

Abstract
Phenotypes are the foundation for clinical and genetic studies of disease risk and outcomes. The growth of biobanks linked to electronic medical record (EMR) data has both facilitated and increased the demand for efficient, accurate, and robust approaches for phenotyping millions of patients. Challenges to phenotyping using EMR data include variation in the accuracy of codes, as well as the high level of manual input required to identify features for the algorithm and to obtain gold standard labels. To address these challenges, we developed PheCAP, a high-throughput semi-supervised phenotyping pipeline. PheCAP begins with data from the EMR, including structured data and information extracted from the narrative notes using natural language processing (NLP). The standardized steps integrate automated procedures reducing the level of manual input, and machine learning approaches for algorithm training. PheCAP itself can be executed in 1-2 days if all data are available; however, the timing is largely dependent on the chart review stage which typically requires at least 2 weeks. The final products of PheCAP include a phenotype algorithm, the probability of the phenotype for all patients, and a phenotype classification (yes or no).
Keywords: electronic health records, phenotype, classification, natural language processing, machine learning
INTRODUCTION
Electronic medical record (EMR) data are a rich resource for clinical research studies ranging from pharmacovigilance to genetic association studies1–4. The growth of large biobanks with novel biologic data linked with EMR data has increased the demand and urgency for efficient, accurate, and robust approaches for phenotyping millions of patients5–9. The majority of studies using EMR data employ algorithms to classify patients with specific phenotypes of interest such as coronary heart disease or rheumatoid arthritis. The most common method for phenotyping using EMR data is a rule-based approach, applying combinations of structured EMR data such as International Classification of Disease (ICD) billing codes and medication prescriptions. While these approaches are simple to use, they are challenging to scale across multiple conditions because each phenotype algorithm typically requires a large degree of manual input to identify the potential features of interest and to create sufficient gold standard labels for training and validation. The performance of these algorithms can also vary as the accuracy of the codes varies across conditions and institutions10, 11.
Since the goal of many large-scale EMR-biobank studies is to perform in-depth studies of specific phenotypes, there was a large unmet need for scalable, standardized, efficient, and portable approaches to develop phenotype algorithms with high accuracy. To address this challenge, we developed, tested, and validated a common semi-supervised approach for phenotyping, PheCAP for use across multiple phenotypes and institutions12–15. The product of PheCAP is an algorithm that can be applied to a large dataset to classify patients with a specific disease or condition. Those who are classified can be included into an EMR based cohort for in-depth studies on risk factors or outcomes. For the clinical readership of these cohort studies, information on the performance characteristics such as positive predictive value (PPV) is important to understand the degree of misclassification in the cohort being studied. Thus, PheCAP, in contrast to unsupervised approaches, requires chart review defined gold standards.
The basic framework of the PheCAP approach was developed as part of the NIH Informatics for Integrating Biology and the Bedside (i2b2) project16. This approach incorporates natural language processing (NLP) and machine learning, enabling a scalable phenotyping pipeline applicable to most common conditions. This goal contrasted with existing rule-based approaches which developed phenotype algorithms one at a time, using project-specific methodologies. It is also important to note that there are several other robust approaches using NLP or machine learning for phenotyping using EMR data7–9, 17–19. In this protocol, we describe PheCAP as an option for investigators interested in using a standardized semi-supervised input from the clinical expert as part of the approach for phenotyping. Additionally, the accuracy of the phenotype algorithm is known. PheCAP has been tested at several institutions to define phenotypes for cohorts in studies on risk factors and outcomes15, 20–22. The standardized steps in PheCAP improve efficiency compared to current approaches and facilitate data checks as well as replication across institutions. As part of this protocol, we have developed R packages and either anonymized datasets or links to freely available datasets to allow the user to understand the details and underlying methods for developing a phenotype algorithm using PheCAP (https://celehs.github.io/PheCAP/).
Application of PheCAP for Clinical and Translational Studies
The most common use of phenotype algorithms is to provide an approach to identify patients with certain conditions among the millions of patients in the EMR to develop a cohort for further study. Operationally, this requires a relational database containing EMR data. The data can then be extracted for the features of interest on all patients to train and validate the phenotype algorithm. Ultimately, the algorithm classifies patients as either having or not having the phenotype. Patients with the phenotype can be included in cohort studies on risk factors and outcomes23–25; additionally, the phenotypes themselves can be used as outcomes in epidemiologic studies. Further, algorithms developed as part of this protocol can be ported across institutions to define subjects in the same manner, allowing for multi-center association studies9, 21.
EMR cohorts are increasingly linked to biorepositories where genetic and biomarker studies can be performed. This EMR research platform has facilitated novel markers of risk or replication of GWAS studies3, 26–28. Prospective cohort studies often take years to decades to recruit sufficient subjects for studies. This issue is particularly apparent when investigating low prevalence conditions. The ability to efficiently develop an algorithm to establish an EMR research platform with linked clinical, biomarker, and genetic data, provides an alternative dataset to perform these studies, particularly for phenotypes for uncommon conditions where there are few existing traditional cohort studies.
The final products of PheCAP provide other advantages compared to other phenotyping approaches using EMR data. One output of the algorithm is a probability of the condition, in addition to the traditional binary yes or no classification. The simplest use of this output is selecting a threshold probability above which subjects are considered to have a phenotype. The threshold itself can be tailored to the study. For example, a genetic association study may have improved power at a specificity of 95%, with a “cleaner” phenotype, compared to a pharmacovigilance study where a specificity of 90% but higher sensitivity is desired. Additionally, the actual probability output can be used in the association study, which can improve the power of genetic association studies29.
Comparison with other methods
Several unsupervised machine learning approaches for algorithm development exist. The learn and anchor approach uses expert-curated “anchors” as silver standard labels to assist with assigning potential phenotypes30. XPRESS31 and APHRODITE7 replace annotated labels with noisy silver standard labels such as the free text count of the phenotype name. PheNorm models key predictors such as the ICD and NLP counts of the phenotype as Gaussian mixture distributions32. All these unsupervised methods rely on silver standard labels for training the phenotyping algorithms. As such, they have varying degrees of accuracy depending on the quality of the silver standard labels. These unsupervised methods cannot provide a binary classification rule to define the cases or to assess the prediction performance without gold standard labels for validation, both important when defining a cohort for study.
Overview of PheCAP
The basic framework of EMR phenotyping starts with the EMR data (Figure 1). A filter provided by the clinical expert, such as the presence of one or a group of ICD billing codes associated with the phenotype of interest, is applied to identify all possible subjects with the condition. This excludes patients with an extremely low probability of having the condition. All subjects who pass the filter are included into a “data mart” containing the de-identified patient information. Next a set of potentially informative features are constructed using both structured data and information extracted from the unstructured narrative data using NLP. The NLP features are curated from online knowledge sources in an automated fashion and selected using a data driven method described in more detail in the NLP dictionary step below.
Figure 1. PheCAP Overview.
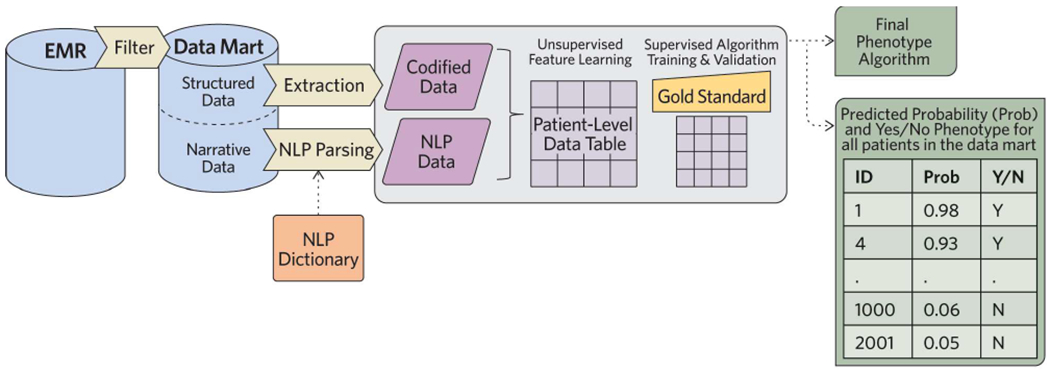
Starting with all EMR data, a sensitive filter (Procedure step #1) such as a diagnosis code is used to create a data mart (Procedure step #2) containing all patients who may potentially have the phenotype. Codified data, such as diagnoses codes or medication prescriptions related to the phenotype are extracted from the data mart (Procedure steps #5-6). Additionally, concepts or terms related to the phenotype are extracted using natural language processing (NLP) (Procedure steps #10-15). The NLP dictionary can be developed manually or using an automated process. These data are combined into a patient level data table (Procedure step #7). In parallel, a random sample of patients is selected for chart review to provide gold standard labels (Procedure steps #3-4). Sparse machine learning is applied in two steps: an unsupervised (Procedure steps #28-35) and a supervised step (Procedure steps #36-41) to identify the important features of interest. The output of the pipeline is a phenotype algorithm, a probability of the phenotype for all subjects in the data mart, and a classification of the phenotype for each subject (yes/no) (Procedure steps #42-43).
