MC, Davidson S, Tull DL, McConville MJ, Cook AD, Murphy AJ, Hamilton JA, Fleetwood AJ, 2019. Autocrine IFN-I inhibits isocitrate dehydrogenase in the TCA cycle of LPS-stimulated macrophages. J. Clin. Invest 10.1172/JCI127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Patron M, Rizzuto R, 2014. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta - Mol. Cell Res. 10.1016/j.bbamcr.2015.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, Nojima T, Munnich A, Schiff M, de Almeida CR, Rehwinkel J, Dziembowski A, Szczesny RJ, Proudfoot NJ, 2018. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 10.1038/s41586-018-0363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Brand MD, 2011. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda). 10.1152/physiol.00046.2010 [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, Andreyev AY, Bowman CE, Caradonna K, Dranka BP, Ferrick DA, Liesa M, Stiles L, Rogers GW, Braas D, Ciaraldi TP, Wolfgang MJ, Sparwasser T, Berod L, Bensinger SJ, Murphy AN, 2018. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 10.1016/j.cmet.2018.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M, 2014. Analysis and interpretation of microplate-based oxygen consumption and pH data, in: Methods in Enzymology. 10.1016/B978-0-12-801415-8.00016-3 [DOI] [PubMed] [Google Scholar]
- Dolan SK, Welch M, 2018. The Glyoxylate Shunt, 60 Years On. Annu. Rev. Microbiol 10.1146/annurev-micro-090817-062257 [DOI] [PubMed] [Google Scholar]
- Duchen MR, 2000. Mitochondria and calcium: From cell signalling to cell death. J. Physiol 10.1111/j.1469-7793.2000.00057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoury R, Kemppainen KK, Dufour E, Szibor M, Jacobs HT, Rustin P, 2014. Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: State of the art and perspectives. Br. J. Pharmacol 10.1111/bph.12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Van Der Windt GJW, Freitas TC, Chott R, Yarasheski KE, Pearce EL, Pearce EJ, 2012. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 10.1182/blood-2012-03-419747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, McAtee-Pereira AG, Guo Y, Trenary I, Hernandez A, Fults JB, Williams DL, Sherwood ER, Bohannon JK, 2018. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J. Immunol 10.4049/jimmunol.1800085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, 2011. Signal transduction by reactive oxygen species. J. Cell Biol. 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván-Peña S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, Covarrubias S, Nadin A, James AM, Haneklaus M, Carpenter S, Kelly VP, Murphy MP, Modis LK, O’Neill LA, 2019. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat. Commun 10.1038/s41467-018-08187-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaude J, Acín-Pérez R, Martínez-Cano S, Enamorado M, Ugolini M, Nistal-Villán E, Hervás-Stubbs S, Pelegrín P, Sander LE, Enríquez JA, Sancho D, 2016. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol 10.1038/ni.3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garedew A, Moncada S, 2008. Mitochondrial dysfunction and HIF1α stabilization in inflammation. J. Cell Sci. 10.1242/jcs.034660 [DOI] [PubMed] [Google Scholar]
- Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA, 2017. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front. Immunol 10.3389/fimmu.2017.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, Dahan R, Harris RA, Rantalainen M, Klevebring D, Sund M, Brage SE, Fuxe J, Rolny C, Li F, Ravetch JV, Karlsson MCI, 2016. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 10.1016/j.celrep.2016.04.084 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, Wolfgang MJ, 2017. Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am. J. Physiol. - Endocrinol. Metab 10.1152/ajpendo.00408.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, 2016. Phagocytosis: The Legacy of Metchnikoff . Cell. 10.1016/j.cell.2016.08.017 [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR, 2005. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- Grazioli S, Pugin J, 2018. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol 10.3389/fimmu.2018.00832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Dubois DC, Boje KMK, Free SJ, Almon RR, 1999. Quantification of iNOS mRNA with reverse transcription polymerase chain reaction directly from cell lysates. Nitric Oxide - Biol. Chem 10.1006/niox.1999.0240 [DOI] [PubMed] [Google Scholar]
- Harris RA, Hawes JW, Popov KM, Zhao Y, Shimomura Y, Sato J, Jaskiewicz J, Hurley TD, 1997. Studies on the regulation of the mitochondrial α-ketoacid dehydrogenase complexes and their kinases, in: Advances in Enzyme Regulation. 10.1016/S0065-2571(96)00009-X [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, Amir S, Lubec G, Park J, Esterbauer H, Bilban M, Brizuela L, Pospisilik JA, Otterbein LE, Wagner O, 2012. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 10.1016/j.cmet.2012.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Miao FJP, Lin DCH, Schwandner RT, Wang Z, Gao J, Chen JL, Tlan H, Ling L, 2004. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 10.1038/nature02488 [DOI] [PubMed] [Google Scholar]
