

Abstract
Objective:
In gout, autoinflammatory responses to urate crystals promote acute arthritis flares. Tophi, chronic synovitis, and erosion pathogeneses are less well understood. Defining epigenomic immunity training can reveal novel pathogenesis factors and biomarkers. Here, gout epigenome-wide analyses seminally probed differential DNA methylation.
Methods:
San Diego gout cohort (n=16, n=14 healthy controls) peripheral blood mononuclear cell (PBMC) methylome data were processed with ChAMP package in R. Methods for Transcription Factor (TF)-gene network analyses used ENCODE data and Taiji. New Zealand Māori whole blood DNA provided an independent gout validation cohort (n=13, n=16 controls).
Results:
Differentially methylated loci clearly separated gout from controls by hierarchical clustering and principal component analyses. Multiple differentially methylated gout risk genes included IL23R, which mediates granuloma formation and cell invasion. Differential methylome pathway enrichment was detected for B and T cell receptor signaling, Th17 differentiation and IL-17 signaling, and convergent longevity regulation, circadian entrainment, and AMP-activated kinase signaling that impact inflammation via IGF1R, PI3K-AKT, NF-κB, mTOR signaling, and autophagy. Cohorts overlapped for 37 of the 70 (52.9%) TFs with hypomethylated sequence enrichment, and 30 of 38 (78.9%) enriched KEGG pathways identified via TFs. Shared differentially methylated gout TF-gene networks, including NF-κB activation-limiting TFs MEF2C and NFATC2, identified osteoclast differentiation as the most strongly weighted differentially methylated pathway that overlapped in both gout cohorts.
Conclusion:
Seminal findings of differential DNA methylation of networked signaling, transcriptional, innate and adaptive immunity, and osteoclastogenesis genes and pathways suggest novel therapy targets for flares, tophi, chronic synovitis, and bone erosion in gout.
Keywords: AMPK, Longevity regulation, osteoclastogenesis, MEF2C, NFATC2
Introduction:
Acute gouty arthritis is promoted by NLRP3 inflammasome-driven autoinflammatory responses to urate crystals (1,2). Gout flares are characteristically superimposed on urate crystalline macroaggregates including in tophi, the majority of whose volume is composed of variable foci of fibrosis, chronic synovitis, and granulomatous changes (3,4). B and T lymphocytes, and plasma cells are among the cells surrounding crystals in tophi (5). Osteoclastogenesis also is prominent at the tophus-bone interface, and linked with bone erosion (3). Enhanced osteoclastogenesis in vitro of RANKL- and M-CSF-treated PBMCs in severe, tophaceous disease has suggested molecular programming in PBMCs in advanced gout (6). However, major knowledge gaps remain for mechanisms promoting chronic synovitis, tophus development and associated granulomatous changes, and bone erosion in gout (1–3,5,6).
Altered innate and adaptive immunity in gout could reflect dietary and genetic factors (1), including effects of common genetic variants linked to ion transport, and purine, carbohydrate, or lipoprotein metabolism (7–9). However, gout-associated gene variants generally have weak effects on gene expression, or transcript processing or stability (7). Epigenetic changes regulate gene expression, and cell differentiation and function, in response to diet, lifestyle, and other factors (10,11). Epigenetic training of both adaptive and innate immunity, including by reversible histone modifications (12,13), regulates inflammation phenotypes. Notably, hyperuricemia modulates inflammatory responses of monocytes, and increases murine urate crystal-induced inflammation in vivo (13). Repeated leukocyte exposure to urate crystals, IL-β, and other inflammatory mediators also could modify the epigenome (12,13).
Epigenetic changes include DNA methylation, defined by covalent methyl group binding to a 5′ carbon cytosine nucleotide, typically followed by guanine (termed CpG)(11). This relatively stable epigenetic change regulates myeloid and lymphoid lineage development (14) and inflammatory gene expression to environmental stressors, aging, pathogens, endogenous danger signals, and many cytokines (11). DNA methylome changes are implicated in obesity and type 2 diabetes (15), co-morbidities frequently associated with gout (16). Moreover, chemokine CCL2 promoter hypomethylation was reported in gout (17). Here, we analyzed gout epigenome-wide DNA methylation.
Methods
Subject recruitment
Work was UC San Diego (SD) IRB pre-approved, and informed consents were obtained from all subjects. Gout outpatients, meeting ACR/EULAR criteria (18), and controls were recruited by rheumatologist referral, invitation via health record search, and solicitation from healthy subject pools. Hemodialysis or stage 4 CKD subjects were excluded. Gout patients (n=17) were recruited first, followed by individually matching 16 healthy controls, without past gout, to 16 of the 17 gout patients, for sex, age (≤9 years difference), and BMI less or greater than 30. Sixteen gout and 14 controls agreed to DNA testing.
All subjects had one visit, with limited clinical lab studies (serum urate, serum creatinine), and examination for palpable tophi. No SD subjects were smokers. Upon blood sampling, gout subjects had to be ≥ 7 days removed from corticosteroid or colchicine use, and ≥ 14 days from resolution of last gout flare.
NZ subjects from a NZ Health and Disability Ethics Committee IRB-approved diabetes-focused sub-study of n=40 males and n=40 female Māori of Aotearoa/NZ (19) gave informed consent for genomic DNA drawn from whole blood. Forty self-reporting type 2 diabetes, and 40 controls, were matched by sex, age, and BMI. Selection was prioritized to controls with normal fasting glucose, or secondarily to normolipidemic controls without evident heart disease. In all, 30 gout (23 male, 7 female) and 23 without gout (16 male, 7 female) subjects meeting 1977 ACR gout classification criteria were considered for inclusion. After those missing smoking and CKD status were excluded, 13 with gout and 16 without gout were analyzed.
PBMCs and DNA isolation and methylation assay and data pre-processing
SD cohort PBMCs were isolated from EDTA-anticoagulated blood using Ficoll-Paque Plus (GE Healthcare, Chicago, IL). DNA was isolated from washed PBMC pellets via DNeasy Blood Kit (Qiagen), and concentrated using Amicon Ultra (Millipore, Temecula, CA). NZ cohort whole blood DNA was isolated by a GuCl-guanadinium-based method. DNA was converted by sodium bisulfite modification and hybridized to Infinium MethylationEPIC BeadChip arrays (Illumina, San Diego, CA). Samples were randomized across plates and chip positions.