In parallel, a training and validation set are selected randomly from the data mart and clinical domain experts review the records to assign gold standard labels for whether or not a patient has the phenotype. The list of codified features such as ICD, procedure codes and medication prescriptions relevant to the phenotype can be provided by clinical experts.
Also performed in parallel is the creation of an NLP Dictionary (Figure 2). We developed an automated process to extract narrative data using a pipeline which includes the use of the Unified Medical Language System (UMLS)33 and NLP. The extracted codified and NLP data are compiled to provide a broad list of potential features for the algorithm.
Figure 2. Creating an NLP dictionary.

Automated process to generate an NLP dictionary by processing knowledge sources using NLP (Procedure steps #10-14).
Unsupervised learning methods are then applied to identify the most informative features for the phenotype from the combined codified data and NLP list (Figure 3). Using a sparse regression model trained against the surrogate features obtained from the EMR database, the final set of features predictive of the phenotype are selected for the final algorithm. The final model is an equation where each feature is assigned a weight. The algorithm is applied to a dataset containing the information for each feature on all patients. When the algorithm is deployed, a predicted probability of having the phenotype is assigned to each individual in the EMR data mart.
Figure 3. Unsupervised Feature Learning.
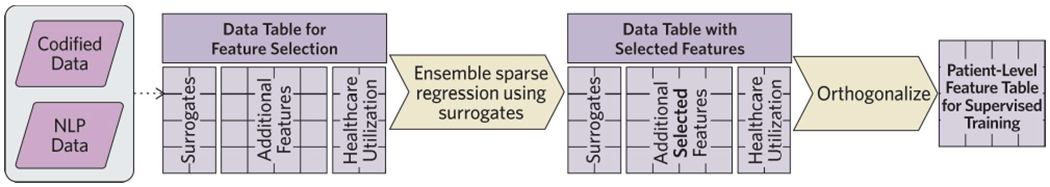
Steps to identify informative codified and NLP features for the algorithm prior to supervised training of the algorithm with gold standard labels (Procedure steps #28-35).
The pipeline we describe here incorporates several innovations since our publication in 201514. These innovations automate previously manual, time-intensive steps. As shown in Figures 1 and 2, we developed a parallel process using online medical knowledge sources and NLP, to assist the investigator in creating a broad list of potentially relevant features to extract from the narrative notes14 Additionally, we have added an intermediate step to prune the list of potential features prior to training the final algorithm against the gold standard. This step uses silver standards in the “unsupervised feature learning” step13 (Figure 3). Finally, a “denoising” step is applied by orthogonalizing the structured and NLP data before training the algorithm against the gold standards, with the aim to create a parsimonious algorithm (Figure 4).
Figure 4. Detailed flow of PheCAP protocol.
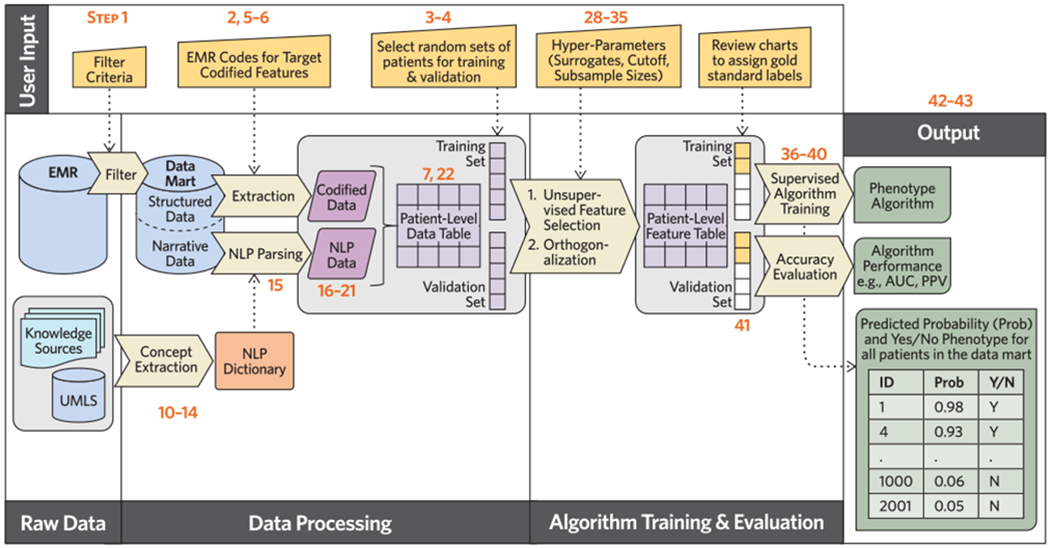
User input required at various steps in the PheCAP protocol are specified at the top of the figure as the protocol moves from data extraction, data processing, through algorithm training and validation, and the final outputs: a phenotype algorithm, a probability of the phenotype for all subjects in the data mart, and a classification of the phenotype for each subject (yes or no). Numbers in the figure correspond to Procedure steps.
Chart review to create gold standard labels is the major rate-limiting step for algorithm development. The automated steps outlined above simultaneously improve efficiency and reduce the number of un-informative and potentially noisy features, thus reducing the number of gold standard labels required for training. Overall the innovations improve the efficiency of the algorithm development process. PheCAP has been tested across over 20 different phenotypes and 4 EMR systems15, 20–22 as well as Veterans Affairs EMR data which covers approximately 170 health centers across the US6 (Table 1).
Table 1.
Institutions where the PheCAP framework has been tested.
| Institutions | Location | Type of EMR | Patients at time of studies (n) | Research EMR data | Related publications |
|---|---|---|---|---|---|
| Partners Healthcare: Brigham and Women’s Hospital &Massachusetts General Hospital | Boston, MA | Internally developed; EPIC after 2015 | 4.5 million; 9 million | Enterprise Data Warehouse | 11, 15, 20, 22, 27, 42–45 |
| Northwestern | Chicago, IL | EpicCare (outpatient) Cerner PowerChart (inpatient) |
2.2 million | Enterprise Data Warehouse | 21 |
| Vanderbilt | Nashville, TN | Internally developed | 1.7 million | De-identified image of EMR (Synthetic Derivative) | 21, 27, 46 |
| Boston Children’s Hospital | Boston, MA | Internally developed | 1.8 million | Enterprise Data Warehouse | 47 |
| VA Healthcare System | Nationwide; 170+ VA Medical Centers | Internally developed | 22 million | Enterprise Data Warehouse | Manuscripts in preparation |
Experimental Design
Here we provide more detail on the key steps mentioned above for the development and validation of an EMR phenotype algorithm starting with the data mart (Figure 1).
Patient consent.
The phenotyping studies performed by our team to date using de-identified EMR data were considered minimal risk to patients, and individual informed consent of millions of patients was not feasible. Thus, our Institutional Review Board (IRB) considered the consent obtained at routine clinical visits pertaining to studying patient EMR data sufficient. Patients who do not wish to participate are flagged in the system and their data are not available for research studies. As phenotypes and the goal for phenotyping projects can vary, and national guidelines regarding using of EMR data have changed over time, investigators should consult with their IRB or ethical boards before initiating a study. At our institutions, an IRB approved protocol is a required part of all requests for any type of research using EMR data.
Manual Annotation of Gold Standard Labels.
