

Abstract
Specific applications of CRISPR/Cas genome editing systems benefit from chemical modifications of the sgRNA. Herein we describe a versatile and efficient strategy for functionalization of the 3′-end of a sgRNA. An exemplary collection of six chemically modified sgRNAs was prepared containing crosslinkers, a fluorophore and biotin. Modification of the sgRNA 3′-end was broadly tolerated by Streptococcus pyogenes Cas9 in an in vitro DNA cleavage assay. The 3′-biotinylated sgRNA was used as an affinity reagent to identify IGF2BP1, YB1 and hnRNP K as sgRNA-binding proteins present in HEK293T cells. Overall, the modification strategy presented here has the potential to expand on current applications of CRISPR/Cas systems.
Keywords: click chemistry, nucleic acids, protein engineering, proteomics, RNA
Introduction
The CRISPR/Cas9 genome editing system uses guide RNAs (gRNAs) and the Cas9 endonuclease to scan and cleave target DNA with high specificity.[1,2] Cas9 derives its specificity by first scanning the DNA for a protospacer adjacent motif (PAM).[3] Once the PAM is located, Cas9 stalls and begins interrogation by local DNA melting, then probing for gRNA–DNA base complementarity.[4,5] If full complementarity exists between the DNA and the gRNA, R-loop formation occurs, and the two nuclease domains are engaged resulting in double-stranded breaks (DSBs).[6,7] Naturally a part of the bacterial adaptive immune system, CRISPR-Cas9 has been repurposed for genome editing.[8] Improvements to editing efficiencies or repurposing Cas9 for new applications have largely been accomplished by engineering Cas9.[9–13] The first significant modification of the gRNAs involved fusing the CRISPR RNA (crRNA) and the trans-activating crRNA (trRNA) via a stem loop creating the chimeric single guide RNA (sgRNA).[1] The sgRNA simplifies the assembly of the ribonuclear protein (RNP) complex as well as the transcription of the gRNA from a delivered plasmid. Essential parts of the sgRNA have been investigated[14] and many studies have been carried out to optimize the sgRNA sequence.[15–17] However, for therapeutic applications of the CRISPR/Cas9 system that involve delivery of an intact RNP, chemical modifications of the sgRNA will be required, similar to other existing therapeutic oligonucleotides (i.e., antisense oligonucleotides, siRNAs).[18–21] Notably, chemically synthesized sgRNAs have been shown to have increased gene editing efficiency relative to in vitro transcribed versions.[22,23] Additional chemical modifications to the sgRNA could increase metabolic stability, help tissue-specific delivery and improve editing efficiencies directly or indirectly through interactions with Cas9. Importantly, modifications to the sgRNA could also prevent unintended sgRNA interactions with endogenous proteins if the sgRNA is orphaned from Cas9.
While chemically modified gRNAs have been explored in some detail, most studies investigate modifications to the sugar or use the dual guide RNA system (e.g., crRNA + trRNA).[21,22,24–31] Generally, these modifications (e.g., 2′-O-methyl) are intended for increasing or maintaining on-target potency while reducing off-target gene editing or sgRNA stability to nucleases[21,22,27,28,31] and less envisioned for application-based experiments although some examples exist.[24–26] Hence, many of these modifications have been incorporated into the 5′-end target sequence. Furthermore, introduction of chemical modifications to the sgRNA have heavily relied on the limited set of commercially available chemical modifications[27] such as 2′-O-methyl or phosphorothioate linkages, amongst others. Although some modifications have been reported to be incorporated in the 5′-end of the crRNA (e.g., strained cyclooctyne or fluorophore),[24,25] modification at this location could result in disruption of DNA unwinding and ultimately disable cleavage of the target DNA. Thus, we sought a single modification strategy for the sgRNA at the 3′-end that was high yielding and versatile enough to be compatible with a variety of different functional groups. Here, we describe a strategy for the efficient and adaptable modification of the sgRNA 3′-end. We rationalize the 3′-end being more suitable for a variety of modifications that were introduced including a fluorophore, covalent crosslinkers and a 3′-end biotin. We describe two different approaches for covalent crosslinking Cas9 to its sgRNA. In addition, the biotinylated sgRNA was used along with mass spectrometry to identify endogenous human proteins capable of sgRNA binding. This provides a powerful new method for identifying cellular factors that may affect sgRNA stability or availability.
Results
Chemical modification of sgRNA at the 3′-end
sgRNAs are typically 90–115-nt in length and challenging to produce in high yield and purity via solid-phase synthesis alone, hence we aimed to synthesize the sgRNA in two parts and use splint ligation to form our desired sgRNA.[24,29,30,32] To accomplish this, we chemically synthesized a 30-nt fragment of the sgRNA modified at the 3′-end with a primary amine (Figure 1A and Table S1 in the Supporting Information). The amine was then allowed to react with several different functionalized NHS esters to produce chemically modified 3′ fragments of the sgRNA (Figure 1B). The amine/NHS ester reactions proceeded efficiently, and identity of product was confirmed by MALDIMS (Table S2). To produce the full-length sgRNA, the functionalized 30-nt RNA bearing a 5′-phosphate and a chemically synthesized 51-nt RNA was hybridized to a complementary DNA and ligated using T4 DNA ligase. It should be noted that the 51-nt fragment contains a 20-nt target sequence complementary to the promoter region of the SNURF1 gene previously described.[33] The ligation reaction proceeded efficiently with minimal starting materials observed for all sgRNAs produced (Figure S1). The modification reaction and ligation efficiently created a collection of six chemically modified sgRNAs that have a variety of functional groups including iodoacetamide, maleimide, strained cyclooctyne, fluorophore and biotin (Figure 1C).
Figure 1.
