

Summary
Background
While antiretroviral regimens containing integrase inhibitors rapidly suppress HIV viral load in non-pregnant adults, few published randomized trial data have compared the safety and efficacy of any integrase inhibitor to efavirenz (EFV) when initiated during pregnancy.
Methods
A randomized, open-label trial was conducted at 19 hospitals/clinics in Argentina, Brazil, South Africa, Tanzania, Thailand, and the US. Antiretroviralnaïve pregnant women (20–36 weeks gestation) living with HIV were assigned to antiretroviral regimens containing either RAL (400mg twice daily) or EFV (600 mg each night), plus lamivudine 150 mg/zidovudine 300 mg twice daily (or approved alternative backbone regimen), using a web-based, permuted-block randomization stratified by gestational age and backbone regimen. The primary efficacy outcome was plasma HIV viral load below 200 copies per mL at (or near) delivery.
The primary efficacy analysis included all women with a viral load measurement at (or near) delivery who had pre-treatment viral load > 200 copies per mL and no pre-treatment genotypic resistance to any study drugs; secondary analyses eliminated these pre-treatment exclusion criteria. The primary safety analyses included all women who received study drug, and their infants. (Clinicaltrials.gov: NCT01618305)
Findings
From September 2013 to December 2018, 408 women were randomized (206 RAL, 202 EFV) and 394 delivered on-study (200 RAL, 194 EFV); 307 were included in the primary efficacy analysis (153 RAL, 154 EFV). 144 RAL arm women (94%) and 129 EFV arm women (84%) achieved the primary efficacy outcome [P=0·0015; absolute difference 10% (95% CI 3 to 18%)]; the difference primarily occurred among women enrolling later in pregnancy (interaction P=0·040). Severe or life-threatening adverse event frequencies were similar among mothers (30% in each arm; 61 RAL, 59 EFV) and infants (25% in each arm; 50 RAL, 48 EFV), with no treatment-related deaths.
Interpretation
These results support initiating raltegravir in pregnant women living with HIV presenting late for care.
Funding
The study was sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Allergy and Infectious Diseases (NIAID). Study drugs were provided by Merck and Company, Bristol Myers Squibb, and ViiV Healthcare.
Introduction
In 2018, an estimated 1·3 million pregnant women living with HIV worldwide required antiretrovirals both for their own health and to prevent transmission of HIV to their infants1. Strategies for prevention of perinatal transmission of HIV prioritize antiretroviral therapy (ART) during pregnancy to rapidly achieve HIV viral load suppression sustained through delivery2. Since the risk of perinatal transmission is greatly affected by the timing of ART initiation3, women who present late in pregnancy need potent ART to suppress viral load quickly and minimize the risk of HIV transmission.
The non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz has been extensively studied in non-pregnant adults and has been demonstrated to effectively reduce HIV RNA viral load. Raltegravir is an integrase inhibitor, which is a newer class of antiretrovirals that have been shown to be extremely potent antiretrovirals that produce a rapid HIV viral load reduction in non-pregnant adults who are treatment-naïve and those who are treatment experienced with triple-class resistant virus. Integrase inhibitors such as raltegravir have also been examined in pre-clinical, phase II, and phase III studies in adult patients, and have been generally well-tolerated. Pregnant women living with HIV, especially those presenting for care late in pregnancy, could potentially benefit from effective and well-tolerated integrase inhibitor based ART regimens that produce more rapid viral load suppression, potentially reducing the risk of vertical transmission of HIV to their infants. While integrase inhibitors have become first line antiretroviral therapy for non-pregnant adults, there have been few studies from large randomized studies comparing the safety and efficacy during pregnancy of integrase inhibitor-based ART with the first-line efavirenz-based ART regimen4–6 recommended for pregnant women by the World Health Organization (WHO) at the time the study was completed.
Methods
Study Design
NICHD P1081 was a Phase IV multicenter, randomized, open-label trial comparing the virologic response, safety, and tolerability of raltegravir, the first licensed integrase inhibitor, with efavirenz, a non-nucleoside reverse transcriptase inhibitor (NNRTI), in combination with two nucleoside reverse transcriptase inhibitors (NRTI) in ART-naïve pregnant women initiating ART from 20 to 36 weeks gestation. The trial enrolled participants at 19 clinic and hospital sites in Brazil (7), Tanzania (1), South Africa (1), Thailand (3), Argentina (2), and the US (5).
P1081 was originally developed by the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) as a three-arm randomized trial (raltegravir vs efavirenz vs lopinavir/ritonavir based ART) for women 28 to <37 weeks gestation, and began enrollment in September 2013, but was closed to accrual in December 2014 due to slow enrollment. Eunice Kennedy Shriver National Institute of Health (NICHD) assumed responsibility for the trial, which was modified to a two-arm trial (raltegravir vs efavirenz ART) with a smaller sample size and inclusion of data from the 14 IMPAACT P1081 participants in the two continuing arms in final study analyses. The six women in the LPV/r arm continued scheduled study evaluations through six months postpartum but were excluded from all data analyses. Enrollment resumed in July 2015, was expanded to include women 20 to <37 weeks gestation in August 2016 after 22% of the target sample was enrolled, and was completed in February 2018.
All versions of the trial protocol and amendments were approved by the institutional review boards (IRBs) or regulatory entities (RE) of each site or country.
Participants
Pregnant women living with HIV infection with gestational age between 20 and <37 weeks (determined by last menstrual period and/or ultrasound) who were ART-naïve or who had received short-course zidovudine (maximum of eight weeks) in previous pregnancies to prevent perinatal transmission of HIV were eligible for enrollment. Pregnant women living with HIV who were referred to HIV care clinics and hospitals and met the inclusion criteria were offered participation in the study. Those who agreed to participate gave oral and written consent.
Participants were required to be of the legal age of consent as defined by the country. Principal inclusion criteria: Naïve to ART or have received ART with short course zidovudine (maximum of 8 weeks) for prevention of perinatal HIV transmission in previous pregnancies; documentation of HIV-1 infection defined as positive results from two samples collected at different time points; viable pregnancy with gestational age of ≥ 20 to ≤ 36 weeks based upon last menstrual period and/or ultrasound; intent to continue pregnancy; Exclusion criteria: active labor; use of antiretroviral drugs during current pregnancy; chemotherapy for active malignancy; HIV genotypic resistance, to efavirenz or raltegravir or to NRTIs that will be included in the antiretroviral regimens; serious active opportunistic infection known allergy/sensitivity to any study drugs or their formulations or sulfonamide allergy; the following laboratory values within 30 days of enrollment: hemoglobin ≥ Grade 3, absolute neutrophil count ≥ Grade 2, alanine aminotransferase (ALT) or aspartate aminotransferase (AST) ≥ Grade 2, serum creatinine ≥ Grade 1, platelet count ≥ Grade 3; evidence of pre-eclampsia; receipt of disallowed medications per protocol. The complete list of inclusion and exclusion criteria is available in the protocol (in the Appendix p. S1).
