

Abstract
Biomimetic hydrogels have emerged as the most useful tissue engineering scaffold materials. Their versatile chemistry can recapitulate multiple physical and chemical features to integrate cells, scaffolds, and signaling molecule for tissue regeneration. Due to their highly hydrophilic nature hydrogels can recreate nutrient-rich aqueous environments for cells. Soluble regulatory molecules can be incorporated to guide cell proliferation and differentiation. Importantly, the controlled dynamic parameters and spatial distribution of chemical cues in hydrogel scaffolds are critical for cell-cell communication, cell-scaffold interaction, and morphogenesis. Herein, we review biomimetic hydrogels that provide cells with spatiotemporally controlled chemical cues as tissue engineering scaffolds. Specifically, hydrogels with temporally controlled growth factor-release abilities, spatially controlled conjugated bioactive molecules/motifs, and targeting delivery and reload properties for tissue engineering applications are discussed in detail. Examples of hydrogels that possess clinically favorable properties, such as injectability, self-healing ability, stimulus-responsiveness, and pro-remodeling features, are also covered.
Keywords: biomimetic hydrogels, spatial- and temporal-controlled, tissue engineering, drug-delivery, biomolecule conjugation
Graphical Abstract

Biomimetic hydrogels work as tissue engineering scaffolds by recapitulating chemical cues and mimicking spatiotemporal characteristics of the native extracellular matrix.
1. Introduction
Tissue engineering involves the most advanced concepts and technologies to repair pathological tissues or create tissue substitutes. Engineered tissues have been successfully used for many applications including drug screening, disease modeling, and tissue regeneration. Currently, over 114,000 people in the US alone are on the waiting list for a life-saving organ transplant1. Tissue engineering may be the most promising solution to the key problems associated with organ transplant including donor organ shortages, rejection, and disease transmission.
The three basic components of tissue engineering are cultured cells, signaling molecules, and scaffolds (Figure 1). Although scaffold-free and cell-free strategies are being heavily explored, constructs with all three components still hold the most important place in tissue engineering research and applications. Scaffolds work as native extracellular matrix (ECM) mimics, which provide mechanical support and integrate chemical cues to promote tissue regeneration. Chemical cues are essential for cell signaling and cell-environment interaction. They provide cell adhesion sites, regulate cell functions, and maintain tissue homeostasis. More specifically, it is well established that many cell activities are anchorage dependent2. Without adhesion sites, cells may not be able to respond appropriately to other stimuli. Native ECM components can guide cell differentiation and regulate cell function. For example, hyaluronic acid (HA), a major component of cartilage, has been shown to increase chondrocyte proliferation and GAG secretion3, 4. Numerous soluble signaling molecules, nutrients, and ions physically embedded in ECM ensure normal cell viability and functionality. In addition, some growth factor-like sequences within ECM molecules can also facilitate tissue regeneration upon degradation of ECM5, 6. As a result, scaffolds should include chemical cues such as cell adhesion site, growth factors, and other signaling molecules to enhance scaffold bioactivity and help maintain homeostasis.
Figure 1.

Biomimetic hydrogels for tissue engineering
Furthermore, the natural cell environment is heterogeneous and dynamic. ECM physical and chemical properties vary with location and staging of development and turnover. For instance, collagen Type IV is mainly found in basement membrane while collagen Type I is generally distributed in most connective tissue ECM7, 8. Another example is that roughly one third of the HA in the human body is degraded naturally via hyaluronidase and resynthesized every day9. In fact, starting from the earliest stages of development, embryonic cells produce and reorganize their ECM to build biological structures10. Additionally, wound healing is associated with the dynamics of chemical signaling, where different cytokines emerge to initiate, promote, and alleviate inflammation reactions and tissue remodeling11. Therefore, inspired by nature, mechanisms that can control spatiotemporal parameters of chemical cues need to be incorporated when designing scaffolds.
Hydrogel based scaffolds are one of the most important scaffold materials due to their readily modifiable chemical structures, minimally invasive delivery methods, and high biocompatibility. Formed from controlled crosslink of hydrophilic polymers, hydrogels can absorb a large amount of water while maintaining their 3-dimensional (3-D) network structures. Chemical features of the native cell environment can be recapitulated by hydrogels so that encapsulated cells can be coaxed to behave as if in their native ECM. The hydrogel network can physically trap signaling molecules to promote tissue regeneration. In addition, hydrogel polymers are amenable to chemical modification to include specific biochemical cues in a highly organized network structure.
Previously, many excellent reviews have summarized hydrogel chemistry and fabrication techniques to spatiotemporally control hydrogel chemical and physical properties12–20. Herein, we dissect the need for chemical cues in tissue engineering applications and review work that has achieved temporal control of growth factor release, spatial control of bioactive motifs, targeted delivery, and drug-reload using hydrogel systems. We highlight the significance of mimicking spatial and temporal manipulation of chemical factors for engineering artificial constructs with structural integrity and biological functionality. Future directions in the field will be addressed at the end.
2. Hydrogel scaffolds with drug release properties
Biomimetic drug-releasing hydrogels can provide cells with soluble chemical cues that exist in the native cell environment. Molecules such as mitogens, growth factors, and morphogens are critical to directing cells for tissue regeneration. Major signaling molecules and their recent applications for tissue engineering are shown in Table 1. Unlike oral or intravenous drug administration, implanting growth factor-loaded hydrogels can release signaling molecules directly to the target, resulting in a therapeutic local drug concentration and minimal systematic side-effects. The rate and duration of the release can be controlled by systematically modulating hydrogel physical and chemical features as shown in Figure 2. This section focuses on using hydrogels to maintain optimized growth factor release patterns for tissue engineering purposes.
Table 1.
Common growth factors and cytokines for tissue engineering applications.
| Name of the growth factors | Main functions | Tissue engineering examples |
|---|---|---|
| Angiopoietin (Ang) | Promote blood vessel formation and cardiomyocyte survival. | 21–24 |
| Bone morphogenetic protein (BMP) | Promote bone and cartilage formation and treat large bone defects. | 25, 26 |
| Brain-derived neurotrophic factor (BDNF) | Promote neural and axonal regeneration and treat neuron damage. | 27–29 |
| Ciliary neurotrophic factor (CNTF) | Promote motor neuron survival and outgrowth. | 30, 31 |
| Epidermal growth factor (EGF) | Promote mesenchymal stem cell (MSC) proliferation and motility and increase proliferation of fibroblast, epithelium, and neural stem progenitor cells. | 32, 33 |
| Erythropoietin (EPO) | Recruit endothelial progenitor cells (EPCs) and work as a neurotrophic factor. | 34–36 |
| Fibroblast growth factor (FGF) | Improve cardiac function, promote fibroblast growth and facilitate angiogenesis. | 37–39 |
| Glial cell line derived neurotrophic factor (GDNF) | Promote the survival and differentiation of dopaminergic neurons. | 40, 41 |
| Growth differentiation factors (GDFs) | Increase expression of tenogenic-specific gene. | 42 |
| Hepatocyte growth factor (HGF) | Enhance myoblast survival and work as neurotrophic and neuroregenerative factors. | 43, 44 |
| Insulin-like growth factor 1 (IGF-1) | Enhance tenocyte proliferation and promote ligament and cartilage regeneration. | 45–47 |
| Interleukins | Regulate immune response. | 48–50 |
| Keratinocyte growth factor | Promote epithelization. | 51 |
| Nerve growth factor (NGF) | Promote neurite outgrowth and promote sympathetic and sensory neuron survival and maintenance. | 52 |
| Placenta growth factor (PlGF) infused growth factors | Bind to native ECM with very high affinities for regenerative medicine. | 53 |
| Platelet-derived growth factor (PDGF) | Increase new bone formation. | 54–57 |
| Transforming growth factor β (TGF-β) | Induce macrophage to M2 phenotype and chondrogenesis of human MSCs (hMSCs). | 39, 42, 58–60 |
| Vascular endothelial growth factor (VEGF) | Promote endothelial survival, migration, and proliferation, induce vessel formation, and stimulate lymphangiogenesis. | 21, 22, 47, 50, 61 |
Figure 2.

Mechanisms that regulate drug release in hydrogels
2.1. Growth factor delivery by passive release
2.1.1. Diffusion and intermolecular force-induced drug retention
Passive drug release driven by free diffusion remains the most commonly used strategy to provide cells with signaling molecules in tissue engineering applications due to its ease of implementation. As shown in Figure 2, physical factors, such as surface area, pore size, and mesh size directly regulate drug diffusion. Since hydrogels can be easily formed into different shapes or sizes, a hydrogel architecture with a larger surface to volume ratio (e.g. microgels) releases drug faster. For example, Hao et al.62, 63 developed hydrogen peroxide (H2O2) releasing dopamine methacrylamide (DMA) hydrogel and microgel for antimicrobial applications. Although the two hydrogels shared the same chemistry, the microgel released approximately one order of magnitude higher H2O2 than the bulk hydrogel due to the much higher (around 100 times higher) surface-to-volume ratio. Additionally, the presence of micropores in the hydrogel allows for faster drug transport within the hydrogel. The space between hydrogel polymer chains, called mesh size, can also influence drug release. Larger hydrogel mesh size gives rise to less resistance to diffusion64. Therefore, increasing crosslink density and polymer concentration to decrease mesh sizes remains the most common method in practice. Hollyway et al.65 developed maleimide-modified HA (MaHA) hydrogels that could be loaded with BMP-2 for bone tissue engineering. HA crosslink density was controlled by the concentration of thiol-containing crosslinker. Their results showed that the hydrogel degradation rates increased with the decrease of the initial crosslink density and MaHA concentration. And BMP-2 release was slowed by increasing hydrogel initial crosslink density, bringing about a beneficial effect in promoting new bone formation in a rat calvarial defect model.
In addition to physical factors, chemical properties of a scaffold influence the diffusion. When additional intermolecular interactions, such as electrostatic forces and hydrophobic interactions, exist between the hydrogel and the drug, longer drug retention can be achieved. For example, hydrogels with different protonation state show different charge states and different pH when hydrated within the same aqueous solution. Based on this, HA hydrogels with different pH have been fabricated by using acidic and neutral dialysis buffers after synthesis66. Carbohydrazide and aldehyde moieties were incorporated to form carbohydrazide-HA and aldehyde-HA, respectively, which were mixed to form hydrazone crosslinked HA hydrogels with controlled pH. The final products, acid (pH=4.5) and neutral (pH=7) hydrogels, were used to deliver BMP-2 to an in vitro BMP-responsive mouse stromal cell model and an in vivo rat ectopic model for bone regeneration. The results showed that the acidic hydrogel, which interacts with BMP-2 through both electrostatic and Van der Waals forces, displayed much slower release compared to the neutral gel, which constrained BMP-2 by Van der Waals interactions only. The slow release increased alkaline phosphatase activity of mouse stromal cell from Day 1 to Day 28, whereas the effect of the neutral hydrogel mainly manifested in the first 7 days. In addition, more osteoblasts and mineralization induced by acidic hydrogel were observed.
2.1.2. Release accelerated by hydrolytic degradation
The breakdown of polymers by water, hydrolysis, causes disruption of hydrogel structure and faster release of drugs. Esters, amides, acyl halides, anhydrides, and thioesters are commonly seen functional groups susceptible to hydrolysis. Proteins and degradable synthetic polymers including poly(glycolic acid) (PGA), polylactic acid (PLA), polyanhydride, poly(ε-caprolactone) (PCL), and polyurethane (PU) contain these groups (Figure 3, labeled in red). They can also be integrated into nondegradable polymer networks to fabricate degradable scaffolds. For example, poly(ethylene glycol) dimethacrylate (PEGDM) and poly(ethylene glycol) diacrylate (PEGDA) which contain ester groups have been widely used67, 68. Aryl-thiol terminated poly(ethylene glycol) PEG can also undergo hydrolysis through the ester groups in between the PEG backbone and the terminal groups (Figure 3)69.
Figure 3.