From the data mart, select a random set of approximately 200 subjects for chart review to assign gold standard labels. This set will be further divided into a training set, used for algorithm development, and a validation set to evaluate the accuracy of the algorithm. We recommend initiating chart reviews once the data mart has been created since obtaining gold standard labels from chart reviews is the most rate-limiting step in phenotyping algorithm development.
Clearly defining the phenotype is a crucial stage prior to chart reviews. We recommend that at the outset the domain expert or end-users of this algorithm write down the phenotype definition in a manner that could be replicated by an investigator at another institution. When available, components of validated diagnostic or classification criteria for the phenotype are also extracted during chart review to provide face validity. However, we typically do not require that patients labeled as having the phenotype meet all published classification criteria, as criteria are often designed for research purposes and these data may not be recorded in the EMR as part of usual care.
We recommend assigning the following categories during chart review: definite, possible, or no phenotype present. Depending on the ultimate application of the algorithm, one may assign the “possible” labels to either “yes” or “no” for algorithm training. Grouping “possible” with “no” can ensure that the algorithm classifies cases that are highly likely at the expense of having a smaller number of identified cases. Grouping “possible” with “yes” can ensure that the algorithm increases the yield of identified cases at the expense of a lower positive predictive value (PPV). The unsupervised feature learning and supervised training will identify patterns in the data based on the training labels. The upper limit of accuracy for the algorithms is directly related to how well two independent investigators can identify the phenotype and agree on the presence or absence of the phenotype on chart review.
Feature Curation.
For most phenotypes, there are typically many potential features that can be informative for the phenotyping algorithm. Using coronary artery disease as an example, potential features include the number of ICD codes for myocardial infarction, cholesterol levels from laboratory testing, procedure codes for cardiac catheterization, or narrative information in a report about perfusion abnormalities on a cardiac stress test. PheCAP leverages both structured data and information extracted using NLP from narrative notes. Feature curation includes identifying and extracting the possible features from the EMR data, as well as reducing the potential features to those that are most informative for the phenotype of interest.
The codified features can be provided by domain experts when feasible. When limited resources are available to curate codified features, one may identify ICD codes corresponding to the phenotype of interest from databases such as the Monarch Disease Ontology34 (MONDO) or the PheWAS catalog2. The investigator can then use the total count of the ICD codes for the phenotype, denoted by main ICD code, as the codified feature.
To form an initial set of candidate NLP features, we have developed a pipeline to extract medical concepts from publicly available knowledge sources including Wikipedia, Medscape eMedicine, Merck Manuals Professional Edition, Mayo Clinic Diseases and Conditions, and MedlinePlus Medical Encyclopedia (Figure 2). Clinical terms are extracted from relevant articles from the knowledge sources using named entity recognition (NER) and are mapped to Concept Unique Identifiers (CUIs) in UMLS14. Briefly, NER is a process that identifies clinical terms in the narrative text and maps the terms to the UMLS concepts. To remove potentially uninformative CUIs, only CUIs that appear in more than half of the source articles are retained, a step we call “Majority Voting” (Figure 2). These CUIs, along with their relevant clinical terms, are used as the NLP dictionary for processing the EMR notes.
For most common phenotypes, online knowledge sources including Wikipedia provide sufficient detail to create a robust dictionary. When there are multiple potential articles from the same source, they can be merged into a single file to ensure broader coverage to avoid missing critical concepts. Alternative sources such as detailed clinical notes from the EMR or paragraphs of review articles can also be used to supplement when online sources are insufficient or not available for a specific phenotype.
Unsupervised Feature Learning.
The previous “Feature Curation” step focuses on generating a broad list of potentially relevant features for the algorithm. Once this broad list is created, the next step is to determine which features among this list are most informative in identifying the phenotype of interest. Reducing the feature space is important because the more features that need to be evaluated in the supervised training step, the more gold standard labels are needed to create a robust algorithm. Thus, the first step is to exclude features that are present <5% of the time in the narrative notes that contain positive mentions of the target phenotype14, a step we call “frequency control.” Next, we pass the features first through an unsupervised feature learning step to further prune the list of features prior to the supervised training step where gold standard labels are used (Figure 3).
To pare down the list of features, the list is regressed against a surrogate, or the “silver standard label”, such as the main ICD code(s) or the main concept extracted using NLP13. The “main ICD” code corresponds with the ICD code(s) that would be used to diagnose the condition. The main NLP concept would be the CUI(s) associated with the phenotype of interest. For example, in a phenotype algorithm for rheumatoid arthritis, the “main NLP” would be C0003873, the CUI for “rheumatoid arthritis.” With the additional candidate features selected, an orthogonalization step is also performed by regressing each candidate feature against the main surrogate features as well as the healthcare utilization level (such as the total number of notes) to obtain the residual as the new representation of the features (Figure 3). This step ensures that the additional candidate features are only included in the final algorithm if they provide information about the disease phenotype above and beyond the main surrogate features. The goal of this step is to reduce the model complexity.
Although the default surrogates include the number of main ICD codes, main NLP, or a combination of the main ICD+NLP for the disease, alternative surrogates can be selected based on domain knowledge. For example, in type II diabetes, laboratory values such as glucose or hemoglobin a1c lab tests can be used as surrogates instead of or in addition to billing codes. The goal of this unsupervised feature learning step using surrogates is to identify features that are associated with the condition of interest, and thus this step may select other related conditions. When hba1c is used as a surrogate for a Type II diabetes algorithm, Type I diabetes codes were selected as a potentially important feature to be assessed for the final supervised training step. In this scenario, when the algorithm is trained against a gold standard in the supervised step, Type I diabetes remained in the algorithm for Type II diabetes as an informative negative predictor.
Supervised Algorithm Training and Validation.
A supervised machine learning algorithm is then trained with the gold standard labels and the candidate features that passed the feature selection step. The default algorithm is the logistic regression with adaptive Elastic-Net penalty which typically yields a sparse regression model. The output is a predicted probability of having the phenotype for each patient.
The algorithm is then validated against gold standard labels in the validation set. Predictive accuracy measures including the area under the Receiver Operating Characteristic curve (AUC), sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) across different probability threshold values for defining a case. Patients in the EMR who did not pass the filter are assigned a probability of zero.
If the ultimate goal is to have a binary classification yes or no for the phenotype, a threshold value for probability can be determined based on a desired level of sensitivity, specificity or PPV. Those with a probability above the threshold value are considered as cases for subsequent studies. For example, cases can be defined at a specificity of 95% for which the corresponding threshold probability is π+ . Thus, all subjects with probability of π+ or higher are defined as cases and included in a phenotype cohort.
A wide range of machine learning algorithms in addition to penalized logistic regression can be used in the supervised training step with gold standard labels. Examples include support vector machine and random forest. In testing different approaches, we found that the penalized logistic regression generally works well with comparable or superior performance to the more complex algorithms with training sets of moderate size (n=200 to 500 subjects).
Expertise Needed to Implement
Developing EMR phenotyping algorithms require collaborative efforts from different backgrounds including NLP, domain knowledge, biostatistics, machine learning, and database programming. Domain expertise is required to clearly define the phenotype of interest, provide gold standard labels and define the downstream application for the algorithm. Additionally, the domain experts can provide information on candidates for the codified features, verify the knowledge source articles for the phenotype, and determine whether the default surrogate features for the unsupervised training step are reasonable. Both the NER applied to the source articles and NLP of the notes require NLP experts. Team members with statistical expertise are needed to perform the analytical steps of the algorithms including surrogate assisted feature selection and the algorithm training and validation, as well as apply the machine learning approaches. Database programming expertise is crucial for extraction and management of the data.
Limitations
This pipeline was developed for phenotypes where there is a universally agreed upon definition among clinicians. Further adaptation of the protocol would be needed to apply this pipeline to less well-defined phenotypes where there is no consensus regarding the clinical definition or where no ICD code exists. Additionally, the majority of the phenotypes tested had a prevalence of 1% or higher in the source EMR population and performance of the algorithms for phenotypes <1% has not been rigorously tested. The phenotyping algorithms can only perform as well as the ability of domain experts to define the phenotype using the available EMR data; the upper limit of performance for the algorithm is directly related to how well two independent reviewers can agree on the presence or absence of a phenotype. Thus, uncommon phenotypes that are poorly documented will be challenging to study. Finally, the PheCAP algorithm can be used in other registry databases where features may only include codified data but gold standard labels are available through existing studies. Deploying PheCAP in claims databases would require linking claims data with registry or EMR cohorts where gold standard labels are available.
