

Abstract
Alcohol use disorder (AUD) is a serious public health problem that results in tremendous social, legal and medical costs to society. Unlike other addictive drugs, there is no specific molecular target for ethanol (EtOH). Here, we report a novel molecular target that mediates EtOH effects at concentrations below those that cause legally-defined inebriation. Using the patch-clamp recording of human α6*-nicotinic acetylcholine receptor (nAChR) function when heterologously expressed in human epithelial cells (SH-EP1 cells), we find that 0.1–5 mM EtOH significantly enhances α6*-nAChR-mediated currents in effects that are influenced by both EtOH and nicotine concentrations. EtOH exposure increases both whole-cell current rising slopes and decay constants. This EtOH modulation is selective for α6*-nAChRs since it does not affect α3β4-, α4β2-, or α7-nAChRs. In addition, 5 mM EtOH also increases the frequency and amplitude of DA transients in mouse brain nuclear accumbens slices, and these are blocked by an α6-nAChR antagonist, α-conotoxin MII, suggesting a role for native α6*-nAChRs in low-dose EtOH effects. Collectively, our data suggest that α6*-nAChRs are sensitive targets mediating low dose EtOH effects through a positively allosteric mechanism, which provides new insight into mechanisms involved in pharmacologically-relevant alcohol effects contributing to AUD.
Keywords: nicotinic acetylcholine receptor, alpha 6 subunit, alcohol, ethanol, patch-clamp, SH-EP1 cells
1. Introduction
Alcohol is the most widely used drug in society, and alcohol abuse is by far the most common form of substance abuse. Alcohol use disorder (AUD), a serious public health problem worldwide, affects approximately 17 million Americans and results in tremendous social, legal, and medical costs estimated at >$249 billion per year (Sacks et al., 2015). As a result, more than 700,000 people in the United States receive alcoholism treatment in either inpatient or outpatient settings (Fuller and Hiller-Sturmhofel, 1999). The Centers for Disease Control and Prevention (CDC) report that there are approximately 80,000 deaths attributable to excessive alcohol use each year in the United States. Based on a WHO report, alcohol consumption is the world’s third largest risk factor for disease and disability. Excessive drinking accounted for 1 in 10 deaths among working-age adults in the United States (Stahre et al., 2014). However, mechanisms involved in AUD remain obscure, and therapeutics for AUD are still limited.
Unlike other addictive drugs (e.g., morphine, cocaine or nicotine) that have specific molecular targets on neurons, no specific molecular target has been validated or accepted by the alcohol research community. The prevailing view is that glutamatergic (GLUergic) and GABAergic receptors and GLU and GABA synaptic transmission are possible targets for ethanol (EtOH), but often the EtOH concentrations needed to modify glutamate or GABA synaptic transmission are physiologically/pharmacologically relevant only in the context of toxic loss of consciousness and death. For example, EC50 values are 37 mM for EtOH modulation of L-type Ca2+ channels (Mullikin-Kilpatrick and Treistman, 1994), 30–100 mM for NMDA receptors (Lovinger et al., 1989; Weight et al., 1991), and 220 mM for kainate/quisqualate-activated channels is 220 mM (Weight et al., 1993). 20% enhancement of GABA-induced chloride channels is achieved at 30 mM EtOH (Nishio and Narahashi, 1990), 40% enhancement of serotonin-induced current occurs at 50 mM EtOH (Lovinger, 1991), and the EC50 for functional blockade of α4β2-nicotinic acetylcholine receptors (nAChRs) are 75 mM (Zuo et al., 2002). In human drinkers, however, blood alcohol levels are 6–13 mM for impairment of attention and onset of relaxed and joyous behavior, 17 mM (0.08%) for most legal definitions of alcohol intoxication (triple risk for an accident; impaired reasoning, perception and reaction time), 20–22 mM for sedation and ataxia, 50 mM for loss of consciousness, and 110 mM or higher for death (Little, 1991). Therefore, there remains a gap in our understanding of mechanisms and molecular targets involved at lower doses (<10 mM) of EtOH that are important to reward and dependence and to treatment of AUD.
The mesolimbic dopamine (DA) system is implicated in pleasure, reward, and mood control and in drug reward and dependence, including for nicotine (NIC) and EtOH. It includes the midbrain ventral tegmental area (VTA) and its projections to structures such as the nucleus accumbens (NAc) and prefrontal cortex [PFC; (de Rover et al., 2002)]. The VTA receives a variety of inputs, including cholinergic innervation not just on DA neurons, but also on VTA GABAergic neurons (Fiorillo and Williams, 2000; Garzon et al., 1999). Neurons within the VTA express a wide variety of nAChRs (Mansvelder et al., 2002; Mansvelder and McGehee, 2000; Wooltorton et al., 2003; Yang et al., 2009a). NIC can activate both DA and γ-aminobutyric acid (GABA) neurons (Mansvelder et al., 2002; Yin and French, 2000) via an increase in activation, and perhaps desensitization of specific nAChRs, suggesting roles for nAChRs not just in NIC dependence, but also in EtOH dependence and in NIC and EtOH modulation of pleasure, mood, and reward (Mansvelder et al., 2002; Mansvelder and McGehee, 2000; Wu et al., 2004). In addition to α4β2- and homomeric α7-nAChRs, there is a considerable expression of heteromeric nAChRs containing α6 subunits (α6*-nAChRs; the * signifies that there are or may be additional nAChR subunits that co-assemble with the indicated subunit) in the VTA (Azam et al., 2002; Klink et al., 2001). α6*-nAChRs have been implicated in DA transmission and NIC dependence (Brunzell et al., 2010; Drenan et al., 2010; Drenan et al., 2008; Exley et al., 2008; Gotti et al., 2010; Jackson et al., 2009; Pons et al., 2008; Sanjakdar et al., 2015; Yang et al., 2009c). α6*-nAChRs are located on GABA terminals on VTA DA neurons, and their activation by acetylcholine (ACh) enhances GABAergic synaptic inhibition to VTA DA neurons (Drenan et al., 2008; Yang et al., 2011). Functional and pharmacological properties of α6*-nAChRs are largely unknown, there are no selective α6*-nAChR agonists, and α6 subunits sometimes are co-expressed with nAChR α4subunits to form natural α4α6*-nAChRs, which could mask properties of α6*(no α4)-nAChRs. Moreover, the ability to heterologously express functional α6 nAChRs has been challenging (Dash et al., 2014).
Recently, we have found that low concentrations of EtOH (1–10 mM) enhance GABAA receptor (GABAAR)-mediated spontaneous and evoked inhibition of IPSCs via processes blocked by the α6*-nAChR-selective antagonist α-conotoxin MII (α-Ctx MII) and that there is lowered EtOH sensitivity and reward in nAChR α6 subunit knock-out (KO) mice (Steffensen et al., 2017). Moreover, we have established functional, heterologous expression of α6*-nAChRs by co-transfection of an α6/3 subunit chimera together with β2 and β3 subunits into the human SH-EP1 cell line (Chen et al., 2018). This transfected α6*-nAChR exhibits robust function and has been used for drug screening, and use of the chimera avoids the enhanced agonist potency and efficacy effects associated with including a 9’S mutant subunit (Letchworth and Whiteaker, 2011). In the present study, we used the α6/α3*-nAChR cell model to demonstrate that low dose EtOH selectively enhances α6*-nAChR-mediated currents in both EtOH- and nicotine-dependent manners, suggesting a positive allosteric modulation, and we extended our work to show modulation of NAc DAergic transients via block of putative, natïve α6*nAChRs.
2. Methods
2.1. Expression of human neuronal α6/α3β2β3-nAChR in human SH-EP1 cells
Construction of the cell line expressing α6Nα3Cβ2β3-nAChR was first described by Breining et al. and Letchworth et al. (Breining et al., 2012; Letchworth and Whiteaker, 2011). α6Nα3C (α6/3) denotes a chimeric subunit composed of the extracellular, ligand-binding domain of the human α6 subunit fused to the first transmembrane domain and following sequence of the human α3 nAChR subunit (see 3D structure in Supplemental Fig. 3A), this approach reproducibly increases expression compared to that seen for native α6 subunits while retaining α6-like pharmacology (Kuryatov et al., 2000). More detailed information of the process of expression of this α6Nα3Cβ2β3-nAChR and maintenance of SH-EP1-α6/3 cells was as previously described (Chen et al., 2018).
2.2. Patch-clamp recordings
Conventional patch-clamp whole-cell current recordings coupled with a computer-controlled fast drug application and removal were implemented as previously described (Wu et al., 2006). Briefly, cells plated on 35-mm culture dishes were placed on the stage of an inverted microscope (Olympus IX7, Lake Success, NY, USA) and continuously superfused with standard external solution (2 ml/min). Glass microelectrodes (1.5×100 mm, Narishige, East Meadow, NY, USA) were made in two steps using a vertical electrode puller (P83, Narishige, East Meadow, NY, USA). Electrodes with a resistance of 3–5 MΩ were used to form tight seals (>1 GΩ) on the cell surface until suction was applied to break the membrane and convert to conventional whole-cell recording. After that, the recorded cell was lifted of the culture dish surface to allow more efficient and thorough perfusion with applied ligands and then voltage-clamped at a holding potential (VH) of –40 mV (unless specifically mentioned), and ionic currents in response to application of nicotinic ligands were measured (Axopatch 200B amplifier, Axon Instruments, Foster City, CA, USA). Whole-cell access resistance was less than 20 MΩ before series resistance compensation and monitored throughout the experiment. If access resistance varied by more than 20%, data were discarded. Both pipette and whole-cell current capacitances were minimized, and series resistance was routinely compensated to 80%. Typically, current output was filtered at 2 kHz, displayed and digitized at 10 kHz on-line (Digidata 1550 series A/D board, Axon Instruments, Foster City, CA, USA), and streamed to disk. Data acquisition and analyses of whole-cell currents were done using Clampex v10.2 (Axon Instruments, Foster City, CA, USA), and results were plotted using Origin 8.0 (Microcal, North Hampton, MA, USA) or Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Concentration-response curves were fit to the Hill equation. nAChR acute desensitization was analyzed for decay time constant (τ), peak current (Ip), and steady-state current (Is) using fits to the single exponential function: I = [(Ip − Is) et/τ] + Is, or to its double-exponential variant as appropriate, using data from 90% to 10% of the period between the peak amplitude of the inward current and the termination of the typical 1-sec period of agonist exposure. Replicate determinations of τ (a measure of the rate of acute desensitization), the current rising slope and peak current amplitude are presented as means ± standard error of the mean (SEM), and were analyzed for statistical significance using Student’s t test (paired or independent). All experiments were performed at room temperature (22 ± 1°C).