Illumina Methylation Beadarray 850K flatform data were processed with ChAMP package (20) in R. Probes were filtered based on the following conditions (champ.filter function): (i) probes with detected P-value > 0.01; (ii) probes with < 3 beads in ≥5% of samples examined; (iii) all non-CpG probes; (iv) all SNP-related probes; (v) all multi-hit probes; (vi) all probes in chromosome X and Y. The masking list included probes with SNPs close to the 3’end, probes with a SNP in the extension base that cause a color channel switch from the designated annotation, and probes with reason to be mapped (21). SNP probes on the array were also removed. Methylation at each locus was reported as β values, from 0 (un-methylated) and 1 (fully methylated). BMIQ (champ.norm function) was applied for normalization. PCA analysis showed top PCs significantly correlated with slide and array (champ.SVD function), and batch effects were corrected for normalized data (champ.runCombat function).
Cell type composition adjustment, and identification of gout-associated differential methylation loci (DML) and mapping to genes
Cell type composition is a co-factor in DNA methylation from blood samples. We downloaded healthy donor leukocyte type-selective DNA methylation data from GEO-NCBI dataset GSE110554 accession number, as reference for DNA methylation measured on the 850K platform. Healthy donor cell types (n indicating number of reference donors) were neutrophils (Neu, n=6) for whole blood in the NZ cohort, and, in both cohorts, monocytes (Mono, n=6), B lymphocytes (Bcells, n=6), CD4+ T cells (CD4T, n=7), CD8+ T cells (CD8T, n=6) and natural killer cells (NK, n=6). Automatic selection in minfi package was used to identify probes specific to each cell type. The top 50 hyper- and hypomethylated probes were selected for each cell type. In total, 600 probes were used to estimate cell type composition. We estimated cell type distribution as described (22), and added cell type proportion to the linear regression to adjust their effects in both cohorts as described (23). Other co-factors corrected were age and smoking (Supplementary Figure 1). For the SD cohort, ethnicity was included in the linear regression. DML related to gout were identified using limma package (24) in R, for differential analysis of microarray, RNA-seq or Methylation using linear models. For both cohorts, the Benjamini-Hochberg procedure (i.e., false discovery rate (FDR)) was used for multiple hypothesis testing correction. Two conditions were used to weight DMLs: (i) FDRs <0.05 and (ii) differences of average β values > 99% quantile. To adjust for effects of allopurinol, we fitted a linear regression using data from gout cases only. Two criteria were applied to identify DML related to allopurinol: (i) P-value<0.05 (due to the small sample size, a liberal significance threshold was used); (ii) selection of the top 1% probes with the largest difference of beta value between taking and not-taking allopurinol. For the downstream analyses, we used the DML sets derived after removing the overlapping DML related to allopurinol.
Mapping from DML to gene was based on “MethylationEPIC_v-1–0_B4.csv” provided by Illumina. We classified DML-genes based on overall DML changes and locations of DML. “Hypermethylated” designated sites where gout cases had more DNA methylation, and “hypomethylated” to sites where gout cases had less DNA methylation. DML-genes with only one differential methylated probe were one of promoter-hypo, promoter-hyper, body-hypo or body-hyper. DML-genes with ≥ 2 differential methylated probes were classified based on each DML. For example, if there were 2 hypo DML within a gene, the classification was “promoter-hypo;body-hypo”. Based on UCSC_RefGene_Group from annotation file, TSS200 and TSS1500 were defined as promoter. Other features were defined as gene-body.
Hierarchical clustering, PCA and pathway enrichment analysis, TF binding sites and TF-gene networks, and statistics
Based on DML, hierarchical clustering was employed via the heatmap.2 function in R with “maximum” as distance function and “ward.D2” as agglomeration method. Principal component analysis (PCA) was performed for the visualization of DML (plotMDS function). Because neither hierarchical clustering nor PCA tested a null hypothesis, no P-values were used to test significance. KEGG pathway enrichment analysis was performed based on DML-genes set, using ConsensusPathDB (25) (http://consensuspathdb.org/)). For enriched pathways: (i) overlaps between DML-genes and pathway genes were >2; (ii) q-value was < 0.01. R package KEGGprofile was used for visualizing KEGG pathways. Keywords “GWAS” and “gout” were used for searching the literature related to GWAS results, with 110 GWAS gout genes tallied (Supplementary Table 1). For TF binding sites in DML regions, sequences from 250bp upstream to 250bp downstream of DML position were extracted; 500bp-sequences were divided into hypo- and hyper- sets based on DML status. AME (26) from MEME suite was employed to find motifs enriched in these two sets, separately, with motif set downloaded from JASPAR (JASPAR2018_CORE_vertebrates_non-redundant.meme file)(27). We ran AME with parameters: --verbose 1 --scoring avg --method fisher --hit-lo-fraction 0.25 --control --shuffle-- --kmer 2. Motifs were enriched in sequence set if corresponding E-value was below 1e-10.
For TF-gene network analyses comparing PBMCs from the SD and NZ cohorts, ATAC-seq data were downloaded, from the ENCODE project, for B cells, T cells, CD14+ monocytes, CD4+ T helper cells, and CD8+ alpha-beta T cells. For each cell type, a TF-gene regulatory network was constructed by Taiji (28) to assess global influence of each TF from network perspective. The top 10% of edges by weights from Taiji were kept. Then networks were combined, into which the 335 DML-genes falling into common classes to both cohorts were mapped (including two TFs with differential methylation and hypo-methylated sequence enrichment in both cohorts), leaving 224 genes and 1,232 edges. Modules were identified using (Affiliation Graph Model) AGMfit algorithm to detect network communities (i.e. modules) by fitting a probabilistic generative model for given networks using maximum likelihood estimation. Fourteen TFs were identified on the basis of membership in all 5 different modules, and as DML genes in both cohorts. Since genes were ranked by number of modules, without hypothesis testing, no P-values were used to test significance. For the DMLs, estimates, standard errors, and P-values in the two cohorts are listed in Supplementary Material Appendix Tables 1–2. Supplementary Material Appendix Excel File 1 provides data for relationships between GWAS genes, CpG sites, SNP sites, and linkage disequilibrium (LD) blocks in different populations.
Results
Assessment of the gout DNA methylome signature
Most SD gout subjects were on stable allopurinol, with serum urate overall comparable to healthy controls (Supplementary Table 2). Study workflow is charted in Supplementary Fig. 2. Epigenome-wide DNA methylation covered 865,859 loci. After filtering low quality probes, 755,223 probes remained, with 5,438 promoter or gene body DML. Of detected DML, 96.7% were hypomethylation changes in gout, mostly (80.3%) in open sea regions (i.e. >4kbp away from CpG islands). The fewest DML (3.1%) were in CpG islands (typically located at promoters with CG:GC ratio >0.6) and N Shelf ( 2–4 kb upstream of CpG island)(Fig. 1C ). Gout DML were mapped to 2,488 genes (DML genes), with 70.8% demonstrating gene body hypomethylation, which modulates gene expression, depending on the context and whether or not the specific CpG site is in an enhancer region that is relevant to that specific gene or neighboring genes. DML allowed clear separability of gout from controls, shown by hierarchical clustering (Fig. 1A), and PCA (Fig. 1B).