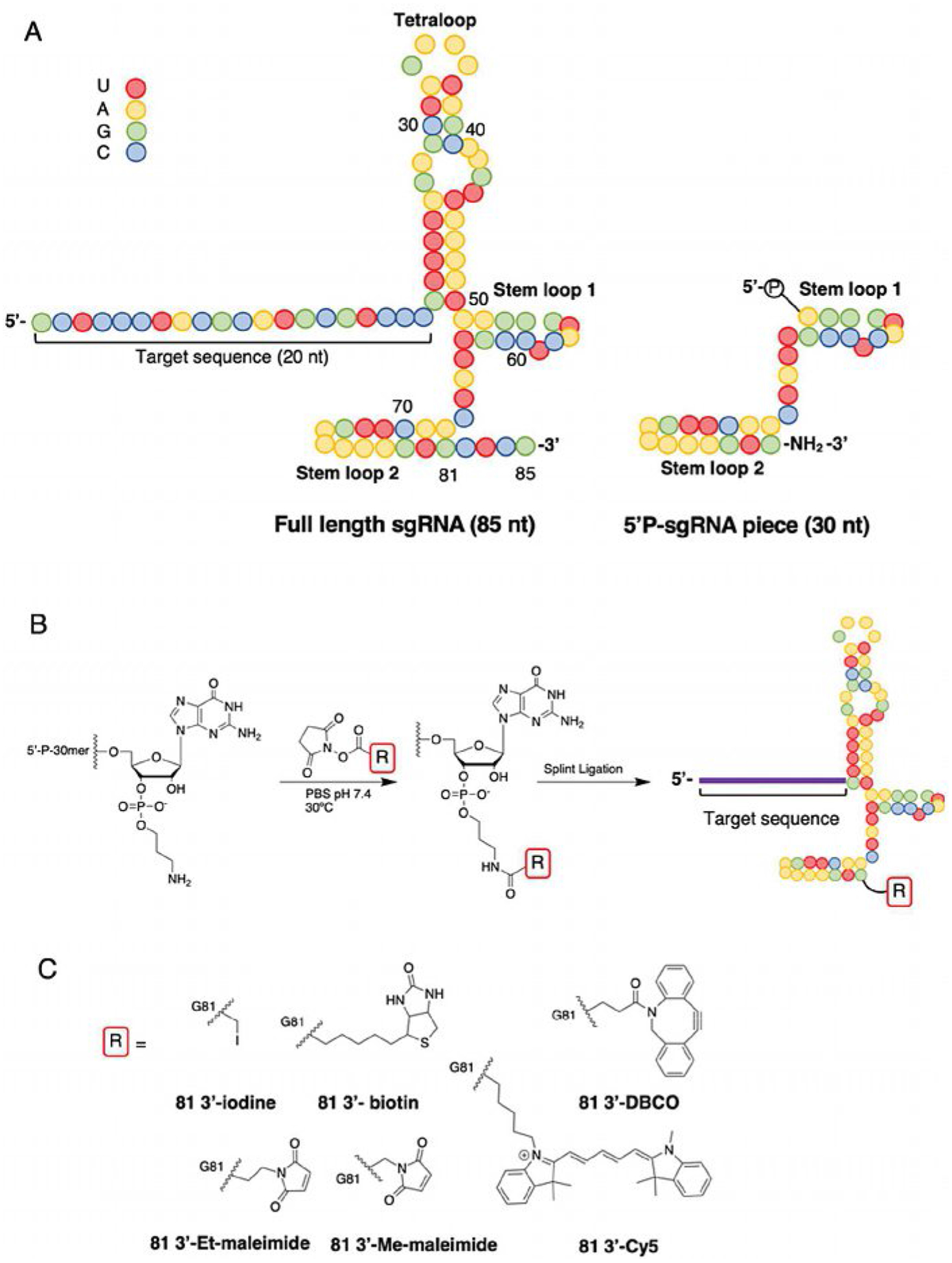
Structures of sgRNA and reaction scheme for synthesis of modified sgRNA. A) Full-length sequence and secondary structure for in vitro transcribed sgRNA (IVT 85; left). Amine-modified 30-nt RNA bearing 5′ phosphate used for modification of the sgRNA (right). Colored spheres represent specific bases (red=U, yellow=A, green =G, blue= C). B) Reaction between amine-modified 5′-P-30-nt fragment with library of functionalized NHS esters. Full-length sgRNA can be obtained by splint ligation of the modified 30-nt and 51-nt RNA. C) Chemical structures and names for the library of the 81-nt sgRNAs generated in this study (DBCO=dibenzocycloocytne, Cy5=cyanine 5, Et-maleimide=ethyl maleimide, Me-maleimide=methyl maleimide).
Impact of 3′-end modifications of sgRNA on Cas9 cleavage efficiency in vitro
We wished to determine whether these sgRNA modifications were detrimental for Cas9-mediated cleavage of a dsDNA target. End-point in vitro cleavage was performed with purified Streptococcus pyogenes NLS-Cas9 protein[1] that was preincubated with sgRNA and then mixed with a dsDNA that contained a single target site. The asymmetric cleavage products were resolved by gel electrophoresis and percent cleavage was determined (Figure 2A and B). We demonstrated that only sgRNA bearing the free amine and the BMPS modified sgRNA decreased the overall cleavage reaction relative to the transcribed sgRNA standard (Figure 2B). To our surprise, the 3′-iodine modification of the sgRNA had a small stimulatory effect. Overall, results from the in vitro cleavage assay indicated these modifications were well tolerated at the sgRNA 3′-end.
Figure 2.
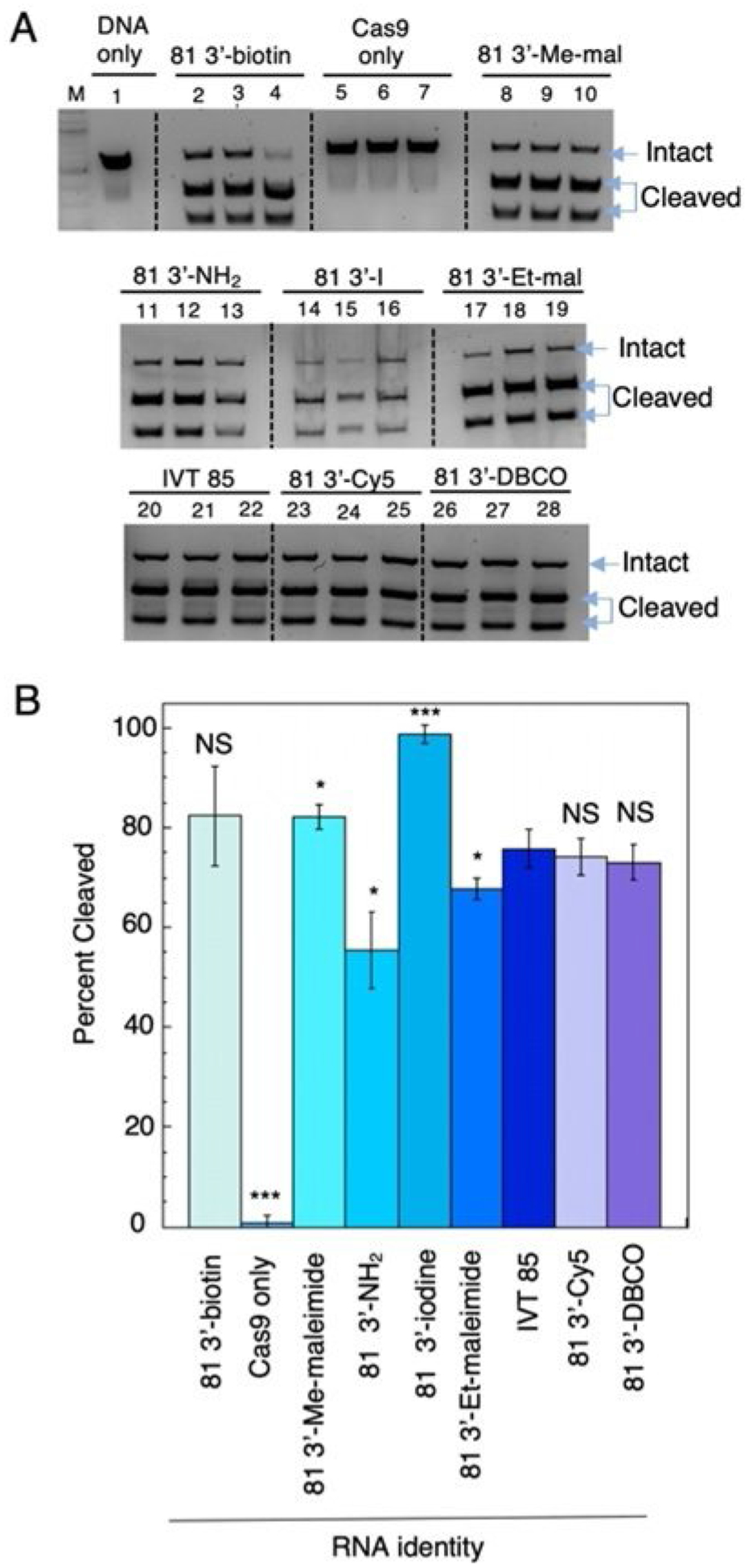
In vitro cleavage results for 643 bp Snurf1 (S1) DNA target (see Experiment Section in the Supporting Information)[33] by Cas9 and sgRNAs. A) Cleavage products resolved using a 1.2% agarose gel prestained with SYBR gold for all sgRNAs used in this study; lane M=DNA ladder, lane 1= DNA only, lanes 2–4=81 3′-biotin, lanes 5–7=Cas9+DNA, lanes 8–10=81 3′-Me-maleimide (3′-Me-mal), lanes 11–13=81 3′-NH2 (unconjugated free amine), lanes 14–16=81 3′-iodine, lanes 17–19=81 3′-Et-maleimide (3′-Etmal), lanes 20–22=in vitro transcribed (IVT) 85, lanes 23–25=81 3′-Cy5, lanes 26–28=81 3′-DBCO. Intact S1 DNA target (623 bp) and cleavage products (257 and 385 bp) indicated by blue arrows. B) Percent cleavage vs. RNA identity graphed, each bar represents triplicate data; *p< 0.05, **p<0.01, ***p<0.001 two-tailed unpaired t-test relative to the IVT 85 sample (NS=not statistically different).
Crosslinking modified sgRNA to Cas9