2.3. Solutions and drug application
The standard external solution contained 120 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 25 mM D-glucose, 10 mM HEPES, pH 7.4 with Tris-base. In some experiments using ACh as an agonist, 1 μM atropine sulfate was added to the standard solution to exclude any possible influences of muscarinic receptors. The pipette solutions used for conventional whole-cell recording were (in mM): Tris phosphate dibasic 110, Tris base 28, EGTA 11, MgCl2 2, CaCl2 0.1, Na-ATP 4, pH 7.3. This “Tris+” electrode solution was used by Huguenard & Prince (Huguenard and Prince, 1992) to prevent nAChR receptor functional rundown (Zhao et al., 2003)
To initiate whole-cell current responses, under constant superfusion of the recording chamber, nicotinic drugs were rapidly delivered to the recorded cell using a computer-controlled ‘U-tube’ application system, in which the applied drug surrounded the recorded cell within 20 msec. Intervals between drug applications (20–60 sec) were adjusted specifically to ensure the stability of nAChR responsiveness (absence of functional rundown), and the selection of pipette solutions used in most of the studies described here was made with the same objective. Drugs used in the present study were: (−) nicotine, ACh, EtOH (Sigma Chemical Co., St. Louis, MO, USA). α-conotoxin MII was a gift from Dr. Michael McIntosh.
2.4. Fast Scan Cyclic Voltammetry Recordings
Slices obtained from the nucleus accumbens core (NAc) of C57BL6/J mice (28–42 PND) were transferred to the recording chamber and perfused with ACSF (34 °C) at a rate of ~1.8 mL/min. Voltammetry recordings were performed and analyzed using Demon Voltammetry and analysis software (Yorgason et al., 2011). Carbon fiber electrodes used in voltammetry experiments were made in-house. Carbon fibers (7 μm diameter, Thornel T-650, Cytec, Woodland Park, NJ) were aspirated into borosilicate glass capillary tubes (TW150, World Precision Instruments, Sarasota, FL). Electrodes were then pulled on a PE-2 vertical pipette puller (Narishige, Amityville, NY) and cut so that 100–150 μm of carbon fiber protruded from the tip of the glass. The electrode potential was linearly scanned by a triangular waveform from −0.4 to 1.2 V and back to −0.4 V (Ag vs. AgCl) with a scan rate of 400 V/sec, repeated every 100 msec. Carbon fibers were advanced completely into the tissue at a 20° angle with the tip positioned ~85 μm below the slice surface. Dopamine transients were measured in the presence of 4-AP (30 μM) to facilitate detection (Yorgason et al., 2011).
2.5. Analysis of Dopamine Transients
Dopamine release was analyzed using Demon Voltammetry software and was measured at peak oxidation currents as described previously (Yorgason et al., 2017). Briefly, a running subtraction on recordings was performed to reduce drift and aliasing noise. Post-subtracted data was then compared across time against known cyclic voltammograms for DA, with a low threshold r2 value for initial detection (r2>0.3). Each detected event (legitimate and spurious) was then verified manually using the original background subtraction (non-running) data to remove spurious events and quantify release amplitude for each event. Signals smaller than the limit of detection (calculated by multiplying the median standard deviation for each file by 3) were automatically rejected. Detected events were simultaneously examined for evidence of a false positive caused by drift and aliasing noise coinciding with the oxidation potential.
Dopamine release concentrations were calculated from calibration values (at 1 μM DA). Baseline frequency of DA transients was measured during a 10 min period before drug application. Group data from experiments where a drug was applied was measured across the 10 min period where the drug was applied. NCSS 8 (NCSS; Kaysville, UT) and Prism 5 (GraphPad; La Jolla, CA) were used for statistical analysis. Statistical significance was determined for groups of 2 variables using a two-tailed Student’s t-test. Experiments with >2 groups, but only one factor, were tested for significance using a one-way ANOVA. For experiments that examined multiple factors, and possible interactions, a two-way ANOVA was used. Tukey’s HSD (for one-way ANOVAs where comparisons were not planned), and Dunnett’s (for one-way ANOVAs when comparisons against control values were planned) correction methods were used for post-ANOVA analysis. Bonferroni correction methods were used for post-ANOVA analysis of two-way ANOVAs only.
2.6. Homology modeling
The homology model of the α6/α3 chimera was built using the sequences described previously (Chen et al., 2018) without signal peptide submitted to the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). One of the five top scored models was used for presentation. The 3D structural presentation of the α6α3 chimera and α6 mutant were made using Discovery Studio Visualizer 4.0 (Accelrys, San Diego, CA).
2.7. Data analysis and statistics
Data were given as mean ± SEM with numbers shown in parentheses (n). A probability level of p<0.05 was considered to be statistically significant. Significant differences were determined using the two-tailed Student’s t-test or One-way ANOVA as appropriate with the software of GraphPad Prism 8.01 (GraphPad Software, Inc, La Jolla, CA).
3. Results
3.1. Effects of acute exposure to low dose EtOH on α6*-nAChR-mediated currents
To examine acute effects of low dose EtOH on human α6*-nAChR-mediated inward whole-cell current responses, we applied 1 μM NIC to cultured SH-EP1 cells expressing α6*-nAChRs. Applications were repeated until NIC-induced responses became stable. Then, we co-applied 1 μM NIC and 0.1 mM EtOH to cells to define EtOH effects, and we followed with another agonist challenge after washout of EtOH. Nicotine-induced currents where enhanced by 0.1 mM EtOH co-application (Fig. 1A). After washout of EtOH for 2–3 min, this enhancement was reversed. To evaluate the effects of 0.5 mM EtOH on 1 μM NIC-induced currents with different pre-treatment times, we compared the effects of EtOH on NIC currents with pre-treatments for 0, 20, 40, 60, 80 ms (Supplemental Fig. 1A), and found no difference of EtOH’s effects with different pre-treatment times (Supplemental Fig. 1B). Thus, all of the following experiments used the EtOH and NIC co-application protocol. We also demonstrated stable recording of α6*-nAChR-mediated currents under our recording experimental conditions (Supplemental Fig. 1C). Figure 1B summarizes pooled data from 20 cells and demonstrates a consistent potentiation of NIC-induced inward current amplitude by 0.1 mM EtOH. Figure 1C shows normalized results of 1 μM NIC-induced responses before, during, and after acute EtOH exposure. One-way ANOVA analysis demonstrated that effects of EtOH on α6*-nAChR-mediated currents were significant (F(2,57) = 32.57, p < 0.001, Tukey’s comparison showed that control vs. EtOH, p < 0.001, and control vs. washout, p = 0.989). These results indicate that acute low dose EtOH enhances α6*-nAChR-mediated currents in human SH-EP1 cells.
Fig. 1. Effects of low dose EtOH on α6*-nAChR-mediated whole-cell currents.