Figure 1. Overview of differential DNA methylation in the SD gout cohort.

(A) Hierarchical clustering separated case and control into two groups. DNA origins from cases and controls are labelled with red and blue, respectively. Methylation probes are shown in rows, and samples in columns. (B) Principal component (PC) analysis also strongly separated gout cases and controls. Proportion of Variance: PC1 64.15%; PC2: 6.50%. In total, PC1 and PC2 contain 70.65 % of the variance. PC1: the first principle component. PC2: the second principle component. (C) Genomic locations of the 5,438 differential methylation loci (DML). Shore = 0–2 kb from island. Shelf = 2–4 kb from island. N = upstream (5’) of CpG island. S = downstream (3’) of CpG island.
Enrichment of GWAS genes in gout DML-genes and identification of differentially methylated pathways
We found 22 DML-genes overlapped to known gout risk genes from past GWAS (Fig. 2A, Supplementary Table 3), with methylation level box plots shown in Fig. 2D. Several loci encode gene products involved in vascular function, or ion transport (eg., SLC2A9, ABCC9). Other loci modulate innate and/or adaptive immunity, such as PRKG2, and BDKRB2 that encodes a bradykinin receptor supporting neutrophil recruitment in experimental gout (29). The PPARGC1A product drives mitochondrial biogenesis, inhibits insulin resistance and is anti-inflammatory. The TRPA1 product mediates pain and neuroinflammation. The probe cg13841979 of SLC2A9 and previous GWAS-identified rs1014290 were in the same linkage disequilibrium (LD) block, as were cg05205932 and rs7688672, rs6837293 for PRKG2 (not shown). DML-genes with the DML and gout-associated SNPs in the same LD block also included A1CF, CFTR, ABCC9, MYL2, PDZK1 and B4GALT1 (not shown). These findings buttressed potential functional impact of GWAS SNPs in gout.
Figure 2. Differentially methylated loci (DML) genes and the associated pathways in the SD gout cohort.
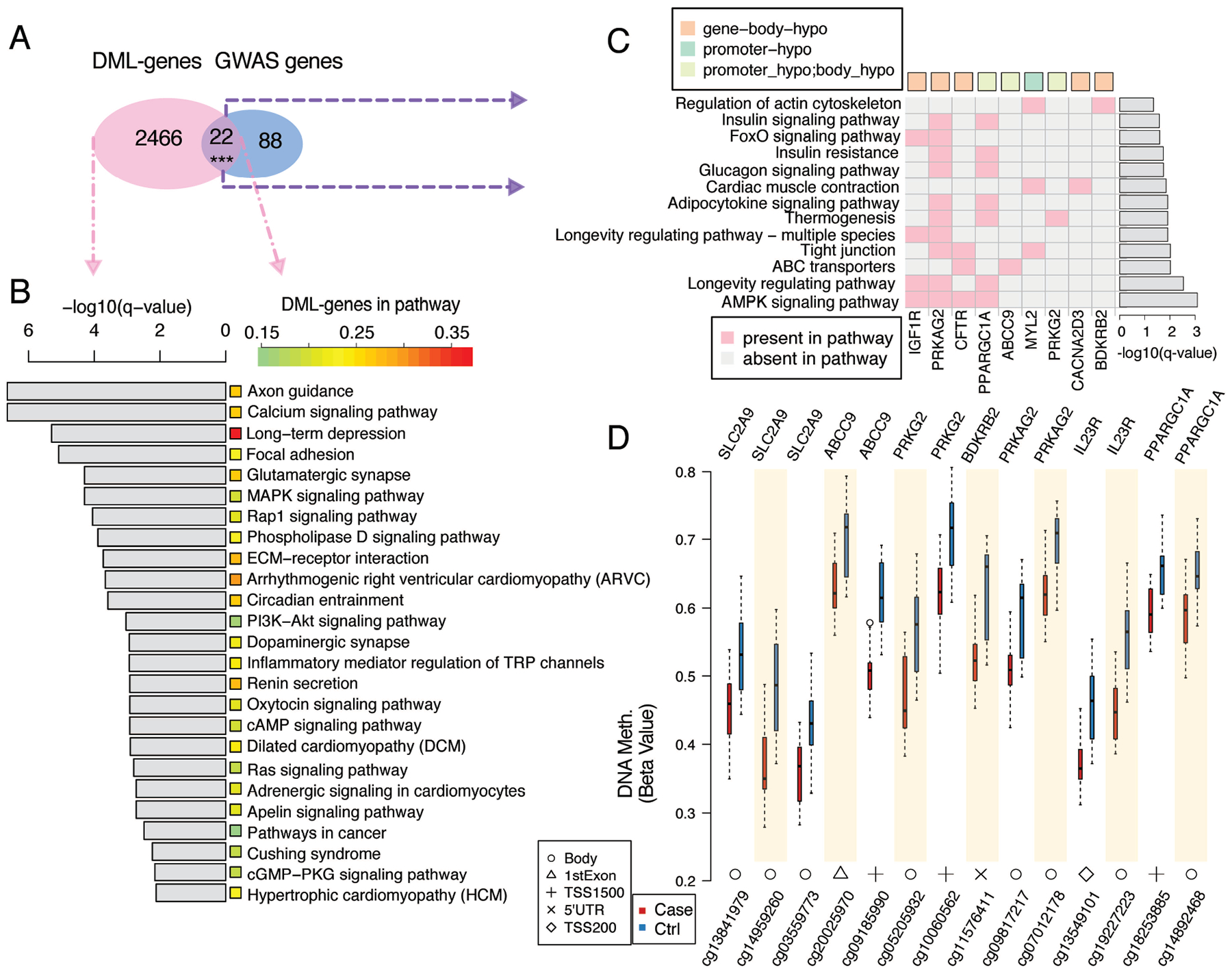
(A) Number of DML genes and overlaps with gout genetic risk loci genes identified from previous GWAS studies. ***P-value = 3.3e-04 from hypergeometric distribution. (B) Signaling pathways (right) strongly associate with the DML genes. For each pathway, the significance is shown as the grey bar on the left and the percentage of DML-genes shown by a color bar in the middle. (C) Pathways associated with the 9 DML genes that overlapped with the gout genetic risk loci genes from past GWAS data. Green and orange squares represent hypomethylation at promoter or gene body regions respectively. Light green represents DML-genes with multiple DML. (D) Methylation levels of the CpG sites in 7 DML-genes in cases and controls. Locations of these CpG sites are shown at the bottom of the boxplots. TSS200 = 0–200 bases upstream of the transcriptional start site (TSS). TSS1500 = 200–1500 bases upstream of the TSS. P-values for pathways in (b) and (c) were calculated using the hypergeometric distribution and transformed into q-value for the multiple hypothesis testing correction.
