

Abstract
Cytomegalovirus (CMV) has been proposed to play a role in cancer progression and invasiveness. However, CMV has been increasingly studied as a cancer vaccine vector and multiple groups, including ours, have reported that the virus can drive anti-tumor immunity in certain models. Our previous work revealed that intratumoral injections of WT murine (M)CMV into B16-F0 melanomas caused tumor growth delay in part by using a viral chemokine to recruit macrophages, which were subsequently infected. We now show that MCMV acts as a STING agonist in the tumor. MCMV infection of tumors in STING-deficient mice resulted in normal recruitment of macrophages to the tumor, but poor recruitment of CD8+ T cells, reduced production of inflammatory cytokines and chemokines, and no delay in tumor growth. In vitro, expression of type I IFN was dependent on both STING and the type I IFN receptor. Moreover, type I IFN alone was sufficient to induce cytokine and chemokine production by macrophages and B16 tumor cells, suggesting that the major role for STING activation was to produce type I IFN. Critically, viral infection of wild-type macrophages alone was sufficient to restore tumor growth delay in STING-deficient animals. Overall, these data show that MCMV infection and sensing in tumor associated macrophages through STING signaling is sufficient to promote anti-tumor immune responses in the B16-F0 melanoma model.
Introduction
As research continues to focus on discovering novel cancer therapies, the approach of utilizing pathogens to alter the immune microenvironment has emerged as an exciting new strategy. With the FDA approval of the oncolytic herpes simplex 1 virus (HSV-1) T-VEC in 2015, pathogens such as Vaccinia, Semliki Forrest virus, Toxoplasma gondii, and Salmonella, and cytomegalovirus (CMV), have all been tested in preclinical models and have the potential to directly lyse tumor cells and/or alter the tumor environment(1–12). In addition to using pathogens to alter the tumor microenvironment experimentally, research has also focused on how the naturally occurring microbiome in human tumors may positively and negatively alter patient outcomes(13–15). Indeed, both viruses and bacteria have been shown to naturally alter the tumor microenvironment in addition to being used as a potential immunotherapy.
Human (H)CMV is a ubiquitous beta-herpesvirus that has been repeatedly identified in human tumors including glioblastoma, non-melanoma skin cancer, colorectal adenocarcinoma, rhabdomyosarcoma, and ovarian cancer(16–20). It is not thought that HCMV initiates tumor development(21). However, the impact of HCMV on tumor growth has been widely debated and studies conflict as to whether HCMV may have a positive or negative impact on tumor progression(16–20, 22, 23). Thus, it is important to evaluate the effect of CMV in experimental tumor models and define the mechanisms by which CMV alters tumor behavior.
In addition to its potential to alter tumor progression, CMV has also been proposed as a vaccine vector for infectious diseases and cancer(8, 24–41). CMV naturally generates a massive CD8+ T cell response in the host which can inflate over time due to blips of reactivation(42). In multiple models including melanoma, prostate cancer, and head and neck squamous cell carcinoma, murine (M)CMV has been successfully used as a vaccine to generate immunity toward tumor antigens encoded in the viral backbone(8, 34–41). However, the potential for such a therapy depends heavily on how CMV naturally alters the tumor environment. Indeed, there has been experimental evidence that latent MCMV may promote tumor growth and invasiveness in models of breast cancer and glioblastoma with both of these studies citing increased angiogenesis as partially responsible for the effect (43, 44). In contrast, MCMV infection in models of melanoma and lymphoma can cause tumor regression(8–12). We recently showed that MCMV, when injected intratumorally (i.t.) into a growing B16-F0 melanoma, slowed progression of the lesion, altered the immune compartment of the tumor, and synergized with blockade of the immune checkpoint PD-L1 to clear established tumors and promote long-term immune memory(8). The efficacy of MCMV approximately correlated with the period of time during which the virus is active in the tumor and could be extended by increasing the number of viral injections(9). The virus was found to recruit macrophages via a viral chemokine and infect the macrophages in the tumor. In vitro work also demonstrated that MCMV increased inflammatory transcripts in anti-inflammatory or M2-like macrophages. However, this work did not address the mechanism by which MCMV infection alters tumor-associated macrophages, nor did we show that infection of tumor infiltrating macrophages per se was important for the therapeutic effect.
Although the mechanisms by which MCMV activates macrophages have not been determined in the mouse model, it has been shown that human (H)CMV can affect the inflammatory state of the human monocytes through activating the PI3K pathway upon entry into cells(45, 46). Additionally, HCMV is known to activate STING in monocytes to promote type I IFN production(47). In the current study, we show that MCMV activates macrophages through the STING pathway. Expression of STING was critical for MCMV to delay the growth of B16 melanomas, an effect that was most likely the result of type I IFN production in the tumor leading to chemokines that recruited T cells to the tumor environment. Most importantly, we found that infected macrophages were sufficient to recruit CD8+ T cells and delay tumor growth in a STING-dependent manner. These data demonstrate that an active MCMV infection delays tumor growth in this model through activation of STING.
Materials and Methods
Mice.
C57BL/6J, Tmem173gt/gt (C57BL/6J-Tmem173gt/J, referred to as STING gt//gt), and Tmem173−/− (B6(Cg)-Tmem173tm1.2Camb/J, referred to as STING −/−) were purchased from Jackson laboratories. All mice, on a B6 background, were bred at Thomas Jefferson University from original breeders obtained from different sources and used at an age of 6–16 weeks. Ifnar1−/− mice back- crossed to B6(48) were a gift from Dr. Thomas Moran (Mount Sinai School of Medicine, New York, NY). Tlr9−/− (B6.129-Tlr9tm1Aki/Obs) mice were produced by Dr. S. Akira (Osaka University, Japan)(49) and generously provided by Dr. Robert Finberg (University of Massachusetts, Worcester, MA). A mix of male and female mice were used for all studies. Due to differences in breeding schedules, data from STING−/− mice (Tmem173−/−) and STINGgt/gt (mice lacking functional STING, Tmem173gt/gt) were combined to form the STING-deficient groups for some experiments. This is noted in the figure legends. No notable differences in tumor growth, survival, or doubling time were observed between the STING−/− and STINGgt/gt animals (Supplemental Figure 1). The Institutional Animal Care and Use Committee at Thomas Jefferson University reviewed and approved all protocols.
Macrophage culture, viruses, and in vitro infection.
Bone marrow derived macrophages were harvested from the femur and tibia of C57BL/6J, Tlr9−/−, Ifnar1−/−, STINGgt/gt, and/or STING−/− animals by flushing the bones with bone marrow macrophage media consisting of DMEM with 10% L929-conditioned media, 10% fetal bovine serum, and 1% penicillin-streptomycin. The L929-conditioned media was produced by plating 7.2x105 L929 cells in T-150 flasks. Supernatant was harvested after 7 days, replaced with fresh media and harvested again after 14 days. All L929 conditioned media was combined and frozen for later use. Bone marrow was strained through a 70μm filter after harvest and plated onto non-tissue culture treated petri dishes to facilitate macrophage recovery from the plastic. After 7 days, macrophages were re-plated at 6x105 cells/well in a 6 well plate. M2-like macrophages were polarized using 40 ng/mL IL-4 on day 8. On day 9 half of the IL-4-treated BMDMs were infected with K181 MCMV or the GFP+ SL8-015 MCMV (as noted in the figure legend) at a multiplicity of infection (MOI) of 5 for 24 hours. M1-like macrophages were also polarized on day 9 by adding 20 ng/mL of IFN-γ and 1 μg/mL LPS. M0 wells were left untreated. 24 hours after infection, all macrophages were harvested for analysis. All WT-MCMV (K181 and SL8-015) was grown as previously described (50).
IFN-I treatment
Bone marrow derived macrophages from C567BL/6J, STINGgt/gt, and Ifnar1−/− animals were harvested and grown as described above. After 7 days, cells were re-plated and treated with 40 ng/mL IL-4 to produce M2 macrophages or left untreated to produce M0 macrophages. After 24 hours, M0 and M2 macrophages were treated with a 1:1 mix of IFNα and IFNβ in increasing concentrations from 0 to 500 units without removing initial polarizing conditions (Supplemental Figure 2) to determine the optimal dose of 500 units. The cells were treated with IFN for 24 hours before processing the samples for RNA using the Qiagen RNeasy Plus Mini Kit. The experiment was repeated with the optimal 500 unit dose of type I IFN (Supplemental Figure 2).
When B16-F0s were treated with IFN, 3x105 B16-F0s were plated in 6 well plates 24 hours prior to treatment with IFN. Cells were either treated with 500 units of IFN or left untreated. After 24 hours with IFN, the live B16-F0s were counted manually using trypan blue and a hemocytometer and harvested for RNA using the Qiagen RNeasy Plus Mini Kit.
Tumor implantation and intratumoral injections.
B16-F0 cells were purchased from ATCC, grown in DMEM with 1% penicillin-streptomycin and 10% FBS and frozen in a large batch of aliquots between passage 5 and 13. In all experiments, cells derived from this frozen batch were thawed, passaged once more, and implanted within seven days. Injected cells were always pigmented and a batch was certified negative for mycoplasma and other pathogens by IMPACT III testing on 10/30/2017 by IDEXX BioResearch. For tumor implantation, B16-F0s were resuspended in HBSS and injected subcutaneously in the shaved right flank of the animal as described previously(51). Tumor growth was monitored by measuring length and width with a 6-inch digital caliper (Neiko). When tumors reached 20mm2 in area they were directly injected with spread-defective ΔgL-MCMV or PBS as a control using an insulin syringe with 5x105 pfu of virus in 50-70 μL of PBS, every other day for a total of three injections. Spread-defective MCMV (ΔgL, based on the K181 backbone) has been previously characterized and was grown as previously described(52). After IT injections, tumors were monitored until they reached 100 mm2.
For bone marrow derived macrophage transfers, macrophages from C57BL/6 animals were harvested and cultured as described above. For timing purposes, bone marrow was generally harvested the same day or the day before tumors were first implanted into STINGgt/gt animals. When tumors had reached about 20mm2, BMDMs were polarized to M2-like macrophages for 24 hours and then infected with spread-defective MCMV (ΔgL) or left untreated. After another 24 hours, 4x104 M2 or infected M2 macrophages were transferred intratumorally into the B16-F0 melanomas in STINGgt/gt animals. The number of macrophages for transfer were based on macrophages counts in immunofluorescent images extrapolated to the whole tumor volume. After the single injection of macrophages, tumors were monitored to an endpoint of 100mm2.
Tumor processing for cell sorting.