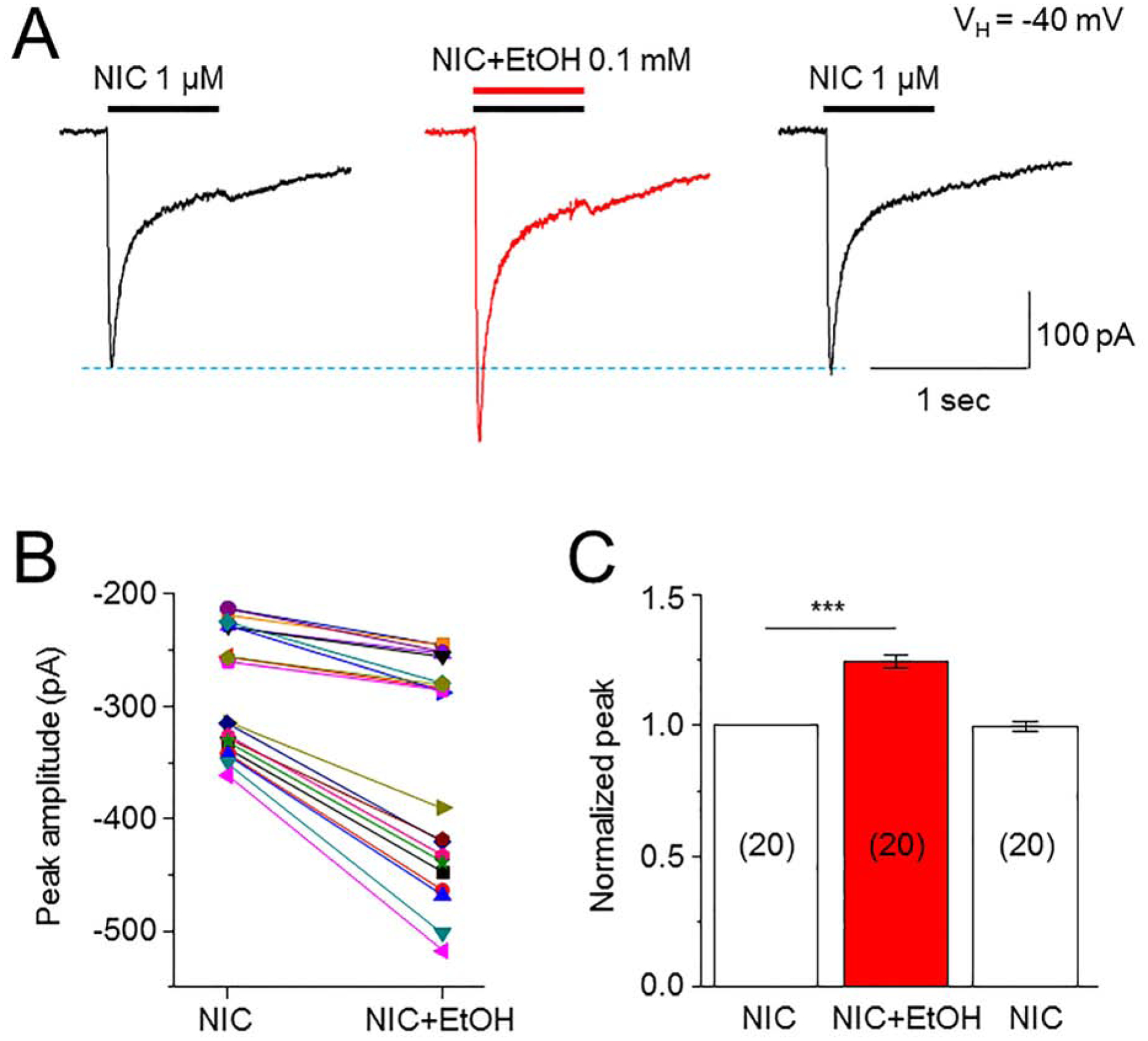
A: Typical traces demonstrate 1 μM NIC-induced inward current before, during and after washout of 0.1 mM EtOH. EtOH and NIC were co-applied. All three traces were recorded from the same cell. B: Comparison of peak amplitudes of NIC-induced inward currents before and during EtOH exposure. C: Bar graph summarizes the normalized peak amplitude of NIC-induced current before, during and after washout of EtOH. In this and all following figures the number inside each column indicates the number of cells tested, *** indicates p<0.001.
3.2. EtOH effects on α6*-nAChR-mediated currents are concentration dependent
We next defined effects of different concentrations of EtOH (from 0.01 to 50 mM) on α6*-nAChR-mediated currents. At a range of concentrations between 0.1 and 5 mM, EtOH enhanced 1 μM NIC-induced whole-cell current amplitude (Fig. 2A), while at either 0.01 or 50 mM, EtOH failed to enhance NIC-induced currents. (Fig. 2B). Thus, there is a bell-shaped concentration-response relationship for EtOH potentiation of NIC-induced currents with no potentiation at the higher or lower end of the range of EtOH concentrations (0.01 and 50 mM), but functional potentiation at intermediate EtOH levels. One-way ANOVA analysis demonstrated that the differences of NIC responses modulated by different EtOH concentrations are significant (F(4,85) = 8.156, p < 0.001). Tukey’s comparison showed that compared to normalized NIC alone (1.0, indicated by a blue horizontal dashed-line), EtOH’s effects were dependent on EtOH concentrations: EtOH 0.01 mM = 1.082 ± 0.047, n = 10, p = 0.367; EtOH 0.1 mM = 1.244 ± 0.037, n = 20, p < 0.001; EtOH 0.5 mM = 1.175 ± 0.033, n = 30, p < 0.001; EtOH 5 mM = 1.138 ± 0.047, n = 10, p = 0.020; and EtOH 50 mM = 0.983 ± 0.374, n = 20, p = 0.994. Thus, EtOH potentiates NIC current at a range of EtOH concentrations (from 0.1 to 5 mM). This suggests that there are the conditions where EtOH acts as a positive allosteric modulator to enhance α6*-nAChR function.
Fig. 2. Effects of different concentrations of EtOH on 1 μM NIC-induced currents.
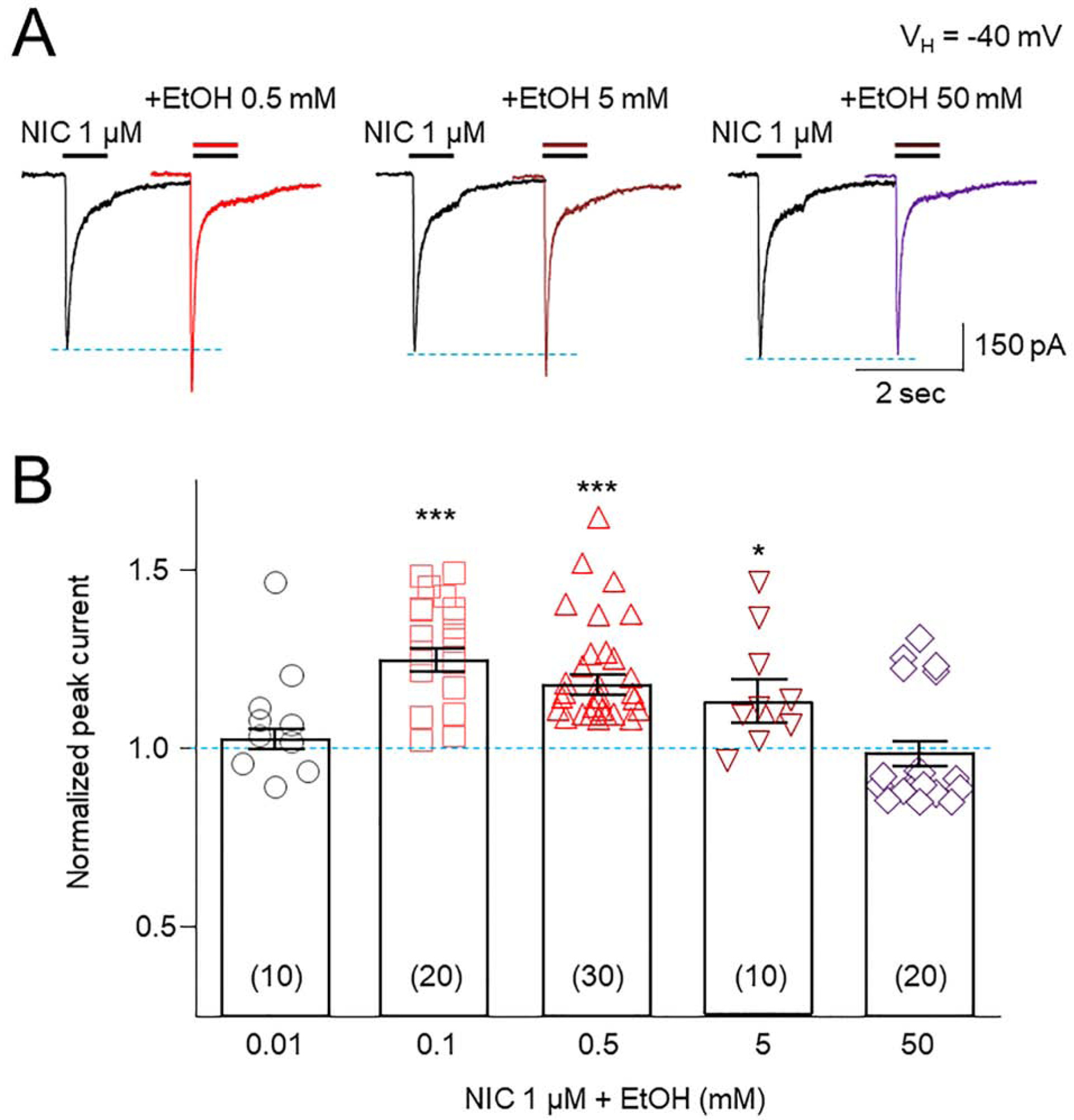
A: Representative traces showing effects of 0.5, 5 and 50 mM EtOH on 1 μM NIC-induced currents. B: Statistical analysis of effects of different concentrations of EtOH on normalized peak amplitudes of NIC-induced currents.
3.3. Kinetic analysis of EtOH-induced potentiation of α6*-nAChR-mediated currents
To understand the nature of EtOH effects on α6*-nAChR function, we assessed influences of EtOH at select concentrations on whole-cell current rising slope and decay (Fig. 3). Figure 3A shows typical 1 μM NIC-induced current traces (black) before and after 0.1 (left and middle panels) or 50 mM (right panel) EtOH exposure. EtOH treatment at 0.1 mM increased peak amplitude (red trace, left panel) and current rising slope but did not alter current decay time constant (blue trace, middle panel). EtOH exposure at 50 mM accelerated current decay constant (green trace) compared to control NIC current (black trace; Fig. 3A, right panel showed that). Pooled data (Fig. 3B) confirmed the elevation in 1 μM NIC-induced, α6*-nAChR-mediated peak current in the presence of 0.1 or 5, but not 50 mM EtOH (left panel), faster rise to peak current in the presence of 0.1 mM EtOH (middle panel), but also faster decay of current amplitude in the presence of 50 mM EtOH (right panel). One-way ANOVA analysis demonstrated that effects of EtOH at each concentration on current peak amplitude (Fig. 3B left panel) were significant (F(2,26) = 8.375, p = 0.002). Tukey’s comparison showed that normalized to current levels elicited by NIC alone (1.0, indicated by a red horizontal dashed-line), peak current levels at the indicated concentration of EtOH were: 0.1, 1.341 ± 0.049, n = 11 (p < 0.001); 0.5 mM, 1.163 ± 0.049, n = 11, (p = 0.006); 50 mM, 1.082 ± 0.056, n = 7 (p = 0.336). Effects of EtOH at each concentrations on current rising time (Fig. 3B middle panel) were significant (F(2,26) = 8.812, p = 0.001). Tukey’s comparison showed that normalized to effects of NIC alone, (1.0), rising times at the indicated concentrations of EtOH were: 0.1 mM, 1.333 ± 0.055, n = 11, (p < 0.001); 0.5 mM, 1.058 ± 0.055, n = 11, (p = 0.593); 50 mM, 1.119 ± 0.062, n = 7 (p = 0.158). Also, effects of each of the EtOH concentrations on current decay time constant (Fig. 3B right panel) were significant (F(2,26) = 7.949, p = 0.002). Tukey’s comparison showed that normalized to decay time constants for current in response to NIC alone (1.0), current decay time constants at the indicated concentration of EtOH were: 0.1 mM, 0.916 ± 0.055, n = 11, (p = 0.306); 0.5 mM, 0.885 ± 0.055, n = 11, (p = 0.109); 50 mM, 0.641 ± 0.062, n = 7 (p < 0.001). Collectively, at low concentration, EtOH accelerates rising time but has lesser effects on decay of NIC-induced currents, whereas at higher concentrations (e.g., 50 mM), EtOH accelerates current decay, which neutralizes peak current enhancement. These results may explain, at least in part, the underlying mechanism of the bell-shaped EtOH concentration-effect relationship.