We generated Cas9 mutants (Figure S2) that positioned a reactive handle near the RNA-protein interface such that covalent crosslinking of the sgRNA to the protein could occur (Figure 3A). With the sgRNAs generated, we envisioned that this crosslinking could occur between a cysteine on Cas9 and the sgRNA bearing an iodoacetamide or maleimide (Figure 3B). Similarly, Cas9 functionalized with azido-phenylalanine could react with a sgRNA containing a strained cyclooctyne (Figure 3B). To decrease unwanted crosslinking, the two native cysteines present in S. pyogenes Cas9 were mutated to serine (C80S, C574S) and the resulting mutant was used as a control in crosslinking experiments. Two triple mutants of a maltose binding protein (MBP)-Cas9 fusion were generated. Both had the two native cysteines mutated to serine, but one triple mutant had a K31C mutation and the other had K31Stop mutation for incorporation of the unnatural amino acid p-azido-l-phenylalanine (pAzF) by stop codon suppression in the presence of a pAzF-charged tRNA.[35] To assay for crosslinking between the sgRNA and Cas9, the two components were incubated in order for Cas9 to bind the sgRNA. The sgRNA-Cas9 complex was then allowed to incubate at room temperature overnight. After incubation, the reaction was quenched, and the products were resolved by denaturing SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis). A protein-RNA crosslink was observed only when the triple mutants of Cas9 were in the presence of an appropriately modified sgRNA (Figure 3C). The extent of crosslinking varied among the sgRNAs tested (Figure 3D). The sgRNA modified with AMAS produced the highest extent of crosslinking giving about 29%. However, this was the highest yield obtained despite further attempts at optimization.
Figure 3.
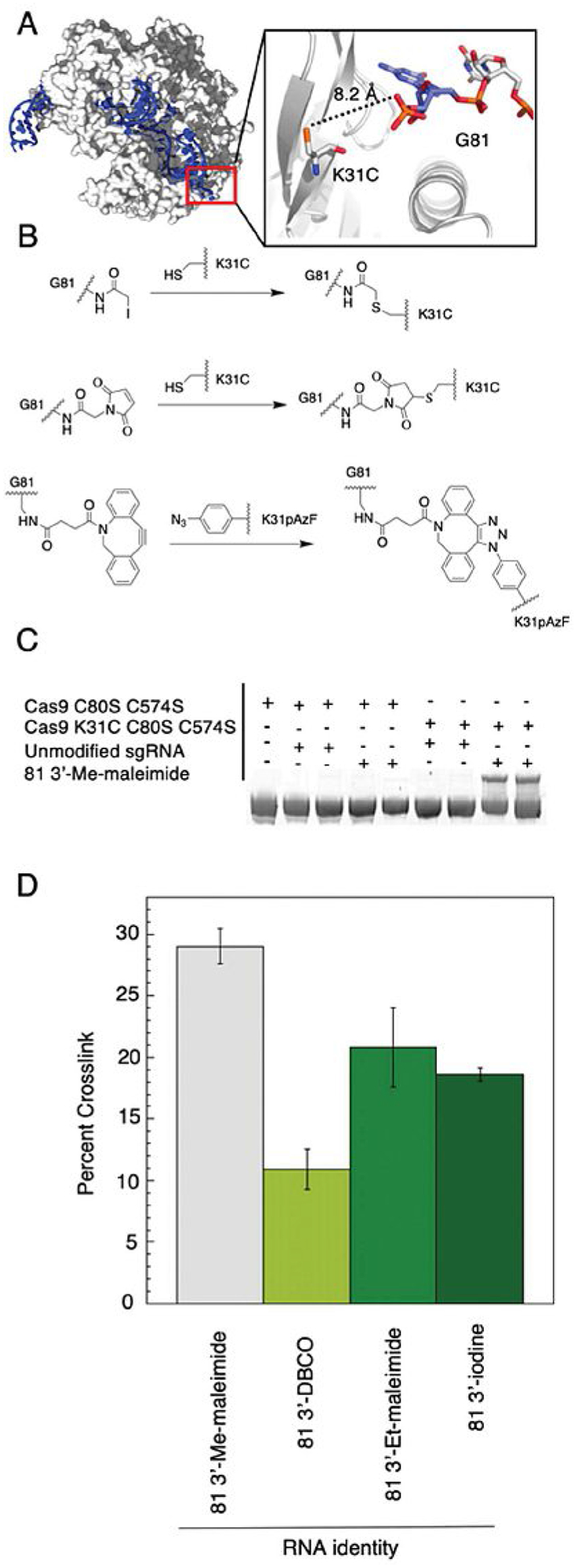
Crosslinking rationale, scheme and results. A) Crystal structure of Cas9:sgRNA complex (PDB ID: 4UN3).[34] Close up view of RNA–protein interface around G81. Distance between 3′ phosphate to a K31C mutation represented by dashed line. B) Crosslinking reaction scheme between K31C and iodoacetamide (SIA; top) or K31C and maleimide (AMAS; middle) or K31pAzF and cyclooctyne (DBCO; bottom) to form a covalently crosslinked Cas9:sgRNA complex. C) Crosslinking products resolved by SDS PAGE. A gel shift occurs only when the modified sgRNA is in the presence of nucleophilic mutant (K31C, C80S, C574S) of Cas9. (For images of other combinations, see Figure S3.) D) Crosslinking percentage vs. RNA identify plotted. Plotted values correspond to averages±standard deviation (n=3).