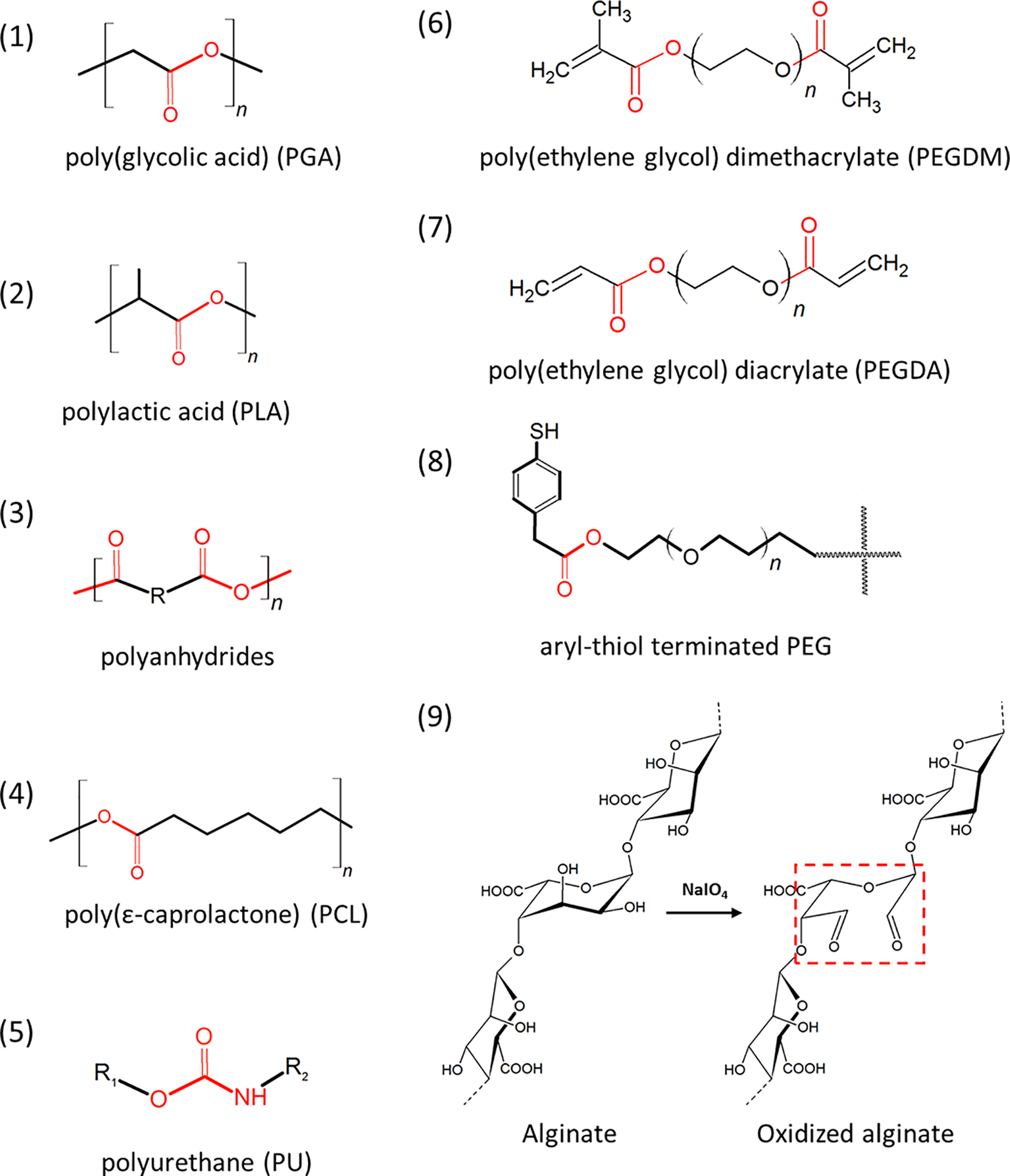
Frequently used degradable hydrogel structures. Groups susceptible to hydrolysis are indicated by red color.
Hydrolytic degradation requires the exposure of hydrolyzable groups to water molecules. Therefore, factors that affect water accessibility such as pore size and crosslink density can modulate hydrogel degradation rate. For example, Singh and coworkers70 designed a hydrogel composed of hyperbranched poly(glycidol) (HPG) and PEG. They found that increasing the HPG content resulted in a faster degradation through increasing hydrogel pore size and the number of hydroxyl groups, which can be easily protonated for the initiation of hydrolysis. In addition, increasing crosslink density decreases degradation rate due to the increased number of bonds that must be cleaved to fully disrupt the 3-D network.
Natural hydrogels normally show inconsistent degradation behaviors. Proper chemical modification can make their degradation more controllable. For example, Boontheekul et al.71 developed a degradable alginate with tunable degradability by partially oxidizing uronate residues on an alginate backbone (Figure 3). Oxidative agents cause the ring opening of uronate residues and conformation changes of the polymer chain. The opened uronic-acid residues are susceptible to hydrolytic scission under both basic and acid conditions72. The degradation rate varies with the degree of oxidation. However, excessive oxidation may compromise hydrogel biocompatibility due to the formation of copious aldehyde groups. A similar hydrogel system was used for VEGF delivery73. New blood vessel formation in the 3-D scaffolds is critical to prevent necrosis, because blood vessels supply oxygen, nutrients, and growth factors for cells and allow stem cells and progenitors to enter the scaffold for new tissue formation. Since VEGF is a well-characterized angiogenic regulator, supplying VEGF is a common practice in tissue engineering. However, due to the short half-life of VEGF, administrating soluble VEGF normally results in a transient high level of VEGF and demonstrates limited beneficial effect. To ensure cells experience a long-term physiological level of VEGF, Silva and Mooney73 proposed a binary partially oxidized alginate hydrogel that slowly released VEGF. Native alginate is hard to degrade in the human body. Low molecular weight alginate and partially oxidized alginate was introduced to optimize the degradation kinetics and VEGF release rate. When the hydrogel was implanted into a mouse ischemic model, it significantly enhanced angiogenesis in ischemic limbs compared to controls (animals treated with bolus VEGF delivery). The angiogenic effect highly correlated with the long-term retention of VEGF in the ischemic tissue, proving that constant long-term VEGF exposure is crucial for tissue regeneration.
2.2. Growth factor release triggered by physical factors
2.2.1. Temperature responsive hydrogel
Many polymers demonstrate sol-to-gel transition above (lower crystal solution temperature (LCST)) or below (upper critical solution temperature (UCST)) a certain temperature. This is because in a homogenous polymer solution, polymers interact with both solvent molecules and polymers themselves. When temperature changes, polymer-polymer interaction (or solvent-solvent interaction) may become dominant, causing separation of the polymer-rich and polymer-poor phases74. Based on this property, polymers with LCST or UCST that are close to body temperature have been used for drug delivery and cell encapsulation75, 76. The most commonly studied thermosensitive material is poly(N-isopropylacrylamide) (PNIPAm). A homogeneous aqueous PNIPAm solution undergoes phase separation when the temperature is increased, which is driven by the hydrophilicity-hydrophobicity shift within PNIPAm molecules77. Specifically, aqueous PNIPAm solutions are formed when PNIPAm polymers and water form hydrogel-bonds (H-bonds). The hydrated PNIPAm polymer takes a coiled conformation (dissolved form) and is highly hydrophilic. Increased temperature results in disruption of these H-bonds, expelling water from the polymer networks. The less hydrated polymer collapses to a globule conformation (crystalline form) and is stabilized by the hydrophobic interactions among the polymers. Thereby, PNIPAm exhibits a LCST at around 31 °C, where a sharp phase transition occurs upon change of molecular hydrophilicity.
Various PNIPAm based polymers or surface modification techniques have been developed to generate intact cell sheets for tissue repair78. A PNIPAm grafted culture dish has been used to transfer cultured hepatocytes, which are extremely sensitive to the standard enzymatic treatments (Figure 4)79. Specifically, cells were cultured on the PNIPAm surface-treated culture dishes to confluence at 37 °C and then culture dishes were transferred into cold media (4 °C or 20 °C water). The PNIPAm layer partially dissolved and the whole cell sheet was released. Since this method preserves cell junctions, cell adhesive proteins, and the local ECM structures, it has been used to engineer myocardial80 and epithelial cell sheets81 for myocardial and corneal tissue regeneration.
Figure 4.

Intact cell sheet generated by using PNIPAm surface-coated tissue culture dishes. Decreasing PNIPAm density and chemically modifying PNIPAm were used to enhance cell adhesions. Reproduced with permission from reference 79, copyright 2011, American Chemical Society.79
Incorporating PNIPAm building blocks into other biocompatible hydrogel molecules is an attractive means of formulating injectable and reversibly crosslinked hydrogels. The presence of other functional groups also improves PNIPAm gel biocompatibility. For example, inserting a biodegradable peptide such as MMP-sensitive crosslinker into PNIPAm polymer backbone can make the polymer biodegradable82. Incorporation of hydrophilic acrylic acid group preserves more water and stabilizes H-bonds in PNIPAm hydrogel. Thus, the hydrogel mechanical properties can be improved for application in liver tissue engineering83. Poly(N-isopropylacrylamide-co-acrylic acid) copolymer has also been used to encapsulate chondrocytes and deliver transforming growth factor beta-3 (TGF-β3) for cartilage regeneration84. Similarly, polyethylene glycol (PEG)-PNIPAm copolymers show robust mechanical properties for fabricating nucleus pulposus substitutes85. It has also been used to deliver TGF-β2 and chondrocytes for engineering cartilage86.
Other commonly used hydrophilic thermosensitive polymers for tissue engineering applications include poloxamer (Pluronic)87, poly(D,L-lactic-co-glycolic acid)-b-methoxy poly(ethylene glycol) (PLGA-mPEG)88, 89, and hydroxybutyl chitosan90, 91. Growth factors can be incorporated into these hydrogels during the sol-to-gel transition. For example, Yin et al.92 used butane diisocyanate (BDI) functionalized Pluronic and collagen Type I to prepare thermosensitive hybrid hydrogels that hardened above 25 °C. These hydrogels supported growth of tendon stem/progenitor cells (TSPCs), preserved the spindle shape of TSPCs, and induce human umbilical vein cells (HUVECs) to form tubular structures. Chen et al.93 developed an injectable temperature sensitive hydrogel composed of PLGA-mPEG copolymer microspheres. The system was used to deliver VEGF and endothelial cells (ECs) to a rabbit femoral head necrosis model to enhance local neovascularization and bone formation.
2.2.2. Mechanical stimuli responsive hydrogel
When mechanical forces are applied to a hydrogel, the energy needs to be dissipated by either deformation of the hydrogel network or breaking of hydrogel crosslinks94. As a result, growth factors physically trapped in or weakly bound to a mechanoresponsive hydrogel can be released on-demand up on mechanical stimulation. Lee et al.95 developed the first mechanical stimulus responsive hydrogel by taking the advantage of the electrostatically-based interactions between VEGF and alginate. This intermediate interaction is strong enough for incorporating VEGF into the alginate matrix and sensitive to mechanical stimulation96, 97. The reversible binding allows for a constant and slow release of VEGF passively under physiological conditions and a strain-dependent burst release to promote granulation tissue formation and neovascularization. Since mechanical stimulation may compromise hydrogel structural integrity, supramolecular and dynamically crosslinked self-healing hydrogels have arisen as propitious biomaterials for tissue engineering. Different from the conventional chemical crosslinks, these hydrogels are crosslinked via non-covalent interactions, such as hydrogen bonding, electrostatic forces, π–π stacking, host–guest interactions, hydrophobic interactions, or metal coordination98, or dynamic chemistry, such as Schiff base reaction, reversible Diels–Alder reaction, oxime chemistry, and disulfide bond99. When the crosslink is disrupted by mechanical deformation, hydrogels can reconstruct their network using these transient interactions. This unique characteristic offers opportunities to generate injectable and seal-healing hydrogels with controlled viscoelastic properties. Hou et al.100 developed a self-healing supramolecular hydrogel that was used to encapsulate BMP-2 and cells (chondrocytes and bone marrow stem cells (BMSCs)) for cartilage-bone tissue complex regeneration (Figure 5a). Supramolecular bonding motif ureidopyrimidinone (UPy) was grafted to the dextran (DEX) backbone. Hydrogel formation was based on the quadruple H-bond array between two UPy units, which can be interrupted under shear force and recover after the removal of externally applied stress.