MATERIALS
EQUIPMENT
Storage space and server: will vary depending on the number of patients and depth of EMR data available. In general, we recommend performing these analyses using data stored in a relational database, e.g. SQL.
R (version 3.3.0 or newer; https://www.r-project.org/) The complete R codes for PheCAP and a test data set are available at: https://celehs.github.io/PheCAP/.
Java Runtime Environment (JRE) version 8 or newer
- NLP software: The UMLS database33 is needed for extracting information from the narrative text for phenotyping.
- ▲CRITICAL The NLP software may come with a built-in dictionary. However, UMLS is generally needed for a comprehensive coverage of clinical concepts.
- NLP software needed for NER and semantic analysis. There are a number of biomedical information extraction systems (see below for a non-exhaustive list), each implementing various core text processing components that can be used for phenotyping.
- APACHE Clinical Text Analysis and Knowledge Extraction system (cTAKES), http://ctakes.apache.org/35
- Health Information Text Extraction (HiTEX), https://www.i2b2.org/software/projects/hitex/hitex_manual.html36
- MetaMAP, https://metamap.nlm.nih.gov/38
- Narrative Information Linear Extraction (NILE), https://celehs.github.io/PheCAP/articles/NLP-NILE.html39
- OBO annotator, http://www.usc.es/keam/PhenotypeAnnotation
- Stanford CoreNLP, https://stanfordnlp.github.io/CoreNLP40
Equipment setup
Obtaining UMLS data33
First Download UMLS data from https://www.nlm.nih.gov/research/umls/licensedcontent/umlsknowledgesources.html.
Follow instructions on the website to load the content into a MySQL database: https://www.nlm.nih.gov/research/umls/implementation_resources/scripts/README_RRF_MySQL_Output_Stream.html
-
The current recommended version of MySQL version 5.5 for use with UMLS is (https://dev.mysql.com/doc/refman/5.5/en/installing.html). Follow instructions from your NLP software’s website to use the UMLS database as the dictionary.
▲CRITICAL Some users have reported disk space issues with early versions of MySQL 5.6 due to default database settings.
Obtaining EMR data elements
Obtain a data mart containing EMR data on all patients of interest. These data are broadly characterized into structured or codified data, and unstructured data. From the data mart, extract the potential features for the algorithm. Codified features include diagnostic codes, procedure codes, laboratory tests, medication codes as well as demographic information. Extract NLP features for all patients from narrative notes via NLP.
Randomly select a subset of patients from the data mart and review charts and assign gold standard labels, for example: yes, possible, no presence of phenotype. Our labeling is typically performed by a domain expert via manual chart review. ▲CRITICAL Prior to extracting EMR data elements, the study should be reviewed by an institutional or ethical review board.
PROCEDURE
▲CRITICAL A detailed overview of the protocol is provided in Figure 4.
Creating EMR data mart [TIMING ~24h, varies depending on size of data and infrastructure]
CAUTION: Please consult with your institutions’ ethical review board regarding patient informed consent before initiating the data request.
-
1
Select a filter criterion with high sensitivity and negative predictive value. The goal of the filter is to identify patients with some chance of having the disease (prevalence filter) with sufficient data in the EMR (information filter). We typically use ≥1 ICD code for the phenotype of interest as the filter. Patients who do not fulfil the criteria, termed the filter negative set, have a near zero probability of having the disease; patients in the filter positive set typically have a reasonable prevalence (e.g. ≥20%). We also apply an information filter of ≥2 notes with more than 500 characters each. This ensures that patients have sufficient information documented in the EMR for classification.
?TROUBLESHOOTING
-
2
Next, export all possible data elements of interest from the EMR from the filter positive patients to create a data mart. Examples of data elements include demographics, diagnosis codes, medications, procedures codes, vital signs, laboratory test codes and results, and narrative clinical notes. Having a data mart provides a static version of the EMR data and facilitates reproducibility and quality control checks. If the selected filter population is too large to query, a random subsample can be used. Ideally, a database programmer creates a relational database and data are loaded from the EMR into the database.
Our studies utilized the i2b2 platform (https://www.i2b2.org/) which provides the ability to query a data warehouse for a specific population and then create a data mart, also in i2b2 format. Each project has its own data mart separate from the EMR data warehouse. A project-specific data mart facilitates both protection and sharing of study data. For example, some studies require access to identified data. Rather than granting access to all projects, having separate data marts enables access only to the project(s) where identified data are needed.
For most phenotypes, information from the narrative notes regarding clinical concepts are informative for the phenotype definition. In our studies, we perform NLP on all types of narrative texts including progress notes, discharge summaries, radiology notes, cardiology reports, for all the patients in the data mart and who are filter positive, i.e. passed both the prevalence and information filter.
▲CRITICAL Prior to data processing, we recommend performing general checks on the data to ensure that the upload/download of data were successful. For example, the total number of patient encounters and total number of notes should increase in the same order of magnitude in successive years.
Conduct chart review and obtain gold standard labels [Timing 1 week, depending on availability of domain expert]; this step can be done in parallel with steps 5 through 36 but we recommend initiating immediately after creation of the data mart as this is the most time-consuming step.
-
3
Randomly select at ≥200 subjects from the data mart with ≥100 for training and ≥100 for validation.
-
4
Perform manual chart review for all patients. When multiple reviewers are available, select a subset of patients for which chart review is performed by multiple reviewers to assess whether raters agree. We suggest providing the following labels for the phenotype of interest: definite, possible/probable, or no phenotype present.
▲CRITICAL Develop a set of criteria for defining a case prior to chart review such that the guidelines can be replicated by an independent reviewer; for multiple reviewers perform review of charts for at least 10 patients to determine inter-rater reliability. If there is disagreement between the reviewers, we recommend discussing the cause for discrepancies and updating the phenotype definition.
▲CRITICAL We strongly recommend performing a check to ensure that the random selection for chart review yielded a subset of subjects with characteristics similar to the data mart. Specifically, we recommend selecting 5 random sets and comparing common characteristics across the 5 sets and the data mart, e.g. mean age, gender, ICD’s of interest and common ICDs and selecting the random set most similar to the data mart.
▲CRITICAL If patients outside of the data mart, i.e. those do not satisfy the filter criteria, are of interest, additional chart review can be performed on a small subset of patients sampled from the filter negative set to examine the NPV of the filter.
Identify and extract codified data features from the structured EMR data for the algorithm [TIMING ~12h]
-
5
For the initial list, get the clinical domain experts to generate a list of terms or concepts associated with the phenotype. These features may include the target phenotype, competing diagnoses, relevant medications, procedures, and laboratory tests.
-
6
Next, identify the corresponding codes in the EMR for each identified term or concept. Then map the clinical concepts to specific EMR codes. We use standard ontologies to map terms and concepts to codes. These include diagnosis codes (ICD-9, ICD-10, DRG, PheCodes), procedure codes (CPT-4, ICD-10), medications (RxNorm, NDF-RT, NDC) and laboratory tests and vital signs (LOINC). In addition, sites often have institution specific codes, e.g. codes for problem lists, that can be included in the mapping. Depending on the phenotype of interest, the domain experts can provide the ICD codes or online resources can be used for mapping to codes. These include but are not limited to: the Monarch Disease Ontology (MONDO) (https://www.ebi.ac.uk/ols/ontologies/mondo)34 and the PheWAS catalogue (https://phewascatalog.org/phecodes)2. To map to the codes of medications, procedures, and laboratory tests, knowledge of the institutions’ codes of preference is needed. If the hospital’s EMR uses a standardized coding system, such as RxNorm for medications or CPT for procedures, one can use the online UMLS Terminology Services - Metathesaurus Browser (https://uts.nlm.nih.gov/home.html) to find the corresponding codes. To map to institution specific codes, input from the institution’s hospital is needed.
?TROUBLESHOOTING
-
7
Create a large table with one row per patient containing each codified data as a column. Get the EMR database programmer to create an analysis file from the data mart using the mapping of EMR codes and custom SQL scripts. Typical columns include patient ID, total number of main ICD codes as well as counts of other codes. When counting the total number of ICD codes, we only count each unique ICD code once per day.