Fig. 3. Effects of EtOH on whole-cell current kinetics of NIC-induced currents.
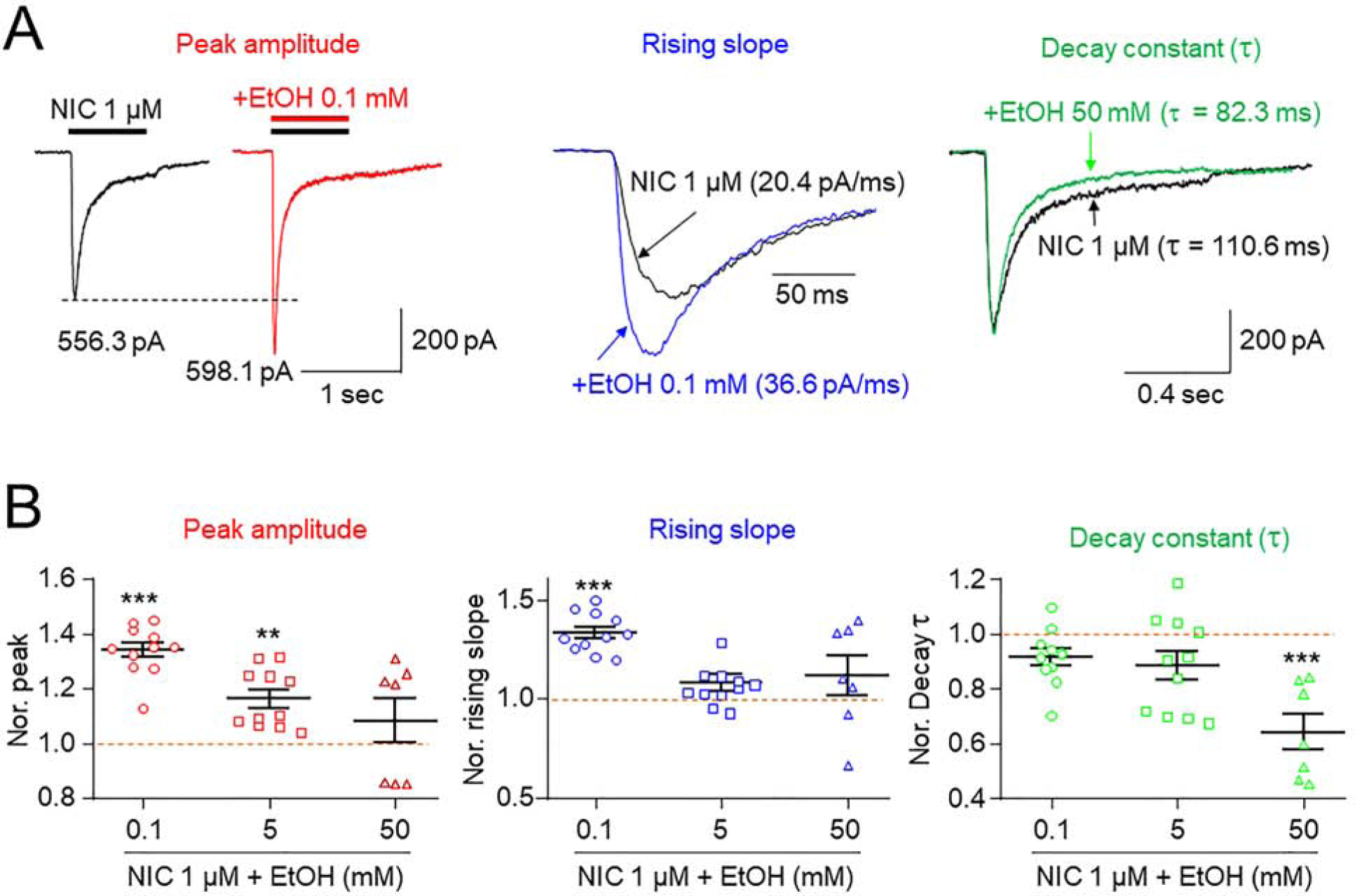
A: Comparison of the effects of 0.1 mM EtOH on peak amplitudes, current rising times and current decay times of 1 μM NIC-induced currents. NIC-induced currents before and during EtOH exposure are superimposed in middle panel. Right panel compared effects of 1 μM NIC-induced currents decay constant with (green trace) and without (black trace) 50 mM EtOH. B: Summary of pooled data to compare alterations of peak currents, rising times and decay times of NIC-induced currents during exposure to different concentrations of EtOH. * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001.
3.4. NIC concentration dependence of α6*-nAChR functional potentiation by EtOH
Studies were done to determine effects of 0.5 mM EtOH exposure at different concentrations of NIC on α6*-nAChR function (Fig. 4). Results showed that 0.5 mM EtOH potentiation of NIC-induced currents was dependent on NIC concentration (Fig. 4A). One-way ANOVA analysis demonstrated that the effects of 0.5 mM EtOH at different NIC concentrations are significant (F(3,37) = 6.377, p<0.001). Relative to effects of NIC alone (t-test), peak current potentiation in the presence of 0.5 mM EtOH at the indicated concentration of NIC was: 10 nM, 1.661 ± 0.143, t(7) = 4.347, p = 0.003; 100 nM, 1.313 ± 0.148; t(6) = 2.596, p =0.040; 1 μM, 1.281 ± 0.148, t(6) = 3.306, p = 0.016; 10 μM, 1.034 ± 0.155, t(6) = 0.321, p = 0.759, respectively (Fig. 4B). Therefore, NIC concentration dependence of EtOH effects is consistent with a positive allosteric modulation mechanism for EtOH potentiation of α6*-nAChR function. Supplemental Fig. 2 further supported this idea by showing potentiation in the presence of 0.1, 1, and 10 mM EtOH on 10 nM-induced NIC currents (Supplemental Fig. 2).
Fig. 4. Effects of 0.5 mM EtOH on different NIC-induced currents.
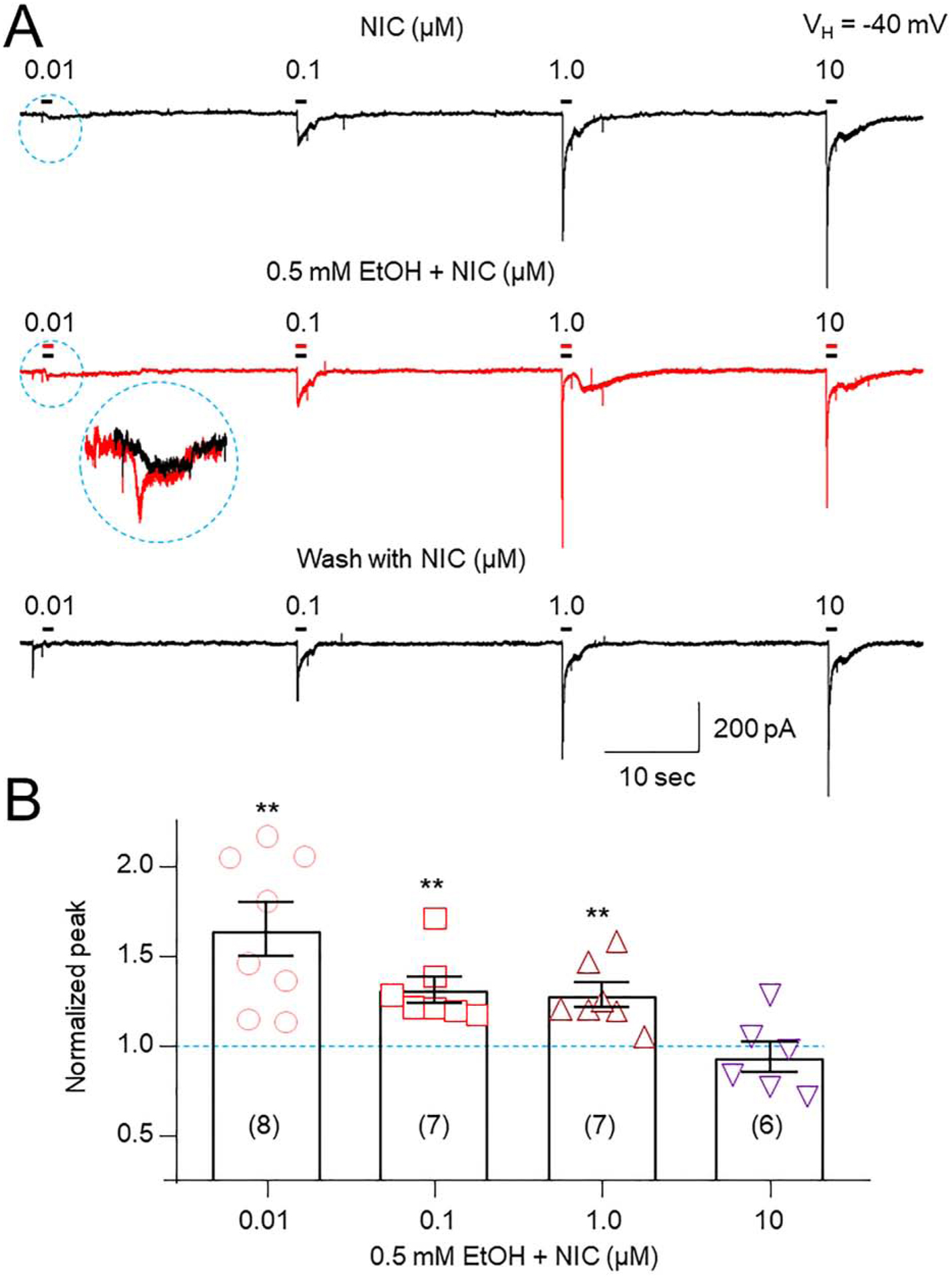
A: Typical traces of different NIC-induced currents from the same recorded cell before (top trace), during (middle trace), and after washout (bottom trace) of 0.5 mM EtOH. Dashed circle indicates magnified responses. B: Bar graph summarizes effects of 0.5 mM EtOH on NIC-induced currents at different NIC concentrations and shows that EtOH-induced enhancement of NIC responses declines as NIC concentrations increase.