- Held TK, Weihua X, Yuan L, Kalvakolanu DV, Cross AS, 1999. Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infect. Immun [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, 2013. Mitochondrial Complex I. Annu. Rev. Biochem 10.1146/annurev-biochem-070511-103700 [DOI] [PubMed] [Google Scholar]
- Hoek JB, Rydstrom J, 1988. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J 10.1042/bj2540001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SCC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF, Artyomov MN, Pearce EL, Pearce EJ, 2014. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol 10.1038/ni.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SCC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ, 2016. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 10.1016/j.immuni.2016.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H, 2009. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. - Endocrinol. Metab 10.1152/ajpendo.00093.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V, 2011. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J 10.1042/BJ20111275 [DOI] [PubMed] [Google Scholar]
- Infantino V, Iacobazzi V, Palmieri F, Menga A, 2013. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun 10.1016/j.bbrc.2013.09.037 [DOI] [PubMed] [Google Scholar]
- Iverson TM, 2013. Catalytic mechanisms of complex II enzymes: A structural perspective. Biochim. Biophys. Acta - Bioenerg. 10.1016/j.bbabio.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM, 2013. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta - Mol. Cell Res 10.1016/j.bbamcr.2012.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN, 2015. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 10.1016/j.immuni.2015.02.005 [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X, 2004. Cytochrome C -Mediated Apoptosis. Annu. Rev. Biochem 10.1146/annurev.biochem.73.011303.073706 [DOI] [PubMed] [Google Scholar]
- Kato M, 1972. Site of action of lipid A on mitochondria. J. Bacteriol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H, 2019. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol 10.1038/s41575-018-0082-x [DOI] [PubMed] [Google Scholar]
- Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoé K, Day NK, Good RA, Ohno H, 2002. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. U. S. A 10.1073/pnas.142206299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenis DS, Medzikovic L, van Loenen PB, van Weeghel M, Huveneers S, Vos M, Evers-van Gogh IJ, Van den Bossche J, Speijer D, Kim Y, Wessels L, Zelcer N, Zwart W, Kalkhoven E, de Vries CJ, 2018. Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 10.1016/j.celrep.2018.07.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ, 2010. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 10.1182/blood-2009-10-249540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SCC, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, Artyomov MN, 2016. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 10.1016/j.cmet.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston PK, Shibata M, Horng T, 2017. Metabolism supports macrophage activation. Front. Immunol 10.3389/fimmu.2017.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaNoue KF, Schoolwerth AC, 1979. Metabolite Transport in Mitochondria. Annu. Rev. Biochem 10.1146/annurev.bi.48.070179.004255 [DOI] [PubMed] [Google Scholar]
- LaNoue KF, Tischler ME, 1974. Electrogenic characteristics of the mitochondrial glutamate aspartate antiporter. J. Biol. Chem [PubMed] [Google Scholar]
- Lee CGL, Jenkins NA, Gilbert DJ, Copeland NG, O’Brien WE, 1995. Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics. 10.1007/BF00172150 [DOI] [PubMed] [Google Scholar]
- Leonardi R, Zhang YM, Rock CO, Jackowski S, 2005. Coenzyme A: Back in action. Prog. Lipid Res. 10.1016/j.plipres.2005.04.001 [DOI] [PubMed] [Google Scholar]
- Leone AM, Palmer RMJ, Knowles RG, Francis PL, Ashton DS, Moncada S, 1991. Constitutive and inducible nitric oxide synthases incorporate molecular oxygen into both nitric oxide and citrulline. J. Biol. Chem [PubMed] [Google Scholar]
- Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, Valeaux S, Gommermann N, Rubic-Schneider T, Junt T, Carballido JM, 2016. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med 10.1084/jem.20160061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, DI Conza G, Cheng WC, Chou CH, Vavakova M, Muret C, Debackere K, Mazzone M, Huang H. Da, Fendt SM, Ivanisevic J, Ho PC, 2017. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 10.1038/ni.3796 [DOI] [PubMed] [Google Scholar]