Randomisation and Masking
Participants were randomly allocated to one of two ART regimens, one raltegravir-based and one efavirenz -based. The trial was open-label with no masking of treatment assignment for participants or clinicians. The web-based, central computer randomization system used permuted block allocation (block size 4) with stratification by gestational age at enrollment (20 to <28 weeks; 28 to <31 weeks; 31 to <34 weeks; 34 to <37 weeks) and NRTI backbone (lamivudine/zidovudine or alternative, locally-supplied NRTI regimen), and dynamic balancing by study site7. The participants, site staff, and the statisticians who analyzed the data were not masked to group assignment.
Trial Procedures
Written informed consent was obtained from maternal participants at an initial screening visit, along with a clinical case history, confirmatory HIV testing, and specimen for HIV-1 genotypic resistance testing. Women were then randomised to receive raltegravir 400 mg twice a day or efavirenz 600 mg each night in addition to lamivudine 150 mg/zidovudine 300 mg twice a day (or an alternative, locally supplied NRTI regimen) from study entry through delivery. Resistance testing results were not required for enrollment or initiation of assigned study drugs; once resistance testing results became available, clinicians could modify ART based on finding resistance, and women with resistance to any study ARV were excluded from the primary efficacy analysis. Maternal evaluations at study visits (screening, entry, weeks 1, 2, 4 and every 2 weeks until delivery, then weeks 2, 6, 16, and 24 postpartum) included an interval medical history, physical examination, hematology and chemistry tests, HIV RNA PCR, and CD4+ T-cell count. Infant evaluations at study visits (birth, and weeks 2, 16, and 24) included clinical history, physical examinations, hematologic and chemistry laboratory evaluations, and HIV nucleic acid testing. Women who had virologic failure and HIV-infected infants had additional laboratory testing. Participants were followed through 24 weeks postpartum.
Laboratory Assessments
Women enrolled to NICHD P1081 had HIV RNA viral load measurements performed using Abbott Real Time assays, with a lower limit of quantification (LLQ) of either 20 or 40 copies per mL. The 14 women enrolled under IMPAACT P1081 who were eligible for inclusion in final analyses may have had their viral load measured using local assays obtained as part of standard of care with an LLQ of 200 copies per mL or lower.
All participating sites and testing labs complied with the IMPAACT DAIDS Manual of Procedures (MOP). Per the MOP, genotyping and viral load assessments were performed at Virology Quality Assurance (VQA)-certified (non-US sites) or Clinical Laboratory Improvement Amendments (CLIA)-certified (US sites) labs. HIV testing labs and hematology and chemistry testing labs were required to maintain College of American Pathologist (CAP) certification. Immunology testing, such as CD4+ T-cell counts, were performed at labs monitored by the DAIDS Immunology Quality Assurance Program. In addition to required certification, sites and labs storing peripheral blood mononuclear cells (PBMC) underwent quarterly testing to evaluate their reliability in cryopreserving PBMC.
Outcome Measures
The primary efficacy outcome measure was maternal plasma HIV-1 RNA viral load <200 copies per mL at or near (within 21 days before delivery) delivery. The primary safety outcome measure for women and infants was a new Grade 3 or 4 adverse event (AE, including a sign or symptom, diagnosis, or hematology/chemistry event) according to the DAIDS Table for Grading the Severity of Adult and Pediatric Adverse Events (V2·0, November 2014)8. “New” AEs were defined as those that occurred on or after randomization, or if present at baseline, AEs that increased in grade from baseline. The primary tolerability outcome measure was remaining on randomized study treatment (raltegravir or efavirenz) through delivery.
Secondary efficacy outcome measures included maternal HIV viral load below the local assay lower-limit-of-quantification at or near delivery, and a composite efficacy-tolerability outcome of both (1) a rapid, sustained viral load decrease, defined as achieving a pre-specified minimum drop in plasma HIV-1 RNA by week 2 (≥2·0 log10 decrease from entry or <200 copies per mL) and (among women who delivered after 28 days on study) maintaining <1000 copies per mL until delivery, and (2) remaining on randomized study treatment through delivery (see the Protocol for further details).
Adverse pregnancy outcome measures included stillbirth, low (<2500 g) and extremely low (<1500 g) birthweight, preterm (<37 weeks) and extremely preterm (<34 weeks) delivery, and major congenital anomaly. All reported congenital anomalies were reviewed centrally and classified as major, minor, or not a defect based on the Metropolitan Atlanta Congenital Defects Program9. Infants were classified centrally as HIV-infected if they had at least two positive HIV nucleic acid test results from different samples and HIV-uninfected if they had at least two negative HIV nucleic acid test results at Week 6, Week 16, and/or Week 24 postpartum, and no positive result at any week.
Statistical Analysis
The target sample size of 334 evaluable women provided 80% power to detect a difference in response proportions of 75% vs. 60% with a two-sided type I error rate of 0·05 and allowing for two interim efficacy analyses. Assuming 5% of women were non-evaluable, and 10% were excluded due to genotypic resistance, target enrollment was 394 women (approximately 197 per arm).
Women eligible for the primary efficacy analysis were those who had a valid viral load result at or near delivery; per DSMB recommendation, evaluable women were those who had plasma HIV-1 RNA viral load at or above 200 copies per mL at screening or entry. The efficacy analysis was performed three ways:
The primary analysis excluded women who had genotypic resistance to, or incomplete resistance results for, any study drugs at screening.
A secondary efficacy analysis included all evaluable women, regardless of genotypic resistance status at screening.
A sensitivity efficacy analysis included all eligible women, regardless of genotypic resistance status or screening/entry viral load.
Pre-specified sensitivity analyses were also performed to assess the potential impact of missing data on the results of the above efficacy analyses. These analyses were planned to be performed in two ways: (a) as an extreme “worst-case” analysis, assuming a missing viral load at delivery would show a successful or unsuccessful viral load decrease in a way that would minimize the difference between arms, and (b), a more plausible analysis assuming a missing viral load would show a successful viral load decrease with probability equal to the estimated probability of achieving the target viral load among women in the same arm who had a viral load at delivery. However, because the “worst-case” analysis (a) showed similar response proportions to the primary efficacy analysis, the more plausible analysis (b) was not needed and not performed.
Women who received at least one dose of study drug (and their infants) were evaluable for the safety and tolerability analyses.
All primary statistical comparisons (efficacy, safety, and tolerability) were performed via Cochran-Mantel-Haenszel test stratified by gestational age at enrollment (but not NRTI backbone). We calculated Wald confidence intervals (with continuity correction) for the difference in response proportions. Subgroup analyses were performed to assess the heterogeneity of treatment effects on the primary efficacy comparison with respect to gestational age stratum at entry, with interaction testing performed via logistic regression. A post-hoc interval-censored analysis based on nonparametric survival analysis for interval-censored data10 compared time to virologic suppression using a generalized log-rank test and a hazard ratio with 95% confidence interval from an interval-censored proportional hazards model. This analysis included all women who received at least one dose of study drug and had a viral load at or above 200 copies per mL at screening or entry (i.e. did not have the event at baseline). Subgroup analyses were again performed to investigate whether the treatment effect differed by gestational age stratum, with interaction testing performed via an interval-censored proportional hazards model. The exact date of virologic suppression during pregnancy is interval-censored because it occurred in the interval between two viral load measurements (the last one above or equal to 200 copies per mL and the first one below 200 copies per mL). An interval-censored survival plot shows the estimated cumulative probability of achieving virologic suppression according to number of weeks since randomization, with dotted lines indicating intervals where probability estimates are undefined11. Approximate numbers of women at risk and censored at each time point were calculated using Kaplan-Meier analysis. Comparisons of pregnancy outcomes and infant infections were performed using Fisher’s exact test.