Tumors were harvested on day 5 and homogenized in RPMI using a gentleMACS Octo Dissociator (Miltenyi Biotec) on m_lung_01_01 and m_lung_02_01settings. Cells were separated with Lymphoprep (Stem Cell Technologies) according to the manufacturer’s instructions and stained for sorting with Zombie UV Fixable Viability Dye (BioLegend) as well as antibodies specific for CD45.2 (clone 104; Biolegend), CD11b (clone M1/70; Biolegend), and Gr-1 (clone RB6-8C5; Biolegend), CD4 (clone RM4-5; Biolegend), CD19 (clone 1D3; eBiosciences), and NK1.1 (clone PK136; Biolegend). Representative FACS plots from sorting are shown in Supplemental Figure 4.
Quantitative PCR.
RNA was extracted from treated BMDMs, B16-F0s, whole tumor homogenate, or cells sorted from the tumor (as indicated in the figure legends). Tumor homogenate was processed by pushing tumors through a 70 μm filter to obtain a single cell suspension before lysing cells for RNA extraction. RNA from all samples was isolated using the RNeasy Mini Kit (QIAGEN) and cDNA was produced using a high capacity cDNA reverse transcription kit (Applied Biosystems). Transcripts were detected using iTaq Universal SYBR Green with the following primers: Gapdh: tgtccgtcgtggatctgac and cctgcttcaccaccttcttg, Cxcl9: cttttcctcttgggcatcat and gcatcgtgcattccttatca, Cxcl10: gctgccgtcattttctgc and tctcactggcccgtcatc, Tnf: tcttctcattcctgcttgtgg and ggtctgggccatagaactga, Il6: tctaattcatatcttcaaccaagagg and tggtccttagccactccttc, Ifn-nona4: aagctgtgtgatgcaacaggt and ggaacacagtgatcctgtgg, and IFNG: gcaaaaggatggtgacatga and ttcaagacttcaaagagtctgaggta. Transcript reported as Ifna was collected using the IFN-nonα4 primers, which amplify all subtypes of IFN-α except for IFN-α4 due to deviations in sequence homology. All samples were run on the Bio-Rad CFX96 Touch Real-Time PCR Detection System. In macrophage studies, transcripts were normalized to the housekeeping gene GAPDH and compared to uninfected M2 macrophages to obtain a ΔΔCT value. Data are expressed as fold change over M2 macrophages using 2-(ΔΔCT). For whole tumor homogenates, transcript concentrations were normalized to GAPDH in each sample and compared to the averages of the PBS treated samples in each experiment to obtain a ΔΔCT value. For sorted cells, transcript concentrations were normalized to GAPDH and displayed as relative expression.
Phagocytosis assay.
BMDMs were harvested from C57BL/6, STINGgt/gt, and Ifnar1−/− animals and cultured as above. After resting in culture for 7 days, 100,000 BMDMs were plated in triplicate for each treatment condition in a 96 well black microplate. BMDMs were polarized to M2-like macrophages using 40 ng/mL IL-4 for 24 hours. After 24 hours, M2-like BMDMs were infected with GFP-expressing SL8–015 MCMV at an MOI of 5 or left untreated. M1-like macrophages were also polarized at this time using 20 ng/mL of IFN-γ and 1 μg/mL LPS. After 24 hours, phagocytic potential of the cells was determined using pHrodo Red E. coli Bioparticles (Invitrogen) using the protocol provided. After 1 hour, the bioparticles were removed and the cells were gently washed once with PBS. 560/585 readings were taken to determine phagocytic potential of the cells using a Molecular Devices Spectromax M2 microplate reader. Immediately after, wells were imaged using a Nikon eclipse Ti confocal microscope to view both phagocytosis and infection percentage in the cells. Images were analyzed with ImageJ (https://fiji.sc/)(53).
Immunofluorescence
Tumors were rapidly frozen in O.C.T. compound and cut tumors into 15-20 μM sections using a Leica CM3050 S cryostat. Sections were placed in cold acetone for 10 minutes, rehydrated with Tris-buffered saline (TBS) for 20 minutes, and blocked with blocking buffer (TBS + 3% BSA and 0.1% Tween-20) for 20 minutes. Sections were stained in blocking buffer for one hour with antibodies from BioLegend specific for F4/80 (clone BM8), CD11b (clone M1/70), CD8α (clone 53-6.7), and CD45.2 (clone 104) as well as DAPI as indicated in the figure legends. Samples were imaged using the Nikon A1R fluorescent confocal microscope. Macrophage number per mm2 image was calculated from the count of F4/80+, CD11b+ macrophages per image from multiple images per tumor and 2-3 tumors per treatment, as indicated in the figure legend. CD8+ T cell number per mm2 image was calculated from the count of CD8+, CD45.2+, CD11b−, or CD8+, CD11b− cells per image in multiple images per tumor and multiple tumors per treatment as indicated in the figure legends. Images were analyzed with ImageJ (https://fiji.sc/)(53).
Statistical analysis.
Statistics were performed using GraphPad Prism v6 or R (v3.3.1 R-project.org). If normally distributed (as assessed by the Kolmogorov-Smirnov test), data were analyzed using a two-tailed t test. If data were non-normally distributed, a Mann-Whitney test was performed instead. For tumor doubling times, the tumor measurements were log transformed and a linear regression was used to model the rate of tumor growth as a function of time. Finally, a log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves.
Results
MCMV increases pro-inflammatory cytokine expression in infected macrophages via STING and IFNAR signaling
Previous data from our lab shows that i.t. injections of WT-MCMV can cause tumor growth delay in a B16-F0 melanoma model (9). This anti-tumor effect was dependent on the recruitment of macrophages, many of which were infected in the tumor environment. Additionally, viral infection of the macrophages in vitro increased pro-inflammatory transcripts in M2 polarized macrophages. However, it remains unclear how MCMV altered macrophages and whether this pathway was required for virus-induced tumor growth delay.
Sensing of CMV after infection can occur through multiple pattern recognition receptors, but is most often attributed to TLR9, which senses viral DNA in the endosome, and the cGAS/STING pathway, which senses viral DNA in the cytosol (47, 54–58). Both of these pathways, when activated by CMV, produce a strong type I IFN response (47, 55). However, in human monocytes infected with HCMV, STING is required for induction of type I IFN (47). To test the importance of these pathways in MCMV activation of macrophages, we harvested bone marrow derived macrophages (BMDMs) from Tlr9−/−, STING-deficient (both knockout and golden ticket), and Ifnar1−/− mice along with WT C57BL/6J controls. Macrophages were polarized to M2-like state for 24hrs to mimic the polarization state of newly arrived tumor-associated macrophages (TAM) and then infected with MCMV. In line with our previous data, MCMV induced pro-inflammatory cytokine production in the M2-polarized B6 macrophages, including significant induction of mRNA for Il6, Tnf, Ifna, Cxcl9 and Cxcl10 (Figure 1A). Loss of TLR9 did not alter the production of these cytokines. However, STING-deficient animals were markedly impaired in the production of Il6, Tnf and Ifna upon infection (Figure 1B). Moreover, loss of the type I IFN receptor, IFNAR, phenocopied the loss of STING, suggesting that STING function was dependent on release of type I IFN. Thus, STING and IFNAR signaling play a critical role in MCMV-mediated induction of inflammatory cytokines in macrophages.
Figure 1. Pro-inflammatory cytokine production is not induced after MCMV infection of STING-def or Ifnar1−/− macrophages.
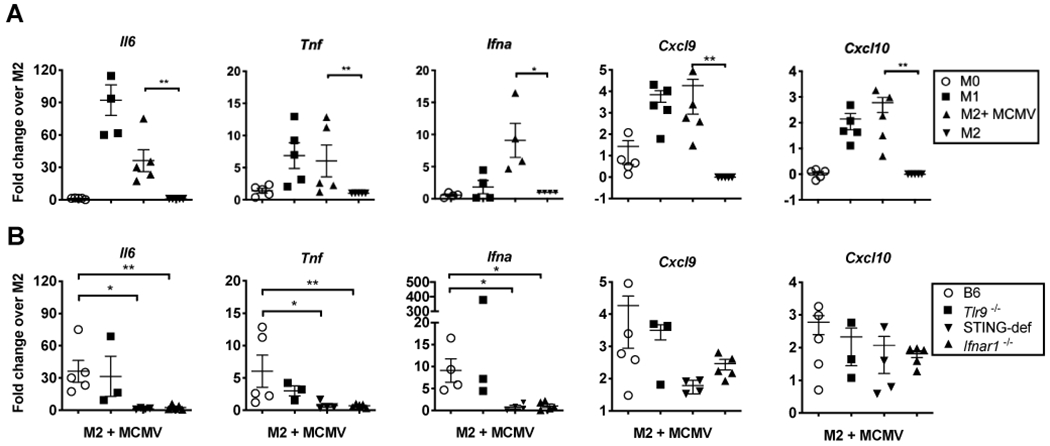
A) BMDMs were harvested from WT B6 mice and left untreated (M0), polarized to M2 or M1 subsets, or polarized to an M2 subset and infected with WT-MCMV for 24 hours (M2+MCMV). Graphs display Il6 (n=5), Tnf (n=5), Ifna (n=4), Cxcl9 (n=3), and Cxcl10 (n=5) transcript as fold change over M2. B) BMDMs were harvested from WT B6 as in A, Tlr9−/− (n=3), Ifnar1−/− (n=5), and STING-def (n=4, from STINGgt/gt and STING−/− combined) animals. All subsets were generated as in A, but only the M2 subsets infected with MCMV (MC+MCMV) are displayed for each genotype. Data is shows as fold change over the relevant M2 subset for each genotype. In all cases, significance was determined using a Mann-Whitney test and is denoted as follows: * p < 0.05, ** p < 0.01, *** p <0.001, **** p <0.0001.
Type I IFN increases inflammatory cytokine expression in uninfected macrophages
The loss of MCMV-induced inflammatory cytokine production from both Ifnar1−/− macrophages and STING-deficient macrophages indicated that IFNAR signaling may be playing a key role secondary to CMV infection. Therefore, we wished to determine if type I IFN alone, via paracrine signaling, could activate production of these cytokines in macrophages. To test this, we added type I IFN alone (a 1:1 mixture of IFN-α and IFN-β) to unpolarized macrophages (M0) or macrophages that had been polarized toward an M2-like state for 24 hours. The concentration of type I IFN was determined by titration experiments, (Supplemental Figure 2). One day after the addition of type I IFN, M0 and M2 macrophages expressed increased levels of Il6, Tnf, Cxcl9 and Cxcl10. In the M0 group, Il6, Cxcl9, and Cxcl10 production was significantly increased while Tnf, Cxcl9, and Cxcl10 production was significantly increased in the M2-polarized group (Figure 2A). STING-deficient BMDMs also followed a similar pattern (data not shown). In contrast however, Ifnar1−/− animals showed no response to the addition of type I IFN, as expected (Figure 2B). Thus, type I IFN alone is sufficient to induce the production of the inflammatory cytokines observed after MCMV infection.