You will often need to decide how to aggregate the important features of interest described in step 2. For example, a clinical concept of body mass index (BMI) might produce multiple columns, for example mean patient BMI, max patient BMI, count of BMI measurements, and count of BMI measurements over 30.
▲CRITICAL The codified data is later combined with the data extracted from narrative notes using NLP (see below) to create the training data for the unsupervised and supervised learning steps.
Prepare analysis environment in R [TIMING <1h]
-
8Install the R package PheCAP by launching R and making sure the Internet connection works prior to running the code below:
- install.packages(“devtools”)
- devtools::install_github(“celehs/PheCAP”)
- install.packages(“PheCAP_1.0.tar.gz”, repos=NULL, type=“source”)
-
9
Download “cui_processing.R” and “main.R” from https://github.com/celehs/PheCAP/tree/master/paper. “cui_processing.R” will be used in Steps 13-14 for performing majority voting and creating the NLP dictionary for note parsing. “main.R” is an example of using PheCAP functions in Steps 23-45 for data preparation, feature selection, and algorithm training and validation.
Concept collection for candidate NLP features [TIMING ~12h]
-
10
Create a list of clinical concepts relevant for the phenotype of interest. Specifically, the concepts should link to a UMLS Concept Unique Identifier (CUI)33. Investigators can also identify concepts starting with a list of codified features and map these features to CUIs using the MRCONSO table in UMLS. The MRCONSO maps CUIs to codes in other coding systems, such as ICD, CPT, and RxNorm.
-
11
Identify text articles describing the phenotype from publicly available knowledge sources including Wikipedia, Medscape, eMedicine, Merck Manuals (Professional Version), Mayo Clinic, and MedlinePlus. Save the articles in plain text (such as .txt, rather than the MS Word’s .doc file). For wikipedia use text from the entire article. For Medscape/eMedicine, click the “Show All” button in the left-hand box and view the entire article on one page. For the Merck Manual select “Professional Version”; the in-site search bar located at the top right corner can also be used. For the Mayo Clinic articles use the content under Basics. Use the Print button to display the entire article. For MedlinePlus, search for the condition of interest, then select the article from the “Medical Encyclopedia.” The page can usually be found in the “External Links” box on the Wikipedia page.
?TROUBLESHOOTING
-
12
Use the NER software along with the UMLS33 to identify clinical terms in each of the articles and record the UMLS CUIs for identified terms.
This can be done using MetaMap. To do this, go to the online MetaMap service (batch mode): https://ii.nlm.nih.gov/Batch/UTS_Required/metamap.shtml. Copy and paste the text of one of the source articles to the text area. In Output/Display Options, check “Show CUIs (-I)”. In “I would like to only use specific Semantic Types,” check the checkbox, and select all the semantic types of which you may want a clinical concept as a feature for the target phenotype. Click “Submit Batch MetaMap.” An email will notify you once MetaMap has finished processing the file. Download “text.out” and rename it, such as “Wikipedia.out”. Create a new folder and save all the output files to the folder.
▲CRITICAL MetaMap only works on ASCII characters and will not run if the text contains non-ASCII characters. One can detect and remove non-ASCII characters with the regular expression “[^\x00-\x7F]”.
-
13
Perform Majority voting. Concepts (CUIs) that appear in ≥50% of articles are considered as potentially important. Denote these concepts as candidates. The occurrence of these concepts in the clinical notes will be assessed at a later step. Next, launch R and open the file “cui_processing.R.” Run line 1. In line 6 of the R code, edit the program to point to the folder where the MetaMap outputs are saved. Run line 6 to extract the CUIs from the output file and identify the features that pass majority voting.
-
14
Extract the terms of the candidate concepts from the UMLS to create a custom dictionary for the note parsing step below. Based on the database connection authentication details, make appropriate changes to lines 13-16 in “cui_processing.R”. Run lines 13-16 to generate the dictionary file.
?TROUBLESHOOTING
Note parsing to obtain NLP feature data [TIMING ~20h+; can vary widely depending on size of dataset and size of NLP dictionary]
-
15
Parse the clinical notes in the data mart using the NLP software. The NLP program should be set to extract information only on non-negated mentions in the notes. Clinical NLP software typically implements a negation analysis module. For example, the default entity extraction pipeline of Apache clinical Text Analysis and Knowledge Extraction System (cTAKES)35 consists of a sentence splitter, context sensitive tokenizer, part-of-speech tagger, a fast dictionary look-up module for entity recognition and terminology/ontology mapping. Each entity is populated with a set of modifiers amongst which are negation41, experiencer (patient, family member, other), uncertainty41, and a conditional value. Although cTAKES has other modules, its default entity extraction pipeline provides all necessary functionalities for the phenotyping task.
If negation is not available, NegEx (https://github.com/chapmanbe/negex) is an option for negation analysis, which can be incorporated into most clinical NLP software. Additionally, mentions of concepts in the family history should be excluded. If the NLP software does not offer an analyzer for family history, one approach is to remove the family history section from the notes.
In practice we exclude negated concepts as potential features because we have found that this reduces the number of gold standard labels needed for training. For example, if a note states that a patient “has no evidence of coronary heart disease,” that note will not be considered to have a mention of coronary heart disease. While including all mentions of a concept, whether or not they are negated may benefit the model, it also doubles the number of features, which in turn increases the number of gold standard labels needed and time for chart review. In practice, we have found that if we include the differential diagnoses of the target phenotype (which are identified from the knowledge sources), also including negated features, for example, “no coronary heart disease” had limited effect on the accuracy. Since chart review is a major rate limiting factor, we do not recommend adding negated features to the model.
The processing time depends on many aspects such as the NLP software, the format of data (e.g. files on a local computer, data tables on databases), the performance of computing environment such as CPU clock speed, RAM and the volume of notes including the number and the average length of notes.
The NLP data used as examples in this manuscript were generated by NILE39, which was developed to identify medical concepts in narrative EMR data. In addition to identifying medical concepts, NILE is able to perform analysis for negation and family history. On a server with multi-core 2.64GHz processors and 64Gb system RAM, the time for processing 62,155 notes is 57,22 seconds by NILE with a single thread of computation using a dictionary with # terms. The average length of each note above is 2523 characters.
-
16
Assemble the NLP output. Represent the identified clinical terms with CUIs, and extract those that have been identified as candidates. Assemble the output in a tabular format, where each row represents a note of a patient. A data row must have a column for the patient ID, and a column for each candidate CUI. The column for candidate CUIs provides the number of positive mentions in the note for a specific concept, not including negated concepts and concepts mention in the family history (Figure 5).
-
17
Perform quality control for the note processing by checking if the number of notes with more than 500 characters matches the row number of result file and manually reviewing a small number of notes to confirm the software extracted all potential concepts listed in the dictionary.
?TROUBLESHOOTING
-
18
Group the drug or chemical concepts into their hierarchical relationship as recorded in the UMLS MRREL table. Concepts grouped by the main active ingredient generally have better predictive power than the individual features. For example, C0699142 Tylenol, C0000970 Acetaminophen, and C0002771 Analgesics may all be candidate CUIs. UMLS provides the relationship between, for example, Tylenol a trade name of acetaminophen, and that acetaminophen is a kind of analgesic. Thus, for this example add the column of Tylenol and all other identified trade names of acetaminophen to the column of acetaminophen, and then add acetaminophen and all other identified analgesics to the column of analgesics. Note that the adding must be done from the bottom moving up the order of the hierarchy.
-
19
Perform frequency control, Part 1 of 2. A clinical concept is unlikely to be informative if it rarely co-occurs with the CUI of the target phenotype. Create a list of CUIs that are mentioned in <5% of the clinical notes that have a positive mention of the target phenotype’s CUI. This list will be used to exclude NLP features at a later step.
-
20
Aggregate the NLP note level data to the patient level such that each patient will have a number of NLP mentions for each concept. The output will be CUI columns representing the count of each concept mentioned in the notes for each patient. For example, patient A has 5 notes. Rheumatoid arthritis is mentioned 3 times in one note and twice in another note. Patient A will have a value of 5 in the CUI column for rheumatoid arthritis.