The trial was monitored semiannually by the US NIAID Division of AIDS (DAIDS) Multinational Data & Safety Monitoring Board (DSMB). The DSMB reviewed study conduct and safety at each meeting, and reviewed two interim efficacy analyses. Because interim efficacy analyses used the conservative Haybittle-Peto stopping boundary (P<0·001), no adjustment for alpha spending was performed in the final analysis, and a nominal two-sided P<0·05 was considered statistically significant. Analyses were conducted with SAS 9.4 (SAS Institute, Cary NC).
The trial was registered at Clinicaltrials.gov (Clinicaltrials.gov number, NCT01618305).
Further details are provided in the statistical analysis plan (for access to the plan, see the Data Sharing Statement at the end of the manuscript).
Role of the Funding Source
Staff of NICHD, which provided the funding for the protocol, were full study team members involved in study design, data collection, data analysis, data interpretation, and writing of the report. Companies that supplied study drug (Merck Company, Bristol Myers-Squibb, and ViiV Healthcare) had no influence on the study design, conduct of the trial, data collection and analysis, or the writing of the manuscript and decision to submit for publication. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
The authors assume responsibility for the accuracy and completeness of the data, as well as the fidelity of the trial to the protocol. Upon acceptance, the protocol will be made available online as per Lancet HIV guidelines, via the NICHD DASH portal (see the Data Sharing Statement at the end of the manuscript).
Results
From September 2013 to December 2018, a total of 408 pregnant women (206 randomized to raltegravir, 202 to efavirenz) were enrolled at sites in Argentina (N=20), Brazil (N=190), South Africa (N=60), Tanzania (N=84), Thailand (N=47), and the US (N=7); 205 (50%) enrolled from 20 to less than 28 weeks gestation, and 203 (50%) from 28 to less than 37 weeks gestation. Baseline characteristics were well-balanced between arms (Table 1). Of the 408 women enrolled, 14 were off study prematurely prior to delivery. Of the 394 on study at delivery, 368 (94%) had either screening or entry plasma HIV-1 RNA viral load ≥200 copies per mL and were thus eligible for the primary efficacy analysis. Among eligible women, 307 had complete genotypic resistance results documenting no resistance to study drug and were evaluable for the primary efficacy analysis. Baseline characteristics were generally well-balanced between evaluable and non-evaluable women, except those that differed by design due to criteria for evaluability (HIV-1 RNA viral load, genotypic resistance status) and race/ethnicity. Baseline characteristics by evaluability status are shown in Table S1 (Appendix, p. S2).
Table 1:
Baseline characteristics of women at randomization$
| EFV (N=202) | RAL (N=206) | Total (N=408) | |
|---|---|---|---|
| CharacteristicΔ,∏ | |||
| Age (years) | |||
| Median | 25 | 27 | 27 |
| IQR | 22 to 31 | 23 to 32 | 22 to 32 |
| Race/ethnicity | |||
| Asian, Pacific Islander | 24/200 (12%) | 23/205 (11%) | 47/405 (12%) |
| Black, Not Hispanic | 74/200 (37%) | 72/205 (35%) | 146/405 (36%) |
| Hispanic, Latino | 101/200 (51%) | 108/205 (53%) | 209/405 (52%) |
| White, Not Hispanic | 1/200 (1%) | 2/205 (1%) | 3/405 (1%) |
| HIV-1 RNA viral load (log10 copies/mL) | |||
| Median | 4·1 | 4·1 | 4·1 |
| IQR | 3·4 to 4·5 | 3·3 to 4·6 | 3·4 to 4·6 |
| Absolute CD4 count (cells/mm3) | |||
| Median | 408 | 389·5 | 395 |
| IQR | 289 to 602 | 240 to 567 | 262 to 574 |
| NRTI background regimen | |||
| Lamivudine and Zidovudine | 170/202 (84%) | 171/206 (83%) | 341/408 (84%) |
| Emtricitabine and Tenofovir disoproxil fumarate | 31/202 (15%) | 33/206 (16%) | 64/408 (16%) |
| Lamivudine and Tenofovir disoproxil fumarate | 1/202 (<1%) | 2/206 (1%) | 3/408 (1%) |
| Gestational age (wks) | |||
| Median | 27 | 28 | 27 |
| IQR | 23 to 31 | 22 to 31 | 23 to 31 |
| 20-<28 weeks | 102/202 (50%) | 103/206 (50%) | 205/408 (50%) |
| 28-<37 weeks | 100/202 (50%) | 103/206 (50%) | 203/408 (50%) |
| Genotypic resistance results†& | |||
| Reverse transcriptase resistance mutation | 14/187 (7%) | 21/197 (11%) | 35/384 (9%) |
| Integrase inhibitor resistance mutation | 0/190 (0%) | 0/192 (0%) | 0/382 (0%) |
| Incomplete genotypic resistance results | 21/202 (10%) | 17/206 (8%) | 38/408 (9%) |
EFV = Efavirenz; RAL = Raltegravir; IQR = Interquartile range
The “baseline” value for each participant refers to the value that was observed or measured closest to (and on or before) their date of randomization.
Continuous variables, including age, HIV-1 RNA viral load, CD4 cell count, and gestational age at entry, are reported as median (interquartile range).
Categorical variables, including race/ethnicity, NRTI background regimen, gestational age at entry, and genotypic resistance test results, are reported as number/total number (%).
Genotypic resistance testing for each woman was performed on a sample taken during the screening visit. Although results from this testing were not available until after entry (i.e. randomization), these results measured resistance prior to randomization.
There were 38 women who had incomplete resistance results; 12 were missing reverse transcriptase results (three in the RAL arm and nine in the EFV arm), 14 were missing integrase results (eight in the RAL arm and six in the EFV arm), and 12 were missing both results (six in each arm).
Overall, 370 (91%) of the 408 women enrolled had complete genotypic resistance results. Among those with resistance results at screening, 9% of women had genotypic resistance to reverse transcriptase inhibitors and none had resistance to integrase inhibitors. Among the 408 women enrolled, five (all randomized to efavirenz) went off study prior to initiating study treatment and were excluded from all post-baseline analyses. Nine additional women (three randomized to efavirenz and six randomized to raltegravir) went off-study prior to delivery and were excluded from the primary efficacy analysis and analyses of pregnancy outcomes (Figure 1).
Figure 1.