Figure 2.
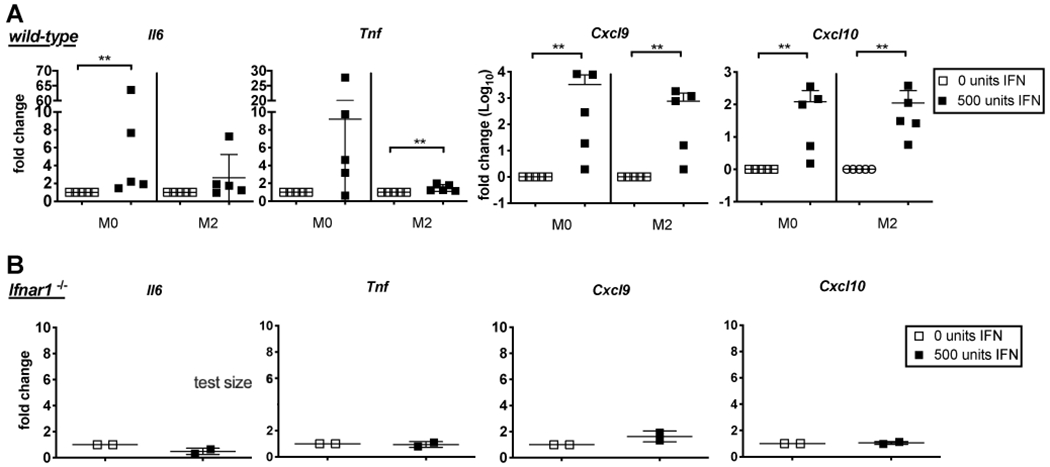
Type I IFN alone increases inflammatory transcript in uninfected macrophages. 500 units of IFN were added to WT B6 BMDMs that were left unpolarized as M0s (n=5) or polarized to an M2 subset (n=5) (A) or Ifnar1−/− BMDMs (n=2) left unpolarized as M0s (B). Il6, Tnf , Cxcl9, and Cxcl10 transcript data is displayed as fold change over untreated control groups (0 units IFN). In all cases, significance was determined using a Mann-Whitney test, ** p < 0.01
MCMV infection enhances macrophage phagocytosis in a STING-independent manner
Since activation of macrophages as well as active effects from cytomegalovirus proteins can lead to changes in phagocytosis and acidification of phagolysozomes (59), we next determined whether STING-deficient and Ifnar1−/− mice displayed altered phagocytosis and acidification after infection. BMDMs were again polarized to an M2-like state and then infected 24 hours later, this time with a GFP-expressing MCMV. Phagocytic capacity was assessed 24 hours later by engulfment of beads coated in a pH sensitive dye that fluoresces upon entering the phagosome. Infection of wild-type B6 macrophages by MCMV enhanced phagocytosis of the beads. However, loss of STING did not alter this effect (Figure 3A and B), indicating that the increased phagocytosis occurred in a STING-independent manner. Interestingly, the Ifnar1−/− macrophages displayed higher phagocytosis activity in the absence of infection and the addition of MCMV did not increase this capacity further. Importantly, the proportion of infected macrophages was unaffected by the loss of STING or IFNAR (Figure 3A and not shown). Thus, differences in infection and phagocytosis also cannot account for the changes in inflammatory cytokine production by B6, STING-deficient, and Ifnar1−/− BMDMs following MCMV infection. Collectively, these data show that MCMV infection enhances inflammatory cytokine production in a STING and type I IFN-dependent manner, but enhances macrophage phagocytosis independently of the STING pathway.
Figure 3.
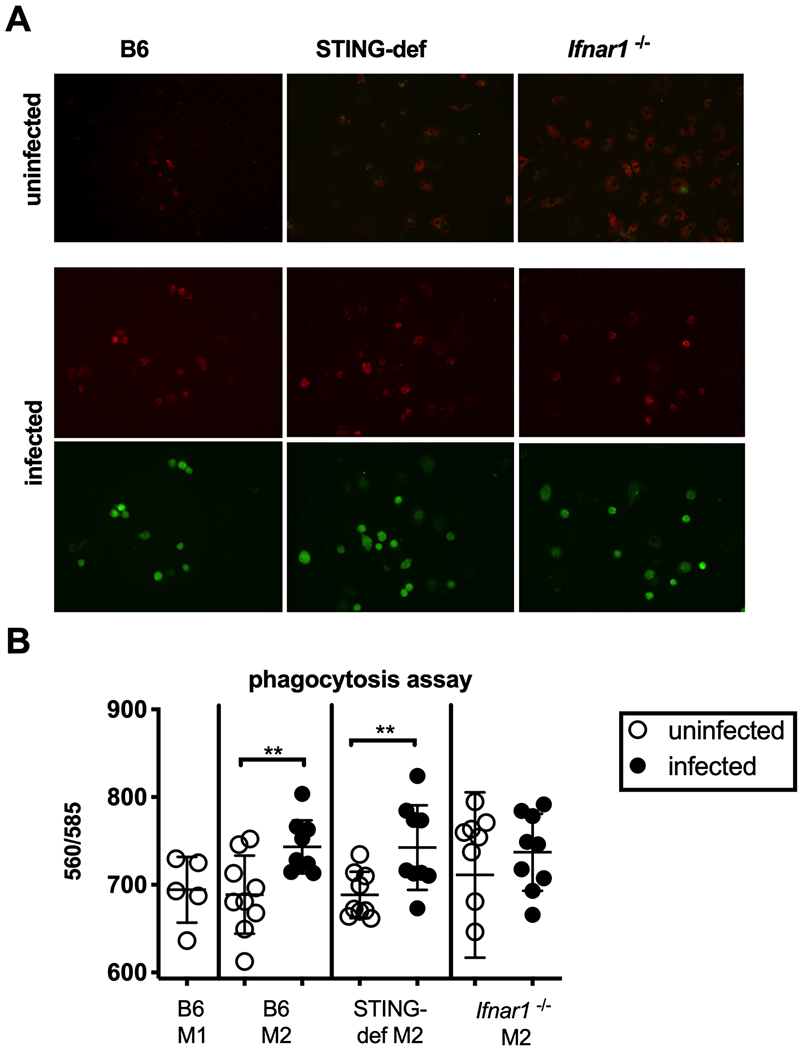
Increased phagocytosis after MCMV infection is STING independent. A) E. coli bioparticles with pH-sensitive red fluorescent dye show phagocytic potential of uninfected macrophages and macrophages infected with a GFP+ WT-MCMV. The assay was done in M2-polarized BMDMs from WT B6, Ifnar1−/−, and STING-def (from STINGgt/gt only) animals. Images are representative of three separate experiments. B) Red fluorescence was quantified from uninfected B6 M1 (2 experiments, n=5), B6 M2 (3 experiments with triplicate values, n=9), Ifnar1−/− M2 (3 experiments with triplicate values, n=9), and STING-def M2 (3 experiments with triplicate values n=9) BMDMs. In all cases, significance was determined using a two-tailed t test, ** p < 0.01
STING signaling is critical for the induction of anti-tumor immunity
Having shown that MCMV activated macrophage inflammatory cytokine production via STING and type I IFN, we wished to determine whether loss of these molecules would impact tumor growth delay in the B16-F0 model. To this end, we implanted B16-F0s subcutaneously into Tlr9−/−, Ifnar1−/− and STING-deficient animals. When tumors had reached 20 mm2, i.t. injections of PBS or spread defective ΔgL-MCMV were administered every other day for a total of three injections. The spread-defective ΔgL-MCMV was used to avoid any enhanced viral replication in the absence of critical viral sensing pathways. Importantly, we have previously shown that ΔgL-MCMV delayed tumor growth equally to wild-type MCMV(9). In agreement with our previous data, injection of ΔgL-MCMV delayed tumor growth (Figure 4A), significantly increased survival (Figure 4B), and significantly increased tumor doubling times (Supplemental Figure 3) when tumors were implanted in wild-type B6 mice. Consistent with our results from above, loss of TLR9 did not alter the ability of the virus to delay tumor growth, increase survival, or increase tumor doubling times. However, lack of STING completely prevented MCMV from delaying tumor growth (Figure 4A), increasing survival (Figure 4B), or increasing tumor doubling time (Supplemental Figure 3). Interestingly, Ifnar1−/− animals displayed some growth delay after injection of ΔgL-MCMV, although it was diminished compared to wild-type B6 animals (Median survival after IT-MCMV treatment: 18 days in B6 mice vs. 11 days in Ifnar1−/− mice). These data show that STING signaling in the host is necessary for MCMV to delay tumor growth in the B16-F0 model.
Figure 4.