- Lu C, Thompson CB, 2012. Metabolic regulation of epigenetics. Cell Metab. 10.1016/j.cmet.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, Oĝrourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB, 2012. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Delproposto JB, Westcott DJ, Saltiel AR, 2008. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 10.2337/db08-0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J, Xie Q, Nathan C, 1997. NITRIC OXIDE AND MACROPHAGE FUNCTION. Annu. Rev. Immunol 10.1146/annurev.immunol.15.1.323 [DOI] [PubMed] [Google Scholar]
- Martin JL, Costa ASH, Gruszczyk AV, Beach TE, Allen FM, Prag HA, Hinchy EC, Mahbubani K, Hamed M, Tronci L, Nikitopoulou E, James AM, Krieg T, Robinson AJ, Huang MM, Caldwell ST, Logan A, Pala L, Hartley RC, Frezza C, Saeb-Parsy K, Murphy MP, 2019. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab 10.1038/s42255-019-0115-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, 2014. The M1 and M2 paradigm of macrophage activation: Time for reassessment . F1000Prime Rep. 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden BA, Purohit S, 1977. Itaconate, an isocitrate lyase directed inhibitor in Pseudomonas indigofera. J. Bacteriol [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Takabayashi Y, Foster DW, 1978. The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J. Biol. Chem [PubMed] [Google Scholar]
- Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K, 2013. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. U. S. A 10.1073/pnas.1218599110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E, O’Neill LAJ, 2014. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 10.1016/j.tcb.2013.11.008 [DOI] [PubMed] [Google Scholar]
- Mills EA, Ogrodnik MA, Plave A, Mao-Draayer Y, 2018. Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front. Neurol 10.3389/fneur.2018.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA, 2016. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, O’Neill LAJ, 2017. Mitochondria are the powerhouses of immunity. Nat. Immunol 10.1038/ni.3704 [DOI] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sévin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O’Neill LA, 2018. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 10.1038/nature25986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD, 2017. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem 10.1074/jbc.M116.774471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP, 2008. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Elías EJ, McKinney JD, 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med 10.1038/nm1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, 2015. Redox modulation by reversal of the mitochondrial nicotinamide nucleotide transhydrogenase. Cell Metab. 10.1016/j.cmet.2015.08.012 [DOI] [PubMed] [Google Scholar]
- Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochem. J 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Hartley RC, 2018. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 10.1038/nrd.2018.174 [DOI] [PubMed] [Google Scholar]
- Murray PJ, 2017. Macrophage Polarization. Annu. Rev. Physiol 10.1146/annurev-physiol-022516-034339 [DOI] [PubMed] [Google Scholar]
- Namgaladze D, Brüne B, 2014. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 10.1016/j.bbalip.2014.06.007 [DOI] [PubMed] [Google Scholar]
- Newsholme P, Curi R, Gordon S, Newsholme EA, 1986. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J 10.1042/bj2390121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ, 2013. Bioenergetics 4, Academic ORess. 10.1017/CBO9781107415324.004 [DOI] [Google Scholar]
- Nickel AG, Von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CRD, Blacker TS, Hall AR, Duchen MR, Kästner L, Lipp P, Zeller T, Müller C, Knopp A, Laufs U, Böhm M, Hoth M, Maack C, 2015. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 10.1016/j.cmet.2015.07.008 [DOI] [PubMed] [Google Scholar]
- Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T, 2016. Fatty acid oxidation in macrophage polarization. Nat. Immunol 10.1038/ni.3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R, Pollard JW, 2014. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. 10.1016/j.immuni.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Artyomov MN, 2019. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol 10.1038/s41577-019-0128-5 [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, Golenbock D, Bowie AG, 2013. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol 10.1038/nri3446 [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, Pearce EJ, 2016. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med 10.1084/jem.20151570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A, 2011. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. Mech. Dis 10.1146/annurev-pathol-011110-130138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A, 2007. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK, 2010. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol 10.1146/annurev-physiol-021909-135846 [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Rutter J, 2013. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 10.1101/gad.229724.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri EM, Baseler WA, Davies LC, Gonzalez-Cotto M, Ghesquiere B, Fan TWM, Lane AN, Wink DA, McVicar DW, 2018. Nitric oxide dictates the reprogramming of carbon flux during M1 macrophage polarization. J. Immunol [Google Scholar]
- Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquière B, McVicar DW, Guarini A, Mazzone M, Castegna A, 2017. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 10.1016/j.celrep.2017.07.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F, 2013. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med 10.1016/j.mam.2012.05.005 [DOI] [PubMed] [Google Scholar]
- Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, Bernabei R, Marzetti E, 2017. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci 10.3390/ijms18050933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleston DJ, Buck MD, Klein Geltink RI, Kyle RL, Caputa G, O’Sullivan D, Cameron AM, Castoldi A, Musa Y, Kabat AM, Zhang Y, Flachsmann LJ, Field CS, Patterson AE, Scherer S, Alfei F, Baixauli F, Austin SK, Kelly B, Matsushita M, Curtis JD, Grzes KM, Villa M, Corrado M, Sanin DE, Qiu J, Pällman N, Paz K, Maccari ME, Blazar BR, Mittler G, Buescher JM, Zehn D, Rospert S, Pearce EJ, Balabanov S, Pearce EL, 2019. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab. 10.1016/j.cmet.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stüve P, Arnold-Schrauf C, Guderian M, Lochner M, Rampertaap S, Romito K, Monsale J, Brönstrup M, Bensinger SJ, Murphy AN, McGuire PJ, Jones RG, Sparwasser T, Berod L, 2018. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab. 10.1016/j.cmet.2018.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS, 2015. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 10.1016/j.ceb.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR, Maréchal A, 2010. The mitochondrial respiratory chain. Essays Biochem. 10.1042/BSE0470001 [DOI] [PubMed] [Google Scholar]
- Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, Murphy MP, 2018. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem 10.1074/jbc.RA118.003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L, 2010. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol 10.4049/jimmunol.0901698 [DOI] [PubMed] [Google Scholar]
- Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwärzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, Carballido JM, 2008. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol 10.1038/ni.1657 [DOI] [PubMed] [Google Scholar]
- Ruetz M, Campanello GC, Purchal M, Shen H, McDevitt L, Gouda H, Wakabayashi S, Zhu J, Rubin EJ, Warncke K, Mootha VK, Koutmos M, Banerjee R, 2019. Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science (80-.). 10.1126/science.aay0934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG, Huang L, VanderVen BC, 2019. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol 10.1038/s41577-019-0124-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, Mills EL, 2019. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab 10.1038/s42255-018-0014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M, Kabat AM, Buck MD, Qiu J, Lawless SJ, Cameron AM, Villa M, Baixauli F, Patterson AE, Hässler F, Curtis JD, O’Neill CM, O’Sullivan D, Wu D, Mittler G, Huang SCC, Pearce EL, Pearce EJ, 2018. Mitochondrial Membrane Potential Regulates Nuclear Gene Expression in Macrophages Exposed to Prostaglandin E2. Immunity. 10.1016/j.immuni.2018.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Frezza C, 2017. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 10.1111/febs.14090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, Pagliarini DJ, Fan J, 2019. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab 10.1038/s42255-019-0083-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E, 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 10.1016/j.ccr.2004.11.022 [DOI] [PubMed] [Google Scholar]
- Shadel G, 2012. MITOCHONDRIAL STRESS SIGNALING IN DISEASE AND AGING. Free Radic. Biol. Med 10.1016/j.freeradbiomed.2012.10.005 [DOI] [Google Scholar]
- Shaw RJ, 2006. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 10.1016/j.ceb.2006.10.005 [DOI] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M, 2012. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 10.1016/j.immuni.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, Ryu DH, Hwang GS, 2011. 1H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J. Proteome Res. 10.1021/pr101054m [DOI] [PubMed] [Google Scholar]