3.5. Effects of EtOH on function of different nAChR subtypes
To determine whether EtOH modulation of α6*-nAChR function is specific, we performed two sets of experiments. First, we compared effects of 0.5 mM EtOH on whole-cell current mediated by other nAChR subtypes including α4β2- and α7-nAChRs heterologously expressed in SH-EP1 cells and α3β4-nAChR naturally expressed by SHSY5Y cells (Liu et al., 2015; Wu et al., 2006; Zhao et al., 2003). Figure 5A shows typical traces of the effects of EtOH on ACh-, NIC-, or choline-induced currents at the EC50 concentrations for those agonists at the indicated nAChR subtype. Ethanol exposure at 0.5 mM did not significantly alter α3β4-, α4β2-, or α7-nAChR-mediated currents (One-way ANOVA statistical analysis showed a F(3,97) = 2.631, p = 0.0544). Second, we expressed a novel type of α6*-nAChR, the α6M211Lα3ICβ2β3. Compared to the α6N/α3C subunit construct (Supplemental Fig. 3A), this variant contains more α6 content (α6 N-terminal and α6 transmembrane domains with α3 second internal loop), and it has been reported that this α6 construct with one point mutation (e.g., M211F or F223L) can assemble to form functional receptors with β2 and β3 subunits (Supplemental Fig. 3B) in Xenopus oocytes (Ley et al., 2014). The resultant α6*-nAChR afforded the ability to assess location of the site of action of low dose EtOH on α6N/α3Cβ2β3-nAChR in SH-EP1 cells. Using two-electrode voltage-clamp recording of responses of receptors expressed in oocytes, we compared effects of different concentrations (0.1– 10 mM) of EtOH on α6M211Lα3ICβ2β3 and on α6N/α3Cβ2β3-nAChR-mediated currents induced by 30 μM ACh (close to the EC50 concentration), and we found that EtOH potentiated ACh-induced currents in α6M211Lα3ICβ2β3 and α6N/α3Cβ2β3 in the same manner (bell-shaped EtOH dose-effect relationship, Supplemental Fig. 3C and D). These results further support that low dose EtOH likely acts on the N-terminal of α6N/α3Cβ2β3.
Fig. 5.
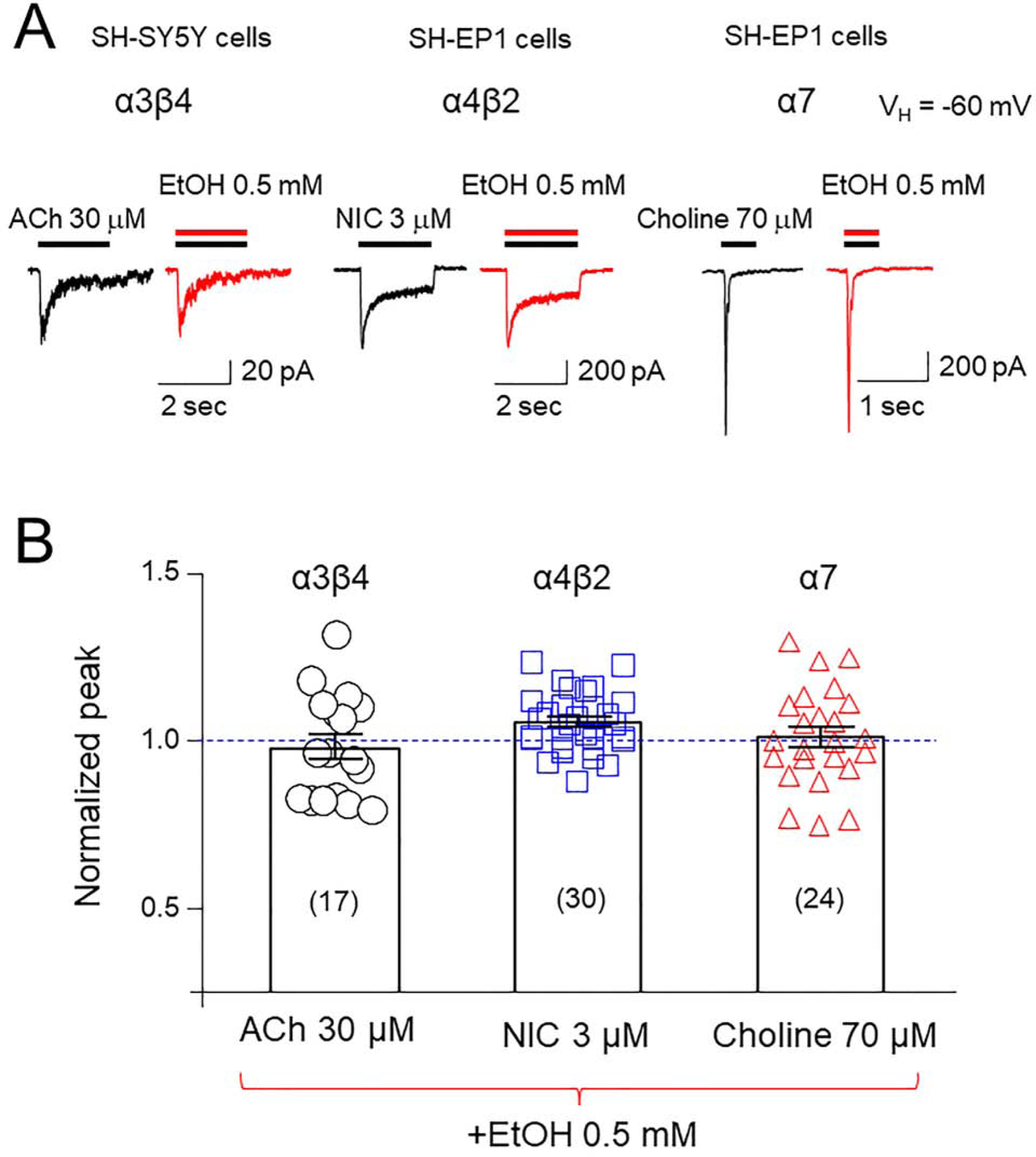
Effects of 0.5 mM EtOH on function of different nAChR subtypes heterologously expressed in SH-EP1 cells (α4β2- and α7-nAChRs) or naturally expressed in SH-SY5Y cells (α3β4*-nAChRs) A: Representative traces of nAChR-mediated current responses to agonists at their EC50 concentrations before and after exposure to 0.5 mM EtOH. When ACh was applied as an agonist (left traces) of α3β4-nAChRs, 1 μM atropine was added in in external solution. B: Summary of the pooled data to show effects of 0.5 mM EtOH on current peak amplitudes of three subtypes of nAChR-mediated currents.
3.6. EtOH enhancement of spontaneous dopamine release in the nucleus accumbens is blocked by the α6*-nAChR-selective antagonist, α-Ctx MII
Since EtOH enhanced α6*-nAChR function when heterologously expressed, we tested whether it would enhance ACh-mediated DA release in a native system. Spontaneous DA transients observed in the striatal slices of mice are dependent on local cholinergic activity mediated by nAChRs expressed on DA terminals (Yorgason et al., 2017). Therefore, the effects of EtOH on spontaneous DA release were tested. In the NAc core, EtOH (1–80 mM) increased the frequency of DA transients (Fig. 6 A–C; One-way ANOVA, F(5,98) = 4.328, p = 0.0013) with greatest enhancement observed at 5 mM EtOH. Administration of the α6*-nAChR-selective antagonist, α-Ctx MII (100 nM), reduced DA transient frequency from 2.53 ± 0.47 to 1.91 ± 0.31 transients/min (t(13) = 2.321, p = 0.0372). Pre-incubation with α-CTx MII (100 nM) prevented EtOH-induced enhancement of DA transient frequency (Fig. 6D; t(24) = 2.349, p = 0.0274). EtOH exposure also increased DA transient amplitude (Fig. 6E,F: F(5,3739) = 15, p < 0.0001), with greatest increases at 5 mM (Fig. 6E). α-Ctx MII pretreatment prevented EtOH (5 mM)-induced increases in spontaneous DA release amplitude (Fig. 6F, : EtOH: F(1,2468) = 1.54, p = 0.214; MII: F(1,2468) =23.59, p < 0.0001; Interaction F(1,2468) = 20.56, p < 0.0001). Thus, α6-nAChRs contribute to transient DA release frequency, and EtOH, most potently at 5 mM, enhances DA transient frequency and amplitude by increasing α6-nAChR activity on DA terminals.