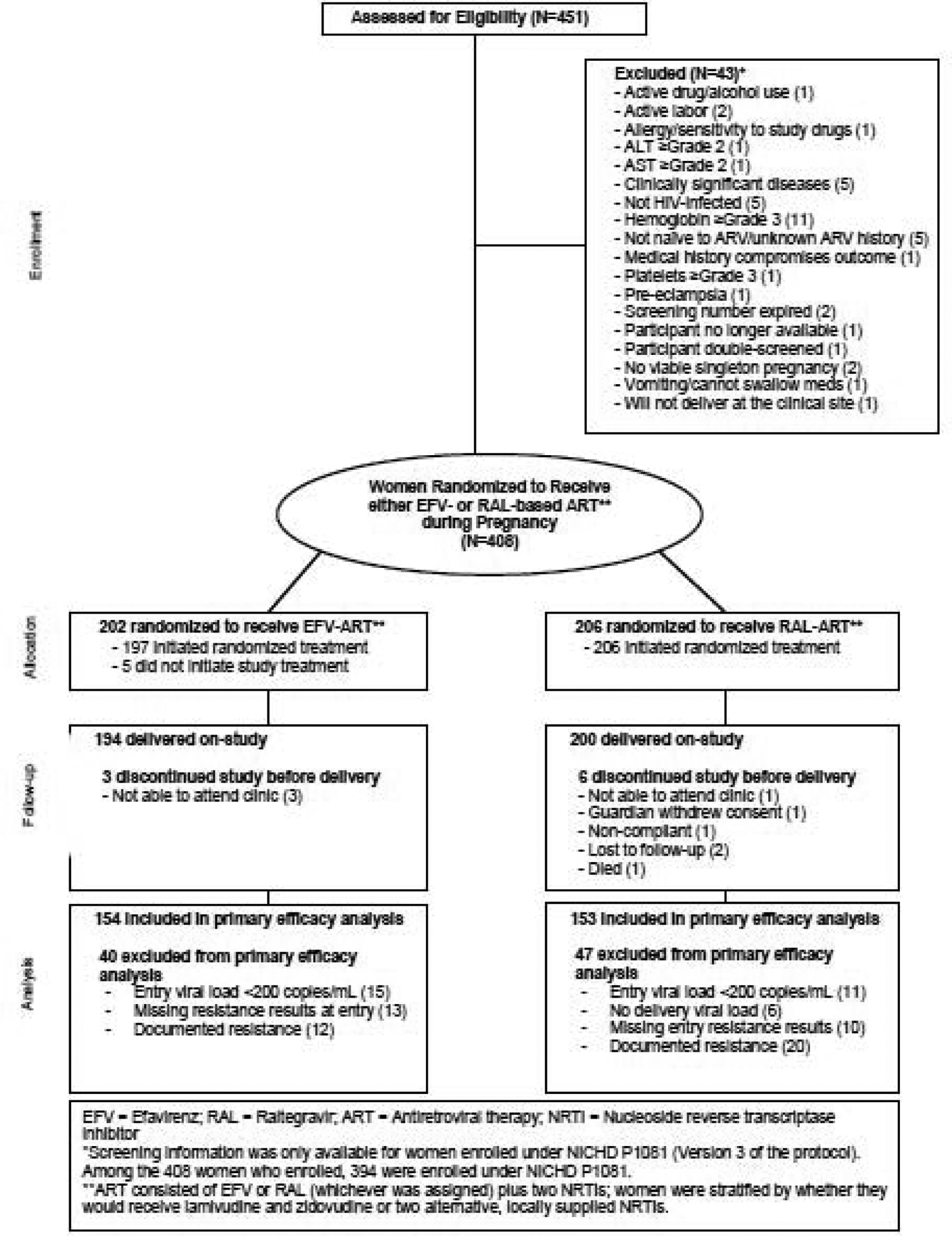
Study profile
In the primary efficacy analysis (Table 2), the proportion of women with viral load <200 copies per mL at or near delivery was significantly higher in the raltegravir arm (94%) than the efavirenz arm (84%) [P=0·0015; absolute difference 10% (95% CI, 3 to 18%)]. The treatment effect differed significantly by gestational age at entry [interaction P=0·040]. Women in each arm who enrolled at 20 to <28 weeks gestation had similar response proportions for the primary efficacy outcome, while among those who enrolled at 28 to <37 weeks gestation, 93% in the raltegravir arm achieved a delivery viral load <200 copies per mL compared with 71% in the efavirenz arm. The results were consistent in both secondary and sensitivity analyses of the primary efficacy outcome measure [Table S2, Appendix, p. S3]. Additionally, similar response proportions were observed in a pre-specified “worst-case” sensitivity analysis (data not shown).
Table 2:
Efficacy
| EFV | RAL | Absolute difference (95% CI) | P-value† | Interactio n P-value‡ | ||
|---|---|---|---|---|---|---|
| Efficacy outcome | Subgroup | N successes/Total (%) | ||||
| PrimaryΩ: Viral Load <200 copies/mL at delivery | Overall | 129/154 (84%) | 144/153 (94%) | 10% (3 to 18%) | P=0·0015 | P=0·040 |
| 20 to <28 weeks gestation | 74/76 (97%) | 68/71 (96%) | −2% (−9 to 6%) | |||
| 28 to <37 weeks gestation | 55/78 [71%) | 76/82 (93%) | 22% (9 to 35%) | |||
| SecondaryΩ: Viral load <LLQ at delivery | 90/154 (58%) | 131/153 (86%) | 27% (17 to 37%) | P<0·0001 | ||
| CompositeΩ: Rapid, sustained viral load decrease + tolerability | Overall | 84/133 (63%) | 124/139 (89%) | 26% (16 to 36%) | P<0·0001 | |
| Rapid viral load decrease | 93/133 (70%) | 129/139 (93%) | 23% (13 to 33%) | |||
| Sustained viral load decrease | 118/124 (95%) | 121/127 (95%) | 0% (−6 to 6%) | |||
| Remained on study treatment through delivery | 129/133 (97%) | 134/139 (96%) | −1% (−6 to 4%) | |||
EFV = Efavirenz; RAL = Raltegravir; LLQ = Lower limit of quantification
Calculated via Cochran-Mantel-Haenszel test stratified by gestational age (20-<28 weeks; 28-<31 weeks; 31-<34; 34-<37 weeks) at entry
Calculated via logistic regression. Gestational age strata were combined due to small sample size and low event rates in the later gestational age strata; two strata (20-<28 weeks; 28-<37 weeks) were considered in interaction testing
See Methods (Outcome measures) for detailed definitions.
Results were similar for the secondary efficacy outcome measures. The proportion of women who achieved a viral load below the local assay lower-limit-of-quantification at or near delivery was significantly higher in the raltegravir arm (86%) than the efavirenz arm (58%) [P<0·0001]. Similarly, the proportion of women who had a successful composite efficacy-tolerability outcome was significantly higher in the raltegravir arm (89%) than the efavirenz arm (63%) [P<0·0001], primarily because a higher proportion (93% vs. 70%) achieved a rapid viral load decrease. Results were consistent in all secondary and sensitivity analyses of both the secondary and composite efficacy outcome measures [Table S2, P<0·0001 for all, Appendix, p. S3]. Overall, six women in the EFV arm and four women in the RAL arm had viral load <200 copies per mL at some point prior to delivery but then had viral rebound by the time of delivery.