We detected hypomethylation of IL23R (cg19227223, cg13549101), SLC2A9, ABCC9, PRKG2, and BDKRB2 (Fig. 2D). Several enriched KEGG pathways (Fig. 2B, C)(eg., renin and insulin secretion) relate to hyperuricemia and hypertension, metabolic syndrome, and type 2 diabetes (16). Others included phospholipase D, regulation of the actin cytoskeleton and Rap1 signaling, cell migration, cell adhesion, extracellular matrix (ECM)-receptor interaction, and MAPK and calcium signaling pathways. Also detected were DML enrichment in circadian entrainment (Fig. 2B), and inflammatory mediator regulation of transient receptor potential (TRP) channels involved in neuroinflammation (Fig. 2B).
AMPK, a nutritional biosensor suppressed by dietary excesses and modulated by inflammation, exerts broad effects on the epigenome, is in turn regulated by epigenetic changes, and limits urate crystal-induced inflammation (30,31). PRKAG2, which encodes an AMPK heterotrimer γ subunit isoform, was gene body hypomethylated (cg09817217, cg07012178) in gout (Fig. 2D). AMPK signaling was among KEGG pathways enriched in both the DML and GWAS sets (Fig. 2C)(q-value 8.1e-04)(Supplementary Fig. 3A.) PPP2R5C (Protein phosphatase 2A regulatory subunit B family) negatively regulates AMPK activity via phosphatase activity for the AMPKα chain catalytic site threonine. In this study, PPP2R5C was also gene body hypomethylated. AMPK-intersecting pathways with DML enrichment in gout included PI3K-AKT, circadian entrainment, and longevity regulation (32) (Fig. 2B, C) (Supplementary Figs. 3B–D).
Transcription factor (TF) motif scanning in DML regions and pathway changes
Transcriptional signaling controls leukocyte development, differentiation, and function. Here, we identified 75 hypomethylated TF motifs, corresponding to 65 TFs, enriched in the 500bp sequences centered at the hypomethylated loci. Examples included circadian clock regulators CLOCK and aryl hydrocarbon nuclear receptor (ARNT) and genes encoding STAT2 and multiple components of AP-1, interferon regulatory transcription factor 1 (IRF1)(33) and Nuclear factor of activated T cells (NFAT)C2 and NFATC3. Among top identified TFs (Fig. 3A), NFATC2 was one of two TFs that themselves were hypomethylated in the gene body region, and all genes regulated by NFATC2 were differentially methylated. MEF2C (34) also had gene-body hypomethylation in gout (Fig. 3A). For NFATC2, one DML was intronic. For MEF2C, one CpG was in an exon region. Specific examples of motifs of NFATC2 and MEF2C showing the conserved DNA binding pattern are shown in Fig. 3B. Detected CpG sites and methylation levels for the CpG sites along the NFATC2 and MEF2C genes in gout, compared to the control cohort, are depicted in Fig. 3C and Fig. 3D, respectively.
Figure 3. Transcription Factor (TF) motifs, CpG sites, and differential DNA methylation in the SD gout cohort.
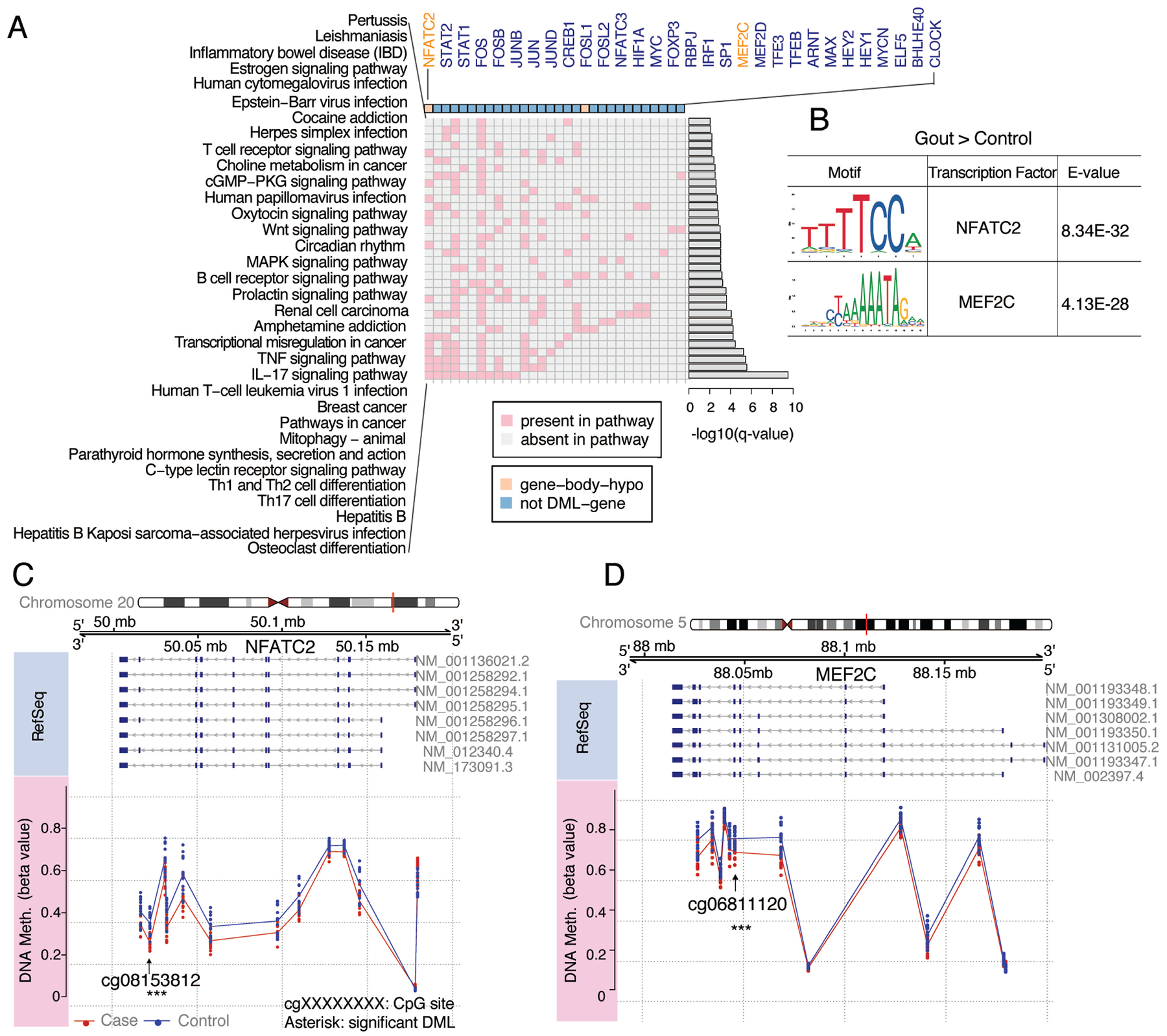
(A) Top biological pathways in gout identified through TF motif scanning in hypo-DML regions. TFs associated with the enriched pathways (rows) are listed in columns. These TFs were either DNA hypomethylated (orange) at the gene body in gout cases or not differentially DNA methylated (blue). P-values for pathways were calculated using hypergeometric distribution and transformed into a q-value (B) Examples of motifs of the significantly enriched TFs in gout cases. E-value is a measurement for significance of a motif. Detection of CpG sites along the TFs NFATC2 (C) and MEF2C (D) is shown. The first track is the chromosome with the gene region highlighted in red. The second track is the genome coordinate. The third track is RefSeq annotation for the gene. NM_XXXXX is the transcript ID for the gene. Exons and introns are labelled by dark blue rectangle and grey line, respectively. The last track is the methylation level for CpG sites along genes in gout and control cohorts. Significant DMLs are labeled with asterisks.