Fig. 6. Effects of low-dose EtOH on spontaneous DA release in the NAc core: Role of α6-nAChRs.
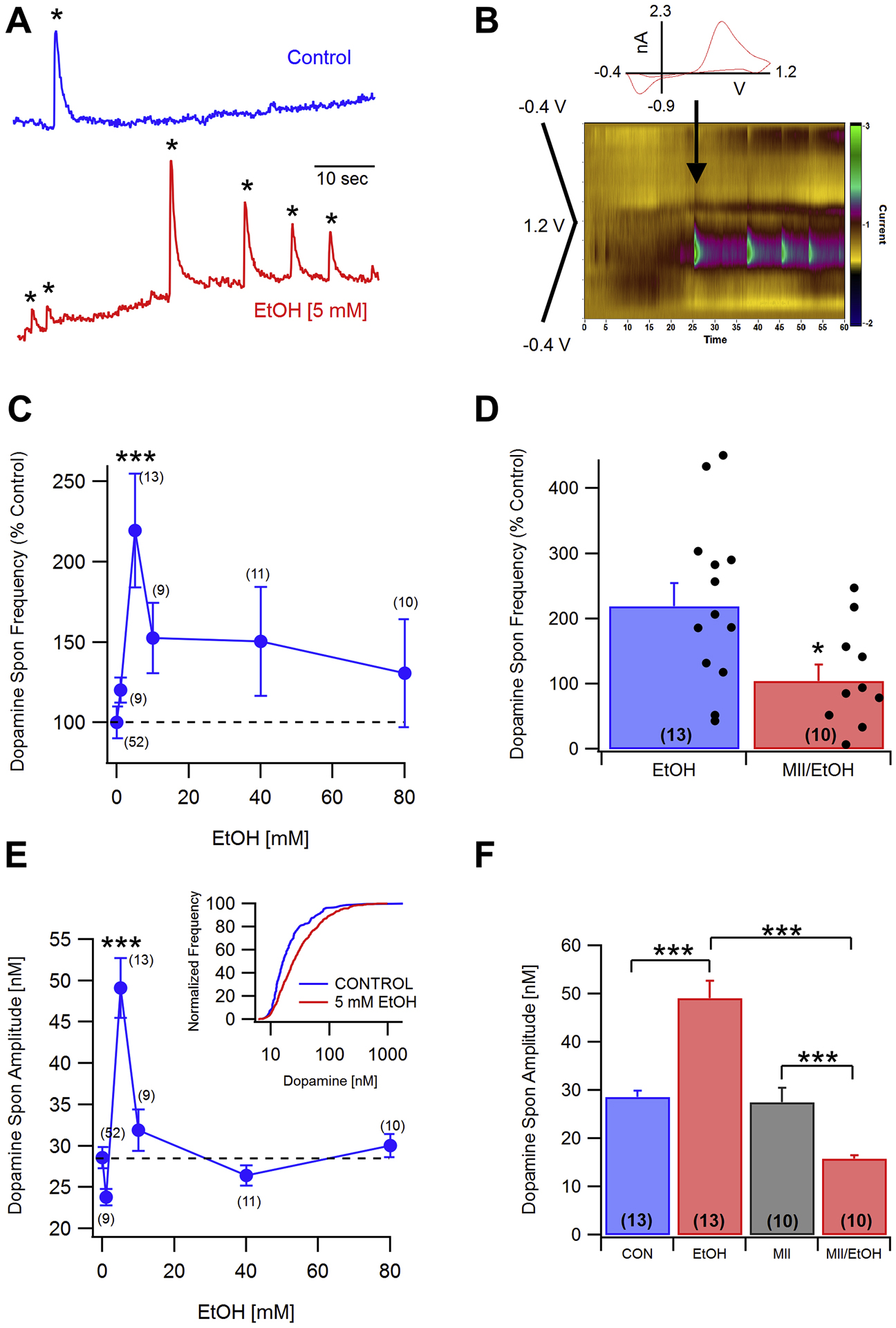
A: Representative 1 min recordings of DA transients under control (top) and 5 mM EtOH (bottom) conditions. Asterisks denote currents corresponding to spontaneous DA release. B: The inset shows a cyclic voltammogram taken at the time indicated by the arrow. Voltammograms were recorded at 10 Hz with a voltage ramp from −0.4 V to 1.2 V to −0.4 V. The color plot shows the DA signal associated with the DA transients in (A, bottom trace). C: Dose-response for EtOH (1–80 mM) effects on DA transient frequency. Ethanol significantly enhanced the frequency of DA transients at the 5 mM concentration. D: Comparisons of exposure to EtOH or to α-conotoxin MII (MII) plus EtOH on DA transient frequency with scatterplots of individual normalized values superimposed on the means ± SEM bars. MII (100 nM) prevented EtOH (5 mM) enhancement of DA transient frequency. E: Dose-response for EtOH (1–80 mM) effects on DA transient amplitude. Ethanol significantly enhanced the amplitude of DA transients at the 5 mM level. The inset shows the DA transient amplitude distribution (normalized for frequency) for control and EtOH (5 mM) exposed slices. F: MII (100 nM) significantly reduced EtOH (5 mM) enhancement of DA transient amplitude without having an effect on its own. There was also a significant difference between MII alone and MII+EtOH. Values in parentheses represent n values. Asterisks *, *** represent significance levels P<0.05 and 0.001, respectively.
4. Discussion
The present study demonstrates that α6*-nAChRs are highly sensitive targets for functionally-relevant, low dose EtOH effects. The EtOH concentration dependence of these effects is bell-shaped, potentiating α6*-nAChR whole-cell current amplitudes in response to 1 μM NIC at 0.1 to 5 mM, but not at lower or higher concentrations of EtOH. Effects also are influenced by the concentrations of NIC, with 0.5 mM EtOH potentiation evident for α6*-nAChR responses to NIC at 0.01–1 μM but not at 10 μM, These results suggest that EtOH serves as a positive allosteric modulator to enhance α6*-nAChR function. Since EtOH (0.5 mM) failed to potentiate other tested subtypes of nAChRs (α3β4, α4β2, α7), the potentiation effect of EtOH on the α6*-nAChR function is specific. Also, low dose EtOH enhanced DA release level in NAc in an α6*-nAChR-dependent manner. Collectively, our results provide innovative knowledge regarding molecular mechanisms of low-dose EtOH effects. This study also provides a significant contribution to the field by forming a foundation for targeting α6*-nAChRs as a novel pharmacological intervention to alleviate alcohol consumption, AUD, and dependence.
nAChRs belong to the ligand-gated ion channel superfamily of neurotransmitter receptors and exist in the vertebrate brain as multiple, diverse subtypes composed as pentamers of unique combinations from a family of genetically distinct subunits. These receptors clearly are involved in nicotine-induced reward and dependence, but they also contribute to effects of other drugs, including alcohol, cocaine, amphetamine, and cannabinoids (Wu and Lukas, 2011). nAChRs are highly expressed in the ventral tegmental area (VTA), and thus play a profound role in modulation of mesolimbic DA system function and response to drug exposure. nAChR α6 subunits are abundant in the VTA, suggesting that α6*-nAChRs could play critical roles in the process of drug reward and dependence (Drenan et al., 2008; Yang et al., 2011; Yang et al., 2009b). α6*-nAChRs are implicated in alcohol reward and dependence based on the ability of the α6*-nAChR-selective antagonist, α-Ctx MII, to inhibit EtOH consumption, EtOH operant responding, and DA release in the nucleus accumbens (NAc) of rats (Kuzmin et al., 2009; Larsson et al., 2004). Also, mice expressing an α6*-nAChR gain of function (α6L9’S) mutation are more highly sensitive to alcohol in several behavioral tests than are wild-type (WT) mice (Powers et al., 2013), whereas EtOH fails to increase VTA DA neuron firing in α6*-nAChR knock-out (KO) mice (Liu et al., 2013). Our new results reported here validate roles for α6*-nAChR subtypes in EtOH-induced behavioral alterations and more specifically illuminate mechanisms involved in effects of low dose EtOH on α6*-nAChRs and on mesolimbic DAergic neuron function. Effects on α6*-nAChR activity are consistent with EtOH action as a positive allosteric modulator and with those actions occurring at EtOH concentrations lower than those associated with EtOH-induced behavioral impairment, consistent with recently-demonstrated, low dose EtOH-induced modulation of synaptic functions in mouse VTA GABA neurons (Steffensen et al., 2017).
Prior investigators have shown that EtOH can serve as a positive allosteric modulator to enhance GABAA receptor function, if the receptor consists of α6, β and γ subunits (Hanchar et al., 2005; Kuner et al., 1993; Liang et al., 2006; Wei et al., 2004) or if it contains δ subunits (Sundstrom-Poromaa et al., 2002; Wallner et al., 2003, 2006; Wallner and Olsen, 2008). The allosteric binding site for EtOH is proposed to be located on the benzodiazepine binding site, and EtOH (<30 mM) affects GABAA receptor function, and also some animal behaviors, through this positive allosteric mechanism (Olsen, 2015). Since this putative high-affinity binding site is different from the transmembrane domain anesthetic site that exhibits lower affinity for EtOH, GABAA receptors containing α4/6, β, and δ subunits represent the brain target for EtOH at intoxicating blood and brain concentrations achieved in humans at 17 mM (Olsen, 2015). Here, we show that EtOH can allosterically potentiate α6*-nAChR function even at concentrations of EtOH below 1 mM, making a6*-nAChRs much more sensitive targets of EtOH. Other studies have shown nAChR-EtOH interactions, but only at much higher EtOH concentrations (Liu et al., 2013). An exception is a report that nAChRs naturally expressed in PC12 cells were found to be super sensitive to even lower EtOH concentrations upon prolonged exposure (“EC50”=88.5 μM) (Nagata et al., 1996). Although the nAChR subtype(s) involved were not identified, sustained EtOH treatment acted to accelerate or to slow decay of ACh-induced currents dependent on the duration of ACh application, induced single channel bursts, but diminished mean channel open times, consistent with increases in receptor desensitization and ACh affinity. PC12 cells express rat α3β4*- and α7-nAChR subtypes (Lukas, 1989), which may differ from the human isoforms of these receptors, which were unaffected by EtOH at the concentrations used in the current study.