In a post-hoc analysis of time to virologic suppression (Figure 2), time to first viral load below 200 copies per mL in pregnancy was shorter in the raltegravir arm than the efavirenz arm [P<0·0001; HR=1·84 (95% CI 1·48 to 2·30)]. In subgroup analyses, time to virologic suppression was shorter in the raltegravir arm in both subgroups of gestational age at enrollment [20 to <28 weeks gestation: HR=1·52 (95% CI 1·13 to 2·07), Figure S1a; 28 to <37 weeks gestation: HR=2·25 (95% CI 1·62 to 3·13), Figure S1b, Appendix, p. S6–7]. The magnitude of the difference in time to virologic suppression between arms was larger among women enrolled from 28 to <37 weeks gestation [interaction P=0·052].
Figure 2. Estimated cumulative probability of achieving virotogic suppression according to time since randomization.
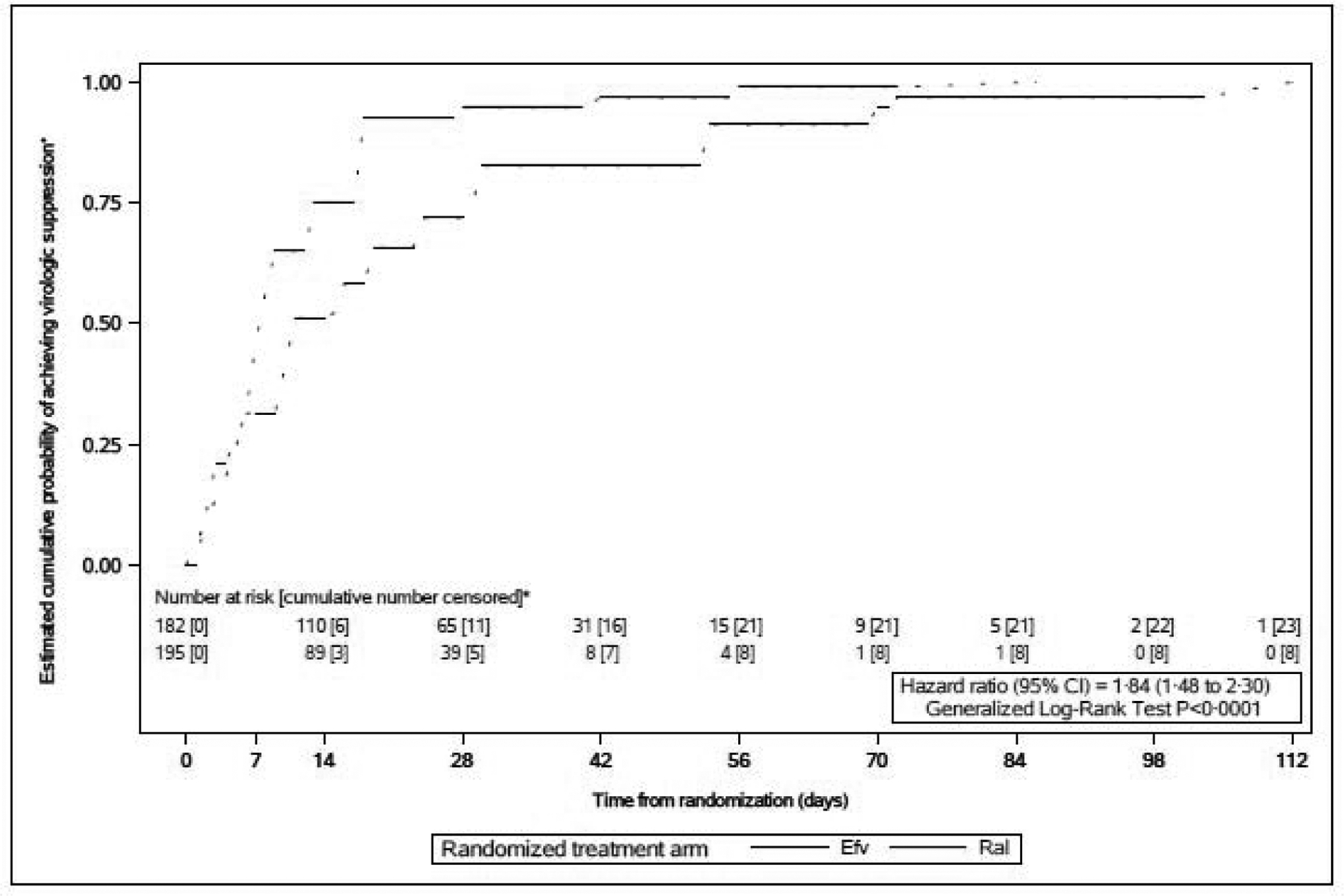
■Virologic suppression was defined as having an HIV-1 RNA plasma viral toad <200 copies/mL.
•This interval-censored survival plot shows the estimated cumulative probability of achieving virologic suppression according to nurrber of weeks since randomization, based on nonparametric survival analysis for interval-censored time-to-event data Dotted lines indicate intervals where probabifty estimates are undefined (unlike Kaplan-Meier survival plots. which are continuous step functions). Interval censoring was used because the exact date of the event of interest, virologic suppression below 200 copies/mL, is not known, however, the event date is known to be in the interval between the last viral toad measurement above or equal to 200 copies/mL and the first one below 200 copies/mL. Participants who did not achieve virologic suppression by delvery were censored at their delivery date (or their off-study date if they discontinued study participation before delivery}. Participants were considered at risk at a time point if they were on study, had not yet deSvered, and had not yet had an observed viral toad <200 copies/mL.
Among the 403 women who initiated study treatment, 397 (99%) received their first dose within one day of randomisation (all 403 received it within one week). Tolerability was high in both arms; 96% of women (97% in the raltegravir arm vs. 95% in the efavirenz arm; P=0·56) stayed on assigned treatment through delivery. The time on assigned treatment until delivery was similar between treatment arms, ranging from one day to 22 weeks (median 11.3 weeks, interquartile range [IQR], 6·9 to 15·6 weeks).
Similar proportions of women in both arms experienced at least one new Grade 3 or 4 AE (30% in each; [P=0·91], Table 3). There were no substantial differences between the arms with respect to specific AEs (Table S3, Appendix, p. S4). The most common laboratory AEs were abnormal hematology values (11% overall), primarily abnormal hemoglobin values. Similar proportions of women experienced a new Grade 3 or 4 clinical diagnosis in both arms, and each diagnosis was infrequently observed (≤2% for all). When grouped by body system, the most common diagnoses were cardiovascular (hypertension in pregnancy/pre-eclampsia) and hematologic diagnoses.