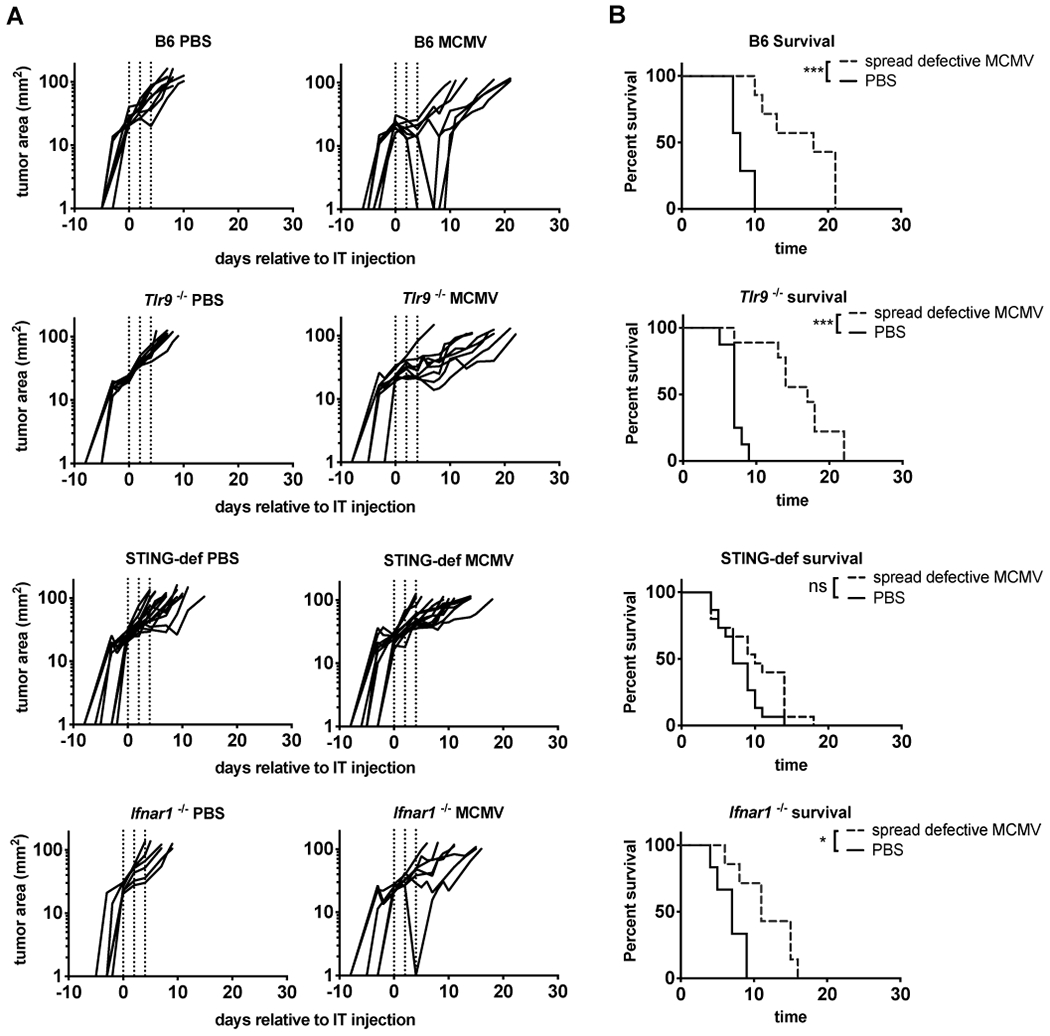
MCMV induced anti-tumor immunity is lost in STING-deficient animals. A) Tracings show B16-F0 tumor growth on a logarithmic scale after PBS or ΔgL-MCMV MCMV was intratumorally injected into WT B6 (PBS n=8, MCMV n=7), Tlr9−/− (PBS n=8, MCMV n=9), Ifnar1−/− (PBS n=6, MCMV n=7), and STING-def (PBS n=15, MCMV n=15, STINGgt/gt and STING−/− combined) animals. Dotted lines indicate the days in which animals received i.t. injections of ΔgL-MCMV or PBS. B) Kaplan-Meier plots show survival in days starting from the initial injection with ΔgL-MCMV or PBS to an endpoint of 100mm2. Any moribund animals sacrificed before the 100mm2 endpoint were excluded from survival curves. In all cases, significance was determined using the log-rank test and is denoted as follows: * p < 0.05, *** p <0.001, ns = not significant.
Type I IFN increases inflammatory cytokine expression in B16-F0 tumor cells
Since tumor growth delay was completely lost in STING-deficient mice, but only partially impaired in Ifnar1−/− mice, we speculated that type I IFN, which would still be produced in Ifnar1−/− mice, might directly affect the B16 tumor cells. To test this, we added 500 units of type I IFN to B16-F0 cells in vitro. Indeed, Cxcl9 and Cxcl10 were significantly increased in B16-F0 cells after exposure to type I IFN with an upward trend in Il6 and Tnf production as well (Figure 5A). Importantly, we observed no obvious impact on B16 growth or survival after 24 hours of type I IFN exposure (Figure 5B). Thus, we speculate that type I IFN can still alter the inflammatory environment of the tumor by directly affecting the B16-F0 tumor cells.
Figure 5.
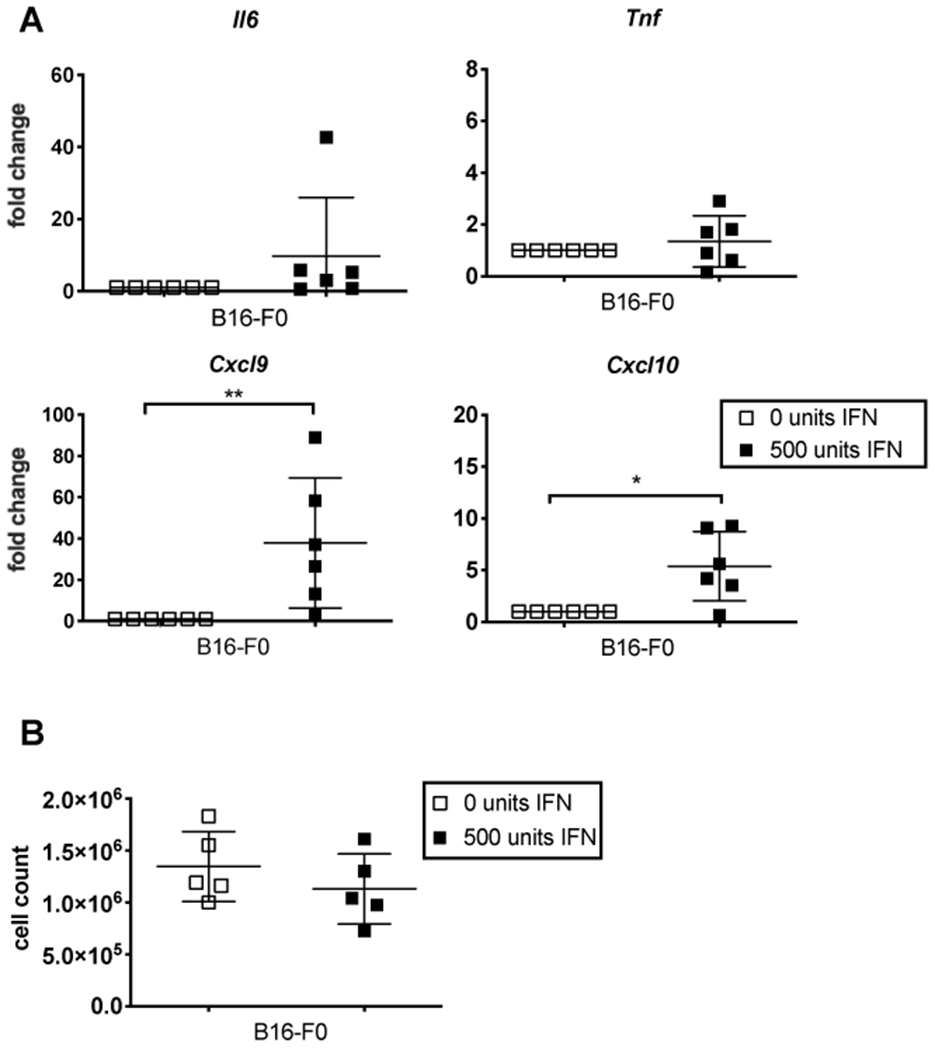
Type I IFN increases inflammatory cytokine production in B16-F0 cells in vitro. A) B16-F0 cells were cultured in the presence or absence of type I IFN (two experiments, n=5). Il6, Tnf, Cxcl9, and Cxcl10 transcript are shown as fold changed over untreated B16-F0s (0 units IFN). Significance was determined using a Mann-Whitney test. B) Cells shown in A were counted prior to processing. No significant difference between cell numbers from untreated and IFN treated cells was observed after 24 hours. In all cases, significance was determined using a two-tailed t-test and significant differences are denoted as follows: * p < 0.05, ** p < 0.01.
STING deficiency impairs the production of Cxcl10 by macrophages in the tumor and the recruitment of CD8+ T cells to the tumor.
Our data suggest that STING-induced type I IFN was important for inducing inflammatory cytokine production. To test whether loss of STING and IFNAR altered the cytokine environment in vivo, tumors were harvested one day after the final i.t. control PBS or MCMV injection (day 5 overall) from B6, STING-deficient, and Ifnar1−/− animals. Transcripts for Il6, Tnf, Ifng, and Cxcl9 were increased in all tumors after MCMV infection indicating their independence from STING or IFNAR in vivo (Figure 6A). In contrast, STING-def animals failed to upregulate transcripts for Cxcl10 indicating that production of this chemokine required STING in the host. Transcripts for type I IFN itself were difficult to detect consistently in these samples, perhaps reflecting the fact that samples were assayed 5 days after the 1st MCMV injection (Figure 6A and not shown). Importantly, macrophages were recruited normally to all tumors regardless of the presence or absence of STING (Figure 6B–C). This result is consistent with our recent study showing that MCMV recruits macrophages to the tumor via the viral chemokine MCK2, which should be independent of host-derived inflammatory cytokines and chemokines(9, 60–62). Next, we sorted CD11b+Gr-1+ monocytic phagocytes from pooled tumors implanted in wild-type or STING-deficient mice and assessed their expression of transcripts for Tnf, Ifna, and Cxcl10. In each case, transcript levels were further compared to those expressed by lymphocytes sorted from the same tumors. Notably, we did not observe significant differences in the expression of Ly6C or MHC-II between wild-type and STING-deficient monocytic phagocytes at this timepoint (data not shown), both of which were similar to data shown in our recent report (9). As in the total tumor (Figure 6A), sorted monocytic phagocytes expressed comparable levels of Tnf, irrespective of STING and Ifn transcripts were again inconsistently detectable (Figure 6D). However, Cxcl10 transcripts were significantly reduced in the absence of STING, implying a reduction in the amount of Type I IFN available in the tumor (Figure 6D). Interestingly, Cxcl10 transcription in STING-deficient monocytic phagocytes was still significantly increased over the tumor-infiltrating lymphocytes (CD4+, CD19+, NK1.1+) from the same animals, indicating some amount of production of this chemokine. PBS-treated tumors could not be used for comparison due to a general lack of leukocyte infiltration into the lesions.
Figure 6.