Beyond these EtOH and α6*-nAChR interactions at the molecular level, there are considerations at cellular, regional and circuit levels about how those interactions affect mesolimbic function and behavior as well as EtOH reward and dependence. On one hand, EtOH could work in parallel with presynaptic α6*- or other nAChR subtypes mediating, cholinergic modulation of GABA releases or on any somatodendritic nAChR subtypes more directly controlling DA release. On the other hand, EtOH could directly affect nAChR levels via stabilization or internalization of functional receptors or via allosteric modulation or desensitization of nAChR function (Dopico and Lovinger, 2009), thereby contributing to EtOH enhancement of mesolimbic DA signaling relevant to reward and dependence. Other in vivo studies have demonstrated that the selective α6*-nAChR antagonist, α-Ctx MII, administered in the VTA was able to reduce EtOH induced NAc DA release (Larsson et al., 2004) and locomotor activity (Jerlhag et al., 2006). α-Ctx MII perfusion into the VTA also blocked recognition of EtOH-associated cues (Lof et al., 2007) and voluntary EtOH drinking in rodents (Larsson et al., 2004). The present findings show an increase in DA transient frequency after EtOH exposure. This increase is prevented by α-Ctx MII, suggesting that the increases observed in vivo may be attributed in part to enhanced α6*-nAChR activity on DA terminals. Striatal cholinergic interneurons are spontaneously active and drive DA release through direct activation of nAChRs on DA terminals. Striatal DA terminals express α6 subunits (Azam and McIntosh, 2005; Klink et al., 2001; Salminen et al., 2004; Zoli et al., 2002), and since EtOH enhances α6*-nAChR channel currents, EtOH is likely increasing α6*-nAChR conductance on DA terminals, resulting in overall DA terminal excitability, enhancing DA transient frequency and amplitude. It has been demonstrated previously that 4-aminopyridine increases both frequency and amplitude of DA transients (Yorgason et al., 2017), suggesting increased depolarization spread within the DA varicosity, which may result in additional release from nearby sites. EtOH may act similarly. Since DA transients are dependent upon local cholinergic activity (Yorgason et al., 2017), EtOH may enhance nAChR conductance resulting in greater influx of cations, and thus greater depolarization spread during an ACh release event. However, voltammetry cannot spatially differentiate between single and multiple varicosities and it is possible that EtOH is not just enhancing DA release from individual sites. Another important factor that was not tested here is whether EtOH is having effects on cholinergic terminals that may involve other local circuitry influenced by α6*-nAChRs, including potentially VTA projection GABA terminals, which are known to innervate cholinergic interneurons and may express α6*-nAChRs at GABA terminals (Brown et al., 2012; Steffensen et al., 2017). Interestingly, when α-Ctx MII was combined with EtOH, we found a significant decrease in DA transient amplitude, but not frequency, suggesting that EtOH has effects on non-α6*-nAChRs. It should be noted that EtOH has diverse effects on this system that are heavily concentration dependent. For instance, while low concentrations in the present study show clear enhancement of DA release, EtOH has also been shown to inhibit evoked DA release at higher concentrations (>40mM) (Schilaty et al., 2014; Yorgason et al., 2014), an effect that is also blocked with α-Ctx MII. These higher concentrations also likely involve other neurotransmitters and their corresponding receptors, as well as downstream effects on nAChRs and, are likely more important in EtOH’s impairing effects on conditioned learning. Importantly, eEtOH’s effects on α6*-nAChRs are also concentration dependent, with robust increases in α6*-nAChR conductance at low concentrations (0.1–1 mM) but no effect at higher concentrations (10–50 mM). Higher concentrations of EtOH (50 mM) decreased the decay time constant in our heterologous expression system, suggesting that EtOH can potentiate α6*-nAChR desensitization. Therefore, the high concentration (>40mM) EtOH-induced decreases in stimulated DA release reported previously may be due to increased α6*-nAChR desensitization, and subsequent decreases in cholinergic activity on DA terminals (Schilaty et al., 2014; Yorgason et al., 2014). It is important to note that there are inherent differences between evoked DA release in the previous studies (Schilaty et al., 2014; Yorgason et al., 2014) and transient DA release in the current study. For instance, electrical stimulation is inherently non-specific, whereas DA transients are entirely dependent on cholinergic interneuron activity (Yorgason et al., 2017) and therefore more physiologically relevant. This underscores the relevancy of α6* nAChR on low-dose EtOH effects on DA transmission in the mesolimbic reward system.
Supplementary Material
HIGHLIGHTS:
A novel heterologous expression system for human α6 subunit-containing nicotinic acetylcholine receptors (α6*-nAChR) is used to evaluate acute effects of alcohol exposure.
Under patch-clamp whole-cell recording conditions, bath-applied low doses of alcohol (0.1–5 mM) potentiates α6*-nAChR-mediated currents, but not α3β4-, α4β2- or α7-nAChR function.
Effects of alcohol on α6*-nAChRs are influenced by both alcohol and nicotine concentrations, suggesting that alcohol exerts effect through a positive allosteric mechanism.
5 mM EtOH increases both frequency and amplitude of spontaneous DA transients in mouse brain slices containing nucleus accumbens core part, which was blocked by conotoxin MII, suggesting a role for α6-nAChRs in low-dose ethanol effects.
Therefore, α6*-nAChR is a sensitive target to mediate low dose EtOH effects through a positive allosteric mechanism.
Acknowledgments:
We thank Dr. Huang, Yuanbin for maintenance of cell cultures and for some data analysis.
Funding Sources:
Work toward this project was supported by NIH R01 DA035958 to SCS, NIH R21 DA026627 to PW, Barrow Neurological Foundation and a Philips Morris External Research Grant to JW, and the Department of Education of Guangdong Province (2017KTSCX069), China, to FG. Production of the cell line was sponsored by Targacept.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES OF INTERSEST
Dr. Gao, FF reports no disclosures
D. Chen, DJ reports no disclosures
Mr. Xiaokuang Ma reports no disclosures
Dr. Sudweeks reports no disclosures
Dr. Yorgason JT reports no disclosures
Dr. Ming Gao reports no disclosures
Mr. Dharshaun Turner reports no disclosures
Dr. Eaton, JB reports no disclosures
Dr. McIntosh, JM reports no disclosures
Dr. Lukas RJ reports no disclosures
Dr. Whiteraker, P reports no disclosures
Dr. Chang, YC reports no disclosures
Dr. Steffensen SC reports no disclosures
Dr. Wu, J reports no disclosures
References
- Azam L, McIntosh JM, 2005. Effect of novel alpha-conotoxins on nicotine-stimulated [3H]dopamine release from rat striatal synaptosomes. J Pharmacol Exp Ther 312, 231–237. [DOI] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan UH, Chen Y, Leslie FM, 2002. Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444, 260–274. [DOI] [PubMed] [Google Scholar]
- Breining SR, Melvin M, Bhatti BS, Byrd GD, Kiser MN, Hepler CD, Hooker DN, Zhang J, Reynolds LA, Benson LR, Fedorov NB, Sidach SS, Mitchener JP, Lucero LM, Lukas RJ, Whiteaker P, Yohannes D, 2012. Structure-activity studies of 7-heteroaryl-3-azabicyclo[3.3.1]non-6-enes: a novel class of highly potent nicotinic receptor ligands. J Med Chem 55, 9929–9945. [DOI] [PubMed] [Google Scholar]
- Brown MT, Tan KR, O’Connor EC, Nikonenko I, Muller D, Luscher C, 2012. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492, 452–456. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Boschen KE, Hendrick ES, Beardsley PM, McIntosh JM, 2010. Alpha-conotoxin MII-sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology 35, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DJ, Gao FF, Ma XK, Shi GG, Huang YB, Su QX, Sudweeks S, Gao M, Dharshaun T, Eaton JB, Chang YC, McIntosh JM, Lukas RJ, Whiteaker P, Steffensen SC, Wu J, 2018. Pharmacological and functional comparisons of alpha6/alpha3beta2beta3-nAChRs and alpha4beta2-nAChRs heterologously expressed in the human epithelial SH-EP1 cell line. Acta Pharmacol Sin 39, 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash B, Li MD, Lukas RJ, 2014. Roles for N-terminal extracellular domains of nicotinic acetylcholine receptor (nAChR) beta3 subunits in enhanced functional expression of mouse alpha6beta2beta3- and alpha6beta4beta3-nAChRs. J Biol Chem 289, 28338–28351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rover M, Lodder JC, Kits KS, Schoffelmeer AN, Brussaard AB, 2002. Cholinergic modulation of nucleus accumbens medium spiny neurons. Eur J Neurosci 16, 2279–2290. [DOI] [PubMed] [Google Scholar]
- Dopico AM, Lovinger DM, 2009. Acute alcohol action and desensitization of ligand-gated ion channels. Pharmacol Rev 61, 98–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, Marks MJ, Miwa JM, Lester HA, 2010. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci 30, 9877–9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA, 2008. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron 60, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ, 2008. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology 33, 2158–2166. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT, 2000. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci 20, 7855–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RK, Hiller-Sturmhofel S, 1999. Alcoholism treatment in the United States. An overview. Alcohol Res Health 23, 69–77. [PMC free article] [PubMed] [Google Scholar]