Analyses of gout DNA methylome changes in certain TFs pointed to differential DNA methylation of multiple biological pathways (Fig. 3A), including osteoclast differentiation (Fig. 3A, Supplementary Fig. 4). Multiple KEGG pathways central to adaptive immunity, specifically B cell receptor signaling, Th1 and Th2 cell differentiation, Th17 cell differentiation, and the IL-17 signaling pathway, also were linked to differential DML and DNA methylated TFs in PBMCs (Fig. 3A).
Validation of DNA methylation signature of genes, pathways, and TFs in an independent cohort
The NZ cohort (Supplementary Table 4), previously selected to examine type 2 diabetes, was exclusively of NZ Māori and Cook Island Māori (East Polynesian) descent, and enriched for obesity and hypertension (19). Using the same cutoff as for the SD cohort, 4845 DML were identified, corresponding to 2530 genes. Hierarchical clustering and PCA analysis clearly separated NZ gout and controls (Fig. 4A, B).
Figure 4. Analysis of the DNA methylome in the NZ gout validation cohort.

(A) Heatmap plot using all DMLs in the NZ cohort. The hierarchical clustering separated gout cases and controls. (B) The PCA plot also shows clear separation between gout cases and controls in the NZ cohort. (C)-(F) Venn diagrams illustrate sharing of DMLs, and related changes, between the SD cohort and NZ cohort. (C) Enriched pathways based on DML-genes. P-value: 2.7e-14 (D): Enriched pathways based on the overlaps between DML-genes and GWAS genes. (E): TFs whose motifs are enriched in hypomethylated DML regions. P-value ≤ 5e-324.(F): Enriched pathways based on TFs. P-value ≤ 5e-324. ***P-value is calculated from hypergeometric distribution.
To probe for the most fundamental methylome changes in gout, we assessed and ranked sharing of methylome findings by looking at DML genes, pathways enriched in DMLs, and TFs and related gene networks. Both cohorts shared 656 of 4,362 DML genes (P-value ≤ 5e-324 from hypergeometric distribution) (Fig. 5A) and 17 of 40 KEGG pathways (Fig 4C, Fig. 5B). The average of -log10(q-value) in both cohorts indicated overlap strength. Shared pathways included circadian entrainment, MAPK signaling, extracellular matrix-receptor interaction, focal adhesion. When assessing overlaps between NZ DML genes and gout GWAS genes, 15 KEGG pathways were enriched, with 5 shared with the SD cohort (Fig. 4D, Fig. 5C and Supplementary Fig. 5A). Strikingly, SD and NZ overlapped for 37 of the 70 (52.9%) identified TFs with motifs enriched in hypo-methylated sequences (Fig. 4E), and 30 of 38 (78.9%) enriched KEGG pathways overlapped based on TFs (Fig. 4F, Fig. 5D and Supplementary Fig. 5B). The strongest overlap was for osteoclast differentiation (Fig. 5D), and differential methylome changes in Th17 cell differentiation, Th1 and Th2 cell differentiation and B cell receptor signaling pathways were supported by the overlap findings (Fig. 5D).
Figure 5. Concordance between San Diego (SD) and New Zealand (NZ) gout sample sets differential DNA methylation findings.

(A) Venn diagrams for DML-genes between the SD and NZ cohorts. ***P ≤ 5e-324 from hypergeometric distribution. (B) Enriched KEGG pathways using DML-genes of the NZ cohort. P-value for each pathway was obtained using the hypergeometric distribution and transformed into a q-value. Sub-class for each pathway is indicated by the colored square. Blue color for pathway names indicates a pathway common to the SD and NZ cohorts. Italics indicates a NZ-cohort-specific pathway. Overlap weight for common pathways is average of the -log10(q-value) in SD and NZ cohorts. (C) Enriched pathways identified from overlaps between DML-genes of the NZ cohort and GWAS genes. (D) Enriched pathways in gout, identified from specific TFs by motif scanning in hypo-DML regions of the NZ cohort.
Integration of gout-related DML with TF-gene regulatory networks
By analyses employing Taiji methodology, NFATC2 and MEF2C, and 12 other TFs, including several involved in retinoid or E26 transformation-specific (ETS) transcriptional programming, exerted effects propagated throughout the entire leukocyte TF-gene regulatory network (Fig. 6). Known biologic effects of these 14 TFs pertinent to gout and/or gout co-morbidities (individually described in Supplementary Table 5) included modulation of myeloid differentiation, inflammation and immunity.
Figure 6. Shared TF-gene regulatory networks between both San Diego and New Zealand cohorts from integration of common gout-related DML-genes.