Capture of sgRNA-binding proteins with 3′-biotinylated sgRNA
The sgRNA bearing a 3′ biotin provided a reagent useful for studying sgRNA-binding proteins present in human cells. Thus, we designed a pull-down assay using HEK293T cell lysate (see the Experiment Section in the Supporting Information) and the 3′-biotin-modified sgRNA (Figure 4A).
Figure 4.
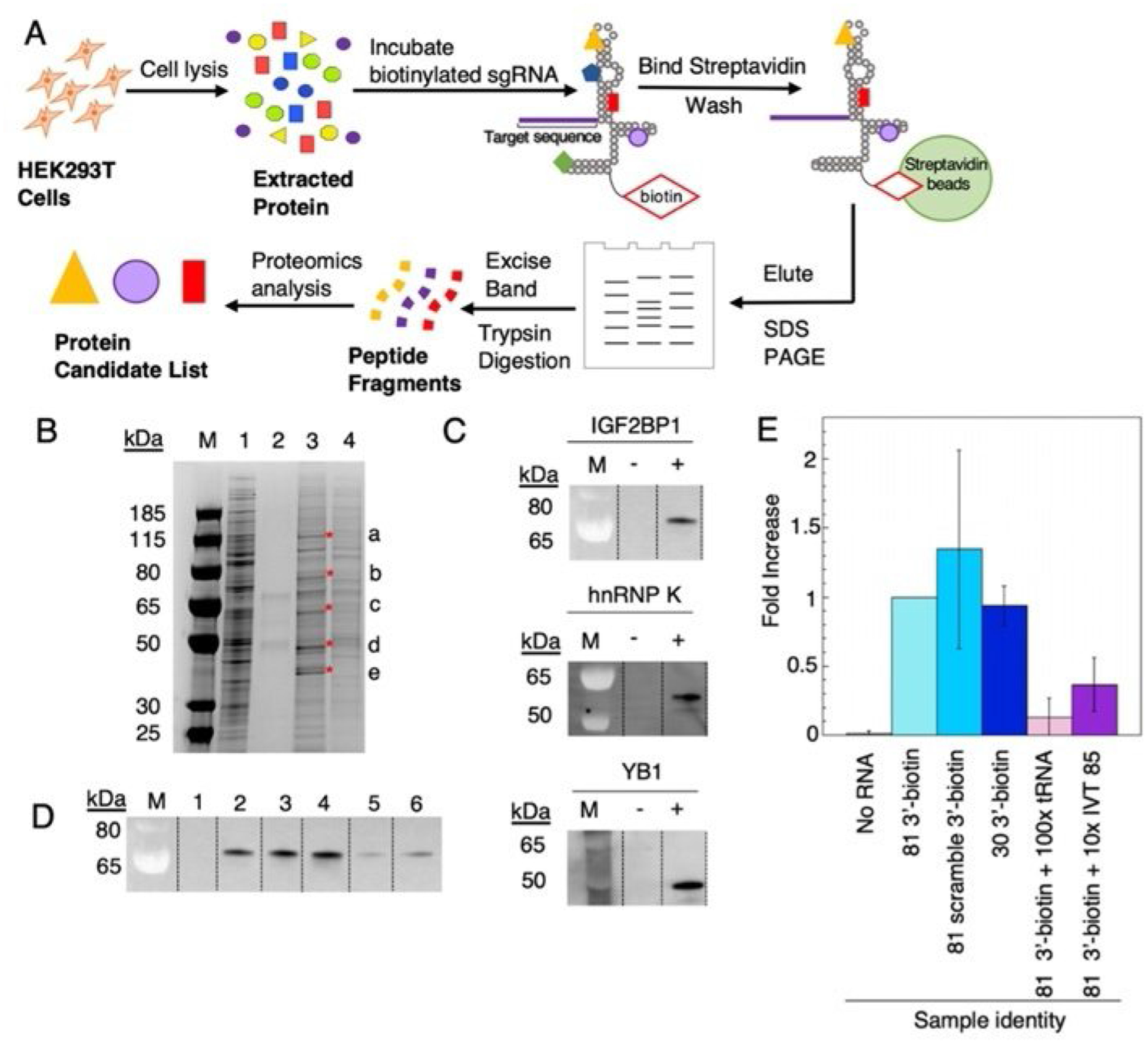
Identification of endogenous human sgRNA-binding proteins. A) Workflow scheme for identification of candidate endogenous human sgRNA binding proteins from HEK 293T cell lysate. B) SDS-PAGE gel stained with Imperial protein stain. Lanes: M=ladder, 1=5 μL of HEK 293T cell lysate, 2=no RNA control, 3= 81 3′-biotin pulldown, 4=81 3′-biotin +100-fold excess tRNA. Excised bands are indicated by a, b, c, d, e and red asterisks. C) Western blot verification of IGF2BP1, hnRNP K, YB1 present in elution from streptavidin beads. D) IGF2BP1 binding to sgRNA variants. Lanes: 1=no 81 3′-biotin, 2=81 3′-biotin, 3=81 3′-biotin with scrambled S1 target sequence (Table S1), 4= 5′P-30 nt-3′-biotin fragment, 5=81 3′-biotin+100-fold excess tRNA, 6=81 3′-biotin+ tenfold excess of IVT 85. Data in plot represents triplicate. E) Fold increase vs. RNA identity plotted. Plotted values correspond to averages±standard deviation (n=3).