Figure 5.

Mechanoresponsive hydrogels for tissue engineering applications. a) Illustration of the transient supramolecular crosslinks under shear force in the DEX-UPy hydrogel and its self-healing property for cartilage and bone tissue engineering, adapted with permission from reference 100, copyright 2015, John Wiley and Sons100, b) ultrasound-inducible basic FGF (bFGF) delivery from the fibrin gel to promote angiogenesis by using the ultrasound-responsive bFGF-loaded emulsion, reprint with permission from Ref.101 with permission from Elsevier, c) magnetic field induced temporally-controlled BMP-2 delivery for osteogenic differentiation of mouse MSCs (mMSCs) by using Fe3O4 nanoparticles, reproduced with permission from reference 102, copyright 2020, American Chemical Society.102
In addition to conventional mechanical stimuli, i.e. compression, tension, and shear, ultrasound can also trigger displacement of materials on micrometer scales. Since focused ultrasound can be applied to deep tissues in a non-invasively and spatiotemporally controlled way, acoustically-responsive hydrogel is an attractive material for controlled growth factor delivery. Moncion et al.101 developed a fibrin-based hydrogel scaffold doped with growth factor-loaded sonosensitive perfluorocarbon (PFC) emulsions (Figure 5b). Basic FGF (bFGF) was loaded into the micro-sized PFC emulsions using microfluidic devices. Gas bubbles were formed within the hydrogel under ultrasound stimulation, a process termed acoustic droplet vaporization, causing vaporization of the PFC and collapse of the micro-emulsions structure to release bFGF. To further slow-down bFGF release, small amount of heparin was incorporated into the fibrin gel. Since bFGF binds to heparin with a high affinity103, the released bFGF will be retained in the scaffold. Their data showed superthreshold megahertz-range ultrasound resulted in a 12.6-fold increase in bFGF release in vitro. And the in vivo study using a mouse subcutaneous model demonstrated up to 3.3-fold greater perfusion and 1.7-fold greater new blood vessel formation compared with the fibrin controls. A similar acoustically-responsive gel-in-gel construct was developed to quantitatively study how bFGF affected angiogenic sprouting in vitro104. The inner fibrin gel contained a bFGF-loaded emulsion, while HUVEC-coated microbeads and normal human dermal fibroblasts were embedded in the outer fibrin gel. bFGF release was initiated by ultrasound at different time points and locations. The results indicated bFGF release increased with an increased ultrasound wave and promoted vessel formation. Note that ultrasound also generates micro-sized pores in the hydrogel. Gel porosity and mechanical properties may be altered to further facilitate cell infiltration and the remodeling process.
Magnetic components undergo magnetization, the alignment of their magnetic moments, when a magnetic field is present105. During this process, magnetic forces are generated. Incorporated into a hydrogel matrix, these magnetic components can exert mechanical forces to cause hydrogel deformation for growth factor delivery. Iron oxide Fe3O4 magnetic nanoparticles have been widely used as a magnetic component for biomedical applications due to their low toxicity. Madani et al.102 developed a composite hydrogel composed of MSC-loaded gelatin and Fe3O4/BMP-2 loaded alginate ferrogel (Figure 5c). The system was used to investigate the osteogenic differentiation of mouse MSC (mMSC) responsive to BMP-2 delivery in both 2-D and 3-D models. Interestingly, Fe3O4 nanoparticles have also been found to greatly enhance hydrogel mechanical properties for cell adhesion and proliferation, indicating its potential for load-bearing tissue engineering applications such as cartilage and tendon repair106, 107.
2.2.3. Photo-cleavable hydrogel
Light works as an excellent stimulus as its parameters can be precisely controlled remotely. These parameters can include intensity, wavelength, and exposure duration and location. Hydrogels with photoreactive components can be used to fabricate complex features for drug delivery and tissue engineering. Currently, o-nitrobenzyl (o-NB) derivatives are the most widely applied photocleavable groups. Kharkar et al.69 developed a light sensitive PEG hydrogel with a photosensitive group o-NB ether. It was cleaved by 365 nm and 400–500 nm wavelength light with degradation half-lives of 2 and 32 min, respectively. Hu et al.108 designed a thermo- and photo-degradable hydrogel formed from four-arm amine-terminated PEG crosslinked by two types of diazene-based crosslinkers. Both crosslinkers can be cleaved at temperature above 44 °C. Due to their specific side groups, Type 1 crosslinker degrades under UV light, while Type 2 crosslinker is sensitive to near infrared radiation (NIR) when upconversion nanoparticles (UCNPs) are present (Figure 6). These hydrogels can be triggered by NIR to locally deliver variable amounts of the anti-cancer drug doxorubin (Dox) on-demand in rat models.
Figure 6.

Preparation and degradation of the four-arm amine-terminated PEG. The thermo-degradable linkers are sensitive to NIR with the presence of UCNP; the degradable hydrogel can be triggered to release anti-cancer drug Dox on-demand. Reproduced with permission from reference 108, copyright 2017, Elsevier.108
2.3. Growth factor release triggered by chemical signals
2.3.1. pH and ionic strength sensitive hydrogel
There are three main strategies to design a pH sensitive hydrogel. The first method relies on the cleavage of pH-sensitive linkages between the drug and the hydrogel such as hydrazone linkage109–111. Hydrazone linkages undergo hydrolytic breakdown at acidic pH112, which has been widely used to deliver anticancer drugs111. For the second strategy, polymers are ionized by losing or receiving H+ when weak acid and basic functional groups are present accordingly in the molecules. Commonly used materials include polyvinyl amine (PVAm)113, poly(acrylic acid) (PAA)114, chitosan115, HA116, cellulose-based hydrogel117, and maleic acid crosslinked hydrogel118. Change in pH works as a switch to cause swelling or deswelling of these hydrogels to deliver the encapsulated drugs. For example, Zhang et al.119 presented an injectable chitosan-graft-poly(N-isopropylacrylamide)-glycidyl methacrylate hydrogel sensitive to both pH and temperature. When pH is low, more H-bonds can be formed to reinforce the hydrogel structure and preserve drug loads. At higher pH, -COOH becomes -COO−. The electrostatic repulsion causes the increase of hydrogel mesh size and facilitates drug release. The third strategy involves undermining hydrogel network by interfering the relatively weak non-covalent crosslinks. For example, UPy undergo pH-driven reversible self-assembly due to the formation and disruption of the H-bonds between UPy units120. Similarly, benzene-1,3,5-tricarboxamides (BTA) fold into fibrous structure driven by the combination of H-bonding and π-π stacking110. These two moieties have been incorporated into conventional hydrogel molecules to synthesize pH responsiveness hydrogels110, 121, 122. Bastings et al.121 used pH sensitive UPy-alkyl-urea-PEG hydrogel, which underwent sol-to-gel transition based on the formation of transient supramolecular networks when solution pH changed from basic to neutral to encapsulate HGF and IGF-1. This formula was applied to a porcine myocardial infarction model as an injectable cardiac patch. The HGF/IGF-1 delivered through the hydrogel significantly reduced collagen content in the infarct scar tissues and preserved myocardium viability in the vicinity of the implant compared with controls, the pristine hydrogel and HGF/IGF-1 in saline.
Similar to pH-responsive polymers, ion-sensitive polymers allow for the exchange of their mobile ions with competitive ions present in the environment, causing change in the crosslink density, porosity, or hydrophilic/hydrophobic balance. This type of material, such as acrylic-based polymer123 and natural polysaccharide-based polymers124–127, has been widely used for oral delivery of drugs112, 124. Since the drug loading and releasing properties of ion-sensitive polymers specifically require oppositely charged groups in drug molecules that can form electrostatic interactions with the charged polymers, these polymers have limited applications in general growth factor delivery.
It is important to note that pH and ionic strength normally remain stable under physiological conditions. Such triggers are not ideal for delivery control in tissue engineering applications. Instead, these hydrogels allow for easy drug encapsulation and demonstrate other useful properties such as injectability and self-healing properties. For example, alginate was grafted onto HA molecules to obtain a calcium triggered injectable hydrogel for chondrocyte delivery for cartilage repair in a mouse model128. Another example is the PEG-glycine/poly(acrylamide-co-acrylic acid) (PEG/PAMAA) double network hydrogel129. There were two types of reversible crosslinks in this system, ionic crosslinks based on coordination between Fe3+ and acrylic acid/glycine and H-bonds between acrylic acid and acrylamide. This hydrogel demonstrated strong mechanical and self-healing properties that are promising for constructing artificial skin.
2.3.2. Redox-responsive hydrogel
Some weak covalent bonds can be cleaved by redox reaction, causing structural degradation and drug release. For example, the formation of disulfide bonds and products of Michael–addition crosslink can be reversed under a physiologically relevant reducing condition, which allows for the controlled gelation and degradation by manipulating redox states. This is useful for preparing reversibly crosslinked hydrogels and controlled drug release systems. For example, thiol presenting polymers crosslink under oxygenated conditions through the formation of disulfide bonds, which can be cleaved by reducing agents such as glutathione (GSH) and dithiothreitol (DTT)130. Crosslinking of maleimide-terminated PEG and thiol-terminated PEG was reported to be reversible through GSH-triggered retro Michael-type addition reaction, although the rate of the reverse reaction is much slower (Figure 7)69.
Figure 7.
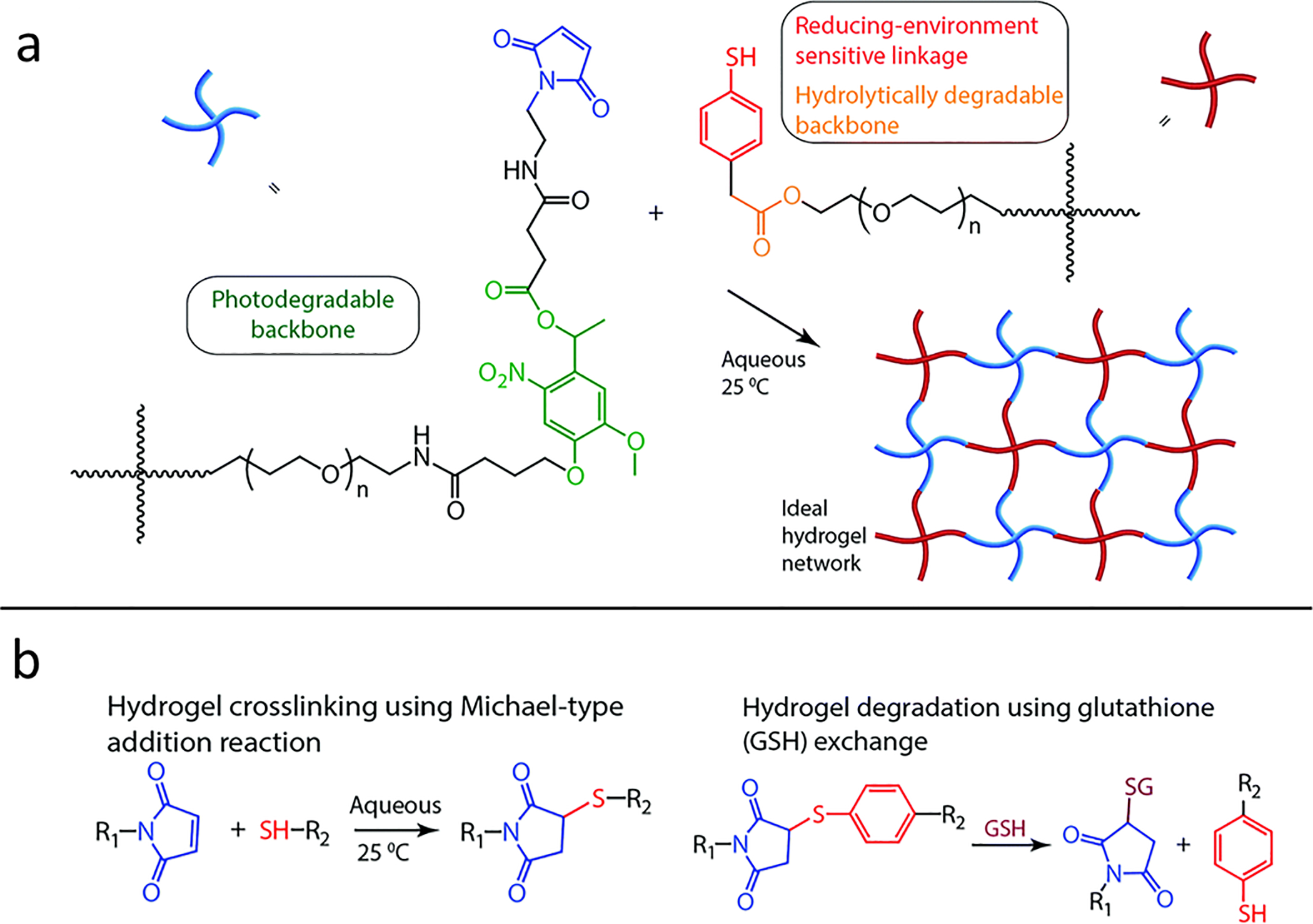
Redox mediated PEG hydrogel degradation. a) Structure of maleimide-terminated PEG and thiol-terminated PEG, b) hydrogel crosslinking through Michael-type addition reaction and hydrogel degradation through GSH-mediated retro Michael-type reaction. Reproduced with permission from reference 69, copyright 2015, Royal Society of Chemistry.69
2.3.3. Enzymatically-responsive hydrogel
Many ECM proteins, such as collagen, elastin, and fibronectin, contain specific amino active sequences that are susceptible to enzymes such as collagenase, elastase, and matrix metalloproteinase (MMP)131. The degradation of these sequences can be regulated by enzymes secreted by cells according to their metabolic status. Naturally derived ECM molecule-based scaffolds possess high biocompatibility. However, they suffer from rapid uncontrollable enzymatic degradation, resulting in inconsistent mechanical properties. For instance, scaffolds for cutaneous tissue engineering must have extended degradation times of up to 8 weeks for adequate tissue healing and growth, while a conventional collagen scaffold can be more than 95% resorbed by 7 days132. To this end, Zhao et al.133 created a photo-cross-linkable gelatin hydrogel terminated with methacrylamide groups (GelMA). Mechanical and degradation properties were controlled by changing GelMA concentration and crosslink density. This allows the hydrogel to mimic ECM with a wide range of mechanical properties needed to support keratinocyte adhesion and proliferation.