- Garzon M, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM, 1999. Cholinergic axon terminals in the ventral tegmental area target a subpopulation of neurons expressing low levels of the dopamine transporter. J Comp Neurol 410, 197–210. [DOI] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, Zanardi A, Rimondini R, Mugnaini M, Clementi F, Chiamulera C, Zoli M, 2010. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci 30, 5311–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M, 2005. Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat Neurosci 8, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA, 1992. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci 12, 3804–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, McIntosh JM, Brunzell DH, Sanjakdar SS, Damaj MI, 2009. The role of alpha6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J Pharmacol Exp Ther 331, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Grotli M, Luthman K, Svensson L, Engel JA, 2006. Role of the subunit composition of central nicotinic acetylcholine receptors for the stimulatory and dopamine-enhancing effects of ethanol. Alcohol Alcohol 41, 486–493. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP, 2001. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21, 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R, Korpi ER, 1993. Ethanol inhibits glutamate-induced currents in heteromeric NMDA receptor subtypes. Neuroreport 5, 297–300. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J, 2000. Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology 39, 2570–2590. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Jerlhag E, Liljequist S, Engel J, 2009. Effects of subunit selective nACh receptors on operant ethanol self-administration and relapse-like ethanol-drinking behavior. Psychopharmacology (Berl) 203, 99–108. [DOI] [PubMed] [Google Scholar]
- Larsson A, Jerlhag E, Svensson L, Soderpalm B, Engel JA, 2004. Is an alpha-conotoxin MII-sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol 34, 239–250. [DOI] [PubMed] [Google Scholar]
- Letchworth SR, Whiteaker P, 2011. Progress and challenges in the study of alpha6-containing nicotinic acetylcholine receptors. Biochem Pharmacol 82, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley CK, Kuryatov A, Wang J, Lindstrom JM, 2014. Efficient expression of functional (alpha6beta2)2beta3 AChRs in Xenopus oocytes from free subunits using slightly modified alpha6 subunits. PLoS One 9, e103244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I, 2006. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J Neurosci 26, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little HJ, 1991. Mechanisms that may underlie the behavioural effects of ethanol. Prog Neurobiol 36, 171–194. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Tapper AR, 2013. Nicotinic acetylcholine receptors containing the alpha6 subunit contribute to ethanol activation of ventral tegmental area dopaminergic neurons. Biochem Pharmacol 86, 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xie X, Emadi S, Sierks MR, Wu J, 2015. A novel nicotinic mechanism underlies beta-amyloid-induced neurotoxicity. Neuropharmacology 97, 457–463. [DOI] [PubMed] [Google Scholar]
- Lof E, Olausson P, Debejczy A, Stomberg R, McIntosh JM, Taylor JR, Soderpalm B, 2007. Nicotinic acetylcholine receptors in the ventral tegmental area mediate the dopamine activating and reinforcing properties of ethanol cues. Psychopharmacology (Berl) 195, 333–343. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, 1991. Ethanol potentiates ion current mediated by 5-HT3 receptors on neuroblastoma cells and isolated neurons. Alcohol Alcohol Suppl 1, 181–185. [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF, 1989. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243, 1721–1724. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, 1989. Pharmacological distinctions between functional nicotinic acetylcholine receptors on the PC12 rat pheochromocytoma and the TE671 human medulloblastoma. J Pharmacol Exp Ther 251, 175–182. [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS, 2002. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33, 905–919. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS, 2000. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27, 349–357. [DOI] [PubMed] [Google Scholar]
- Mullikin-Kilpatrick D, Treistman SN, 1994. Ethanol inhibition of L-type Ca2+ channels in PC12 cells: role of permeant ions. Eur J Pharmacol 270, 17–25. [DOI] [PubMed] [Google Scholar]
- Nagata K, Aistrup GL, Huang CS, Marszalec W, Song JH, Yeh JZ, Narahashi T, 1996. Potent modulation of neuronal nicotinic acetylcholine receptor-channel by ethanol. Neurosci Lett 217, 189–193. [PubMed] [Google Scholar]
- Nishio M, Narahashi T, 1990. Ethanol enhancement of GABA-activated chloride current in rat dorsal root ganglion neurons. Brain Res 518, 283–286. [DOI] [PubMed] [Google Scholar]
- Olsen RW, 2015. Allosteric ligands and their binding sites define gamma-aminobutyric acid (GABA) type A receptor subtypes. Adv Pharmacol 73, 167–202. [DOI] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W, 2008. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci 28, 12318–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MS, Broderick HJ, Drenan RM, Chester JA, 2013. Nicotinic acetylcholine receptors containing alpha6 subunits contribute to alcohol reward-related behaviours. Genes Brain Behav 12, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD, 2015. 2010 National and State Costs of Excessive Alcohol Consumption. Am J Prev Med 49, e73–e79. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR, 2004. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65, 1526–1535. [DOI] [PubMed] [Google Scholar]
- Sanjakdar SS, Maldoon PP, Marks MJ, Brunzell DH, Maskos U, McIntosh JM, Bowers MS, Damaj MI, 2015. Differential roles of alpha6beta2* and alpha4beta2* neuronal nicotinic receptors in nicotine- and cocaine-conditioned reward in mice. Neuropsychopharmacology 40, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilaty ND, Hedges DM, Jang EY, Folsom RJ, Yorgason JT, McIntosh JM, Steffensen SC, 2014. Acute ethanol inhibits dopamine release in the nucleus accumbens via alpha6 nicotinic acetylcholine receptors. J Pharmacol Exp Ther 349, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahre M, Roeber J, Kanny D, Brewer RD, Zhang X, 2014. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev Chronic Dis 11, E109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Shin SI, Nelson AC, Pistorius SS, Williams SB, Woodward TJ, Park HJ, Friend L, Gao M, Gao F, Taylor DH, Foster Olive M, Edwards JG, Sudweeks SN, Buhlman LM, Michael McIntosh J, Wu J, 2017. alpha6 subunit-containing nicotinic receptors mediate low-dose ethanol effects on ventral tegmental area neurons and ethanol reward. Addict Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstrom-Poromaa I, Smith DH, Gong QH, Sabado TN, Li X, Light A, Wiedmann M, Williams K, Smith SS, 2002. Hormonally regulated alpha(4)beta(2)delta GABA(A) receptors are a target for alcohol. Nat Neurosci 5, 721–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW, 2003. Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A 100, 15218–15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW, 2006. Low-dose alcohol actions on alpha4beta3delta GABAA receptors are reversed by the behavioral alcohol antagonist Ro15–4513. Proc Natl Acad Sci U S A 103, 8540–8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Olsen RW, 2008. Physiology and pharmacology of alcohol: the imidazobenzodiazepine alcohol antagonist site on subtypes of GABAA receptors as an opportunity for drug development? Br J Pharmacol 154, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Faria LC, Mody I, 2004. Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci 24, 8379–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weight FF, Lovinger DM, White G, 1991. Alcohol inhibition of NMDA channel function. Alcohol Alcohol Suppl 1, 163–169. [PubMed] [Google Scholar]
- Weight FF, Peoples RW, Wright JM, Lovinger DM, White G, 1993. Ethanol action on excitatory amino acid activated ion channels. Alcohol Alcohol Suppl 2, 353–358. [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA, 2003. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23, 3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, George AA, Schroeder KM, Xu L, Marxer-Miller S, Lucero L, Lukas RJ, 2004. Electrophysiological, pharmacological, and molecular evidence for alpha7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J Pharmacol Exp Ther 311, 80–91. [DOI] [PubMed] [Google Scholar]
- Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, St John PA, Lukas RJ, 2006. Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4-containing nicotinic receptors. J Physiol 576, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lukas RJ, 2011. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol 82, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Buhlman L, Khan GM, Nichols RA, Jin G, McIntosh JM, Whiteaker P, Lukas RJ, Wu J, 2011. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci 31, 2537–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Hu J, Lucero L, Liu Q, Zheng C, Zhen X, Jin G, Lukas RJ, Wu J, 2009a. Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol 587, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Jin GZ, Wu J, 2009b. Mysterious alpha6-containing nAChRs: function, pharmacology, and pathophysiology. Acta Pharmacol Sin 30, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Jin GZ, Wu J, 2009c. Mysterious alpha6-containing nAChRs: function, pharmacology, and pathophysiology. Acta Pharmacol Sin 30, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, French ED, 2000. A comparison of the effects of nicotine on dopamine and non dopamine neurons in the rat ventral tegmental area: an in vitro electrophysiological study. Brain Res Bull 51, 507–514. [DOI] [PubMed] [Google Scholar]
- Yorgason JT, Espana RA, Jones SR, 2011. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 202, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Ferris MJ, Steffensen SC, Jones SR, 2014. Frequency-dependent effects of ethanol on dopamine release in the nucleus accumbens. Alcohol Clin Exp Res 38, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Zeppenfeld DM, Williams JT, 2017. Cholinergic Interneurons Underlie Spontaneous Dopamine Release in Nucleus Accumbens. J Neurosci 37, 2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Kuo YP, George AA, Peng JH, Purandare MS, Schroeder KM, Lukas RJ, Wu J, 2003. Functional properties of homomeric, human alpha 7-nicotinic acetylcholine receptors heterologously expressed in the SH-EP1 human epithelial cell line. J Pharmacol Exp Ther 305, 1132–1141. [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C, 2002. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 22, 8785–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Kuryatov A, Lindstrom JM, Yeh JZ, Narahashi T, 2002. Alcohol modulation of neuronal nicotinic acetylcholine receptors is alpha subunit dependent. Alcohol Clin Exp Res 26, 779–784. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.