First, we wanted to identify possible sgRNA binding protein candidates from human lysates. To accomplish this, HEK293T cell lysate was incubated in the presence of the 3′-biotinylated RNA before mixing with streptavidin magnetic beads. The beads were washed extensively to remove any nonspecific binding proteins and were further eluted by heating in SDS loading buffer under reducing conditions. The eluted samples were resolved by SDS-polyacrylamide gel electrophoresis and protein bands were visualized. To avoid identifying abundant HEK293T proteins, cell lysate was used as a negative control. Similarly, protein cell lysate treated in the absence of biotinylated RNA was used to avoid proteins that interact with the streptavidin beads. Lastly, a sample was prepared with 100-fold excess of tRNA to determine which protein bands corresponded to generic RNA-binding proteins. In total, five bands were excised from the gel (Figure 4B) and subjected to mass spectrometric analysis for protein identification (see the Experiment Section in the Supporting Information).
From the mass spectrometric analysis, insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1), heterogenous nuclear ribonucleoprotein K (hnRBP K), heterogeneous nuclear ribonucleoprotein C1/C2 (hnRBP C1/C2), Y-box-binding protein 1 (YB1), and Matrin 3 (MATR3) were determined to be candidates (Table S3). To confirm these proteins were present in the final elution, the pull-down assay was repeated, and bound proteins were detected by western blot analysis with commercially available antibodies to the candidate proteins. Of the five candidates, three were confirmed (IGF2BP1, hnRBP K, and YB1; Figure 4C). As the RNA binding requirements for IGF2BP1 binding are not well understood, we tested the effect of scrambling the 20-nt targeting sequence on IGF2BP1 binding. However, we found that scrambling the targeting sequence did not lead to an appreciable change in IGF2BP1 detected by the western blot analysis (Figure 4B). Similarly, we wished to determine whether the 30-nt biotinylated fragment of the sgRNA was sufficient for detection of IG2BP1. Interestingly, we found that this fragment was sufficient for significant IGF2BP1 binding (Figure 4D). Furthermore, IGF2BP1 could be competed with excess tRNA or an in vitro transcribed 85-nt sgRNA (Figure 4D). Overall, these experiments identified IFG2BP1, YB1, and hnRBP K as human sgRNA-binding proteins and indicate that the IFG2BP1 binding determinants reside in the 3′ fragment of the sgRNA.
Discussion
In this paper, we report a simple, efficient and versatile method for chemically modifying the 3′-end of the sgRNA. Given the buried location of the 5′-end of the sgRNA within the complex with Cas9, we suspected that modification of the 5′-end could have unintended effects on Cas9 cleavage activity or sgRNA binding. Therefore, we chose to modify the sgRNA at the 3′-end. This method allowed for the creation of a small collection of six chemically modified sgRNAs, generated from a single 3′-propylamine-modified 30-nt fragment. Although only a sample library of bifunctional NHS-esters was used here, we expect a wide variety of functionalized NHS-esters would be compatible with this method. This work expands the types of modifications that can be attached to the sgRNA and location of these functional groups.
Once our modified sgRNAs were generated, we sought to determine if the location or type of modification was detrimental to Cas9′s ability to cleave a double stranded DNA target. For this purpose, we used an sgRNA bearing an RNA scaffold that had been shown previously to support cleavage and was used for structural studies of Cas9 complexes.[34] We chose to study the impact of modification at this location using the in vitro DNA cleavage assay,[36] which is known to show a strong correlation with cleavage in a cellular context.[29,37,38] None of the chemically modified sgRNAs disabled Cas9′s ability to cleave the target DNA in vitro, suggesting this location is suitable for a wide variety of modifications. However, the 3′-amine modified sgRNA showed a small decrease in cleavage efficiency, possibly due to repulsion with positively charged amino acids at the RNA binding interface (i.e., K30, K31, K33). Smaller functional groups like 3′-iodine and 3′-methyl-maleimide showed a small increase in cleavage, whereas larger and more flexible appendages were not significantly different from the unmodified, transcribed sgRNA. We believe that the slight stimulation of the 3′-iodine modified sgRNA could arise from an interaction between the adjacent lysine residues and the haloalkyl group, however further studies are needed to validate this hypothesis.
Tight regulation of Cas9 expression and activity is highly desirable in genome editing so unwanted off-target edits do not occur.[39] For this reason, delivery of an intact RNP containing Cas9 and sgRNA is considered potentially advantageous over continuous expression from a plasmid or mRNA.[40–42] Expression of Cas9 from a transfected plasmid also poses spatial and temporal control issues. If the timing and location of Cas9 and sgRNA expression are not tightly coupled, free Cas9 might bind other endogenous RNAs, which could have unintended effects. Likewise, unbound sgRNA might interact with endogenous RNA-binding proteins and affect their function or be degraded by RNase present in the cell. Assembly of the RNP in vitro, followed by delivery, would mitigate these issues and artificially stabilizing the Cas9–sgRNA complex could be beneficial in this context. As a potential means of advancing this idea, we sought to introduce a covalent crosslink into the Cas9–sgRNA complex. To do so, we positioned a reactive amino acid (K31C or K31pAzF), near the 3′-end of the sgRNA modified with a compatible reactive handle. Only when the appropriately modified sgRNA was incubated in the presence of the Cas9 mutant with the reactive amino acid positioned near the 3′-end was a crosslinked band observed. We demonstrated crosslinking using a Cas9 cysteine mutant and different electro-philes including iodoacetamide and maleimide. We also showed that a Cas9 protein expressed with the unnatural amino acid pAzF crosslinked to sgRNA bearing a cyclooctyne. The 81 AMAS sgRNA combined with the K31C mutant gave the highest yield of crosslinked Cas9. However, this yield never surpassed ≈30% with additional attempts at optimization. Further improvements in covalent crosslinking yields will be needed for this to be a practical approach to stabilizing a sgRNA-Cas9 RNP. While the 3′-end of the sgRNA was fairly amendable to the crosslinking reaction, the close proximity of the sgRNA to the protein at this location might influence the fluorescence of 81 3′-Cy5 and could be a potential limitation to imaging inside cells.