Table 3:
Maternal Safety, Pregnancy Outcomes, and Infant Safety
| EFV | RAL | Total | P-value* | |
|---|---|---|---|---|
| Maternal Adverse events | N=197 | N=206 | N=403 | |
| Any Grade ≥3 AE | 59/197 (30%) | 61/206 (30%) | 120/403 (30%) | P=0·91 |
| Grade ≥3 Sign/Symptom | 14/197 (7%) | 11/206 (5%) | 25/403 (6%) | |
| Grade ≥3 Lab AE | 34/197 (17%) | 33/206 (16%) | 67/403 (17%) | |
| Hematologic | 22/197 (11%) | 24/206 (12%) | 46/403(11%) | |
| Chemistry | 13/197 (7%) | 9/206 (4%) | 22/403 (5%) | |
| Grade ≥3 Diagnoses | 31/197 (16%) | 34/206 (17%) | 65/403 (16%) | |
| Deaths (any reason)# | 0/197 (0%) | 1/206 (<0.5%) | 1/403 (<0.5%) | |
| Adverse pregnancy outcomes | ||||
| Stillbirth† | 1/194 (1%) | 3/200 (2%) | 4/394 (1%) | P=0·62 |
| Preterm birth (<37 weeks) | 20/190 (11%) | 24/195 (12%) | 44/385 (11%) | P=0·63 |
| Extremely preterm birth (<34 weeks) | 6/169 4%) | 4/171 (2%) | 10/340 (3%) | P=0·54 |
| Low birthweight (<2500 g) | 24/193 (12%) | 25/197 (13%) | 49/390 (13%) | P>0·99 |
| Extremely low birthweight (<1500 g) | 0/193 (0%) | 1/197 (1%) | 1/390 (<0.5%) | P>0·99 |
| HIV transmissions | ||||
| HIV-infected infantΔ | 6/184 (3%) | 1/190 (1%) | P=0·064 | |
| Infant Adverse Events | N=194 | N=199 | ||
| Any Grade ≥3 AE | 48/194 (25%) | 50/199 (25%) | 98/393 (25%) | P=0·94 |
| Grade ≥3 Sign/Symptom | 15/194 (8%) | 15/199 (8%) | 30/393 (8%) | |
| Grade ≥3 Lab AE | 22/194 (11%) | 17/199 (9%) | 39/393 (10%) | |
| Hematologic | 13/194 (7%) | 6/199 (3%) | 19/393 (5%) | |
| Chemistry | 11/194 (6%) | 11/199 (6%) | 22/393 (6%) | |
| Grade ≥3 Diagnosis | 32/194 (16%) | 34/199 (17%) | 66/393 (17%) | |
| Deaths (any reason)## | 1/194 (1%) | 1/199 (1%) | 2/393 (1%) |
EFV = Efavirenz; RAL = Raltegravir; AE = Adverse event
The p-value for the primary safety comparisons (proportion that experienced any grade ≥3 AE) was calculated via Cochran Mantel Haenszel test stratified by gestational age at entry. P-values for comparisons of adverse pregnancy outcomes and HIV transmissions were calculated via Fisher’s Exact test.
The maternal death was ruled a murder, attributed by the reporting site as unrelated to study treatment or conduct. The primary cause of death was liver-rupture due to trauma.
One woman in the raltegravir arm had discordant outcomes. Because one of the outcomes occurred prior to study entry (intra-uterine fetal demise), only the outcome for the infant who was delivered on-study (live birth) is included (see Results for further details).
Excluded infants (ten in the efavirenz arm and 9 in the raltegravir arm) had no positive HIV-infection test result, but also did not have sufficient negative test results to meet the definition of “HIV-uninfected”.
Both infant deaths were attributed by the reporting site as not related to study treatment or conduct. The primary causes of death were neonatal necrotizing enterocolitis (efavirenz arm) and presumed sudden infant death syndrome (raltegravir arm).
Of the 394 women who remained on study through delivery, 390 had live-births and four had stillbirths (one in the efavirenz arm and three in the raltegravir arm) (Table 3). One mother who had discordant outcomes (one intrauterine fetal demise (IUFD) at 18 weeks gestation and one live-birth on-study) was considered to have had a live-birth singleton pregnancy because the IUFD occurred prior to study entry and initiation of study treatment. There was no significant difference between arms in the proportion of stillbirths [Fisher’s exact P=0·62].
There were three pairs of live-born twins, making a total of 393 live-birth infants delivered on-study. There were no significant differences between arms in the proportions of infants with (extremely) preterm or (extremely) low birth weight [P>0·5 for all, Table 3].
There was no significant difference in the proportion of infants who experienced at least one new Grade 3 or 4 AE (25% in both arms; [P=0·94], Table 3). There were also no substantial differences in the proportions of infants experiencing individual AEs (Table S4, Appendix, p. S5).Congenital anomalies were infrequently observed and were balanced between arms.
Seven (1·9%; 95% CI, 0·8 to 3·8%) infants were HIV-infected, one [0·5%; 95% CI, <0·1 to 2·9%] in the raltegravir arm and six [3·3%; 95% CI, 1·2 to 7·0%] in the efavirenz arm [P=0·064; absolute difference −2·7% (95% CI −5·5 to <0·1%)]. Each HIV-infected infant had their first positive HIV test within 48 hours of birth, suggesting all infections occurred in utero, and all infected infants were born to mothers who enrolled from 28 to <37 weeks gestation.
Discussion
In this large randomized trial comparing the treatment of ART-naïve pregnant women living with HIV infection with the integrase inhibitor raltegravir to efavirenz, the proportion of women with viral load below 200 copies per mL at or near delivery was significantly greater in the raltegravir arm, primarily among those enrolling in the third trimester. Both regimens were safe and well-tolerated. An even larger difference was observed when comparing virologic suppression below the local assay lower-limit-of-quantification. In a composite analysis of efficacy and tolerability, the difference in outcomes between raltegravir and efavirenz was even more pronounced, and was driven by the faster viral load reduction with raltegravir.
Integrase inhibitors are potent, well-tolerated antiretrovirals that are recommended as first-line ART regimens in nonpregnant adults living with HIV12. The integrase inhibitors raltegravir and dolutegravir are also recommended as preferred regimens for use during pregnancy by the US Perinatal HIV Guidelines Panel due to their efficacy, tolerance, and pharmacokinetic (PK) profiles.
The Dolphin-2 trial compared efavirenz to dolutegravir in late-presenting pregnant women6,13. The safety analysis included 268 women and the efficacy analysis 237. Virologic response defined as the percentage of women with less than 50 copies was 72% in the dolutegravir arm vs 43% in the efavirenz group. P1081 are data are in keeping with what is known from the Dolphin trial insofar as we found that the integrase inhibitor raltegravir had greater response than efavirenz.
Another open-label trial involving late-presenting pregnant women comparing RAL and LPV/r found that, of 33 patients, 76·5% in the RAL group and 25% in the LPV/r group achieved virologic suppression at delivery18. Observational “real-life” studies have reported rates of virologic suppression ranging from 70 to 86% among women who received raltegravir during pregnancy. These studies, which included participants with considerable variability in the length of ART exposure, observed a rapid decline in levels of plasma HIV RNA among women receiving raltegravir, and a large proportion quickly achieved suppression of HIV replication9,14. In our population, a larger proportion of women receiving raltegravir achieved a 2·0 log decrease by week two compared to those receiving efavirenz, which is consistent with previous observational studies15–18.