-
21
Perform frequency control, Part 2 of 2. Remove the CUIs listed in Step 19 from the aggregated patient level NLP data created in Step 20.
-
22
Create the feature for healthcare utilization (H). Create a table with one row per patient and two columns. It should contain a column for patient ID and a column for measurement of the patient’s healthcare utilization, denoted by H. Healthcare utilization is an important feature that can significantly improve the prediction. The H feature can be the total number of unique billing codes, the total number of visits, or the total number of notes. Based on our previous experience, the results are not sensitive to which specific definition is used to represent healthcare utilization.
Figure 5.

Clinical terms identified by MetaMAP along with their mapped CUIs from a Wikipedia article on coronary artery disease (example of results obtained from Procedure step #13).
Load EMR data into R [Timing: <1h]
-
23
Launch R and open the script “main.R”.
-
24
Run line 1 in “main.R”.
-
25
Modify the path in line 4 in “main.R” to reflect the actual location of the PheCAP data file(s). Alternatively, uncomment line 7 to use the sample dataset in PheCAP.
-
26
Specify the variable name for the H feature, the variable name for the label, and the proportion of data reserved as the validation set in line 12 in “main.R”.
-
27
Load the data into R by running lines 3-13 in “main.R”. Verify that the data summary (number of observations, number of variables, and so on) is consistent with your knowledge of the data.
?TROUBLESHOOTING
Perform surrogate assisted feature selection (SAFE) [Timing <1h]
-
28
Specify a set of surrogate variables that are expected to be highly predictive of the phenotype status when their values are in extreme tails. For most phenotypes, good surrogates include the total ICD count of the phenotype (denoted by SICD), the total NLP mentions of the phenotype (SNLP), as well as SICDNLP=SICD+SNLP. Alternative surrogates can be used to replace or augment the ICD or NLP based on procedure codes or laboratory measurements. For example, measurements of fasting glucose or HbA1c can be used as alternative or additional surrogates for type II diabetes.
-
29
For each surrogate Sk, specify lower (lk) and upper (uk) cutoff values to define silver standard labels. Patients with Sk value higher than uk are assigned with Sk*=1; those with value below the lower cutoff value are assigned with Sk*=0; and those between the lower and upper cutoff values are excluded.
-
30
Based on steps 28 and 29, modify lines 22-31 in “main.R” accordingly. Then run these lines.
-
31
For each surrogate Sk, sample 500 patients with Sk*=1 and another 500 patients with Sk*=0. I.e. a total of 1000 patients. To change this number, add the argument subsample_size=<number> within phecap_run_feature_extraction in line 34 in “main.R”.
▲CRITICAL Steps 31 to 35 can be conducted by running lines 33-35 in “main.R”. The selected features will be printed to the R console.
-
32
Perform penalized logistic regression for those sampled patients with S* being the response variable and all features other than S as predictors to obtain regression coefficients for the features. Assign a coefficient of 1 to the surrogate itself.
-
33
Repeat steps 31 and 32 200 times for each choice of the surrogate.
-
34
Calculate the percentage of times each feature receives a non-zero coefficient across all replications and all surrogates.
-
35
Select features with non-zero frequency higher than 50% as potential features for supervised algorithm training. The selected feature set is denoted by X+=(X+1,...,X+J)
?TROUBLESHOOTING
Supervised algorithm training [Timing <1h]
▲CRITICAL Steps 36-39 can be done via lines 37-40 in “main.R”.
-
36
Create the list of features for the supervised algorithm training. These include the surrogates used in the unsupervised feature selection step (denoted by S), H, and the candidate features selected in the previous steps: unsupervised feature selection (denoted by X+). The default program uses SICDNLP as a candidate feature in the supervised algorithm. For example, if SICD, SNLP and SICDNLP are used as surrogates in the unsupervised algorithm training, and X+ are selected from the unsupervised feature selection, the final list of candidate features for the supervised training step would be {SICD, SNLP, SICDNLP, H, X+}; using this full set of features typically produces the best prediction model and is the default in the PheCAP R package. However, if the goal is to have interpretable coefficients in the algorithm, SICDNLP should be excluded from the list of potential features for supervised training. This is because SICDNLP is created from the combination of SICD and SNLP and can be potentially collinear leading to coefficients that may be difficult to interpret.
-
37
For each feature X+j in X+, orthogonalize it against S and H by performing a linear regression of X against S and H and taking the residual from the fitting to obtain X+j*. Assemble S, H and all { X+1*,...,X+J* } to create the feature set F for algorithm training.
-
38
Fit a supervised machine learning algorithm with F as the predictors and the annotated labels as the response. The default algorithm in the R package is the penalized logistic regression with the tuning parameter selected via the cross validation. Alternative algorithms such as the support vector machine and random forest can also be considered.
?TROUBLESHOOTING
-
39
Obtain the initial estimates of the model prediction performance, including the AUC, sensitivity, specificity, PPV, and NPV, on the training set via cross-validation.
-
40
The supervised algorithm training process also generates an output containing the probability of having a phenotype for each subject in the data mart (Figure 7). This can be performed by running lines 42-43 in “main.R”.
Figure 7.
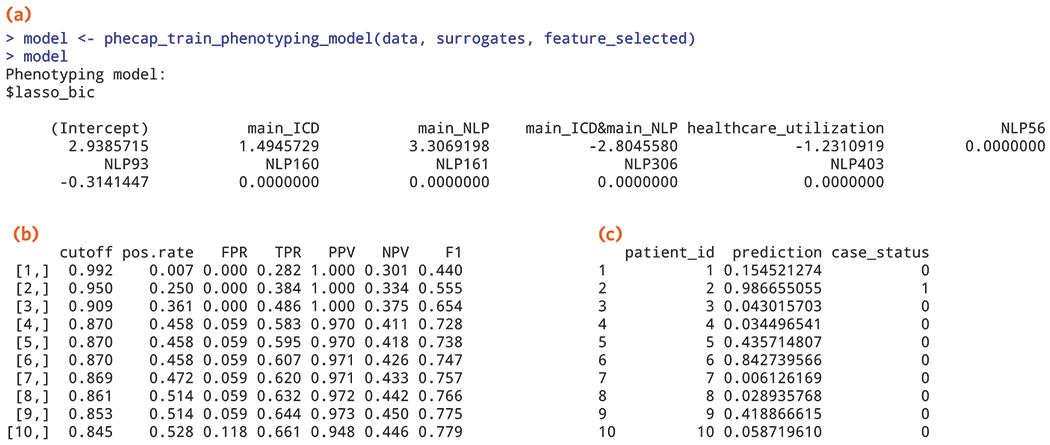
Output from the supervised algorithm training step depicting (a) SAFE selected features with coefficients from the supervised training step (example of results obtained from Procedure step 38); (b) estimated percent of patients classified as cases (pos.rate), false positive rate (FPR), true positive rate (TPR), PPV, NPV, and F-score over a range of cut-off values from validating the algorithm (example of results obtained from Procedure step 41); (c) predicted probability of being a case for patients in the data mart along with their predicted case status, 1=case, 0=non-case (example of results obtained from Procedure steps 42-43).
Algorithm validation [Timing <1h]
-
41
Use the predicted probability, π, and the gold standard labels in the validation set to compute sensitivity, specificity, PPV, NPV across a range of threshold values as well as the AUC to summarize the overall classification performance. Lines 45-49 in “main.R” show how to obtain the AUC on the validation set, as well as how to visualize ROC and related curves.
?TROUBLESHOOTING
Application of the model [Timing <1h]
-
42
Run the function “phecap_predict_phenotype” to generate the predicted probabilities for all subjects.
-
43
Determine the cutoff value for the predicted cases, π+, using the cross-validated accuracy table (“split_roc”) or the validation-set accuracy table (“valid_roc”) created by the “phecap_validate_phenotyping_model” function. Choose π+ to match the desired specificity or PPV. Patients with π greater than or equal to π+ are the predicted cases and labeled as phenotype=yes. Alternatively, the actual predicted probabilities can be directly used for downstream analyses (e.g. genetic association studies) without thresholding.
TROUBLESHOOTING
See Table 3 for troubleshooting guidance.
Table 3.