Although there are reports that sgRNAs that have a 5′-tri-phosphate activate the RIG-I and MDA5 pathways of the innate immune response,[23,43] there are no reports of endogenous human proteins that bind directly to the sgRNA. Moreover, removal of the 5′-triphosphate abolishes inflammatory signaling,[23,43] suggesting that this interaction is a result of the 5′-triphosphate and not specific to the secondary structure of the sgRNA. Therefore, we sought to identify human proteins, not associated with the RIG-I or MDA5 pathway, that are capable of binding to the unique structure of the sgRNA in the absence of a 5′-triphosphate. It is possible that sgRNA orphaned from Cas9 could bind to endogenous proteins causing disruption of normal cellular functions. Similarly, temporal or spatial issues may arise from delivered plasmids that constitutively express Cas9 and the sgRNA component.[40–42] This could result in unbound sgRNA inside cells with the potential to interact with endogenous human proteins. Thus, we used our 3′-biotinylated sgRNA to reveal human sgRNA-binding proteins. From this experiment, we observed five protein bands that were enriched in the presence of the biotinylated sgRNA (Figure 4B). Protein identification by tandem mass spectrometry for these five bands gave five possible protein candidates (Table S3). Protein candidates were further validated by western blot and IGF2BP1, YB1, and hnRNP K were confirmed to be sgRNA-binding proteins (Figure 4C). Interestingly, MATR3 was not confirmed by western blot analysis despite literature evidence that MATR3 and hnRNP K co-immunoprecipitate in the presence of RNA.[44] We decided to further investigate the structural requirements of IGF2BP1 because of reports in the recent literature on this protein.[45,46] These reports demonstrated that IGF2BP1 preferably binds highly structured RNAs,[46] and a consensus sequence was established.[45] Scrambling the target sequence on the sgRNA did not abolish binding, suggesting the sequence and structure in this region is not critical for IGF2BP1 binding to the sgRNA. Interestingly, the 30-nt biotinylated sgRNA 3′ fragment, comprising stem loops 1 and 2, was sufficient for detection of IGF2BP1. Intriguingly, we were able to pull down IGF2BP1 despite the fact that the 30-nt biotinylated 3′ fragment does not contain any known IGF2BP1 consensus sequence.[45,47,48] Because most sgRNAs employed today contain the stem loop 1 and 2 structures, binding affinity for IGF2BP1 is likely a feature common to all these sgRNAs. Additional experiments are needed to determine the precise binding location between IGF2BP1 and the sgRNA. Further studies following the strategy reported here could also help identify other endogenous human sgRNA-binding proteins from other cell lines, tissues or under different conditions (e.g., after inter-feron treatment).
Conclusions
In this paper, we have described a versatile, efficient and generalizable method for functionalizing the 3′-end of the CRISPR sgRNA. We constructed an example collection of six modified sgRNAs and showed that the modifications do not interfere with the cleavage activity of Cas9 in vitro. We demonstrated crosslinking of the sgRNA to Cas9 in a site-specific manner and used a biotinylated sgRNA to identify three endogenous human sgRNA-binding proteins (IGF2BP1, YB1, and hnRNP K). Lastly, we showed the 30-nt 3′ fragment of the sgRNA was sufficient for binding IGF2BP1 indicating this is likely a property of most sgRNAs.
Experimental Section
Modification of 30-nt fragment of sgRNA:
N-α-maleimidoacetoxysuccinimide ester (AMAS), N-β-maleimidopropyloxysuccinimiide ester (BMPS), succinimidyl iodoacetate (SIA), and EZ-link NHS biotin were purchased from Thermo Fisher Scientific. Dibenzocycloocytne-N-hydroxysuccinimidyl ester (DBCO) was purchased from Sigma Aldrich. 3H-Indolium, 2-[5-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)-1,3-pentadien-1-yl]-1-[6-[(2,5-dioxo-1-pyrrolidinyl)-oxy]-6-oxohexyl]-3,3-dimethyl-, tetrafluoroborate (Cy5) was purchased from Lumiprobe. A 30-nt RNA was synthesized with a 5′-phosphate and a 3′-propylamine (N3–3′) from (Horizon-Dharmacon). In a microcentrifuge tube, 30-nt RNA (20 μL, 10 nmol), 10× PBS pH 7.4 (5 μL), and the heterobifunctional NHS-ester (25 μL, 20 mm dissolved in DMSO) were added and thoroughly mixed. The reaction was allowed to incubate at 30°C overnight. To remove excess of the reagents, the reaction was applied to a 3000 MWCO Amicon Ultra 0.5 mL (Millipore) centrifuge filter followed by 400 μL of high-purity nuclease-free water. The reaction was concentrated to ≈100 μL by centrifugation (16200g for 8 min). The flow through was discarded. Nuclease free water (400 μL) was added to the centrifugation unit and concentrated again to ≈ 100 μL by centrifugation (16200g for 8 min). This process was repeated a total of five times and concentrated to a final volume of 50 μL. Reaction completion and identity of the oligonucleotides was confirmed by MALDI-TOF mass spectroscopy. Mass spectra were recorded in negative ion mode and calibrated to an external DNA standard mass (6999.9 Da; Supporting Information, Table S2). We assumed the reaction between the NHS ester and the amine modified RNA proceeded quantitively because an ion corresponding to the unmodified sgRNA was not found in the mass spectrum for any of the modified sgRNAs synthesized.
Splint ligation of modified sgRNAs:
Full-length 81-nt sgRNAs were generated by splint ligation of two RNA fragments: a synthetic 51-nt RNA (Integrated DNA Technologies) and the modified 30-nt RNA bearing a 5′-phosphate. In a microcentrifuge tube, the two RNA fragments (3.5 nmol) and the complementary DNA splint (3.5 nmol, Integrated DNA Technologies) were combined and lyophilized to dryness. The pellet was resuspended in 10× ligation buffer (50 mm Tris·HCl pH 7.5, 10 mm MgCl2, 1 mm ATP) and 90 μL of water. To generate a DNA:RNA hybrid the solution was heated at 95°C for 5 min then slow cooled to 25°C. T4 DNA ligase (10 μL, 400000 UmL−1, New England Biolabs) was added to the solution and the solution was incubated at 30°C overnight. The sample was phenol-chloroform extracted and ethanol precipitated. The sample was lyophilized to dryness. To remove the DNA template, the sample was resuspended in 10× DNase buffer (5 μL, New England Biotechnologies), water (45 μL) and DNase I (5 μL, 2 UμL−1, New England Biolabs) and incubated at 37°C for 1 h. The reaction was quenched in 2× RNA loading dye (New England Biolabs) and heated at 95°C for 5 min. RNA was further purified by 10% denaturing PAGE gel and visualized by UV shadowing and excised. To elute the sgRNA, gel slices were crush and soaked overnight at room temperature in 500 mm NH4OAc and 100 mm EDTA. Polyacrylamide fragments were removed by 0.2-μm filter (Costar) followed by ethanol precipitation and lyophilized to dryness. The modified sgRNAs were resuspended in nuclease free water and stored in −20°C.