Alternatively, exogenous enzymes can be incorporated into a hydrogel if there is a lack of endogenous enzyme. However, enzyme leaching prevents the application of this method. One proposed solution was a crosslinked enzyme aggregate (CLEA) prepared by precipitating free enzymes and crosslinking the precipitated enzyme aggregates134. CLEA has a particle size up to a micrometer and demonstrates high catalytic activity. Therefore, physically embedding CLEA into typical hydrogels is sufficient to immobilize the enzymes. Kunjukunju et al.135 developed an alginate hydrogel that contains a crosslinked enzyme aggregate of alginate lyase (CLEA-AL). By controlling the degree of aggregation, the size of CLEA and alginate degradation kinetics can be modulated. Alginate hydrogels with different CLEA-ALs can be designed to last for 2 to 21 days.
Synthetic bioinert hydrogels can also become enzymatically biodegradable when proteolytically sensitive sequences are incorporated. For example, Sokic and Papavasiliou136 developed PEGDA hydrogels that included several peptides sensitive to proteolysis. In their system, both the hydrogel degradation and fibroblast migration were faster in hydrogels with higher contents of collagenase-sensitive domains. Although enzymatic degradation is sensitive to pH and temperature, these factors normally do not significantly influence degradation in vivo because the in vivo environment is relatively stable. Thus, enzyme accessibility, enzyme concentration, substrate concentration, and enzyme specificity become the main considerations in tissue engineering practices. To study these effects, Lutolf et al.137 developed hydrogels using sulfone-functionalized PEG macromers crosslinked with a series of bis-cysteine peptides that can be cleaved by MMPs at different rates. Their results showed that the presence of MMP-cleavable sites was required for eliciting cell infiltration and hydrogel degradation rate was substrate and crosslink density dependent. Importantly, inserting an enzyme-sensitive sequence into a 3-D hydrogel network may enhance the steric hindrance effect that alters degradation kinetics.
Enzyme responsive sequences can also be used to covalently link drugs to the backbone. Thus, drug release can be initiated by specific enzymes or cells to promote tissue regeneration138. Seliktar et al.139 developed a matrix metalloproteinase-2 (MMP-2) sensitive PEG-based hydrogel that degraded and released growth factors in the presence of MMPs secreted by ECs for vascular regeneration. The hydrogel was crosslinked by MMP-2 cleavable peptides. To enhance reendothelialization, VEGF was chemically linked to the hydrogel backbone and TGF-β1 was physically embedded in the hydrogel matrix (Figure 8a). This biomimetic design allowed for cell-mediated hydrogel degradation that released TGF-β1 in response to ECs infiltration. The results showed that, VEGF enhanced MMP-2 zymogen expression in ECs by 70%. Release of TGF-β1 activated the latent MMP zymogen and facilitated vascular morphogenesis (Figure 8b).
Figure 8.

Promotion of tissue remodeling by using MMP-2 sensitive PEG-based hydrogel. a.) Structure of PEG-based hydrogel, b.) immobilized VEGF enhanced MMP-2 expression (72 kDa band) and TGF-β activated MMP-2 (66 kDa band). Reproduced with permission from reference 139, copyright 2004, John Wiley and Sons.139
2.4. Release agents and composite hydrogels
Numerous nano-/micro-particles have been used as drug carriers due to their large surface area and the multiple functional groups present on particle surfaces. Drug-loaded particles can be mixed with hydrogels to prepare drug-releasing hydrogel composites. The major advantage of this method is that the drug-loading process and hydrogel preparation are relatively independent of each other, so that drug release rate and hydrogel properties can be tuned orthogonally. Interestingly, incorporation of particles to hydrogels may also improve hydrogel mechanical properties140, 141. Both inorganic and organic nanoparticles have shown promising applications. Organic carriers include liposomes, nano-/micro-spheres or capsules fabricated from polyacrylamide (PAM), polyacrylate, PLA, gelatin, cyclodextrins, chitosan, and metal–organic frameworks. For example, temperature sensitive hydrogel PNIPAm can be loaded with growth factors easily. But it lacks the active capacity for cell adhesion and releases drugs in a relatively fast manner. Adibfar et al.61 fabricated PNIPAm nanoparticles by using free radical polymerization and loaded VEGF into the nanoparticles through the temperature sensitive sol-to-gel transition. Then, the VEGF loaded PNIPAm nanoparticles were incorporated into a collagen hydrogel matrix to generate a composite hydrogel that possesses high bioactivity and drug release properties. VEGF release rate was greatly slowed down by approximately 70% when VEGF-PNIPAm nanoparticles were loaded in the collagen hydrogel compared to the VEGF-PNIPAm nanoparticles alone. This system was successfully used to manipulate the balance of angiogenic and osteogenic differentiation of hMSCs to achieve the optimal 20%:80% ratio in vitro, which can potentially provide both pre-vascularized structures and the bone tissue for bone repair. A similar composite comprised of TGF-β3 and parathyroid hormone-related protein (PTHrP) loaded alginate microspheres and HA matrix that mimics the chondrogenic microenvironment for hMSCs differentiation was developed for cartilage repair142. Alginate undergoes a sol-to-gel transition through ionic crosslinking, which was used to embed TGF-β3 and PTHrP. To further slow the release of the encapsulated molecules, alginate microspheres were coated with a layer of self-assembled poly(allylamine hydrochloride) and poly(sodium 4-styrenesulfonate) nanofilm. This hMSC-laden TGF-β3 and PTHrP loaded composite hydrogel significantly enhanced collagen Type II and chondroitin sulfate deposition while reducing calcium content in the implant in a subcutaneous in vivo model, indicating pro-neocartilage formation properties of the sustained release of these two drugs by using microspheres. In addition, inorganic nanoparticles include silica, hydroxyapatite, graphene, and metallic nanoparticles are also good drug carriers143–146. Note that different carriers show specific drug loading efficiency and release rates. Readers are directed to other sources for more information on these studies141, 147–149.
3. Temporal control: biphasic release
In addition to the key signaling molecules, tissue regeneration requires a sequence of biological events to occur in the correct order. As a result, this requires the capacity to mimic these temporal patterns by delivering multiple growth factors from a single scaffold as well as controlling the release profiles of each individual drug element. Currently, a robust universal method of differential drug delivery is still missing. Fortunately, many studies have shown promising results for releasing specific drugs by investigating individual drug and matrix properties.
Kuttappan et al.150 demonstrated a system composed of silica coated nanohydroxyapatite-gelatin reinforced with electrospun PLA for bone tissue engineering. VEGF was used for angiogenesis, while BMP-2 was selected for the mediation of osteogenesis and cartilage formation. During natural regeneration, the expression of VEGF starts in the early phase of healing, while BMP-2 is present in the later phase. To ensure cells receive specific growth factors at the correct stage, release kinetics of each growth factor was optimized. Since the composite is negatively charged, growth factors with higher pI (VEGF pI = 8.5, BMP-2 pI = 8.5–9.2) will be retained longer. Meanwhile, BMP-2 contains more hydrophobic domains that can bind with gelatin more strongly through cumulatively more hydrophobic interactions. These specific properties allow for differential delivery of growth factors i.e., VEGF release lasted from Day 1 to Day 7 in vivo, whereas BMP-2 release remained up to Day 20. This is comparable to the native expression profiles for each growth factor. The system was used to repair critical sized calvarial defects in rats. The differential delivery of multiple growth factors significantly enhanced neovascularization, vascular function, and new bone formation compared to the groups that were only treated with a single type or temporally inappropriate growth factor.
Another important application of biphasic delivery is controlling macrophage differentiation during wound healing. Due to the plasticity of monocytes, different stimuli can lead to distinct macrophage phenotypes151. The classical activation by using lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), or monocyte chemoattractant protein-1 (MCP-1) results in M1 macrophages. This macrophage phenotype is characterized by the secretion of high levels of pro-inflammatory factors and pathogen killing molecules (reactive oxygen species and reactive nitrogen species) to recruit more monocyte cells and remove damaged cells. Conversely interleukin-4 (IL-4) and interleukin-13 (IL-13) can elicit M2 type activation. M2 macrophages produce anti-inflammatory cytokines to support angiogenesis, ECM production, and scar formation in the later stages of regeneration. Therefore, manipulating macrophage phenotype by differential drug release is a useful method to control the transition of the wounds from the inflammatory phase to the resolution or proliferative phase.
Kumar et al.48 developed a novel multidomain peptides (MDP) self-assembled hydrogel that achieved biphasic delivery of two cytokines, MCP-1 and IL-4. Due to the distinct differences in molecular size and charge, MCP-1 was released in the first two to three days, while IL-4 release lasted over weeks. This mimics the natural control of macrophage differentiation at desired time points. The in vitro study showed that MDP hydrogel with MCP-1 and IL-4 induced more localized macrophage adhesion. A similar effect was observed in an in vivo rat model, where more infiltrating cells were found in the scaffolds with MCP-1 after 3 days. On the other hand, distinct blood vessels and red blood cells were only discovered in scaffolds with IL-4 after 7 days, emphasizing the importance of temporal control of different growth factors in tissue engineering.