Troubleshooting table.
| Steps | Problem | Possible reason | Solution |
|---|---|---|---|
| 1 | The prevalence of the phenotype after the filter is much lower than 20% | Suboptimal ICD code selected | Consider either more specific or additional ICD codes |
| 5-6 | Limited resources to extract codified data | Lack of informatics support at some institutions | Reduce the codified list to features such as key medications, procedures or lab results |
| 11 | Failure to process the full articles from website | Incorrect character encoding | Please use UTF-8 for the entire NER or NLP process |
| 14 | Error in R: Failed to connect to database | The UMLS MySQL database has not been set up correctly or the authentication specified in the R code is wrong | Check if the UMLS database is working properly; put correct authentication information in lines 13-16 in “cui_processing.R” |
| 17 | The number of notes with more than 500 characters doesn’t match the number of rows in the result file | Incorrect character encoding of notes | Convert note encoding to UTF-8 |
| Some concepts in the dictionary are not extracted | Possible issue with character encoding in the dictionary file | Convert the dictionary to UTF-8 | |
| 27 | Error in R: <variables> are not found in the dataset | The name for the H feature and the label are not consistent with the column names in the data | Make sure the names specified in line 12 in “main.R” can be matched to the column names of the data |
| 27 | Error in R: ‘feature_transformation’ should be a function or NULL | The transformation given is not a function | By default, the log(1+x) transformation is applied to the ICD counts and CUI counts. To suppress the transformation, use NULL. To use an alternative transformation, for example, add feature_transformation=sqrt in line 12 in “main.R” |
| 35 | Error in R: <surrogate> has too few cases / controls | For rare phenotypes, the patients are so few that one cannot sample as many as 500 patients from each extreme | Sample fewer patients as instructed in step 31. One may also consider changing the lower and upper cutoff values in step 29. However, such changes may lead to fewer features selected in step 35 |
| 35 | Too few features are selected | The subsample size is too small | Instead of sampling 500 patients from each extreme, sample 1000 or more instead. To achieve this, add subsample_size=1000 (or a larger number) in line 34 in “main.R” |
| 38 | Error in R: “Unrecognize specification for method” | Method specified is not included in the options or misspelled | Check the available options for methods and make sure the specified method is correctly spelled |
| 38 | Error in R: “Too few training samples” | The size of training set is too small | Check the percentage of validation labels specified in PhecapData in line 12 in “main.R”, and make sure the percentage of training labels is not too small |
| 38 | Error in R: “Package randomForestSRC not found” | Packages for alternative algorithms have not been installed | Go to R CRAN page, search and install the related packages |
| 41 | The AUC on the validation set differs substantially from the cross-validated estimate of the AUC on the training set | The training and validation labels are created differently, either over different time periods or by different chart reviewers | To resolve this, check the consistency of the labeling process to recalibrate the labels. Alternatively, pool the training and validation sets together and resample, randomly sampling a new training and validation set |
TIMING
Steps 1-2, Creating EMR data mart [TIMING ~24h, varies depending on infrastructure]
Steps 3-4, Conduct chart review and obtain gold standard labels [Timing 1 week, depending on availability of domain expert]
Steps 5-7, Identify and extract codified data features from the structured EMR data for the algorithm [TIMING ~12h]
Steps 8-9, Prepare analysis environment in R [TIMING <1h]
Steps 10-14, Concept collection for candidate NLP features [TIMING ~12h]
Steps 15-22, Note parsing to obtain NLP feature data [TIMING ~20h+; can vary widely depending on size of dataset and size of NLP dictionary]
Steps 23-27, Load EMR data into R [Timing: <1h]
Steps 28-35, Perform surrogate assisted feature selection [Timing <1h]
Steps 36-40, Supervised algorithm training [Timing <1h]
Step 41, Algorithm validation [Timing <1h]
Steps 42-43, Application of the model [Timing <1h]
ANTICIPATED RESULTS
Below, we demonstrate some of the anticipated data from key steps in the phenotype algorithm development process. The key stages include data extraction and curation, algorithm training and validation, and assigning predicted probabilities and case status for all patients of interest.
While data extraction for structured data is more straightforward, we provide anticipated results for the steps involving NLP. In this example, we demonstrate the results of applying MetaMAP on an article for “coronary artery disease” in Wikipedia (Procedure step #13). Figure 5 demonstrates the output from MetaMap indicating that “coronary artery disease” was identified as a clinical term and mapped to three distinct CUIs, C0010054, C0010068, and C1956346. The link to the source article and the coronary artery disease dictionary generated using MetaMap can be found at, https://celehs.github.io/PheCAP/articles/NER-MetaMAP.html.
To generate NLP features, we parse the narrative notes in the EMR to identify and count positive mentions of all CUIs in the dictionary. We provide an example of NLP output using NILE after processing a set of notes (Figure 6). This same set of narrative notes can be found on the i2b2 website, i2b2 NLP Research Data Sets (https://www.i2b2.org/NLP/DataSets/Main.php), “Training: RiskFactors Complete Set 1 MAE” data under “2014 De-identification and Heart Disease Risk Factors Challenge”/. We use “xml_Utils.java” (https://celehs.github.io/PheCAP/articles/NLP-NILE.html) to extract notes from downloaded xml files. We then use the dictionary generated from MetaMap and parse these notes using NILE (Procedure step #15).
Figure 6.

Output from parsing the notes using after processing the i2b2 NLP Research Data Set using NILE (example of results obtained from Procedure step #15).
With the curated feature data along with labels, the algorithm training and validation starts with SAFE (Procedure step 28 - 35) to perform unsupervised feature selection. Next, the coefficients are assigned to the features in the supervised training step using the gold standard labels. Figure 7A shows the coefficients for the selected features after SAFE (Procedure step 38 using the EMR example data available from, https://celehs.github.io/PheCAP/index.html. The sensitivity, specificity, PPV and NPV for a range of probability cut-off values for defining cases are reported in the validation step as part of the R package (Figure 7B). Investigators can choose different threshold values depending on the goal of the study. For example, if the goal for the algorithm is to achieve a PPV >95%, line 9 might be selected as the threshold. In this case, all subjects with a cutoff or a probability of having the phenotype ≥0.853 would be considered a case. The estimated performance of the algorithm at this threshold (p=0.853) corresponds to a false positive rate (FPR), 0.059 or specificity of 94%; true positive rate (TPR) 0.644, or a sensitivity of 64%, and a PPV of 97%. Once the threshold for probability is selected, all subjects with a probability of the threshold or higher are classified as cases. The final output shown in Figure 7C contains predicted probabilities for each patient along with classified case status, 1=yes, 0=no.
Table 2.
Phenotype algorithms developed using the PheCAP framework.
| General clinical categories | Phenotype |
|---|---|
| Cancer | Breast cancer |
| Cardiovascular | Cerebral aneurysms |
| Coronary artery disease | |
| Hypertension | |
| Heart failure | |
| Ischemic stroke | |
| Myocardial infarction | |
| Endocrine | Diabetes Mellitus, Type 1 |
| Diabetes Mellitus, Type 2 | |
| Diabetic neuropathy | |
| Polycystic Ovarian Syndrome | |
| Gastrointestinal | Crohn’s Disease |
| Ulcerative Colitis | |
| Neurology | Multiple sclerosis |
| Epilepsy | |
| Psychiatry | Bipolar Disorder |
| Depression | |
| Schizophrenia | |
| Suicidal ideation in pregnancy | |
| Pulmonary | Asthma |
| Chronic Obstructive Pulmonary Artery Disease | |
| Pediatric pulmonary hypertension | |
| Rheumatology | Axial spondyloarthropathy |
| Rheumatoid arthritis | |
| Systemic Lupus Erythematosus |
EDITORIAL SUMMARY
PheCAP takes structured data and narrative notes from electronic medical records and enables patients with a particular clinical phenotype to be identified.
TWEET
High throughput phenotyping with electronic medical record data
COVER TEASER
High throughput phenotyping with EMR data
RELATED LINKS
Key reference(s) using this protocol
Xia, Z. et al. PLoS One. 8:e78927 (2013) [doi: 10.1371/journal.pone.0078927]
Liao, KP. et al. Ann Rheum Dis. 73, 1170-1175 (2014) [doi: 10.1136/annrheumdis-2012-203202]
Liao, KP. et al. BMJ. 350, h1885 [doi: 10.1136/bmj.h1885]
Ananthakrishnan, AN. et al. Inflamm Bowel Dis. 22:151-158 (2016) [doi: 10.1097/MIB.0000000000000580]
ACKNOWLEDGEMENTS
We would like to thank Rachel Eastwood for her assistance with the figure designs and Zeling He for establishing the PheCAP website.