- Sica A, Mantovani A, 2012. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S, Viney I, Wellen KE, 2018. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci 10.1016/j.tibs.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Laganière J, Mehl IR, Barish GD, Chong L, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, Lee C, Giguère V, Evans RM, 2007. Nuclear receptor ERRalpha and coactivator PGC-1beta are effectors of IFN- gamma -induced host defense. Genes Dev. 10.1101/gad.1553007.oxide [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF, 2011. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc 10.1021/ja2070889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ, Nathan CF, 1989. Nitric oxide: A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med 10.1084/jem.169.5.1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Ikeda S, Niigata K, Tomita M, Sato H, Soga T, 2012. MMMDB: Mouse multiple tissue metabolome database. Nucleic Acids Res. 10.1093/nar/gkr1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, Green DR, 2010. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 10.1038/nrm2952 [DOI] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A, 2009. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U. S. A 10.1073/pnas.0S07694106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, Liu G, 2015. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J. Immunol 10.4049/jimmunol.1402469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LAJ, 2013. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakoli S, Downs K, Short JD, Nguyen HN, Lai Y, Jerabek PA, Goins B, Toczek J, Sadeghi MM, Asmis R, 2017. Characterization of macrophage polarization states using combined measurement of 2-deoxyglucose and glutamine accumulation: Implications for imaging of atherosclerosis. Arterioscler. Thromb. Vasc. Biol 10.1161/ATVBAHA.117.308848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, 2017. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 10.1016/j.tcb.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MCS, Ahmed M, Hoeksema MA, de Vos AF, de Winther MPJ, 2016. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 10.1016/j.celrep.2016.09.008 [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, O’Neill LA, Menon D, 2017. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 10.1016/j.it.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, van der Windt GJW, 2018. Fatty Acid Oxidation in Macrophages and T Cells: Time for Reassessment? Cell Metab. 10.1016/j.cmet.2018.09.018 [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Greaves DR, Murray PJ, Chawla A, 2006. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 10.1016/j.cmet.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan V, Pradhan P, Braud L, Fuchs HR, Gueler F, Motterlini R, Foresti R, Immenschuh S, 2019. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide - A divergent role for glycolysis. Redox Biol. 10.1016/j.redox.2019.101147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A, 2019. The Metabolic Signature of Macrophage Responses. Front. Immunol 10.3389/fimmu.2019.01462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, Paradies E, Scarcia P, Palmieri F, Bouillaud F, Fiermonte G, 2014. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. U. S. A 10.1073/pnas.1317400111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, 2013. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans 10.1042/BST20110773 [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD, Herrmann J, 2018a. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine. 10.1016/j.ebiom.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, Dzeja PP, Herrmann J, 2018b. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 10.1016/j.cmet.2018.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, Chandel NS, 2015. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 10.1016/j.immuni.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S, 2011. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 10.1038/nature09973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, 2017. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol 10.1038/nri.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Pfanner N, 2017. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem 10.1146/annurev-biochem-060815-014352 [DOI] [PubMed] [Google Scholar]
- Wynn TA, Vannella KM, 2016. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 10.1016/j.immuni.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL, 2012. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 10.1101/gad.191056.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang Ping, Kim SH, Ito S, Yang C, Wang Pu, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y, 2011. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Petrovic JM, Callaghan D, Jones A, Cui H, Howlett C, Stanimirovic D, 2006. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1β (IL-1β) in astrocyte cultures. J. Neuroimmunol 10.1016/j.jneuroim.2006.01.014 [DOI] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X. jia, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M, 2018. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 10.1038/s41586-018-0372-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Tian R, 2018. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest 10.1172/JCI120849 [DOI] [PMC free article] [PubMed] [Google Scholar]