Furthermore, many self-assembled hydrogels contain both hydrophilic and hydrophobic domains152.Thus both hydrophilic and hydrophobic drugs can be loaded into a single hydrogel system. In general, hydrophilic drugs release faster due to their high solubility and free diffusion nature under aqueous conditions. Hydrophobic drugs on the other hand are often encapsulated in hydrophobic pockets. The strong hydrophobic interactions between drugs and hydrophobic domains delay drug release (Figure 9a). Accordingly, Li et al.153 designed injectable self-assembled hydrogels to deliver different drugs including anticancer drugs (daunorubicin and SN-38), nonsteroidal anti-inflammatory drugs (diflunisal and etodolac), and antibiotics (levofloxacin and norfloxacin). Hydrophobic drugs (SN-38, diflunisal, and etodolac) showed prolonged release patterns, whereas hydrophilic drugs exhibited burst release patterns. Disruption of the hydrogel structure needs to occur before hydrophobic drugs can release, while hydrophilic drugs diffuse freely across the matrix.
Figure 9.

Biphasic drug release by using self-assembly hydrogels. a) Control of drug release rate by using hydrophilic and hydrophobic domains (adapted with permission from reference 153, copyright 2016, American Chemical Society153), and b) biphasic releasing by using liposome encapsulation and self-assembly hydrogel composite. The figure was adapted with permission from reference 154, copyright 2014, American Chemical Society.154
Wickremasinghe et al.154 combined self-assembling MDPs and phospholipid/cholesterol based liposome to construct a general biphasic release system. Growth Factor 1 (PlGF) was physically entrapped in the MDP hydrogel, while Growth Factor 2 (EGF) was encapsulated in the liposome. The liposome was then embedded in the MDP network to form a composite hydrogel. Due to the large size of the liposome and the protective effect of the lipid layer, Growth Factor 2 exhibited 4–5 days delayed release compared with Growth Factor 1 (Figure 9b). This general system may be used to deliver multiple drugs for tissue engineering applications where timed cascades of biological signals are important.
4. Spatial control of bioactive moiety conjugation
In nature, most growth factor proteins have low stability and a short circulating half-life. Conventional delivery of these molecules requires high doses that are not physiologically relevant and cost-efficient. Ultimately, high signaling molecule concentration may not only result in higher cost, but also other deleterious outcomes including tissue overgrowth, tumor growth, and immune reactions. Additionally, many growth factors are chemotactic. Proper growth factor gradients inside a scaffold can be crucial for cell migration and morphogenesis. To resolve these problems, approaches include using active peptides derived from the morphogenetic proteins or ECM (which are generally more stable than the full-length proteins) and chemically linking the active motifs to the scaffold. The key is to ensure a controlled local concentration and an appropriate concentration gradient of the bioactive cues.
4.1. Click chemistry and biomolecule conjugation
Click chemistry refers to fast and specific reactions that produce high yield of products with well-defined structures. Since many of these reactions can occur at mild conditions without being affected by other functional groups, click chemistry has been widely used when dealing with biological systems. Well-known reaction systems include azide-alkyne reaction, thiol-ene reaction, Diels-Alder reaction, and aldehyde-hydroxylamine reaction. These systems are particularly useful for preparing structurally controlled polymer networks and conjugating bioactive molecules.
Truong et al.155 have developed a dual-click chemistry based one-pot PEG/chitosan double network hydrogel for cell encapsulation. The PEG-based first network was crosslinked by thiol-alkene reaction, whereas the PEG-chitosan-based second network was formed from tetrazine-norbornene reaction (a Diels-Alder reaction) in one phosphate-buffered saline solution (Figure 10a). The reaction system was compatible with the pre-loaded MSCs (Figure 10b). Interestingly, this click chemistry system allowed for the control of mechanical properties and bioactivity of the hydrogel independently. Through adjusting the starting concentration of the functionalized groups, hydrogel crosslink density, the key factor of polymer mechanical properties, can be tuned. Additionally, when there are extra alkene and norbornen groups present in the gel, bioactive molecules with –SH, such as proteins with cysteine, and tetrazine functionalized molecules can be conjugated to the hydrogel backbone (Figure 10c). This orthogonal control of physiochemical variables provides a useful tool to mimic multiple cues in one system for tissue engineering and evaluate the effect of each individual factor independently.
Figure 10.
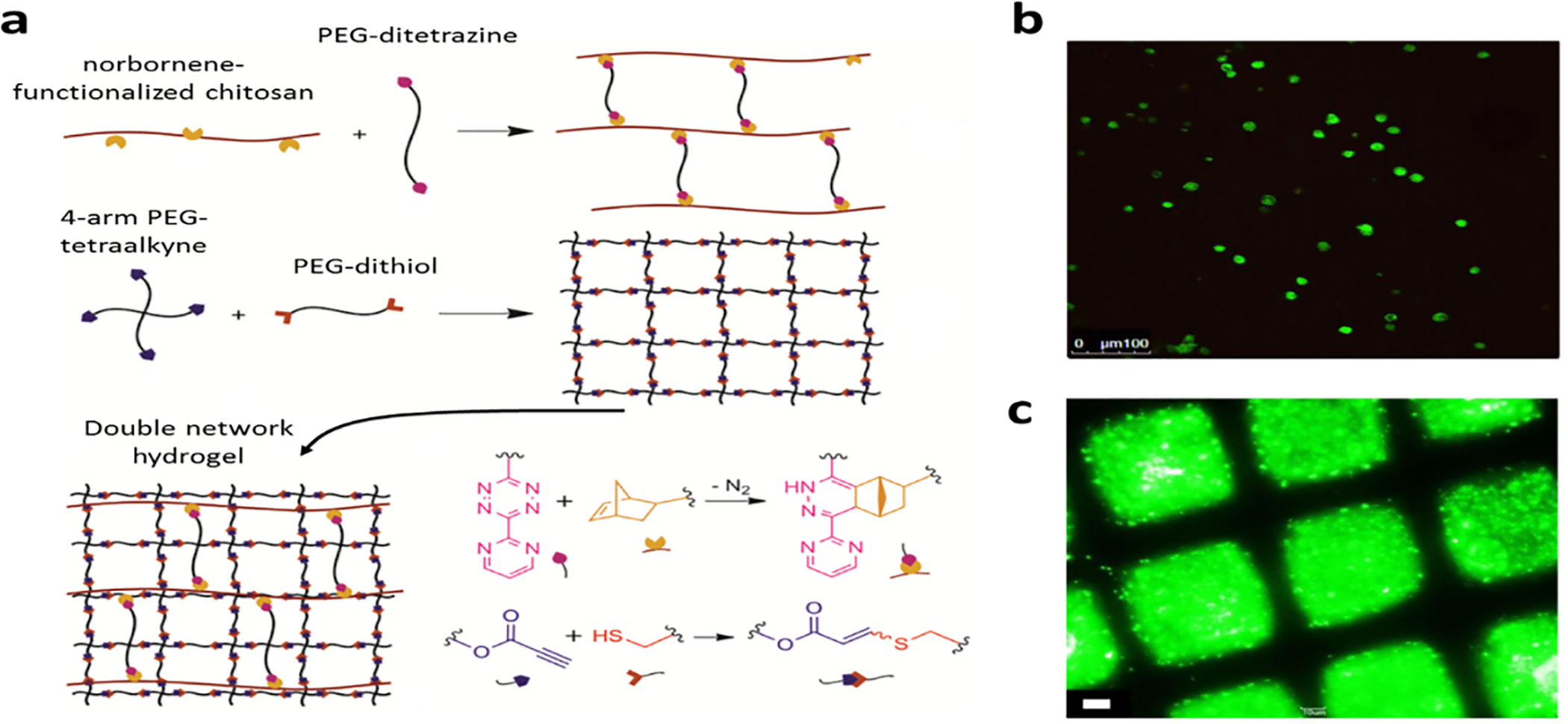
Click chemistry based double network hydrogel. a) Formation of the double network hydrogel by click-chemistry, b) live/dead assay image of MSC encapsulated in the hydrogel, c) conjugation of BODIPY-SH dye to the hydrogel through the reaction between thiol and the pendant alkene groups. Adapted with permission from reference 155, copyright 2015, American Chemical Society.155
4.2. Light-mediated hydrogel modification
Incorporation of light-responsive moieties into hydrogels allows us to adjust hydrogel chemical properties in a highly spatiotemporally controlled manner for morphogenesis in tissue engineering. One major challenge in tissue engineering is guiding cell growth and migration in the 3-D scaffold. Photochemically modifiable hydrogels were developed to graft cell adhesion motifs or growth factors to the predetermined location within the solidified hydrogel structure. Luo et al.156 used photosensitive chemical S-2-nitrobenzyl-cysteine (S-NBC), which decomposes and generates free thiol in aqueous solutions when exposed to UV light. The released free thiols act as the anchoring sites to which maleimide-terminated biomolecules can be chemically linked. More specifically, S-NBC was first chemically linked to an agarose gel through 1,1’-carbonyldiimidazole (CDI) (Figure 11a). The S-NBC modified agarose gel was irradiated with a 325 nm laser beam, generating channels with free thiols (Figure 11b). When the irradiated gel was immersed in the solution of maleimide-terminated GRGDS, a synthetic peptide mimicking the cellular binding site, GRGDS was conjugated to the free thiols (Figure 11c). Any unreacted maleimide-terminated GRGDS was rinsed away. The generated hydrogel demonstrated a desired spatial display of bioactive motifs (Figure 11d). When cells were seeded on the gel surface, they spread and migrated following the photo-irradiated channels. Figure 11e shows the co-localization of the GRGDS-modified channels (green) and the migrating primary rat dorsal root ganglia cells (red).
Figure 11.