Support from this project from the NIH P30 AR 072577 (TC1, CH, JH, TC2, KPL) and the VA Office of Research and Development VA Merit Award I01-CX001025 (KC, YLH, JH, DG, HF, COD, JMG); Past support for i2b2 from NIH U54 LM008748 (ANA, ZX, SYS, VG, VC, EWK, RMP, PS, GS, SC, SNM, IK, TC2, KPL) and current support from R01 HG009174 (VG, VC, SNM); ANA receives support from the Crohn’s and Colitis Foundation, NIH, and Pfizer. ZX receives support from the NIH NINDS098023. SH receives support from T32 AR 007530. KPL receives support from the Harold and DuVal Bowen Fund.
Footnotes
COMPETING INTERESTS
RMP is employed at Celgene, however his contributions to the protocol were performed while at Brigham and Women’s Hospital. The remaining authors declare that they have no competing financial and non-financial interests.
DATA AVAILABILITY STATEMENT
The datasets generated or analyzed in this protocol can be downloaded from: https://celehs.github.io/PheCAP/
CODE AVAILABILITY STATEMENT
The R package and code referenced in this protocol can be downloaded from: https://celehs.github.io/PheCAP/
REFERENCES
- 1.Brownstein JS et al. Rapid identification of myocardial infarction risk associated with diabetes medications using electronic medical records. Diabetes care 33, 526–531 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denny JC et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31, 1102–1110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurreeman F et al. Genetic basis of autoantibody positive and negative rheumatoid arthritis risk in a multi-ethnic cohort derived from electronic health records. American journal of human genetics 88, 57–69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao KP et al. Associations of autoantibodies, autoimmune risk alleles, and clinical diagnoses from the electronic medical records in rheumatoid arthritis cases and non-rheumatoid arthritis controls. Arthritis and rheumatism 65, 571–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canela-Xandri O, Rawlik K & Tenesa A An atlas of genetic associations in UK Biobank. bioRxiv (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaziano JM et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–223 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Banda JM, Halpern Y, Sontag D & Shah NH Electronic phenotyping with APHRODITE and the Observational Health Sciences and Informatics (OHDSI) data network. AMIA Jt Summits Transl Sci Proc 2017, 48–57 (2017). [PMC free article] [PubMed] [Google Scholar]
- 8.Kho AN et al. Electronic medical records for genetic research: results of the eMERGE consortium. Science translational medicine 3, 79re71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirby JC et al. PheKB: a catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc 23, 1046–1052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Malley KJ et al. Measuring diagnoses: ICD code accuracy. Health services research 40, 1620–1639 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao KP et al. Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res (Hoboken) 62, 1120–1127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao KP et al. Methods to develop electronic medical record phenotype algorithms incorporating natural language processing. BMJ, h1885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu S et al. Surrogate-assisted feature extraction for high-throughput phenotyping. J Am Med Inform Assoc 24, e143–e149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu S et al. Toward high-throughput phenotyping: unbiased automated feature extraction and selection from knowledge sources. J Am Med Inform Assoc 22, 993–1000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro VM et al. Validation of electronic health record phenotyping of bipolar disorder cases and controls. Am J Psychiatry 172, 363–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy SN et al. Serving the enterprise and beyond with informatics for integrating biology and the bedside (i2b2). J Am Med Inform Assoc 17, 124–130 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Son JH et al. Deep Phenotyping on Electronic Health Records Facilitates Genetic Diagnosis by Clinical Exomes. American journal of human genetics 103, 58–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen LV et al. Design patterns for the development of electronic health record-driven phenotype extraction algorithms. Journal of biomedical informatics 51, 280–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basile AO & Ritchie MD Informatics and machine learning to define the phenotype. Expert Rev Mol Diagn 18, 219–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ananthakrishnan AN et al. Improving case definition of Crohn’s disease and ulcerative colitis in electronic medical records using natural language processing: a novel informatics approach. Inflamm Bowel Dis 19, 1411–1420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll RJ et al. Portability of an algorithm to identify rheumatoid arthritis in electronic health records. J Am Med Inform Assoc (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Z et al. Modeling disease severity in multiple sclerosis using electronic health records. PLoS One 8, e78927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ananthakrishnan AN et al. Association Between Reduced Plasma 25-Hydroxy Vitamin D and Increased Risk of Cancer in Patients With Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol 12, 821–827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai T et al. The Association Between Arthralgia and Vedolizumab Using Natural Language Processing. Inflamm Bowel Dis 24, 2242–2246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao KP et al. Association between low density lipoprotein and rheumatoid arthritis genetic factors with low density lipoprotein levels in rheumatoid arthritis and non-rheumatoid arthritis controls. Annals of the rheumatic diseases 73, 1170–1175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurreeman FA et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. American journal of human genetics 90, 524–532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada Y et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ananthakrishnan AN et al. Common Genetic Variants Influence Circulating Vitamin D Levels in Inflammatory Bowel Diseases. Inflamm Bowel Dis 21, 2507–2514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinnott JA et al. Improving the power of genetic association tests with imperfect phenotype derived from electronic medical records. Human genetics 133, 1369–1382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halpern Y, Horng S, Choi Y & Sontag D Electronic medical record phenotyping using the anchor and learn framework. J Am Med Inform Assoc 23, 731–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agarwal V et al. Learning statistical models of phenotypes using noisy labeled training data. J Am Med Inform Assoc 23, 1166–1173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S et al. Enabling phenotypic big data with PheNorm. J Am Med Inform Assoc 25, 54–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindberg DA, Humphreys BL & McCray AT The Unified Medical Language System. Methods of information in medicine 32, 281–291 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jupp S, Burdett T, Leroy C & Parkinson HE A new Ontology Lookup Service at EMBL-EBI. SWAT4LS, 118–119 (2015). [Google Scholar]
- 35.Savova GK et al. Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): architecture, component evaluation and applications. J Am Med Inform Assoc 17, 507–513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goryachev S, Sordo M & Zeng QT A suite of natural language processing tools developed for the I2B2 project. AMIA Annu Symp Proc, 931 (2006). [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Wagholikar KB, Siddhartha J & Sohn S Integrated cTAKES for Concept Mention Detection and Normalization. CEUR Workshop Proceedings 1179, http://ceur-ws.org/Vol-1179/ (2013). [Google Scholar]
- 38.Aronson AR Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program. Proc AMIA Symp, 17–21 (2001). [PMC free article] [PubMed] [Google Scholar]
- 39.Yu S & Cai T A short introduction to NILE. arXiv preprint arXiv:1311.6063 (2013). [Google Scholar]
- 40.Manning C et al. The Stanford CoreNLP natural language processing toolkit. Proceedings of 52nd annual meeting of the association for computational linguistics: system demonstrations, 55–60 (2014). [Google Scholar]
- 41.Chapman WW, Bridewell W, Hanbury P, Cooper GF & Buchanan BG A simple algorithm for identifying negated findings and diseases in discharge summaries. Journal of biomedical informatics 34, 301–310 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Castro VM et al. Large-scale identification of patients with cerebral aneurysms using natural language processing. 88, 164–168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro V et al. Identification of subjects with polycystic ovary syndrome using electronic health records. Reprod Biol Endocrinol 13, 116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jorge A et al. Identifying lupus patients in electronic health records: Development and validation of machine learning algorithms and application of rule-based algorithms. Semin Arthritis Rheum (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perlis RH et al. Using electronic medical records to enable large-scale studies in psychiatry: treatment resistant depression as a model. Psychol Med 42, 41–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doss J, Mo H, Carroll RJ, Crofford LJ & Denny JC Phenome-Wide Association Study of Rheumatoid Arthritis Subgroups Identifies Association Between Seronegative Disease and Fibromyalgia. Arthritis Rheumatol 69, 291–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geva A et al. A Computable Phenotype Improves Cohort Ascertainment in a Pediatric Pulmonary Hypertension Registry. J Pediatr 188, 224–231 e225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]