The TF-gene regulatory networks were constructed, using Taiji methodology as described in the Methods, in B-cell, T-cell, CD14+ monocytes, CD4+ helper T cells and CD8+ alpha-beta T cells, respectively, and then combined into one network. After mapping DML-genes and TFs to the network, 224 DML-genes and 1231 edges remained. Five modules were identified using the AGMfit algorithm. Genes were colored based on module membership, with presence in ≥ 2 modules in olive green. Supplementary Table 5 summarizes properties of the 14 genes present in all Modules, depicted here in red. Shape schemes of nodes are: diamond, body-hypermethylation; Ellipse, body-hypomethylation; Triangle, promoter-hypermethylation; V-shape, promoter-hypomethylation; Rounded rectangle: DML-gene with multiple DML.
Discussion
This study probed the gout PBMC DNA methylome, which was clearly separable from controls. In gout, epigenome-wide differential DNA methylation was highlighted by changes in gout genetic risk genes, signaling and transcriptional pathways in innate and adaptive immunity, neuroinflammation, and osteoclastogenesis. Many results from the SD cohort, particularly for differentially methylated TF motifs and related pathways, were validated in the NZ gout cohort. Study design limited potential confounding effects from current hyperuricemia, ongoing acute gouty arthritis, colchicine or corticosteroid use, and age, sex and some comorbidities.
The gout DNA methylome signature could reflect combined effects of inherited genetic variants (1,7–9) and of past hyperuricemia (13), as well as adaptations to age, and diet, lifestyle exposures and co-morbidity stressors, (11,15, 35). Other contributors likely include innate immunity training effects via past bouts of systemic stress triggered by repeated, self-limiting flares of marked, acute inflammatory arthritis (12). Significantly, the circulating monocyte DNA methylome in RA, in a small study of 33 with RA, was separable from 17 healthy controls, reflected differences in RA disease activity, demonstrated plasticity in response to therapy, and was associated with distinct markers of the inflammatory state in RA peripheral blood (36). TNF and interferons can induce some of the monocyte DNA methylome changes in vitro associated with high RA disease activity in RA circulating monocytes (36). Monocyte DNA methylome changes with high disease activity in RA were pinpointed in certain ETS family TFs (36), and effects of two ETS TF-gene regulatory networks were propagated throughout the entire leukocyte TF-gene regulatory network in gout patients in this study.
We saw differential DNA methylation in several gout GWAS genes relevant to gouty arthritis, including IL23R, which modulates bridging of innate and adaptive immunity, and promotes granuloma formation and cell invasiveness. PRKAG2 encodes the γ2 chain of AMPK, a major nutrition biosensor that is a native suppressor of urate crystal-induced inflammation, partly by promoting autophagy and inhibiting NF-κB and NLRP3 inflammasome activation (30). AMPK activation also mediates anti-inflammatory effects of colchicine (30). Through IGF1R and PI3K-AKT signaling, mTOR signaling, autophagy and mitophagy, and NF-κB activation, AMPK signaling intersects with the longevity regulation pathway, also a modulator of innate inflammation (32). AMPK signaling also intersects with circadian rhythm. Circadian entrainment modulates diurnal changes in phagocyte behavior (37) that could promote the nocturnal flare onset so characteristic in gout (38). The PI3K-AKT pathway regulates inflammatory monocyte/macrophage activation by downstream signaling by HIF-1, the product of a gout genetic risk locus, and via intersection with mTOR activity and AMPK in regulating anti-inflammatory autophagy.
Gout DNA methylome changes were detected in multiple pathways fundamental in phagocyte migration and activation, including for cytoskeletal organization, and cell adhesion and migration. The gout DNA methylome also included changes in the TRP ion channels signaling pathway that acts in endothelial cells and leukocytes, and mediates neuroinflammation, including in response to urate crystals in vivo (39).
Many interconnected TF-gene networks and signaling pathways had differential DNA methylation in gout, including osteoclastogenesis (3). Enhanced osteoclastogenesis in vitro of RANKL- and M-CSF-treated gout patient PBMCs and joint fluid mononuclear leukocytes was reported in severe, tophaceous gout, associated with increased circulating RANKL and M-CSF (6). Origins and site(s) of priming of osteoclasts precursors (ie, bone marrow, peripheral blood, synovial lining) that develop into osteoclasts remain to be clarified in gout. Characteristically, bone erosion takes years to be evident by imaging in gout (4). Our results suggest that epigenomic training potentially modulating bone erosion is identifiable even in relatively early gout, and even with reasonable control of serum urate.
This study identified gout-associated enrichment of differential DNA methylation in pathways for B and T cell receptor signaling, Th17 development and IL-17 signaling, and of DMLs for STAT2, IRF1, and NFATC2, and NFATC3. These findings collectively pointed to changes in adaptive immunity programming in gout. Not only IL-1β and IL-17 (40), but also potentially IRF1 and STAT2, could help bridge innate and adaptive immunity in gout. For example, IRF1 activates genes involved in both innate and acquired immune responses, and promotes M1 inflammatory macrophage commitment (33). Moreover, NFATC3 regulates inflammatory macrophage M1 polarization (41). By modulating IRF1, MYC and STAT1, and myeloid cell inflammatory function, and by suppressing NF-κB hyperactivation, NFATC2 regulates both acute and chronic inflammation (42). Whether such methylome changes mediate the presence of B and T lymphocytes, and plasma cells in the corona surrounding urate crystals in tophi (5) is germane to fundamental processes in tissue urate crystal deposition, which might be modulated by crystal-bound immunoglobulin.
This study identified differentially DNA methylated TF-gene regulatory networks in leukocytes. NFATC2 and MEF2C stood out amongst TFs propagated throughout the network; notably, both demonstrated gene body hypomethylation and both NFATC2 (also termed NFAT1 or NFATp) and MEF2C regulate multiple DML-genes. Collective evidence supports NFATC2 and MEF2C potentially acting in the functional regulation cascade modulating gouty inflammation. NFATC2 is a member of an inducible TF family with 5 members, with 4 of these regulated by Ca2+ signaling, itself a pathway differentially methylated in PBMCs in the SD gout cohort. NFATs transduce signaling after ligation of TLR4, CD14, and dectin-1 to stimulate intracellular Ca2+ signaling in myeloid cells. Consequent calcineurin phosphatase activation is permissive for NFAT nuclear translocation, allowing NFATs to act with AP-1 and other TFs, to regulate transcription (43). Calcineurin, partly by acting on NFATC2, limits NF-κB hyperactivation and signaling and suppresses TNF expression in macrophages (43). Conversely, calcineurin inhibition promotes hyperactivation of NF-κB and tolerance to the TLR4 ligand LPS (43). Notably, the calcineurin inhibitor cyclosporine is associated with not only hyperuricemia and gout but also accelerated tophus development, and particularly severe acute and chronic gouty arthritis (44).