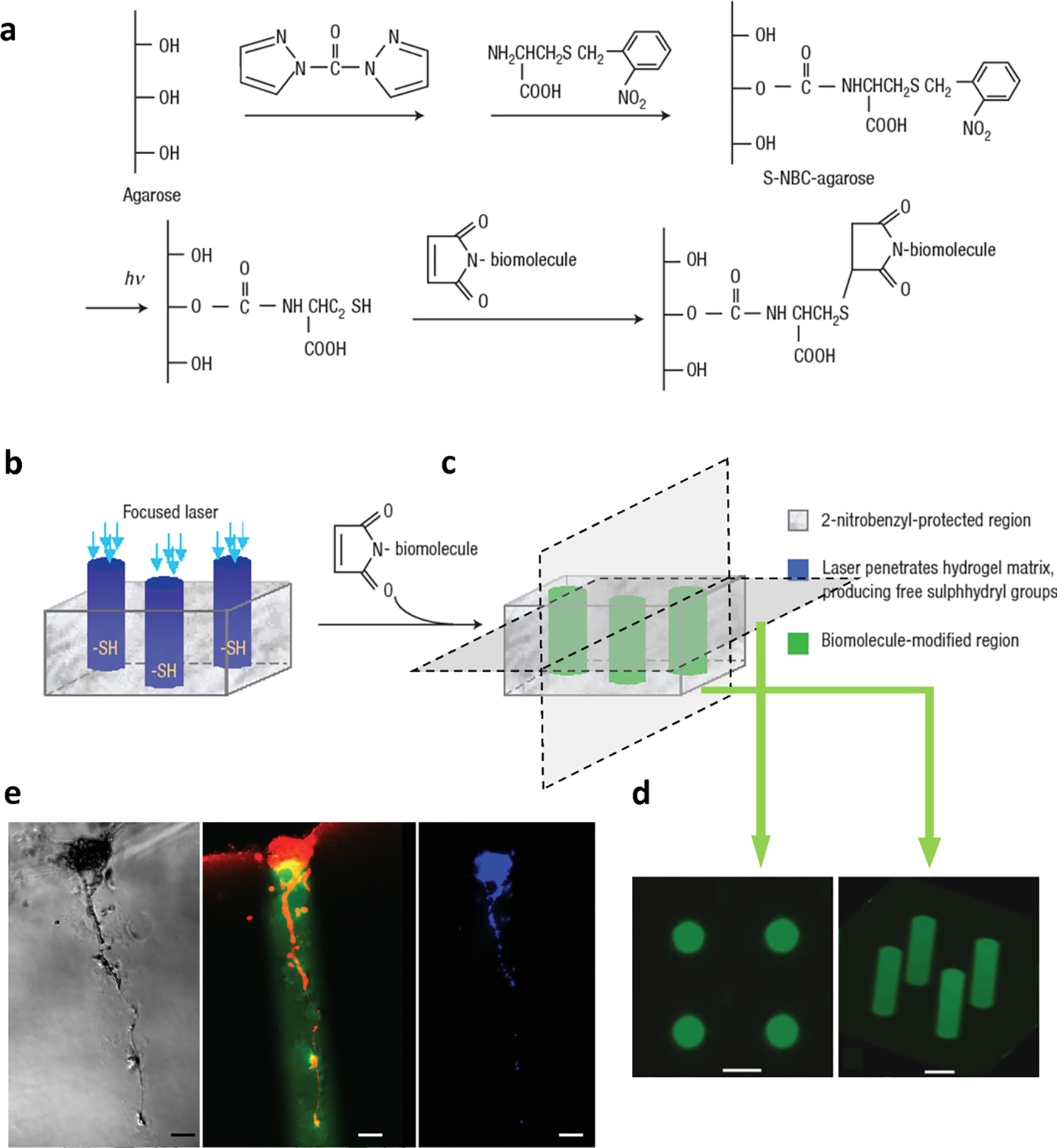
Cell growth and migration guidance in agarose hydrogel with photolabile GRGDS-tagged channels. a) Synthesis of S-NBC-agarose hydrogel, b) photo-fabrication procedures, c) grafting cell adhesive peptide specifically to the irradiated regions, d) the bioactive channels indicated by fluorescently labeled oligopeptide; scale bar = 200 μm, e) colocalization of GRGDS-modified channels and the migrating primary rat dorsal root ganglia, where red indicates cells (rhodamine–phalloidin stained F-actin); green indicates the channels (fluorescein-labeled GRGDS); and blue is from DAPI stain; scale bar = 100 μm. The figure was adapted with permission from reference 156, copyright 2004, Springer Nature.156
Although the abovementioned photo-responsive platform has achieved cell guidance in 3-D scaffolds, two factors limited the working distance of photopatterning: the focus limit of the laser beam and the UV absorbance of the hydrogel. Additionally, UV light is potentially harmful for cells. To resolve these issues two-photo ultrafast irradiation was used to improve z-direction resolution and working depth157. Two-photo irradiation sensitive photocage 6-bromo-7-hydroxy coumarin (Bhc) was used as the mediator. Specifically, Bhc-sulfide derivatives were synthesized from chloride derivative Bhc and tert-butoxycarbonyl-protected mercaptoethylamine. Then Bhc-sulfide derivative was conjugated to agarose molecules in dimethyl sulfoxide (DMSO) with 1,1’-carbonyldiimidazole (CDI) (Figure 12a). After multiphoton laser patterning by using NIR that is harmless to cells (wavelength: 710–800 nm), free thiols were released (Figure 12b) and maleimide functionalized bioactive molecules (GRGDS and VEFG165) can be grafted. Additionally, by controlling the laser power, this system precisely controlled the local concentration of the bioactive motifs. Since VEGF is chemotactic, Aizawa et al.158 created a 3-D scaffold with a VEGF gradient, where the highest VEGF concentration was inside the hydrogel (Figure 12c). When endothelial cells (ECs) were seeded on the hydrogel surface, cells migrated into the hydrogel and tubular-like extensions towards the high VEGF concentrations were observed (Figure 12d–f). By using this platform, they also quantitatively analyzed the chemotactic response. The extension of ECs stopped when VEGF’s local concentration reached 600–800 ng/ml because of the saturation of VEGF receptors. These results indicate the importance of controlling the local concentration of the bioactive molecules.
Figure 12.

EC guidance in multiphoton chemical patterned 3-D hydrogel. a) Synthesis of photo-responsive agarose (Reproduced with permission from reference 157, copyright 2008, American Chemical Society157), b) illustration of multiphoton chemical patterning hydrogels with defined bioactive motif spatial distribution, c) illustration of seeding cell clumps on multiphoton chemical patterned VEGF gradient hydrogel, d-f.) phase-contrast microscope images showing the in-growth of ECs on the VEGF gradient agarose hydrogels; red triangles indicate the tubular-like extensions following the VEGF gradients; d.) VEGF gradient: 1.65 ng/mL/μm, e.) 1.00 ng/mL/μm, and f.) no VEGF as the control. Scale bars = 100 μm. Reproduced with permission from reference 158, copyright 2010, John Wiley and Sons.158
4.3. Self-assembled hydrogels
The well-defined chemical structures of self-assembling molecules make it easy to conjugate bioactive motifs and control their spatial distribution. Webber et al.159 developed a binary peptide amphiphile (PA) system to control local concentration of bioactive peptides. PA is a self-assembling unit that contains a hydrophobic alkyl tail at the N-terminus, a β-sheet forming peptide sequence in the middle, and charged residues for adjusting solubility and forming ionic bridging at the C-terminus. For specific biological applications, biological epitopes can be integrated to the C-terminus. PA molecules can self-assemble in aqueous solutions when triggered by low pH or counterions that screen the charged amino acid residues152, 160. The fibronectin-derived cell adhesion epitope RGDS was conjugated to the N-terminus of PA molecule C16-V3A3K3 to generate the bioactive PA. Another inactive PA molecule C16-V3A3E3 was used as the diluent. To prepare hydrogels with specific RGDS local concentrations, bioactive PA was mixed with the diluent PA in different ratios (Figure 13a). Interestingly, the RGDS epitope was constantly on the surface of the nanofiber after preparation. This method can be used to incorporate even more hydrophobic sequence such as IKVAV on the fiber surface, which ensures that cell receptors can access the epitopes161. In the study, 10% RGDS PA caused the highest bone marrow mononuclear cell (BMNC) retention (Figure 13b), viability, proliferation rate, and differentiation marker expressions. Interestingly, these beneficial effects cannot be reproduced by adding soluble RGDS or increasing bioactive PA components (Figure 13c). The results clearly indicate the importance of controlling spatial distribution and local concentration of the bioactive motifs in tissue engineering applications. Other spatially-controlled self-assembled hydrogels have also shown promising results for tissue engineering. For example, RGD was chemically linked to the C-terminus of self-assembling MDP to enhance human exfoliated deciduous teeth stem cell adhesion and spreading162. MMP-2 consensus cleavage motif LRG was immobilized onto the MDP sequence to induce local hydrogel biodegradation by attracting MSCs, while hydrogel overall mechanical property was largely preserved163. SLanc sequence derived from vascular endothelial growth factor (VEGF) was covalently linked to the C-terminus of MDP to locally promote angiogenesis and scaffold remodeling164, 165.
Figure 13.

10% RGD-grafted PA guidance enhance BMNC encapsulation. a) Illustration of formation of PA fibers assembled from 90% diluent PA and 10% RGD-grafted PA (indicated in yellow), b) luciferase-expressing BMNCs encapsulated within the 10% RGD-grafted PA hydrogel (left), inactive PA (middle), and saline control, c) BMNC proliferation 5 days after encapsulation. Adapted with permission from reference 159, copyright 2010, Elsevier.159
5. Growth factor sequestration and drug-reload
Limited drug payload can often be a problem with a hydrogel delivery system. The native ECM can automatically collect and orchestrate growth factors by sequestering and presenting growth factors to cells. For instance, most ECM proteins such as fibronectin, fibrinogen, vitronectin, and collagen display very strong binding to PlGF-253. Heparin can bind with VEGF, FGF, TGF-β1, BMP-2, EGF, and midkine166–169. HA has a high affinity for PDGF. Therefore, smart hydrogels that can sequester growth factors from the environment can be formulated by using these elements to resolve the drug payload problem. Bragg et al.170 designed a heparin-conjugated gelatin/silk fibroin hydrogel that sequestered bFGF dissolved in solution and slowly released it. Li et al.171 fabricated an ECM-mimetic scaffold by using gelatin and a Eucommia ulmoides-derived polysaccharide, EUP3 (Figure 14a). Due to the high affinity of EUP3 for PDGF-BB172, the hydrogel bonded to and sequestered the endogenous PDGF-BB. Gelatin rendered bioactivity to the hydrogel, so that cells infiltrated into and digested the hydrogel to release the growth factors. Therefore, without addition of exogenous growth factors, the hydrogel garnered PDGF-BB in situ and promoted wound healing in a full-thickness injury model in mice (Figure 14b and c). Jeon et al.173 also developed heparin micropatterned dual-crosslinked alginate hydrogels by using a photo-initiated reaction. Since the spatial distribution of heparin can be precisely controlled by light, growth factors can be immobilized into a designated spot to guide hMSC migration and differentiation.
Figure 14.

Growth factor sequestration and controlled release. a) Illustration of EP3/gelatin hydrogel that can sequester endogenous PDGF-BB and release PDGF-BB locally, b) sequestering PDGF-BB in situ; Ctrl indicates PBS treatment, c) fluorescent images showing growing cells (Ki67) and neovascularization (CD31) induced by EP3/gelatin hydrogel, d) illustration of the aptamer and its complimentary sequence (CS), e) growth factor release initiated by complementary sequence, f) on-demand release controlled by different concentrations of CS. Reproduced with permission from reference 171, copyright 2017, Elsevier; and reference 174, copyright 2014, Elsevier.171, 174
Natural materials normally show affinity for multiple targets, which is not ideal to achieve variable control over active factors. To better achieve control over sequestration of specific growth factors, nucleic acid aptamers, short oligonucleotides screened from large random sequence pools to bond to specific biological molecules175, 176, have been incorporated into hydrogel structures (Figure 14d). Due to the high-affinity and high-specificity binding between aptamers and their targets, aptamer-hydrogels can draw in specific growth factors from the environment and release them slowly. Thereby, aptamer-incorporated hydrogel can create a high local growth factor concentration for the cells encapsulated in the gel. Remarkably, the release rate can be boosted on-demand by supplying the complementary sequence of the aptamer, which competes against the target growth factor for the binding site on the aptamer (Figure 14e). Batting et al.174 demonstrated that their PEG-based aptamer-hydrogel sequestered PDGF and released PDGF slowly without complementary sequence triggers (3% release after 15 days). When complementary sequence was added, PDGF release was increased by up to 175 times and this increase was complementary sequence concentration-dependent (Figure 14f). Later, a modified design was developed to investigate the synergistic effects of VEGF and bFGF release on angiogenesis by using two-aptamer incorporated hydrogels177. The HUVEC migration and chicken chorioallantoic membrane vascularization results demonstrated that the combination of two growth factors promoted more angiogenesis compared with the single growth factor groups.
Click-chemistry offers another opportunity to solve the payload problem. It allows for targeted and repetitive drug refill after hydrogel implantation. Brudno et al.178 prepared chemically modified alginate hydrogels that can selectively collect circulating drugs in both ischemic and non-ischemic animal models. When drugs are administered intravenously or orally, drugs will be distributed all over the body through the circulatory system. If the circulating drugs bind to an implanted hydrogel with high specificity and affinity, drugs can be targeted to the hydrogel. When the hydrogel degrades, or the drug-hydrogel bond is disrupted, drugs can be released locally to avoid the systematic side-effects. Two click chemistries were utilized for this purpose, the reaction between trans-cyclooctene and tetrazine (the TCO-Tz system), and the reaction between dibenzocyclooctyne and azide (the DBCO-Az system) (Figure 15a). Specifically, alginate hydrogel was grafted with either Tz or Az side groups. Drugs of interest were modified with TCO or DBCO. The proof of concept experiment was carried out by using Cy5 fluorescent labeled TCO and Cy7 labeled DBCO as drug surrogates. Alginate–Tz was implanted to the mammary fat pad (the non-ischemic model), while alginate–Az hydrogel was implanted to mouse muscular tissue (hind-limb, the ischemic model). Then a mixture of Cy5–TCO and Cy7–DBCO was injected intravenously. After 48 hours, Cy5 and Cy7 distribution in the body was examined through live animal fluorescence imaging which clearly showed that Cy5 was mainly found in the mammary tissue while Cy7 was targeted to hind-limbs (Figure 15a). Remarkably, since the implanted hydrogels contain excessive ligands, drugs can be repeatedly targeted to the hydrogel (Figure 13b).
Figure 15.