It is important to consider these results in the context of ART resistance profiles, which can vary significantly by country/region. Nine percent of women enrolling in our study had at least one resistance mutation to NNRTIs or NRTIs. In a cohort of ART-naïve pregnant women in Brazil, the rates of resistance to NNRTIs and NRTIs were 9% and 21%, respectively19. Our study found no resistance to integrase inhibitors at study entry, consistent with an observational study conducted in the United States from 2010–2016 that reported very low rates of resistance to integrase inhibitors (0·2%) in ART-naïve non-pregnant adults20. Treatment for late presenting pregnant women living with HIV in regions with comparatively high rates of NNRTI resistance should prioritize integrase inhibitors including raltegravir, particularly when genotype test results cannot be obtained rapidly, because pretreatment genotypic resistance to integrase inhibitors is currently uncommon.
Pregnancy outcomes in this study were consistent with previous studies of initiation of other antiretroviral regimens in pregnancy. The preterm delivery rate was approximately 10% in both arms in our study, lower than the reported preterm delivery rates of 18% among ART-exposed pregnant women and their infants in Botswana and of 20% in NISDI studies in Latin America, but similar to results reported in a trial in HIV-exposed infants whose mothers did not receive ART21–23. The proportion of infants with low birth weights was 13% in our study, similar to the rate (14%) reported in HIV-exposed infants at similar sites24. The most common comorbidity among infants was congenital syphilis (6% in both arms). A study conducted at similar sites in Africa and Brazil found a rate of syphilis of 9·3% in infants whose mothers did not received prenatal care21.
As in our study, most retrospective studies have reported no difference in AEs between integrase inhibitor and NNRTI use in pregnancy22. One previous study in nonpregnant adults reported higher rates of AEs among taking efavirenz compared to raltegravir4. There were few AEs among our study infants, consistent with the results of observational studies that have reported few AEs in infants whose mothers used raltegravir9,15.
While the HIV perinatal transmission rate was not significantly different between treatment arms in our study, substantially fewer infants were infected in the raltegravir arm compared to the efavirenz arm (P=0·064). Previous studies have shown that elevated viral load and late presentation to antenatal care are major causes of perinatal transmission25,26. INSTIs such as raltegravir may be valuable for rapidly reducing high VL, and ultimately an effective option for PMTCT27, but ideally the diagnosis and the treatment cascade should begin before conception.
However, one of the limitations of this study was under-powered to detect a difference in the HIV perinatal transmission rate between study arms. The proportion of infected infants in the raltegravir arm was very low (0·5%) and was comparable to results in previous observational studies9,14,15,17. Limitations of the study include our use of a lamivudine/zidovudine nucleoside reverse transcriptase inhibitor (NRTI) backbone, which may not be applicable to current clinical practice.
Another limitation is that fixed dose combination was not used in the trial which could have led to increased attribution and reporting of specific toxicities in a treatment arm. We attempted to minimize these biases by setting up stringent criteria for toxicity management, participant management including regimen modification, treatment discontinuation, and inadequate virologic response.
A strength of this study is the high quality and completeness of data. Approximately 96% of women who initiated treatment remained on study drug through delivery, and 96% of infants completed all protocol requirements. This high completion rate permitted evaluation of safety in a large, representative group of mothers and infants.
In this multicenter randomized trial comparing an integrase inhibitor with efavirenz in pregnant women living with HIV, the rate of suppression at delivery was higher with raltegravir, and raltegravir was shown to be a potent and well-tolerated antiretroviral. These results support the use of raltegravir in pregnant women who present late for care, particularly those initiating treatment after 28 weeks of gestation.
Supplementary Material
Implications of all the available evidence
In NICHD P1081, all participants’ genotypic resistance testing was performed at screening and ART-experienced women were excluded. On the other hand, participants in the DolPHIN2 trial were permitted to have taken ART >12 months prior to enrollment and resistance testing was not performed. Despite these differences, both studies found that integrase inhibitors were potent and well tolerated, with low HIV vertical transmission. Like trial NCT01854762, in NICHD P1081 RAL was more potent than the comparator regime. However, NCT01854762 compared RAL and LPV, whereas P1081 compared RAL and EFV, and had a larger sample size.
Acknowledgments
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under Award Numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Declaration of interests
Dr. Mirochnick reports grants from NICHD/NIH, during the conduct of the study; grants from Merck & Co, grants from ViiV Healthcare, grants from Gilead Sciences, outside the submitted work. Dr. Moreira reports grants NICHD/IMPAACT during the conduct of the study. Mr. Morrison reports grants from US National Institute of Allergy and Infectious Diseases, non-financial support and other from US Eunice Kennedy Shriver National Institute of Child Health and Human Development via Westat Inc., during the conduct of the study; grants from US National Institute of Allergy and Infectious Diseases, grants from US Eunice Kennedy Shriver National Institute of Child Health and Human Development via University of Michigan, outside the submitted work. Dr. Shapiro reports grants from US National Institute of Allergy and Infectious Diseases, non-financial support and other from US Eunice Kennedy Shriver National Institute of Child Health and Human Development via Westat Inc., during the conduct of the study; grants from US National Institute of Allergy and Infectious Diseases, grants from US Eunice Kennedy Shriver National Institute of Child Health and Human Development via University of Michigan, outside the submitted work. Dr. Violari reports grants from National Institute of Child Health and Development, during the conduct of the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Sharing Statement:
Individual participant data will be available (including data dictionaries). Individual participant data that underlie the results reported in this article will be shared after de-identification (text, tables, figures, and appendices). The study protocol (including sample informed consent form) and statistical analysis plan will also be available. The data will be available beginning 9 months following article publication and will be shared with researchers whose proposed use is approved by the NICHD Data and Specimen Hub (DASH) Data Access Committee as scientifically and ethically appropriate and does not conflict with constraints or informed consent limitations and will be made available for analyses required to achieve aims in the approved proposal. To gain access, data requestors will need to create a free NICHD DASH account, submit a data access proposal, and if approved, sign a data access agreement. Information regarding creating a NICHD DASH account and accessing data may be found at https://dash.nichd.nih.gov/
Evidence before this study
We searched PubMed, Clinicaltrials.gov, Web of Knowledge, and abstracts of the Conference on Retroviruses and Opportunistic Infections and International AIDS Society for clinical trials of integrase strand transferase inhibitors in pregnancy between November 1, 2009 and November 1, 2019. We used the keywords “raltegravir” and “pregnancy” and searched for articles in English, French, and Portuguese. We found two randomized clinical trials supporting the use of integrase inhibitors during pregnancy. The DolPHIN2 trial compared efavirenz (EFV) to dolutegravir (DTG) in late-presenting pregnant women. The safety analysis included 268 women and the efficacy analysis 237. The trial found that virologic response defined as the percentage of women with less than 50 copies was 72% in the DTG arm vs 43% in the EFV group. Trial NCT01854762 randomized 17 late-presenting pregnant women to raltegravir (RAL) and to 16 lopinavir (LPV)-based regimens. Participants in the raltegravir group achieved significantly greater virologic response within two weeks of enrollment (77% vs. 25%). These trials suggest that integrase inhibitors could be a good option for women living with HIV who present late in pregnancy.