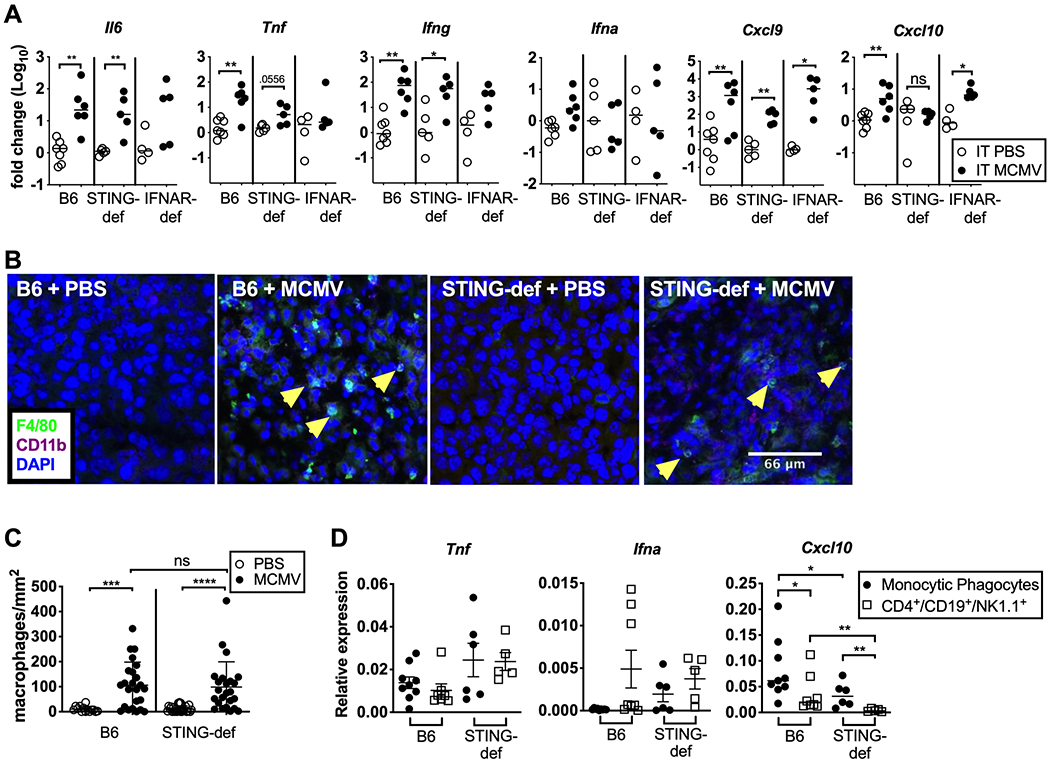
STING signaling is required for Ifna and Cxcl10 production and recruitment of CD8+ T cells in vivo. A) Intratumoral injections of PBS or ΔgL-MCMV were administered every other day for a total of three injections once tumors reached around 20mm2 in WT B6 (PBS n=7, ΔgL-MCMV n=6), Ifnar1−/− (PBS n=6, ΔgL-MCMV n=5), and STING-def (PBS n=4, ΔgL-MCMV n=5, from STINGgt/gt only) animals. Tumor homogenate was taken the day after the third injection (day 5). Fold change in inflammatory cytokines and chemokines for each individual mouse are shown relative to the average value from PBS-treated mice in each experiment (2 independent experiments for each genotype). Statistics were performed using a Mann-Whitney test. Additional tumors were taken on day 5 and imaged for the presence of macrophages (B). C) B6 + PBS (n=2 tumors, 6–8 images per tumor), B6 + MCMV (n=3 tumors, 8 images per tumor), STING-def +PBS (n=3 tumors, 8 images per tumor, STINGgt/gt only), STING-def + MCMV(n=3 tumors, 8 images per tumor, STINGgt/gt only) tumor images were quantified to generate macrophage numbers per mm2 image by counting F4/80+CD11b+ cells with clear DAPI staining. D) Tumors treated with MCMV from B6 (pooled tumors, n=3 repeats) or STING-def (pooled tumors, n=2 repeats) animals were taken the day after the third injection and homogenized. Monocytic phagocytes (CD11b+Gr-1+) or bulk lymphocytes (CD4+CD19+NK1.1+) were sorted from tumor homogenate. Expression of the indicated transcripts is shown relative to PBS using technical replicates (n=3) from each experiment. In all cases, significance was determined using a Mann-Whitney test and is denoted as follows: * p < 0.05, ** p < 0.01, *** p <0.001, **** p <0.0001.
We have previously shown that i.t. MCMV infection results in the accumulation of CD8+ T cells in the tumor and that these T cells are involved in the growth delay (8, 63). Thus, the lack of Cxcl10 expression in the tumor and by tumor-associated macrophages led us to investigate whether CD8+ T cells were recruited to the tumor normally in STING-deficient mice. Indeed, there was a significant reduction in the number of CD8+ T cells recruited to the tumors in STING-deficient mice (Figure 7A–B). These data imply that STING deficiency impairs inflammatory cytokine and chemokine production, resulting in a failure to engage T cells after MCMV infection.
Figure 7.
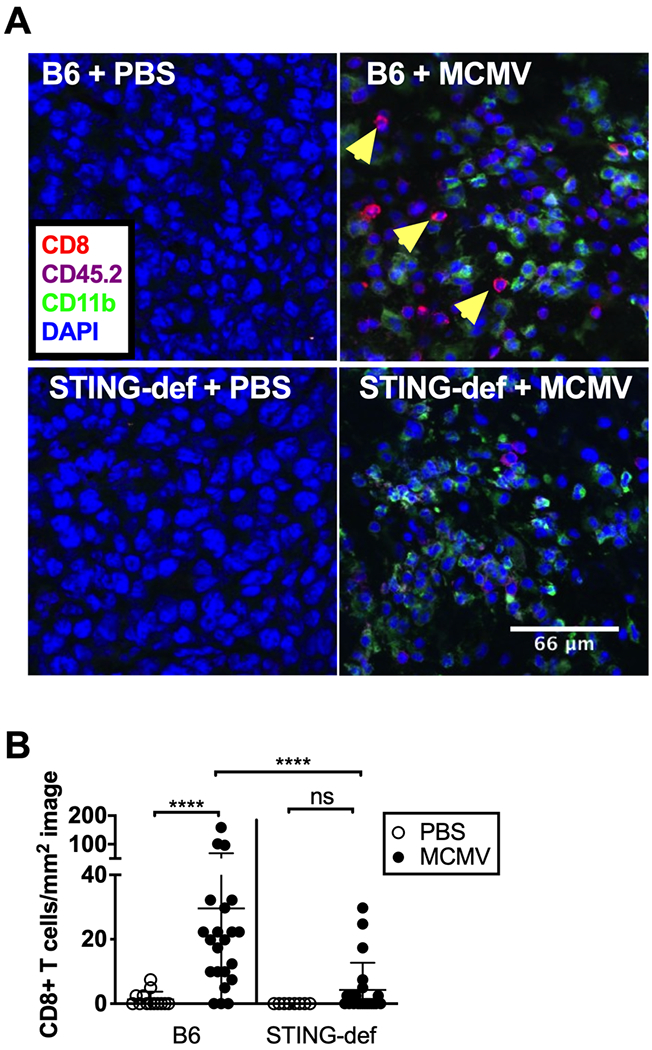
CD8+ T cell recruitment is significantly impaired in STING-deficient tumors A) Tumors were taken on day 5 and imaged for the presence of CD8+ T cells. B) B6 + PBS (n=2 tumors, 5–8 images per tumor), B6 + MCMV (n=3 tumors, 6–8 images per tumor), STING-def +PBS (n=1 tumor, 8 images per tumor, from STINGgt/gt only), STING-def + MCMV(n=3 tumors, 6–8 images per tumor, from STINGgt/gt only) tumor images were quantified to generate CD8 numbers per mm2 image by counting CD8+CD45.2+CD11b- cells with clear DAPI staining. In all cases, significance was determined using a Mann-Whitney test, **** p <0.0001.
Infected wild-type macrophages alone are sufficient to promote anti-tumor responses in STING-deficient animals
To this point, our data show that STING is necessary to induce inflammatory cytokine production by infected macrophages and that STING deficiency prevents MCMV from delaying tumor growth. To definitively test whether STING signaling in MCMV infected macrophages was sufficient to induce tumor growth delay, we infected M2-polarized wild-type or STING-deficient macrophages with ΔgL-MCMV and injected these macrophages into tumors growing in STING-deficient mice. Since ΔgL-MCMV can produce only non-infectious particles in these infected macrophages(52), this protocol restricts infection to the injected cells. Moreover, since the recipients are STING-deficient, any particles engulfed by host macrophages will be unable to activate STING. As a comparison, mice received uninfected M2 macrophages. In all cases, the number of macrophages injected was based on the counts of macrophages obtained from histological images which were then extrapolated to the entire tumor volume (see materials and methods). Critically, transfer of uninfected macrophages did not significantly alter tumor growth (median survival = 7 days) compared to tumors injected with PBS alone (median survival = 8 days, compare Figure 8B to Figure 4A). Strikingly however, a single injection of infected wild-type macrophages delayed the growth of tumors in STING-deficient animals (Figure 8A) and significantly increased survival (Figure 8B) compared to injection of uninfected B6 macrophages. Importantly, this growth delay was lost when tumors received STING-deficient macrophages that were infected by MCMV (Figure 8A and 8B). Moreover, while transfer of infected wild-type macrophages was sufficient to recruit CD8+ T cells into the tumor, this effect was lost when the infected macrophages lacked STING (Figure 8C and 8D). These data show that MCMV infection of macrophages is sufficient to induce CD8+ T cell recruitment and anti-tumor effects in the B16-F0 model via STING signaling and that infection of tumor-associated macrophages alone can tilt the tumor immune environment toward tumor control.
Figure 8.