MEF2C is a checkpoint for precursor commitment to monocyte or neutrophil differentiation; via effects on Jun, MEF2C promotes monocyte differentiation over granulopoiesis (34). MEF2C also enhances activation-induced macrophage apoptosis, and limits NF-κB activation. Furthermore, MEF2C promotes the CD14 expression essential for inflammatory function of classical monocytes (45). CD14 rs2569190 is a gout genetic risk locus in European and New Zealand Polynesian populations (46), and CD14 rs2569190 can modulate transcriptional activity of the CD14 promoter (47). Mononuclear phagocyte CD14, which associates with TLR2 and TLR4, is directly engaged by urate crystals, where CD14 supports crystal-induced NLRP3 inflammasome activation, and IL-1β and CXCL1 release in vitro (48). The extent of neutrophil recruitment in experimental gouty inflammation in mice in vivo is diminished by ~75% in global CD14 knockout mice (48). Hence, multiple recognized effects of both NFATC2 and MEF2C could regulate inflammation in gouty arthritis.
Limitations of this study included studying SD gout patients mostly established on allopurinol, with serum urate not significantly different from healthy controls. There also were differences in the PBMC vs, whole blood source of the genetic material studied in the SD vs. NZ cohort, respectively. Moreover, we studied only small subject numbers. Given these limitations, the results are seminal, but differential DNA methylation in even a small study of gout emphasizes potential utility of the DNA methylome in discriminating gout from non-gout. Controlling for comorbidity severity was not built into the design of this study. That said, the DNA methylome results bring up the question of whether epigenetic training of immunity in gout can impact comorbidities such as obesity, type 2 diabetes, and coronary artery disease. We focused on unseparated PBMCs in this work. Despite our computational adjustments for cell type composition, future single cell DNA epigenetic and transcriptomic analyses would be informative. We cannot exclude that anti-inflammatory medications previously used to control gout flares (11) may have contributed to DNA methylome findings. However, the duration of such a form of epigenetic memory is unknown. Last, gout methylome changes might modulate not simply gouty arthritis, but also changes in urate levels (eg, through SLC2A9, IGF1R), renal function, and metabolism (eg, through PRKAG2, A1CF) pertinent to gout co-morbidities (16).
In summary, we observed grossly altered PBMC DNA methylation in gout. The disease signature, from two distinct cohorts, included involvement of AMPK signaling and multiple intersecting pathways, circadian entrainment, neuroinflammatory TRP ion channel signaling, and osteoclast differentiation. Differential DNA methylation in genes involved in B and T cell signaling, Th17 and IL-17 signaling, and loci including STAT2, IRF1, in combination with IL-23 signaling could bridge innate and adaptive immunity to promote tophaceous granuloma and bone erosion in gout. NFATC2 and MEF2C stood out among PBMC TF-gene regulatory networks potentially involved in gout. Conspicuous absence of the NLRP3 pathway was a notable finding from the DML enrichment analysis. The results suggest a model whereby the DNA methylome can regulate acute and chronic gouty inflammation through signal transduction pathways and transcriptional pathways including for leukocyte differentiation and function and in neuroinflammation (49–52). The DNA methylome potentially acts on immune, metabolic, and other elements (13–15, 30,31) regulating first signal NLRP3 inflammasome priming, and phagocyte behavior following second signal inflammasome activation by phagocytosed urate crystals (53). Future studies to elucidate novel target genes and pathways highlighted here may help advance gout biomarkers and treatment.
Supplementary Material
Acknowledgements:
The project described was conducted in part using clinical research resources supported by NIH Grant UL1TR001442 of CTSA.
Supported by the NIH (P50 AR060772, R21 AR075990 to RT and TM, P30AR073761 to WW, UL1TR001442 to DLB), and VA Research Service (I01BX001660 to RT, I01BX002234 to RLB) and a prior research award to RT and WW from ARDEA/Astra-Zeneca and Ironwood Pharmaceuticals. TM and AP-G were also supported by grant 14/527 from the Health Research Council of New Zealand, and NZ data generation supported by a grant from the Maurice Wilkins Center of New Zealand.
Disclosures: Dr. Terkeltaub has served as paid consultant to Astra-Zeneca, Horizon, Selecta, and SOBI. Dr. Merriman has received research funding from Ardea Biosciences and Ironwood pharmaceuticals. Dr Liu-Bryan holds a research grant from CymaBay.
References:
- 1.Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet 2016;388:2039–2052. [DOI] [PubMed] [Google Scholar]
- 2.Terkeltaub R What makes gouty inflammation so variable? BMC Med. 2017;15:158.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalbeth N, Saag KG, Palmer WE, Choi HK, Hunt B, MacDonald PA, et al. Effects of Febuxostat in Early Gout: A Randomized, Double-Blind, Placebo-Controlled Study. Arthritis Rheumatol. 2017;69:2386–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalbeth N, Pool B, Gamble GD, Smith T, Callon KE, McQueen FM, Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62:1549–56. [DOI] [PubMed] [Google Scholar]
- 6.Dalbeth N, Smith T, Nicolson B, Clark B, Callon K, Naot D, et al. Enhanced osteoclastogenesis in patients with tophaceous gout: urate crystals promote osteoclast development through interactions with stromal cells. Arthritis Rheum. 2008;58:1854–65. [DOI] [PubMed] [Google Scholar]
- 7.Major TJ, Dalbeth N, Stahl EA, Merriman TR. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14:341–353. [DOI] [PubMed] [Google Scholar]
- 8.Köttgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuo H, Yamamoto K, Nakaoka H, Nakayama A, Sakiyama M, Chiba T, et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum Dis. 2016;75:652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20:109–127. [DOI] [PubMed] [Google Scholar]
- 11.Whyte JM, Ellis JJ, Brown MA, Kenna TJ. Best practices in DNA methylation: lessons from inflammatory bowel disease, psoriasis and ankylosing spondylitis. Arthritis Res Ther. 2019;21:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and Adaptive Immune Memory: an Evolutionary Continuum in the Host’s Response to Pathogens. Cell Host Microbe. 2019;25:13–26. [DOI] [PubMed] [Google Scholar]