Click chemistry-mediated drug targeting and repetitive drug refill. a) Targeting two different small molecules to mouse hind-limb (blue) and mammary fat pad (red) by using TCO-Tz and DBCO-Az click chemistry, b) repetitive refill indicated by fluorescent signal (adapted from Ref.178 with permission from John Wiley and Sons), c) in vivo drug release based on the TCO-Tz reaction, d.) intravenous injection and local release of drug for cancer therapy. Reproduced with permission from reference 179, copyright 2016, American Chemical Society.179
This system has been successfully applied to cancer models (Figure 15c and d)179. Specifically, Tz modified alginate was injected into the tumor site. The chemotherapy pro-drug TCO-functionalized doxorubicin, which is significantly less toxic than doxorubicin itself, was injected intravenously. The circulating TCO-doxorubicin bond to Tz-alginate through the inverse-electron demand Diels–Alder (IEDDA) reaction. Additionally, TCO-Tz reaction caused the localized release of the attached active doxorubicin (Figure 15c). As a result, the chemotherapy drug only took effect in the vicinity of the tumors. In a xenograft model of soft tissue sarcoma HT1080, this novel drug delivery system demonstrated significantly higher efficiency in inhibiting tumor volumes compared with the tradition doxorubicin therapy. In addition, animals treated with the new method showed limited side effects. No weight loss across the whole experiment period was observed, whereas over 20% weight loss was detected in the control. Although click chemistry has been widely used in hydrogel preparation, its application in drug targeting for tissue engineering is uncommon. This design offers opportunities to collect tagged drugs of interest that occur systemically to a specific target.
6. Future direction and challenges
Effective tissue engineering depends on a combination of diverse techniques in biology, chemistry, and physics. New insights in tissue regeneration, stem cell biology, and cell-environment interactions will continue to inform us and help mimic biochemical cues. In addition to the direct delivery of growth factors, studies on scaffold-guided gene delivery have been explored as another means for spatiotemporal control of growth factors with the advent of the latest gene engineering techniques180–182. Furthermore, well-defined molecules, such as specific bioactive sequences, uniform building blocks, and highly specific crosslinking mechanisms are preferred for controlling hydrogel structure and quality. Self-assembly or bottom-up assembly techniques will allow scientists to incorporate chemical cues in a highly spatiotemporally controlled manner. Well-defined chemical structures also promote morphogenesis and cell differentiation by controlling physical factors such as biomimetic hierarchical structure160, 183 and dynamic mechanical properties184, 185. In addition, the development of spatiotemporally controlled hydrogels relies on both hydrogel chemistry and up-to-date fabrication techniques. Bioprinting, microfluidic systems, photo-patterning, and computer-aided design and manufacturing systems will be more frequently used in scaffold fabrication, especially for constructing complex functional tissues in the future.
Other critical properties for hydrogel clinical application include: antimicrobial properties, immune regulatory characters, integration with native tissues, and bioadhesive properties. The control over these properties may also greatly promote the translation of hydrogel designs to medical tools. For example, Hao et al.63 used catechol chemistry to offer hydrogel antibacterial and antiviral properties where H2O2 can be released locally in an on-demand model for days without concern to active storage or transport of H2O2, a molecule that needs to be tightly regulated to avoid detrimental dose-dependent effects on cells. Our group has utilized S-nitrosothiol chemistry to develop injectable PEG-fibrin hydrogels that releases the immune regulatory molecule nitric oxide to control inflammation upon thermal and mechanical activation186. Zhao and Gerecht187 developed an injectable and self-healing gelatin-oxidized DEX hydrogel based on a dynamic Schiff base reaction. The crosslinked hydrogel can be injected into a designated tissue due to its self-healing property, while also allowing for better cargo retention and cell morphogenesis. More importantly the hydrogel network reorganizes as new tissue develops without losing scaffold structural integrity. Sani et al.188 engineered methacrylate modified gelatin that can be crosslinked under visible light as a robust and cost-effective bioadhesive for corneal repair. Despite these and other similar studies, the future focus should continue to build towards a quantitative understanding of the mechanistic relationship between hydrogel chemistry and the clinical outcomes that will lead ultimately to better biomimetic hydrogel design.
7. Summary
Hydrogels with spatiotemporally controlled chemical cues work as the state-of-the-art mimics of native ECM for tissue engineering. They provide cells with mechanical support and, more importantly, integrates chemical signals to guide cell growth and maintain cell functions. Drug-releasing hydrogels can mimic the expression profiles of growth factors in native tissue to promote neovascularization and tissue remodeling. Modulating the local distribution of bioactive molecules by using stimuli-responsive hydrogel, supramolecular hydrogel, and bioactive motif-containing hydrogel facilitates morphogenesis. Smart hydrogels that can accumulate specific signaling molecules from the environment and present them to cells provide us with more flexible windows to treat pathological conditions locally. We highlight the significance of the spatiotemporal control of the chemical cues when designing hydrogel scaffolds for tissue engineering. Comprehensive consideration of controlling release rate, spatial distribution of bioactive molecules, and adjustability of spatiotemporal parameters to stimuli is needed for fabricating biomimetic hydrogel for tissue engineering mechanistic research and clinical applications.
Acknowledgement
The authors acknowledge funding from the National Institutes of Health under Awards No. R15GM104846 (B.P.L) and R15GM112082 (R.M.R.), and the Office of the Assistant Secretary of Defense for Health Affairs through the Defense Medical Research and Development Program under Award No. W81XWH1810610 (B.P.L).
Footnotes
Conflict of interest statement: Weilue He, the first author of this paper is an employee of FM Wound Care, LLC, Hancock, MI.
References
- 1.American Transplant Foundation, Facts and myths about transplant, https://www.americantransplantfoundation.org/about-transplant/facts-and-myths/).
- 2.Gospodarowicz D, Delgado D and Vlodavsky I, Proceedings of the National Academy of Sciences, 1980, 77, 4094–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan H, Chu CR, Payne KA and Marra KG, Biomaterials, 2009, 30, 2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levett PA, Melchels FP, Schrobback K, Hutmacher DW, Malda J and Klein TJ, Acta biomaterialia, 2014, 10, 214–223. [DOI] [PubMed] [Google Scholar]
- 5.Appella E, Weber IT and Blasi F, FEBS letters, 1988, 231, 1–4. [DOI] [PubMed] [Google Scholar]
- 6.Krusius T, Gehlsen KR and Ruoslahti E, Journal of Biological Chemistry, 1987, 262, 13120–13125. [PubMed] [Google Scholar]
- 7.LeBleu VS, MacDonald B and Kalluri R, Experimental biology and medicine, 2007, 232, 1121–1129. [DOI] [PubMed] [Google Scholar]
- 8.Shin Y, Kim H, Han S, Won J, Jeong HE, Lee ES, Kamm RD, Kim JH and Chung S, Advanced healthcare materials, 2013, 2, 790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stern R, European journal of cell biology, 2004, 83, 317–325. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Luo X and Leighton J, Biochemistry insights, 2015, 8, BCI. S30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondo T and Ohshima T, International journal of legal medicine, 1996, 108, 231–236. [DOI] [PubMed] [Google Scholar]
- 12.Brown TE and Anseth KS, Chemical Society Reviews, 2017, 46, 6532–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Li Y, Huang G, Zhang X, Pingguan-Murphy B, Gao B, Lu TJ and Xu F, Critical reviews in biotechnology, 2016, 36, 553–565. [DOI] [PubMed] [Google Scholar]
- 14.Seliktar D, Science, 2012, 336, 1124–1128. [DOI] [PubMed] [Google Scholar]
- 15.Uto K, Tsui JH, DeForest CA and Kim D-H, Progress in polymer science, 2017, 65, 53–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosales AM, Vega SL, DelRio FW, Burdick JA and Anseth KS, Angewandte Chemie International Edition, 2017, 56, 12132–12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YS and Khademhosseini A, Science, 2017, 356, eaaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sood N, Bhardwaj A, Mehta S and Mehta A, Drug Delivery, 2016, 23, 748–770. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Wang Z, Lu WW, Zhen W, Yang D and Peng S, Npg Asia Materials, 2017, 9, e435–e435. [Google Scholar]
- 20.Koetting MC, Peters JT, Steichen SD and Peppas NA, Materials Science and Engineering: R: Reports, 2015, 93, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rufaihah AJ, Johari NA, Vaibavi SR, Plotkin M, Kofidis T and Seliktar D, Acta biomaterialia, 2017, 48, 58–67. [DOI] [PubMed] [Google Scholar]
- 22.Smith RJ Jr, Yi T, Nasiri B, Breuer CK and Andreadis ST, The FASEB Journal, 2019, 33, 5089–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reis LA, Chiu LL, Wu J, Feric N, Laschinger C, Momen A, Li R-K and Radisic M, Circulation: Heart Failure, 2015, 8, 333–341. [DOI] [PubMed] [Google Scholar]
- 24.Chiu LL and Radisic M, Biomaterials, 2010, 31, 226–241. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Casanova S, Martin-Saavedra FM, Escudero-Duch C, Uceda MIF, Prieto M, Arruebo M, Acebo P, Fabiilli ML, Franceschi RT and Vilaboa N, Biomaterials, 2020, 241, 119909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma D, An G, Liang M, Liu Y, Zhang B and Wang Y, Materials Science and Engineering: C, 2016, 65, 221–231. [DOI] [PubMed] [Google Scholar]
- 27.Obermeyer JM, Tuladhar A, Payne SL, Ho E, Morshead CM and Shoichet MS, Tissue Engineering Part A, 2019, 25, 1175–1187. [DOI] [PubMed] [Google Scholar]
- 28.Limongi T, Rocchi A, Cesca F, Tan H, Miele E, Giugni A, Orlando M, Donnorso MP, Perozziello G and Benfenati F, Molecular neurobiology, 2018, 55, 8788–8798. [DOI] [PubMed] [Google Scholar]
- 29.Cook DJ, Nguyen C, Chun HN, L Llorente I, Chiu AS, Machnicki M, Zarembinski TI and Carmichael ST, Journal of Cerebral Blood Flow & Metabolism, 2017, 37, 1030–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laughter MR, Bardill JR, Ammar DA, Pena B, Calkins DJ and Park D, ACS biomaterials science & engineering, 2018, 4, 3374–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C, Yu R, Li Z, Feng C, Wang Q, Liu Y and Su Z, International journal of pharmaceutics, 2017, 529, 275–284. [DOI] [PubMed] [Google Scholar]
- 32.Goh M, Hwang Y and Tae G, Carbohydrate polymers, 2016, 147, 251–260. [DOI] [PubMed] [Google Scholar]