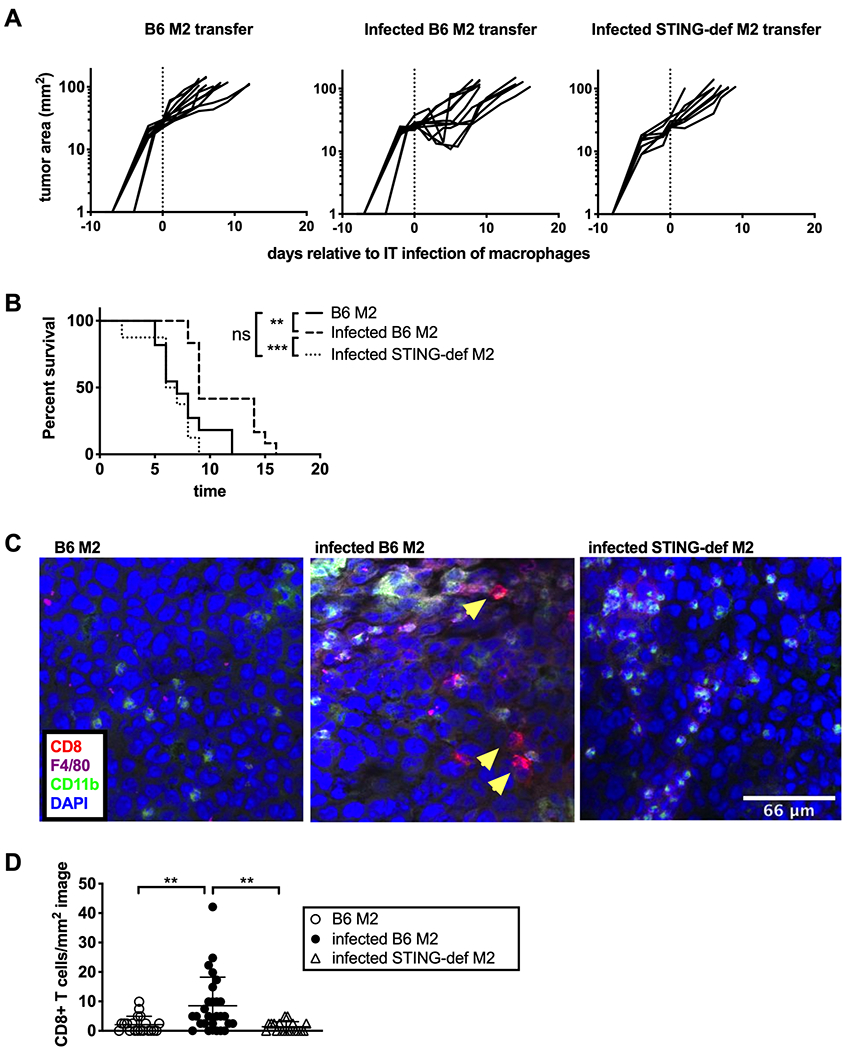
Infected WT macrophages are sufficient to cause tumor growth delay in STING-def animals. A) ΔgL-MCMV infected B6, uninfected B6, or infected STING-def M2-polarized macrophages were transferred intratumorally into established B16-F0 tumors in STING-def (from STINGgt/gt only) animals when the lesions reached around 20mm2. Tracings show tumor growth after B6 M2 transfer (n=11), infected B6 M2 transfer (n=12), or infected STING-def M2 transfer (n=8) on a logarithmic scale after a single injection of macrophages as indicated by the dotted line. B) Kaplan-Meier plots show survival in days following the macrophage transfer to an endpoint of 100mm2. Significance was determined using the log-rank test. C) Additionally, tumors were harvested on day 5 and imaged to assess the CD8+ T cell population. D) Images from tumors receiving infected M2-polarized B6 macrophages (n=2 tumors, 13–14 images per tumor), uninfected M2-polarized B6 macrophages (n=2 tumors, 10–11 images per tumor), and infected M2-polarized STING-deficient macrophages(n=2 tumors, 9 images per tumor) were quantified to generate CD8+ T cell numbers per mm2 image by counting CD8+CD11b- cells with clear DAPI staining. In all cases, significance for all statistics is denoted as follows: ** p < 0.01, *** p <0.001, ns = not significant.
Discussion
CMV infects most cells in the body(64, 65), including tumor cells, and it is has been identified in multiple human tumors(16–20), although the precise cells infected in the tumor and the net effect of the virus are still unknown. It has been hypothesized that the virus may lead to increased tumor aggression(16, 43, 44) which can either enhance or hinder targeted therapeutic approaches(66, 67). Additionally, CMV itself has shown promise as a potential vaccine vector(8, 34–41). Thus, it becomes increasingly important that we understand how the virus interacts with its surroundings. Our data show that an active WT-MCMV infection can engage anti-tumor immune responses by recruiting macrophages to the tumor(9) and acting as a STING agonist. As HCMV is also known to both recruit myeloid cells to the site of infection and activate STING in myeloid cells, this mechanism may also be translationally relevant(47, 68–70).
Despite some successful preclinical studies, STING agonists have not yet been successful in the clinic. The first STING agonist to go through clinical trials, DMXAA, failed due to low specificity for human STING(71). Testing of newer compounds is still under way, alone or in combination with checkpoint inhibitors(72–75). However, the conditions under which such agonists might successfully promote anti-tumor immunity are unclear. Indeed, multiple tumors mutate the STING pathway and the immune environment in the tumor varies greatly, ranging from immune deserts that lack significant immune cell infiltration, to immune replete tumors. Given our previous work showing that intratumoral MCMV therapy depended on viral recruitment of myeloid cells to the tumor(9), our data suggest that the composition of the immune cells in the tumor may greatly influence the success of STING agonists.
It is worth noting that pathogen-based STING agonists have been explored in some settings as well. In fact, inactivated modified Vaccinia Virus Ankara (iMVA) has shown promise as a STING agonist in both murine and human tumor models(7). Although many groups are focused on using vaccinia as an oncolytic agent, inactivating vaccinia removes any effect of viral immune modulatory mechanisms, thereby enhancing the immune stimulation and the anti-tumor effect(7). The ability of both CMV and vaccinia to target the immune response and not the tumor directly may be particularly valuable, because it will not be impacted by tumor-associated mutations within immune stimulatory pathways. Moreover, these approaches may provide synergy with other immune therapies. We previously showed that intratumoral MCMV synergized with a PD-L1 blockade to clear B16-F0 tumors(8), which are normally resistant to blockade of the PD-1 pathway(76). Likewise, the Deng laboratory demonstrated that iMVA was synergistic with blockade of PD-1, PD-L1 or CTLA-4(7). These data fit with other work showing that STING agonists can enhance the effect of checkpoint inhibition (77–85) with one group reporting that STING can sensitize tumors otherwise resistant to PD-1 blockade(86).
The data shown here indicate that STING-sufficient macrophages infected with ΔgL-MCMV produced an anti-tumor response in STING-deficient animals. Moreover, our previous work revealed that the recruitment of myeloid cells via the MCMV-encoded chemokine MCK2 was critical for MCMV to induce anti-tumor immunity(9). Thus, STING in the tumor environment was not sufficient without monocytic phagocytes in the tumor and we infer that the recruitment of myeloid cells by the virus primed the tumor to respond to a STING agonist. Although we explored the impact of infected macrophages on tumor growth in the current study (Figure 8), it is important to note that other phagocytic cells may also contribute to the anti-tumor effects after direct injection of the virus and STING may be necessary in these cells as well. Future work will explore these possibilities.
Other groups have shown that STING promotes anti-tumor responses through production of type I IFN in the immune compartment(87). Our data show that inflammatory cytokine expression was impaired in Ifnar1−/− BMDMs and that type I IFN alone was sufficient to activate chemokine and cytokine production by macrophages and the tumor cells themselves in vitro. Thus, we hypothesize that the effect of type I IFN on the tumor cells is mediating the partial response to therapy in the IFNAR KO mice (Figure 4). However, the complete lack of anti-tumor effects in the absence of STING may argue that STING expression by tumor-associated myeloid cells (or indeed the presence or absence of myeloid cells in the tumor) may be critical for determining whether type I IFN is produced in sufficient quantities after STING activation. Further studies are therefore warranted with STING agonists in the presence or absence of tumor-associated myeloid cells and when the tumor cells themselves express or lack the type I IFN receptor to precisely define the sources and effects of IFN in the tumor.
Interestingly, our data also showed a lack of Cxcl10 transcript in tumors and sorted tumor-associated macrophages in the absence of STING, and an impaired ability of these tumors to recruit CD8+ T cells after MCMV infection. Our previous work has shown that CD8+ T cells are necessary for full therapeutic effect of the virus(8, 9). Therefore, a major long-term goal is to understand the interactions between macrophages and CD8+ T cells in this system. The MCMV infection in this model resulted in rapid tumor growth delay that was typically evident within a few days of viral injection. Such kinetics are not consistent with new priming of tumor-specific CD8+ T cells as a result of activation of professional APCs, but rather of reactivation, recruitment, or retention of anti-tumor T cells that were primed previously when the tumor was injected(88, 89). Therefore, it is possible that STING contributes in three places: (1) STING signaling and type I IFN may serve to induce chemokines that attract pre-existing CD8+ T cells into the tumor where they begin to kill their targets; The CXCL10/CXCR3 axis is an obvious target and we would hypothesize that CXCR3-deficient mice will not receive the benefit from intratumoral MCMV infection; (2) type I IFN may support T cell function upon arrival in the tumor by altering the concentrations of inflammatory and suppressive cytokines and/or improving antigen presentation and APC activation within the tumor environment; (3) STING may contribute to the initial priming of tumor-specific T cells at the tumor after the tumor was implanted, but prior to MCMV infection. These mechanisms are not mutually exclusive and future work will be required to tease apart the mechanisms that govern the interplay between macrophages, CD8+ T cells and the tumor in this system.
In the context of how CMV alters the tumor environment, it is important to note that we only determined the effects of an active MCMV infection in a tumor model in which macrophages were the dominant cell type infected. With multiple groups showing that latent HCMV in the tumor may promote tumor aggression, future work will need to investigate the effects of active or latent CMV infection in multiple models. We hypothesize that CMV may promote different effects depending on the viral activity, tropism, and tumor location in the body(90). We further hypothesize that myeloid cell recruitment and STING activation will only be evident during an active infection, and possibly only when hematopoietic cells are present or recruited and can be infected. In contrast, when HCMV is hypothesized to promote tumor aggressiveness, it is generally thought to be caused by a latent infection or a relatively inactive virus, and it is not clear which cells in the tumor might be infected. This raises the possibility that approaches to reactivate latent virus already in a tumor may lead to STING activation and the promotion of anti-tumor immunity. In addition, because MCMV can increase myeloid cells and CD8+ T cell numbers in the tumor, any infection or reactivation of virus in the tumor could fundamentally alter immunologically “cold” tumors, possibly enabling efficacy of additional immune therapies. In fact, our lab has shown previously that PD-L1 blockade combined with intratumoral MCMV cleared up to 70% of established B16-F0 melanomas, a response rate much greater than either therapy alone(8). However, it must be noted that inflammation in the local tissue may be problematic in some tumors such as glioblastoma.
In summary, our data suggest that an active CMV infection may profoundly alter the tumor microenvironment by recruiting myeloid cells and activating the STING pathway, thereby engaging the adaptive immune system. These basic mechanistic studies raise the question of whether hematopoietic cell infiltration will be critical to the function of STING therapies and shed light on one way that CMV can alter the tumor microenvironment.
Supplementary Material
Key Points:
1) Intratumoral infection of B16 melanomas by MCMV is sensed by STING
2) MCMV activation of STING in host cells induces anti-tumor immunity in B16 tumors
3) MCMV-infected macrophages delay tumor growth and recruit CD8 T cells via STING
Acknowledgements
Immunofluorescent images were captured at the Sidney Kimmel Cancer Center Bioimaging Facility (NCI 5 P30 CA-56036) with assistance from Dr. Maria Yolanda Covarrubias. Additional microscopy assistance was provided by Tiago Monteiro-Brás.