- 13.Crişan TO, Cleophas MCP, Novakovic B, Erler K, van de Veerdonk FL, Stunnenberg HG, et al. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc Natl Acad Sci U S A. 2017114:5485–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gore AV, Weinstein BM. DNA methylation in hematopoietic development and disease. Exp Hematol. 2016;44:783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ling C, Rönn T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab 2019;29:1028–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med. 2012;125:679–687. [DOI] [PubMed] [Google Scholar]
- 17.Li B, Chen X, Jiang Y, Yang Y, Zhong J, Zhou C, et al. CCL2 promoter hypomethylation is associated with gout risk in Chinese Han male population. Immunol Lett. 2017;190:15–19. [DOI] [PubMed] [Google Scholar]
- 18.Neogi T, Jansen TL, Dalbeth N, Fransen J, Schumacher HR, Berendsen D, et al. 2015 Gout Classification Criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol. 2015;67:2557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan M, Major TJ, Topless RK, Dewes O, Yu L, Thompson JMD, et al. Discordant association of the CREBRF rs373863828 A allele with increased BMI and protection from type 2 diabetes in Māori and Pacific (Polynesian) people living in Aotearoa/New Zealand. Diabetologia. 2018;61:1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, et al. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014;30:428–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou W, Triche TJ Jr, Laird PW, Shen H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;46:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011. January;39(Database issue):D712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buske FA, Bodén M, Bauer DC, Bailey TL. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics. 2010;26:860–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46:D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang K, Wang M, Zhao Y, Wang W. Taiji: System-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci Adv. 2019: eaav3262 Published online 2019 Mar 27. doi: 10.1126/sciadv.aav3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw OM, Harper JL. Bradykinin receptor 2 extends inflammatory cell recruitment in a model of acute gouty arthritis. Biochem Biophys Res Commun. 2011;416:266–9. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Viollet B, Terkeltaub R, Liu-Bryan R. AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Ann Rheum Dis. 2016;75:286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez-Lopez E, Zhong Z, Stubelius A, Sweeney SR, Booshehri LM, Antonucci L, et al. Choline Uptake and Metabolism Modulate Macrophage IL-1β and IL-18 Production. Cell Metab. 2019;29:1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeng Y, Nie C, Min J, Liu X, Li M, Chen H, et al. Novel loci and pathways significantly associated with longevity. Sci Rep. 2016; 6: 21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology. 2018;223:101–111. [DOI] [PubMed] [Google Scholar]
- 34.Schüler A, Schwieger M, Engelmann A, Weber K, Horn S, Müller U, et al. The MADS transcription factor Mef2c is a pivotal modulator of myeloid cell fate. Blood. 2008;111:4532–41. [DOI] [PubMed] [Google Scholar]
- 35.Li E, Zhang Y. DNA Methylation in Mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez-Ubreva J, de la Calle-Fabregat C, Li T, Ciudad L, Ballestar ML, Català-Moll F, et al. Inflammatory cytokines shape a changing DNA methylome in monocytes mirroring disease activity in rheumatoid arthritis. Ann Rheum Dis. 2019. August 1 pii: annrheumdis-2019–215355. [DOI] [PubMed] [Google Scholar]
- 37.Heipertz EL, Harper J, Lopez CA, Fikrig E, Hughes ME, Walker WE. Circadian Rhythms Influence the Severity of Sepsis in Mice via a TLR2-Dependent, Leukocyte-Intrinsic Mechanism. J Immunol. 2018;201:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi HK, Niu J, Neogi T, Chen CA, Chaisson C, Hunter D, et al. Nocturnal risk of gout attacks. Arthritis Rheumatol. 2015;67:555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trevisan G, Hoffmeister C, Rossato MF, Oliveira SM, Silva MA, Ineu RP, et al. Transient receptor potential ankyrin 1 receptor stimulation by hydrogen peroxide is critical to trigger pain during monosodium urate-induced inflammation in rodents. Arthritis Rheum. 2013;65:2984–95. [DOI] [PubMed] [Google Scholar]
- 40.Raucci F, Iqbal AJ, Saviano A, Minosi P, Piccolo M, Irace C, et al. IL-17A neutralizing antibody regulatesmonosodium urate crystal-induced gouty inflammation. Pharmacol Res. 2019;147:104351. [DOI] [PubMed] [Google Scholar]
- 41.Hu L, He F, Huang M, Peng M, Zhou Z, Liu F, Dai YS. et al. NFATc3 deficiency reduces the classical activation of adipose tissue macrophages. J Mol Endocrinol. 2018;61:79–89. [DOI] [PubMed] [Google Scholar]
- 42.Chae CS, Kim GC, Park ES, Lee CG, Verma R, Cho HL, et al. NFAT1 Regulates Systemic Autoimmunity through the Modulation of a Dendritic Cell Property. J Immunol. 2017. November 1;199:3051–3062. [DOI] [PubMed] [Google Scholar]
- 43.Bendickova K, Tidu F, Fric J. Calcineurin-NFAT signalling in myeloid leucocytes: new prospects and pitfalls in immunosuppressive therapy. EMBO Mol Med. 2017;9:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamp LK, Chapman PT. Gout and organ transplantation. Curr Rheumatol Rep. 2012;14:165–72. [DOI] [PubMed] [Google Scholar]
- 45.Zheng R, Wang X, Studzinski GP. 1,25-Dihydroxyvitamin D3 induces monocytic differentiation of human myeloid leukemia cells by regulating C/EBPβ expression through MEF2C. J Steroid Biochem Mol Biol. 2015;148:132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinney C, Stamp LK, Dalbeth N, Topless RK, Day RO, Kannangara DR, et al. Multiplicative interaction of functional inflammasome genetic variants in determining the risk of gout. Arthritis Res Ther. 2015;17:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mertens J, Bregadze R, Mansur A, Askar E, Bickeböller H, Ramadori G, Mihm S. Functional impact of endotoxin receptor CD14 polymorphisms on transcriptional activity. J Mol Med (Berl). 2009. August;87(8):815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006;177:6370–8. [DOI] [PubMed] [Google Scholar]
- 49.Immler R, Simon SI, Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest. 2018;48 Suppl2:e12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta. 2015;1851:882–97. [DOI] [PubMed] [Google Scholar]
- 51.Dierickx P, Van Laake LW, Geijsen N. Circadian clocks: from stem cells to tissue homeostasis and regeneration. EMBO Rep. 2018;19:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurotaki D, Sasaki H, Tamura T. Transcriptional control of monocyte and macrophage development. Int Immunol. 2017;29:97–107. [DOI] [PubMed] [Google Scholar]
- 53.Yang Q, Liu R, Yu Q, Bi Y, Liu G. Metabolic regulation of inflammasomes in inflammation. Immunology. 2019;157:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.