Added value of this study
We compared two potent HIV treatment regimens, one with RAL and the other with EFV, for pregnant women who start prenatal care late in the second or third trimester, between 20 and 36 weeks of pregnancy. The goal was to compare how well the regimens reduced the plasma HIV viral load by delivery, and how safe the regimen was and how well women tolerated the regimens. While 94% of women who took the HIV regimen that included RAL had undetectable viral load by delivery, 84% of women who took the HIV regimen that included EFV were undetectable by delivery.
References
- 1.UNAIDS. Fact Sheet - Global AIDS Update 2019. Geneva: UNAIDS; 2019. [Google Scholar]
- 2.AIDSinfo. Recommendations for the Use of Antiretroviral Drugs in Pregnant Women with HIV Infection and Interventions to Reduce Perinatal HIV Transmission in the United States. Washington, DC: US Department of Health and Human Services; 2019. [Google Scholar]
- 3.Mandelbrot L, Tubiana R, Le Chenadec J, et al. No perinatal HIV-1 transmission from women with effective antiretroviral therapy starting before conception. Clin Infect Dis 2015; 61(11): 1715–25. [DOI] [PubMed] [Google Scholar]
- 4.Lennox JL, DeJesus E, Lazzarin A, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet 2009; 374(9692): 796–806. [DOI] [PubMed] [Google Scholar]
- 5.WHO. Updated recommendations on first-line and second-line antiretroviral regimens and post-exposure prophylaxis and recommendations on early infant diagnosis of HIV: interim guidelines Supplement to the 2016 consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection. Geneva: World Health Organization; 2018. [Google Scholar]
- 6.Waitt C, Orrell C, Walimbwa S, et al. Safety and pharmacokinetics of dolutegravir in pregnant mothers with HIV infection and their neonates: A randomised trial (DolPHIN-1 study). PLoS Med 2019; 16(9): e1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zelen M The randomization and stratification of patients to clinical trials. Journal of chronic diseases 1974; 27(7–8): 365–75. [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services NIoH, National Institute of Allergy and Infectious Diseases, Division of AIDS,. Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Corrected Version 2.1. Bethesda: DAIDS Regulatory Support Center; 2017. [Google Scholar]
- 9.Gantner P, Sylla B, Morand-Joubert L, et al. “Real life” use of raltegravir during pregnancy in France: The Coferal-IMEA048 cohort study. PLoS One 2019; 14(4): e0216010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillespie BW, Chen Q, Reichert H, et al. Estimating Population Distributions When Some Data Are Below a Limit of Detection by Using a Reverse Kaplan-Meier Estimator. Epidemiology 2010; 21(4): S64–S70. [DOI] [PubMed] [Google Scholar]
- 11.Guo C, So Y, Johnston G. Analyzing Interval-Censored Data with the ICLIFETEST Procedure. Paper SAS279- 2014. SAS Global Forum Proceedings 2014: 1–16. [Google Scholar]
- 12.AIDSInfo. Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Washington, DC: Department of Health and Human Services; 2019. [Google Scholar]
- 13.Kintu K, Malaba T, Nakibuka J, et al. RCT of dolutegravir vs efavirenz-based therapy initiated in late pregnancy: Dolphin-2. Abstract 40. Conference on Retroviruses and Opportunistic Infections 2019. [Google Scholar]
- 14.Blonk MI, Colbers AP, Hidalgo-Tenorio C, et al. Raltegravir in HIV-1-Infected Pregnant Women: Pharmacokinetics, Safety, and Efficacy. Clin Infect Dis 2015; 61(5): 809–16. [DOI] [PubMed] [Google Scholar]
- 15.Maliakkal A, Walmsley S, Tseng A. Critical Review: Review of the Efficacy, Safety, and Pharmacokinetics of Raltegravir in Pregnancy. J Acquir Immune Defic Syndr 2016; 72(2): 153–61. [DOI] [PubMed] [Google Scholar]
- 16.Brites C, Nobrega I, Luz E, Travassos AG, Lorenzo C, Netto EM. Raltegravir versus lopinavir/ritonavir for treatment of HIV-infected late-presenting pregnant women. HIV clinical trials 2018; 19(3): 94–100. [DOI] [PubMed] [Google Scholar]
- 17.Rahangdale L, Cates J, Potter J, et al. Integrase inhibitors in late pregnancy and rapid HIV viral load reduction. Am J Obstet Gynecol 2016; 214(3): 385.e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westling K, Pettersson K, Kaldma A, Naver L. Rapid decline in HIV viral load when introducing raltegravir-containing antiretroviral treatment late in pregnancy. AIDS Patient Care STDS 2012; 26(12): 714–7. [DOI] [PubMed] [Google Scholar]
- 19.de Lourdes Teixeira M, Nafea S, Yeganeh N, et al. High rates of baseline antiretroviral resistance among HIV-infected pregnant women in an HIV referral centre in Rio de Janeiro, Brazil. International Journal of STD & AIDS 2015; 26(13): 922–8. [DOI] [PubMed] [Google Scholar]
- 20.Menza TW, Billock R, Samoff E, Eron JJ, Dennis AM. Pretreatment integrase strand transfer inhibitor resistance in North Carolina from 2010–2016. Aids 2017; 31(16): 2235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen-Saines K, Watts DH, Veloso VG, et al. Three postpartum antiretroviral regimens to prevent intrapartum HIV infection. The New England journal of medicine 2012; 366(25): 2368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zash R, Jacobson DL, Diseko M, et al. Comparative safety of dolutegravir-based or efavirenz-based antiretroviral treatment started during pregnancy in Botswana: an observational study. Lancet Glob Health 2018; 6(7): e804–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreitchmann R, Li S, Melo V, et al. Predictors of adverse pregnancy outcomes in women infected with HIV in Latin America and the Caribbean: a cohort study. BJOG: An International Journal of Obstetrics & Gynaecology 2014; 121(12): 1501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joao EC, Calvet GA, Krauss MR, et al. Maternal antiretroviral use during pregnancy and infant congenital anomalies: the NISDI perinatal study. J Acquir Immune Defic Syndr 2010; 53(2): 176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewing AC, Ellington SR, Wiener JB, et al. Predictors of Perinatal HIV Transmission Among Women Without Prior Antiretroviral Therapy in a Resource-Limited Setting The Breastfeeding, Antiretrovirals and Nutrition Study. Pediatric Infectious Disease Journal 2019; 38(5): 508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters H, Thorne C, Tookey PA, Byrne L. National audit of perinatal HIV infections in the UK, 2006–2013: what lessons can be learnt? Hiv Medicine 2018; 19(4): 280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.British HIV Association guidelines for the management of HIV in pregnancy and postpartum 2018. HIV Med 2019; 20 Suppl 3: s2–s85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.