Financial support: This work was supported by grants from the American Cancer Society RSG-15-184-01 awarded to C.M.S., PA Department of Health Research Formula Funds SAP 4100072566, NIA/NIH RO1 AG048602, NIAID/NIH AI110457, and NIAID/NIH AI065544, all awarded to L.J.S, NIAID/NIH F32AI129352 awarded to E.W., and NIAD T32 AI134646 awarded to B.M.
References
- 1.Tu D-G, Chang W-W, Lin S-T, Kuo C-Y, Tsao Y-T, and Lee C-H. 2016. Salmonella inhibits tumor angiogenesis by downregulation of vascular endothelial growth factor. Oncotarget 7: 37513–37523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vola M, Mónaco A, Bascuas T, Rimsky G, Agorio CI, Chabalgoity JA, and Moreno M. 2018. TLR7 agonist in combination with Salmonella as an effective antimelanoma immunotherapy. Immunotherapy 10: 665–679. [DOI] [PubMed] [Google Scholar]
- 3.Quetglas JI, Labiano S, Aznar MÁ, Bolaños E, Azpilikueta A, Rodriguez I, Casales E, Sánchez-Paulete AR, Segura V, Smerdou C, and Melero I. 2015. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res . 3: 449–454. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Paulete AR, Teijeira A, Quetglas JI, Rodriguez-Ruiz ME, Sanchez-Arraez A, Labiano S, Etxeberria I, Azpilikueta A, Bolaños E, Ballesteros-Briones MC, Casares N, Quezada SA, Berraondo P, Sancho D, Smerdou C, and Melero I. 2018. Intratumoral immunotherapy with XCL1 and sFlt3L encoded in recombinant Semliki Forest Virus-derived vectors fosters dendritic cell-mediated T cell cross-priming. Cancer Res. canres.0933.2018. [DOI] [PubMed] [Google Scholar]
- 5.Baird JR, Byrne KT, Lizotte PH, Toraya-Brown S, Scarlett UK, Alexander MP, Sheen MR, Fox BA, Bzik DJ, Bosenberg M, Mullins DW, Turk MJ, and Fiering S. 2013. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J. Immunol. Baltim. Md 1950 190: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox BA, Sanders KL, Chen S, and Bzik DJ. 2013. Targeting tumors with nonreplicating Toxoplasma gondii uracil auxotroph vaccines. Trends Parasitol. 29: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai P, Wang W, Yang N, Serna-Tamayo C, Ricca JM, Zamarin D, Shuman S, Merghoub T, Wolchok JD, and Deng L. 2017. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci. Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erkes DA, Xu G, Daskalakis C, Zurbach KA, Wilski NA, Moghbeli T, Hill AB, and Snyder CM. 2016. Intratumoral Infection with Murine Cytomegalovirus Synergizes with PD-L1 Blockade to Clear Melanoma Lesions and Induce Long-term Immunity. Mol. Ther. J. Am. Soc. Gene Ther 24: 1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilski NA, Del Casale C, Purwin TJ, Aplin AE, and Snyder CM. 2019. Murine cytomegalovirus infection of melanoma lesions delays tumor growth by recruiting and re-polarizing monocytic phagocytes in the tumor. J. Virol . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erlach KC, Böhm V, Seckert CK, Reddehase MJ, and Podlech J. 2006. Lymphoma Cell Apoptosis in the Liver Induced by Distant Murine Cytomegalovirus Infection. J. Virol 80: 4801–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlach KC, Podlech J, Rojan A, and Reddehase MJ. 2002. Tumor Control in a Model of Bone Marrow Transplantation and Acute Liver-Infiltrating B-Cell Lymphoma: an Unpredicted Novel Function of Cytomegalovirus. J. Virol 76: 2857–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erlach KC, Reddehase MJ, and Podlech J. 2015. Mechanism of tumor remission by cytomegalovirus in a murine lymphoma model: evidence for involvement of virally induced cellular interleukin-15. Med. Microbiol. Immunol. (Berl.) 204: 355–366. [DOI] [PubMed] [Google Scholar]
- 13.Stern J, Miller G, Li X, and Saxena D. 2019. Virome and bacteriome: two sides of the same coin. Curr. Opin. Virol 37: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, and Franceschi S. 2016. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob. Health 4: e609–e616. [DOI] [PubMed] [Google Scholar]
- 15.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, Scheet P, Xu H, Hanash SM, Feng L, Burks JK, Do K-A, Peterson CB, Nejman D, Tzeng C-WD, Kim MP, Sears CL, Ajami N, Petrosino J, Wood LD, Maitra A, Straussman R, Katz M, White JR, Jenq R, Wargo J, and McAllister F. 2019. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178: 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rådestad AF, Estekizadeh A, Cui HL, Kostopoulou ON, Davoudi B, Hirschberg AL, Carlson J, Rahbar A, and Söderberg-Naucler C. 2018. Impact of Human Cytomegalovirus Infection and its Immune Response on Survival of Patients with Ovarian Cancer. Transl. Oncol 11: 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zafiropoulos A, Tsentelierou E, Billiri K, and Spandidos DA. 2003. Human herpes viruses in non-melanoma skin cancers. Cancer Lett. 198: 77–81. [DOI] [PubMed] [Google Scholar]
- 18.Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, and Cobbs CS. 2002. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet Lond. Engl 360: 1557–1563. [DOI] [PubMed] [Google Scholar]
- 19.Price RL, Bingmer K, Harkins L, Iwenofu OH, Kwon C-H, Cook C, Pelloski C, and Chiocca EA. 2012. Cytomegalovirus Infection Leads to Pleomorphic Rhabdomyosarcomas in Trp53+/− Mice. Cancer Res. 72: 5669–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, and Britt WJ. 2002. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res. 62: 3347–3350. [PubMed] [Google Scholar]
- 21.Herbein G 2018. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bigley AB, Rezvani K, Shah N, Sekine T, Balneger N, Pistillo M, Agha N, Kunz H, O’Connor DP, Bollard CM, and Simpson RJ. 2016. Latent cytomegalovirus infection enhances anti-tumour cytotoxicity through accumulation of NKG2C+ NK cells in healthy humans. Clin. Exp. Immunol 185: 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez-Valentin M, Gras Navarro A, Rahman AM, Kumar S, Retière C, Ulvestad E, Kristensen V, Lund-Johansen M, Lie BA, Enger PØ, Njølstad G, Kristoffersen E, Lie SA, and Chekenya M. 2016. Identification of a Natural Killer Cell Receptor Allele That Prolongs Survival of Cytomegalovirus-Positive Glioblastoma Patients. Cancer Res. 76: 5326–5336. [DOI] [PubMed] [Google Scholar]
- 24.Hansen SG, Jr MP, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Früh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, and Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M, Lifson JD, Nelson JA, Jarvis MA, and Picker LJ. 2009. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med 15: 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Früh K, and Picker LJ. 2013. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340: 1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, Reed JS, Gilbride RM, Ainslie E, Morrow DW, Ford JC, Selseth AN, Pathak R, Malouli D, Legasse AW, Axthelm MK, Nelson JA, Gillespie GM, Walters LC, Brackenridge S, Sharpe HR, López CA, Früh K, Korber BT, McMichael AJ, Gnanakaran S, Sacha JB, and Picker LJ. 2016. Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science 351: 714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Lifson JD, and Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuda Y, Caposio P, Parkins CJ, Botto S, Messaoudi I, Cicin-Sain L, Feldmann H, and Jarvis MA. 2011. A Replicating Cytomegalovirus-Based Vaccine Encoding a Single Ebola Virus Nucleoprotein CTL Epitope Confers Protection against Ebola Virus. PLoS Negl. Trop. Dis 5: e1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuda Y, Parkins CJ, Caposio P, Feldmann F, Botto S, Ball S, Messaoudi I, Cicin-Sain L, Feldmann H, and Jarvis MA. 2015. A cytomegalovirus-based vaccine provides long-lasting protection against lethal Ebola virus challenge after a single dose. Vaccine 33: 2261–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marzi A, Murphy AA, Feldmann F, Parkins CJ, Haddock E, Hanley PW, Emery MJ, Engelmann F, Messaoudi I, Feldmann H, and Jarvis MA. 2016. Cytomegalovirus-based vaccine expressing Ebola virus glycoprotein protects nonhuman primates from Ebola virus infection. Sci. Rep 6: 21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, Gilbride RM, Hughes CM, Ventura AB, Ainslie E, Randall KT, Selseth AN, Rundstrom P, Herlache L, Lewis MS, Park H, Planer SL, Turner JM, Fischer M, Armstrong C, Zweig RC, Valvo J, Braun JM, Shankar S, Lu L, Sylwester AW, Legasse AW, Messerle M, Jarvis MA, Amon LM, Aderem A, Alter G, Laddy DJ, Stone M, Bonavia A, Evans TG, Axthelm MK, Früh K, Edlefsen PT, and Picker LJ. 2018. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med 24: 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beverley PCL, Ruzsics Z, Hey A, Hutchings C, Boos S, Bolinger B, Marchi E, O’Hara G, Klenerman P, Koszinowski UH, and Tchilian EZ. 2014. A Novel Murine Cytomegalovirus Vaccine Vector Protects against Mycobacterium tuberculosis. J. Immunol 193: 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klyushnenkova EN, Kouiavskaia DV, Parkins CJ, Caposio P, Botto S, Alexander RB, and Jarvis MA. 2012. A cytomegalovirus-based vaccine expressing a single tumor-specific CD8+ T cell epitope delays tumor growth in a murine model of prostate cancer. J. Immunother. Hagerstown Md 1997 35: 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dekhtiarenko I, Ratts RB, Blatnik R, Lee LN, Fischer S, Borkner L, Oduro JD, Marandu TF, Hoppe S, Ruzsics Z, Sonnemann JK, Mansouri M, Meyer C, Lemmermann NAW, Holtappels R, Arens R, Klenerman P, Früh K, Reddehase MJ, Riemer AB, and Cicin-Sain L. 2016. Peptide Processing Is Critical for T-Cell Memory Inflation and May Be Optimized to Improve Immune Protection by CMV-Based Vaccine Vectors. PLoS Pathog. 12: e1006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beyranvand Nejad E, Ratts RB, Panagioti E, Meyer C, Oduro JD, Cicin-Sain L, Früh K, van der Burg SH, and Arens R. 2019. Demarcated thresholds of tumor-specific CD8 T cells elicited by MCMV-based vaccine vectors provide robust correlates of protection. J. Immunother. Cancer 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grenier JM, Yeung ST, Qiu Z, Jellison ER, and Khanna KM. 2018. Combining Adoptive Cell Therapy with Cytomegalovirus-Based Vaccine Is Protective against Solid Skin Tumors. Front. Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tršan T, Vuković K, Filipović P, Brizić AL, Lemmermann NAW, Schober K, Busch DH, Britt WJ, Messerle M, Krmpotić A, and Jonjić S. 2017. Cytomegalovirus vector expressing RAE-1γ induces enhanced anti-tumor capacity of murine CD8+ T cells. Eur. J. Immunol 47: 1354–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu G, Smith T, Grey F, and Hill AB. 2013. Cytomegalovirus-based cancer vaccines expressing TRP2 induce rejection of melanoma in mice. Biochem. Biophys. Res. Commun 437: 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu Z, Huang H, Grenier JM, Perez OA, Smilowitz HM, Adler B, and Khanna KM. 2015. Cytomegalovirus-Based Vaccine Expressing a Modified Tumor Antigen Induces Potent Tumor-Specific CD8(+) T-cell Response and Protects Mice from Melanoma. Cancer Immunol. Res 3: 536–546. [DOI] [PubMed] [Google Scholar]