In vitro cleavage assay:
Recombinant S. pyogenes Cas9-NLS was purchased from QB3 Macrolabs (University of California Berkeley). The purchased protein was received at a stock concentration of 40 μm in 20 mm HEPES-KOH pH 7.5, 150 mm KCl, 10% glycerol, 1 mm DTT and stored at −80°C. Prior to the reaction, a sample of Cas9 was thawed on ice and diluted in 1x cleavage buffer (20 mm HEPES-KOH pH 7.5, 100 mm KCl, 5 mm MgCl2, 5% glycerol) to a concentration of 6 μm. sgRNAs were diluted to a concentration of 7.2 μm and heated at 95°C for 5 min then cooled to room temperature. The sgRNA and Cas9 samples (1 μL each) were incubated together at room temperature for 15 minutes. Cleavage reactions were initiated by the addition of the Snurf1 (S1) DNA (the sequence can be found in the Supporting Information), mixed thoroughly and incubated at 37°C for 45 min. Cleavage assays were performed in a final volume of 15 μL with a final concentration of 400 nm Cas9. The final reaction solution contained 150 ng Snurf1 DNA target, 20 mm HEPES-KOH pH 7.5, 100 mm KCl, 5 mm MgCl2, 5% glycerol. Reactions were quenched by the addition of proteinase K (1 μL, 20 mgmL−1, New England Biolabs) and RNase A (0.3 μL, 10 mgmL−1, Thermo Fischer Scientific) and incubated at room temperature for 20 min. Loading dye (6×; New England Biolabs) was added to the reaction, and the cleavage products were separated on a 1.2% agarose gel prestained with SYBR gold (Thermo Fischer Scientific). Cleavage reactions were carried out in triplicate and values reported are averages ±standard deviations.
General crosslinking sgRNAs to Cas9 procedure:
Cas9 mutant (K31C C80S C574S or C80S C574S or K31pAzF C80S C574S) was thawed on ice and then diluted to 6 μm in 1x cleavage buffer containing 20 mm HEPES·KOH pH 7.5, 100 mm KCl, 5 mm MgCl2, 5% glycerol, supplemented with HALT protease inhibitor (Thermo Fischer Scientific). sgRNAs were diluted to 7.2 μm in water, hybridized at 95°C for 5 min and slowly cooled to room temperature. To each reaction was added sgRNA (1 μL, 7.2 μm) and Cas9 mutant (1 μL, 6 μm) in a PCR tube and incubated at room temperature for 15 min. The reaction mixture was then diluted with additional 1fl cleavage buffer to a volume of 10 μL and incubated at room temperature overnight. The reaction was quenched by the addition of 2fl SDS loading buffer and heated at 95°C for 5 min. Covalently crosslinked sgRNA:Cas9 product was resolved from unreacted Cas9 by 4–12% SDS-polyacrylamide gel (200 V, 2 h). The resulting gel was stained in SYPRO Orange stain and visualized on a Typhoon imager (Molecular Dynamics). Cas9 bands were quantified using volume integration using ImageQuant software (Molecular Dynamics). Percent crosslinking was determined by the equation: % cross-linking=100×(volume covalent sgRNA:Cas9/[volume covalent sgRNA:Cas9+volume unreacted Cas9].
Pulldown of endogenous sgRNA binding proteins:
Pulldown reactions were prepared by combining 375 μL of 2flTENT buffer (20 mm Tris·HCl pH 8, 2 mm EDTA, 500 mm NaCl, 1% (v/v) Trion X-100, 7.5 μL 100fl HALT protease, 7.5 μL RiboLock RNase inhibitor, 300 μg of cell lysate, 100 picomoles of biotinylated sgRNA, and 100 μL of water in a total volume of 750 μL. To account for endogenous proteins interacting with the streptavidin beads, a reaction was prepared with no biotinylated RNA. Another reaction was prepared by replacing the volume of water with yeast tRNA (10 mgmL−1, Invitrogen). This created a 100-fold excess of tRNA relative to biotinylated RNA as bait for endogenous proteins. Another sample was prepared with a 10-fold excess of unmodified 85-nt sgRNA. Reactions were mixed by pipetting and incubated at room temperature for 30 min.
Streptavidin-coupled M280 Dynabeads (Thermo Fischer Scientific; 75 μL) were washed three times in 1× TENT buffer (1 mL) and separated using a BD IMag magnetic stand (BD Biosciences). Each RNA-protein mixture prepared above was added to the washed beads and mixed thoroughly by pipetting. The reactions were incubated at room temperature for an additional 30 min, mixing intermittently by tapping the tube every 5–10 min. Tubes were applied to the magnetic stand for 1 min to allow the beads to settle and the supernatant was discarded. The reaction was removed from the magnetic stand and 1 mL of ice-cold 1× TENT buffer was added, resuspending the magnetic beads and applied to the magnetic stand again. In total, the beads were washed three times with 1 mL of ice-cold 1× TENT buffer. To elute proteins, 40 μL of 1× SDS loading buffer supplemented with DTT was added to the beads and heated for 5 min at 95°C. The eluted volume containing the RNA interacting proteins was resolved on a 4–12% BOLT Bis-Tris Plus gel (Thermo Fischer Scientific). Proteins were visualized with Imperial Protein Gel Stain (Thermo Fischer Scientific) following the manufacturer′s protocol for overnight staining. Bands were chosen for further analysis by the intensity of the band and competition with added tRNA (Figure 4B).
Verification of protein candidates by western blot:
Cells were harvested, lysed and pull-down reactions were performed according to the protocol above. Proteins were eluted from the Dynabeads in 1× SDS loading buffer (40 μL). The eluted protein sample (10 μL) was separated by SDS-PAGE. Transferred. Proteins were transferred to an Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore). Blots were blocked in a 5% skim milk solution in Tris·HCl (25 mm, pH 7.4), 3 mm KCl, 140 mm NaCl, 0.05% Tween 20 (TBST, Affymetrix) and then probed with primary rabbit IgG antibodies for IGF2BP1 (18H5L31, 1:5000 dilution, Invitrogen), hnRBP K (D9A8, 1:1000 dilution, Cell Signaling Technology), hnRNP C1/C2 (D6S3N, 1:1000 dilution, Cell Signaling Technology), YB1 (D2B12, 1:1000 dilution, Cell Signaling Technology), MATR3 (A300–591A, 1:1000 dilution, Bethyl Laboratories) respectfully. This was followed by incubation with a secondary goat anti-rabbit IgG antibody (1:5000 dilution; Santa Cruz Biotechnology). PageRuler Plus (10 μL, Thermo Fischer) was loaded into each gel to estimate protein sizes. As a negative control, a pulldown reaction without the biotinylated RNA (10 μL) was loaded onto the gel. The protein candidates were detected using ECF substrate (GE Healthcare) on a Typhoon Trio Variable Mode Imager (GE Healthcare).
sgRNA variants binding to IGF2BP1:
A 51-nt RNA (Integrated DNA technologies) was obtained with the Snurf1 targeting sequence randomly scrambled (Table S1). The scrambled 51-nt RNA was ligated to the 30-nt biotinylated fragment of the sgRNA using the splint ligation protocol above. Cell lysate from three different passages were used as biological replicates. Western blots were conducted according to the same protocol as above. IGF2BP1 was quantified using volume integration and reactions were normalized to the unscrambled Snurf1 targeted 81 3′-biotin sgRNA sample.
Supplementary Material
Acknowledgements
P.A.B. acknowledges support from the US National Institutes of Health (NIH) in the form of grant R01GM08784. C.M.P. acknowledges support from the NIH predoctoral training program T32GM113770. H.O.G. received support from the NIH in the form of grant R21HG010559, and D.J.S. received support from the Innovative Genomics Center. The authors acknowledge the technical assistance of Michelle Salemi at the University of California Davis Proteomics Core. The contents of this publication are the sole responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://doi.org/10.1002/cbic.201900736.
References
- [1].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, Science 2012, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gasiunas G, Barrangou R, Horvath P, Siksnys V, Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Anders C, Niewoehner O, Duerst A, Jinek M, Nature 2014, 513, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA, Nature 2014, 507, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R, Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gong S, Yu HH, Johnson KA, Taylor DW, Cell Rep. 2018, 22, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, Doudna JA, Science 2016, 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Charpentier E, Marraffini LA, Curr. Opin. Microbiol 2014, 19, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hilton IB, D′Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA, Nat. Biotechnol 2015, 33, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, Nature 2016, 533, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR, Nature 2017, 551, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wright AV, Sternberg SH, Taylor DW, Staahl BT, Bardales JA, Kornfeld JE, Doudna JA, Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, et al. , Nature 2015, 523, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Briner AE, Donohoue PD, Gomaa AA, Selle K, Slorach EM, Nye CH, Haurwitz RE, Beisel CL, May AP, Barrangou R, Mol. Cell 2014, 56, 333–339. [DOI] [PubMed] [Google Scholar]
- [15].Wang D, Zhang C, Wang B, Li B, Wang Q, Liu D, Wang H, Zhou Y, Shi L, Lan F, Wang Y, Nat. Commun 2019, 10, 4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Graf R, Li X, Chu VT, Rajewsky K, Cell Rep. 2019, 26, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xu J, Lian W, Jia Y, Li L, Huang Z, Oncotarget 2017, 8, 94166–94171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fellmann C, Gowen BG, Lin P-C, Doudna JA, Corn JE, Nat. Rev. Drug Discovery 2017, 16, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dai W-J, Zhu L-Y, Yan Z-Y, Xu Y, Wang Q-L, Lu X-J, Mol. Ther. Nucleic Acids 2016, 5, e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilson RC, Gilbert LA, ACS Chem. Biol 2018, 13, 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ryan DE, Taussig D, Steinfeld I, Phadnis SM, Lunstad BD, Singh M, Vuong X, Okochi KD, McCaffrey R, Olesiak M, Roy S, Yung CW, Curry B, Sampson JR, Bruhn L, Dellinger DJ, Nucleic Acids Res. 2018, 46, 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, Bacchetta R, Tsalenko A, Dellinger D, Bruhn L, Porteus MH, Nat. Biotechnol 2015, 33, 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim S, Koo T, Jee H-G, Cho H-Y, Lee G, Lim D-G, Shin HS, Kim J-S, Genome Res. 2018, 28, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deng W, Shi X, Tjian R, Lionnet T, Singer RH, Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE, Murthy N, eLife 2017, 6, e25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Slesarev A, Viswanathan L, Tang Y, Borgschulte T, Achtien K, Razafsky D, Onions D, Chang A, Cote C, Sci. Rep 2019, 9, 3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].O′Reilly D, Kartje ZJ, Ageely EA, Malek-Adamian E, Habibian M, Schofield A, Barkau CL, Rohilla KJ, DeRossett LB, Weigle AT, Damha MJ, Gagnon KT, Nucleic Acids Res. 2019, 47, 546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin H, Song C-Q, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, Mou H, Oberholzer A, Ding J, Kwan S-Y, Bogorad RL, et al. , Nat. Biotechnol 2017, 35, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taemaitree L, Shivalingam A, El-Sagheer AH, Brown T, Nat. Commun 2019, 10, 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He K, Chou ET, Begay S, Anderson EM, van Brabant Smith A, Chem-BioChem 2016, 17, 1809–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Basila M, Kelley ML, van B. Smith A, PLoS One 2017, 12, e0188593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moore MJ, C. C. Query in RNA–Ligand Interactions, Part A, Vol. 317 (Eds.: Abelson JN, Simon M, Celander D), Academic Press, San Diego, 2000, pp. 109–123. [Google Scholar]
- [33].O′Geen H, Henry IM, Bhakta MS, Meckler JF, Segal DJ, Nucleic Acids Res. 2015, 43, 3389–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jiang F, Zhou K, Ma L, Gressel S, Doudna JA, Science 2015, 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- [35].Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG, J. Am. Chem. Soc 2002, 124, 9026–9027. [DOI] [PubMed] [Google Scholar]
- [36].Anders C, Jinek M, Methods Enzymol. 2014, 546, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grainger S, Lonquich B, Oon CH, Nguyen N, Willert K, Traver D, Zebrafish 2017, 14, 383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mehravar M, Shirazi A, Mehrazar MM, Nazari M, Avicenna J. Med. Biotechnol 2019, 11, 259–263. [PMC free article] [PubMed] [Google Scholar]
- [39].Senturk S, Shirole NH, Nowak DG, Corbo V, Pal D, Vaughan A, Tuveson DA, Trotman LC, Kinney JB, Sordella R, Nat. Commun 2017, 8, 14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim S, Kim D, Cho SW, Kim J, Kim J-S, Genome Res. 2014, 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen Z-Y, Liu DR, Nat. Biotechnol 2015, 33, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ramakrishna S, Kwaku Dad A-B, Beloor J, Gopalappa R, Lee S-K, Kim H, Genome Res. 2014, 24, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wienert B, Shin J, Zelin E, Pestal K, Corn JE, PLoS Biol. 2018, 16, e2005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Salton M, Elkon R, Borodina T, Davydov A, Yaspo M-L, Halperin E, Shiloh Y, PLoS One 2011, 6, e23882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Biswas J, Patel VL, Bhaskar V, Chao JA, Singer RH, Eliscovich C, Nat. Commun 2019, 10, 4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sanchez de Groot N, Armaos A, GraÇa-Montes R, Alriquet M, Calloni G, Vabulas RM, Tartaglia GG, Nat. Commun 2019, 10, 3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Patel VL, Mitra S, Harris R, Buxbaum AR, Lionnet T, Brenowitz M, Girvin M, Levy M, Almo SC, Singer RH, Chao JA, Genes Dev. 2012, 26, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chao JA, Patskovsky Y, Patel V, Levy M, Almo SC, Singer RH, Genes Dev. 2010, 24, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.