- 41.Benonisson H, Sow HS, Breukel C, Claassens JWC, Brouwers C, Linssen MM, Redeker A, Fransen MF, van Hall T, Ossendorp F, Arens R, and Verbeek S. 2018. FcγRI expression on macrophages is required for antibody-mediated tumor protection by cytomegalovirus-based vaccines. Oncotarget 9: 29392–29402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Berg SPH, Pardieck IN, Lanfermeijer J, Sauce D, Klenerman P, van Baarle D, and Arens R. 2019. The hallmarks of CMV-specific CD8 T-cell differentiation. Med. Microbiol. Immunol. (Berl.) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krenzlin H, Behera P, Lorenz V, Passaro C, Zdioruk M, Nowicki MO, Grauwet K, Zhang H, Skubal M, Ito H, Zane R, Gutknecht M, Griessl MB, Ricklefs F, Ding L, Peled S, Rooj A, James CD, Cobbs CS, Cook CH, Chiocca EA, and Lawler SE. 2019. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J. Clin. Invest 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z, Tang X, Meng G, Benesch MGK, Mackova M, Belon AP, Serrano-Lomelin J, Goping IS, Brindley DN, and Hemmings DG. 2019. Latent Cytomegalovirus Infection in Female Mice Increases Breast Cancer Metastasis. Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan G, Bivins-Smith ER, Smith MS, Smith PM, and Yurochko AD. 2008. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J. Immunol. Baltim. Md 1950 181: 698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan G, Bivins-Smith ER, Smith MS, and Yurochko AD. 2009. NF-κB and Phosphatidylinositol 3-Kinase Activity Mediates the HCMV-Induced Atypical M1/M2 Polarization of Monocytes. Virus Res. 144: 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paijo J, Döring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, and Kalinke U. 2016. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLOS Pathog. 12: e1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moltedo B, Li W, Yount JS, and Moran TM. 2011. Unique Type I Interferon Responses Determine the Functional Fate of Migratory Lung Dendritic Cells during Influenza Virus Infection. PLOS Pathog. 7: e1002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, and Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408: 740–745. [DOI] [PubMed] [Google Scholar]
- 50.Zurbach KA, Moghbeli T, and Snyder CM. 2014. Resolving the titer of murine cytomegalovirus by plaque assay using the M2–10B4 cell line and a low viscosity overlay. Virol. J 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Overwijk WW, and Restifo NP. 2001. B16 as a Mouse Model for Human Melanoma. Curr. Protoc. Immunol. Ed. John E Coligan Al CHAPTER: Unit-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snyder CM, Allan JE, Bonnett EL, Doom CM, and Hill AB. 2010. Cross-Presentation of a Spread-Defective MCMV Is Sufficient to Prime the Majority of Virus-Specific CD8+ T Cells. PLoS ONE 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krug A, French AR, Barchet W, Fischer JAA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, and Colonna M. 2004. TLR9-Dependent Recognition of MCMV by IPC and DC Generates Coordinated Cytokine Responses that Activate Antiviral NK Cell Function. Immunity 21: 107–119. [DOI] [PubMed] [Google Scholar]
- 55.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, and Beutler B. 2004. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A 101: 3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lio C-WJ, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, and Sharma S. 2016. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J. Virol 90: 7789–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X-D, Wu J, Gao D, Wang H, Sun L, and Chen ZJ. 2013. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 341: 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishikawa H, Ma Z, and Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461: 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaworowski A, Cheng W-J, Westhorpe CL, Abendroth A, Crowe SM, and Slobedman B. 2009. Enhanced monocyte Fc phagocytosis by a homologue of interleukin-10 encoded by human cytomegalovirus. Virology 391: 20–24. [DOI] [PubMed] [Google Scholar]
- 60.Daley-Bauer LP, Roback LJ, Wynn GM, and Mocarski ES. 2014. Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 15: 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saederup N, Aguirre SA, Sparer TE, Bouley DM, and Mocarski ES. 2001. Murine Cytomegalovirus CC Chemokine Homolog MCK-2 (m131-129) Is a Determinant of Dissemination That Increases Inflammation at Initial Sites of Infection. J. Virol 75: 9966–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagner FM, Brizic I, Prager A, Trsan T, Arapovic M, Lemmermann NAW, Podlech J, Reddehase MJ, Lemnitzer F, Bosse JB, Gimpfl M, Marcinowski L, MacDonald M, Adler H, Koszinowski UH, and Adler B. 2013. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 9: e1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Erkes DA, Smith CJ, Wilski NA, Caldeira-Dantas S, Mohgbeli T, and Snyder CM. 2017. Virus-Specific CD8+ T Cells Infiltrate Melanoma Lesions and Retain Function Independently of PD-1 Expression. J. Immunol. Baltim. Md 1950 198: 2979–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffiths P, Baraniak I, and Reeves M. 2015. The pathogenesis of human cytomegalovirus. J. Pathol 235: 288–297. [DOI] [PubMed] [Google Scholar]
- 65.Reddehase MJ, and Lemmermann NAW. 2019. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. (Berl.) . [DOI] [PubMed] [Google Scholar]
- 66.Söderberg-Nauclér C, Rahbar A, and Stragliotto G. 2013. Survival in patients with glioblastoma receiving valganciclovir. N. Engl. J. Med 369: 985–986. [DOI] [PubMed] [Google Scholar]
- 67.Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, Bjurman B, Tammik C, Skarman P, Peredo I, and Söderberg-Nauclér C. 2013. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: a randomized, double-blind, hypothesis-generating study. Int. J. Cancer 133: 1204–1213. [DOI] [PubMed] [Google Scholar]
- 68.Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, and Sinclair JH. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol 72 ( Pt 9): 2059–2064. [DOI] [PubMed] [Google Scholar]
- 69.Smith MS, Bentz GL, Alexander JS, and Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol 78: 4444–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ibanez CE, Schrier R, Ghazal P, Wiley C, and Nelson JA. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol 65: 6581–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lara PN, Douillard J-Y, Nakagawa K, von Pawel J, McKeage MJ, Albert I, Losonczy G, Reck M, Heo D-S, Fan X, Fandi A, and Scagliotti G. 2011. Randomized Phase III Placebo-Controlled Trial of Carboplatin and Paclitaxel With or Without the Vascular Disrupting Agent Vadimezan (ASA404) in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol 29: 2965–2971. [DOI] [PubMed] [Google Scholar]
- 72.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, and McWhirter SM. 2018. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity. Cell Rep. 25: 3074–3085.e5. [DOI] [PubMed] [Google Scholar]
- 73.Study of the Safety and Efficacy of MIW815 With PDR001 to Patients With Advanced/Metastatic Solid Tumors or Lymphomas - Full Text View - ClinicalTrials.gov.
- 74.Safety and Efficacy of MIW815 (ADU-S100) +/− Ipilimumab in Patients With Advanced/Metastatic Solid Tumors or Lymphomas - Full Text View - ClinicalTrials.gov.
- 75.Study of MK-1454 Alone or in Combination With Pembrolizumab in Participants With Advanced/Metastatic Solid Tumors or Lymphomas (MK-1454-001) - Full Text View - ClinicalTrials.gov. .
- 76.Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, Rochaix P, Andrieu-Abadie N, Levade T, Meyer N, Colacios C, and Ségui B. 2017. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, and Kim YJ. 2018. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine Nanotechnol. Biol. Med 14: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, Kanne DB, Jaffee EM, Dubensky TW, and Emens LA. 2017. A STING Agonist Given with OX40 Receptor and PD-L1 Modulators Primes Immunity and Reduces Tumor Growth in Tolerized Mice. Cancer Immunol. Res 5: 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, Kim S, Kim G, Chon HJ, and Kim C. 2019. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, Qin Y, Laszewski T, Stover DG, Barrera V, Hutchinson JN, Freedman RA, Mittendorf EA, and McAllister SS. 2019. Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li A, Yi M, Qin S, Song Y, Chu Q, and Wu K. 2019. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol.J Hematol Oncol 12: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pei J, Zhang Y, Luo Q, Zheng W, Li W, Zeng X, Li Q, and Quan J. 2019. STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett. 450: 110–122. [DOI] [PubMed] [Google Scholar]
- 83.Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, Woolman S, Vithayathil T, Glickman LH, Ndubaku CO, McWhirter SM, Dubensky TW, Armstrong TD, Jaffee EM, and Zaidi N. 2018. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ghaffari A, Peterson N, Khalaj K, Vitkin N, Robinson A, Francis J-A, and Koti M. 2018. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 119: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, and Curran MA. 2017. Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol. Res 5: 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW, and Kim Y. 2015. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med 7: 283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, Watkins SC, Sarkar SN, and Okada H. 2014. STING contributes to anti-glioma immunity via triggering type-I IFN signals in the tumor microenvironment. Cancer Immunol. Res 2: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, and Schreiber RD. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kline J, Zhang L, Battaglia L, Cohen KS, and Gajewski TF. 2012. Cellular and molecular requirements for rejection of B16 melanoma in the setting of regulatory T cell depletion and homeostatic proliferation. J. Immunol. Baltim. Md 1950 188: 2630–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilski NA, and Snyder CM. 2019. From Vaccine Vector to Oncomodulation: Understanding the Complex Interplay between CMV and Cancer. Vaccines 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.