- 33.Haddad T, Noel S, Liberelle B, El Ayoubi R, Ajji A and De Crescenzo G, Biomatter, 2016, 6, e1231276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuladhar A, Obermeyer JM, Payne SL, Siu RC, Zand S, Morshead CM and Shoichet MS, Biomaterials, 2020, 119794. [DOI] [PubMed] [Google Scholar]
- 35.Naseri-Nosar M, Farzamfar S, Salehi M, Vaez A, Tajerian R and Azami M, Journal of Bioactive and Compatible Polymers, 2018, 33, 269–281. [Google Scholar]
- 36.Salehi M, Naseri‐Nosar M, Ebrahimi‐Barough S, Nourani M, Khojasteh A, Hamidieh AA, Amani A, Farzamfar S and Ai J, Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2018, 106, 1463–1476. [DOI] [PubMed] [Google Scholar]
- 37.Roberts JJ, Farrugia BL, Green RA, Rnjak-Kovacina J and Martens PJ, Journal of tissue engineering, 2016, 7, 2041731416677132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen G, Ren J, Deng Y, Wu X, Huang J, Wang G, Zhao Y and Li J, Journal of biomedical nanotechnology, 2017, 13, 1660–1672. [DOI] [PubMed] [Google Scholar]
- 39.Fathi‐Achachelouei M, Keskin D, Bat E, Vrana NE and Tezcaner A, Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2019. [DOI] [PubMed]
- 40.Basu S and Yang S-T, Tissue engineering, 2005, 11, 940–952. [DOI] [PubMed] [Google Scholar]
- 41.Tajdaran K, Gordon T, Wood MD, Shoichet MS and Borschel GH, Acta biomaterialia, 2016, 29, 62–70. [DOI] [PubMed] [Google Scholar]
- 42.Tellado SF, Chiera S, Bonani W, Poh PS, Migliaresi C, Motta A, Balmayor ER and van Griensven M, Acta biomaterialia, 2018, 72, 150–166. [DOI] [PubMed] [Google Scholar]
- 43.Chapanian R and Amsden B, Journal of controlled release, 2010, 143, 53–63. [DOI] [PubMed] [Google Scholar]
- 44.Yamane K, Mazaki T, Shiozaki Y, Yoshida A, Shinohara K, Nakamura M, Yoshida Y, Zhou D, Kitajima T and Tanaka M, Scientific reports, 2018, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller RE, Grodzinsky AJ, Cummings K, Plaas AH, Cole AA, Lee RT and Patwari P, Arthritis & Rheumatism, 2010, 62, 3686–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Florine EM, Miller RE, Liebesny PH, Mroszczyk KA, Lee RT, Patwari P and Grodzinsky AJ, Tissue Engineering Part A, 2015, 21, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raimondo TM, Li H, Kwee BJ, Kinsley S, Budina E, Anderson EM, Doherty EJ, Talbot SG and Mooney DJ, Biomaterials, 2019, 216, 119246. [DOI] [PubMed] [Google Scholar]
- 48.Kumar VA, Taylor NL, Shi S, Wickremasinghe NC, D’Souza RN and Hartgerink JD, Biomaterials, 2015, 52, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barthes J, Dollinger C, Muller CB, Liivas U, Dupret-Bories A, Knopf-Marques H and Vrana NE, Frontiers in bioengineering and biotechnology, 2018, 6, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nie L, Chang P, Sun M, Huo H, Zhang C, Ji C, Wei X, Zhou Q, Guo P and Yuan H, Applied Sciences, 2018, 8, 2438. [Google Scholar]
- 51.Geer DJ, Swartz DD and Andreadis ST, The American journal of pathology, 2005, 167, 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faustino C, Rijo P and Reis CP, Pharmacological research, 2017, 120, 68–87. [DOI] [PubMed] [Google Scholar]
- 53.Martino MM, Briquez PS, Güç E, Tortelli F, Kilarski WW, Metzger S, Rice JJ, Kuhn GA, Müller R and Swartz MA, Science, 2014, 343, 885–888. [DOI] [PubMed] [Google Scholar]
- 54.Saik JE, Gould DJ, Watkins EM, Dickinson ME and West JL, Acta biomaterialia, 2011, 7, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bayer EA, Jordan J, Roy A, Gottardi R, Fedorchak MV, Kumta PN and Little SR, Tissue Engineering Part A, 2017, 23, 1382–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao N, Suzuki A, Zhang X, Shi P, Abune L, Coyne J, Jia H, Xiong N, Zhang G and Wang Y, ACS applied materials & interfaces, 2019, 11, 18123–18132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mantovani A, Sica A, Allavena P, Garlanda C and Locati M, Human immunology, 2009, 70, 325–330. [DOI] [PubMed] [Google Scholar]
- 58.Levinson C, Lee M, Applegate LA and Zenobi-Wong M, Acta biomaterialia, 2019, 99, 168–180. [DOI] [PubMed] [Google Scholar]
- 59.Kudva AK, Dikina AD, Luyten FP, Alsberg E and Patterson J, Acta biomaterialia, 2019, 90, 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim H, Park H, Lee JW and Lee KY, Carbohydrate polymers, 2016, 151, 467–473. [DOI] [PubMed] [Google Scholar]
- 61.Adibfar A, Amoabediny G, Eslaminejad MB, Mohamadi J, Bagheri F and Doulabi BZ, Materials Science and Engineering: C, 2018, 93, 790–799. [DOI] [PubMed] [Google Scholar]
- 62.Meng H, Li Y, Faust M, Konst S and Lee BP, Acta biomaterialia, 2015, 17, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meng H, Forooshani PK, Joshi PU, Osborne J, Mi X, Meingast C, Pinnaratip R, Kelley J, Narkar A and He W, Acta biomaterialia, 2019, 83, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J and Mooney DJ, Nat Rev Mater, 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holloway JL, Ma H, Rai R and Burdick JA, Journal of controlled release, 2014, 191, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan HJ, Casalini T, Hulsart-Billström G, Wang S, Oommen OP, Salvalaglio M, Larsson S, Hilborn J and Varghese OP, Biomaterials, 2018, 161, 190–202. [DOI] [PubMed] [Google Scholar]
- 67.Elisseeff J, McIntosh W, Anseth K and Langer R, ACS Publications, 1999.
- 68.Zhu J, Biomaterials, 2010, 31, 4639–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kharkar PM, Kiick KL and Kloxin AM, Polymer chemistry, 2015, 6, 5565–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh D, Huh PH and Kim SC, Journal of Biomedical Materials Research Part A, 2016, 104, 48–56. [DOI] [PubMed] [Google Scholar]
- 71.Boontheekul T, Kong H-J and Mooney DJ, Biomaterials, 2005, 26, 2455–2465. [DOI] [PubMed] [Google Scholar]
- 72.Dalheim MØ, Ulset A-ST, Jenssen IB and Christensen BE, Carbohydrate polymers, 2017, 157, 1844–1852. [DOI] [PubMed] [Google Scholar]
- 73.SILVA EA and MOONEY DJ, Journal of Thrombosis and Haemostasis, 2007, 5, 590–598. [DOI] [PubMed] [Google Scholar]
- 74.Seuring J and Agarwal S, Journal, 2013.
- 75.Boustta M, Colombo P-E, Lenglet S, Poujol S and Vert M, Journal of Controlled Release, 2014, 174, 1–6. [DOI] [PubMed] [Google Scholar]
- 76.Wu S-W, Liu X, Miller II AL, Cheng Y-S, Yeh M-L and Lu L, Carbohydrate polymers, 2018, 192, 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Q, Weber C, Schubert US and Hoogenboom R, Materials Horizons, 2017, 4, 109–116. [Google Scholar]
- 78.Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y and Sakurai Y, Die Makromolekulare Chemie, Rapid Communications, 1990, 11, 571–576. [Google Scholar]
- 79.Takahashi H, Matsuzaka N, Nakayama M, Kikuchi A, Yamato M and Okano T, Biomacromolecules, 2011, 13, 253–260. [DOI] [PubMed] [Google Scholar]
- 80.Miyahara Y, Nagaya N, Kataoka M, Yanagawa B, Tanaka K, Hao H, Ishino K, Ishida H, Shimizu T and Kangawa K, Nature medicine, 2006, 12, 459. [DOI] [PubMed] [Google Scholar]
- 81.Yamato M, Utsumi M, Kushida A, Konno C, Kikuchi A and Okano T, Tissue engineering, 2001, 7, 473–480. [DOI] [PubMed] [Google Scholar]
- 82.Kim S and Healy KE, Biomacromolecules, 2003, 4, 1214–1223. [DOI] [PubMed] [Google Scholar]
- 83.Gan T, Guan Y and Zhang Y, Journal of Materials Chemistry, 2010, 20, 5937–5944. [Google Scholar]
- 84.Na K, Park JH, Kim SW, Sun BK, Woo DG, Chung H-M and Park K-H, Biomaterials, 2006, 27, 5951–5957. [DOI] [PubMed] [Google Scholar]
- 85.Vernengo J, Fussell G, Smith N and Lowman A, Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2008, 84, 64–69. [DOI] [PubMed] [Google Scholar]
- 86.Yasuda A, Kojima K, Tinsley KW, Yoshioka H, Mori Y and Vacanti CA, Tissue engineering, 2006, 12, 1237–1245. [DOI] [PubMed] [Google Scholar]
- 87.Xu H-L, Xu J, Zhang S-S, Zhu Q-Y, Jin B-H, ZhuGe D-L, Shen B-X, Wu X-Q, Xiao J and Zhao Y-Z, Drug Delivery, 2017, 24, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nie L, Zou P, Feng S and Suo J, Journal of Materials Science: Materials in Medicine, 2013, 24, 689–700. [DOI] [PubMed] [Google Scholar]
- 89.Jeong B, Wang L-Q and Gutowska A, Chemical Communications, 2001, 1516–1517. [Google Scholar]
- 90.Wang QQ, Kong M, An Y, Liu Y, Li JJ, Zhou X, Feng C, Li J, Jiang SY and Cheng XJ, Journal of Materials Science, 2013, 48, 5614–5623. [Google Scholar]
- 91.Qu C, Bao Z, Zhang X, Wang Z, Ren J, Zhou Z, Tian M, Cheng X, Chen X and Feng C, International journal of biological macromolecules, 2019, 125, 78–86. [DOI] [PubMed] [Google Scholar]
- 92.Yin H, Yan Z, Bauer RJ, Peng J, Schieker M, Nerlich M and Docheva D, Biomedical Materials, 2018, 13, 034107. [DOI] [PubMed] [Google Scholar]
- 93.Chen D, Zhang C, Huo H, Ji C, Sun M and Nie L, Materials Letters, 2018, 229, 138–141. [Google Scholar]
- 94.Wang J, Kaplan JA, Colson YL and Grinstaff MW, Advanced drug delivery reviews, 2017, 108, 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee KY, Peters MC, Anderson KW and Mooney DJ, Nature, 2000, 408, 998–1000. [DOI] [PubMed] [Google Scholar]
- 96.Jay SM and Saltzman WM, Journal of Controlled Release, 2009, 134, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peters MC, Isenberg BC, Rowley JA and Mooney DJ, Journal of Biomaterials Science, Polymer Edition, 1998, 9, 1267–1278. [DOI] [PubMed] [Google Scholar]
- 98.Saunders L and Ma PX, Macromolecular bioscience, 2019, 19, 1800313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uman S, Dhand A and Burdick JA, Journal of Applied Polymer Science, 2019, 48668. [Google Scholar]
- 100.Hou S, Wang X, Park S, Jin X and Ma PX, Advanced healthcare materials, 2015, 4, 1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moncion A, Lin M, O’Neill EG, Franceschi RT, Kripfgans OD, Putnam AJ and Fabiilli ML, Biomaterials, 2017, 140, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Madani SZM, Reisch A, Roxbury D and Kennedy SM, ACS Biomaterials Science & Engineering, 2020, 6, 1522–1534. [DOI] [PubMed] [Google Scholar]
- 103.Thompson LD, Pantoliano MW and Springer BA, Biochemistry, 1994, 33, 3831–3840. [DOI] [PubMed] [Google Scholar]
- 104.Dong X, Lu X, Kingston K, Brewer E, Juliar BA, Kripfgans OD, Fowlkes JB, Franceschi RT, Putnam AJ and Liu Z, Acta biomaterialia, 2019, 97, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sensenig R, Sapir Y, MacDonald C, Cohen S and Polyak B, Nanomedicine, 2012, 7, 1425–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Silva ED, Babo PS, Costa-Almeida R, Domingues RM, Mendes BB, Paz E, Freitas P, Rodrigues MT, Granja PL and Gomes ME, Nanomedicine: Nanotechnology, Biology and Medicine, 2018, 14, 2375–2385. [DOI] [PubMed] [Google Scholar]
- 107.Zhang N, Lock J, Sallee A and Liu H, ACS applied materials & interfaces, 2015, 7, 20987–20998. [DOI] [PubMed] [Google Scholar]
- 108.Hu J, Chen Y, Li Y, Zhou Z and Cheng Y, Biomaterials, 2017, 112, 133–140. [DOI] [PubMed] [Google Scholar]
- 109.Stals PJ, Smulders MM, Martín‐Rapún R, Palmans AR and Meijer E. e. W., Chemistry–A European Journal, 2009, 15, 2071–2080. [DOI] [PubMed] [Google Scholar]
- 110.Bernet A, Albuquerque RQ, Behr M, Hoffmann ST and Schmidt H-W, Soft Matter, 2012, 8, 66–69. [Google Scholar]
- 111.Sonawane SJ, Kalhapure RS and Govender T, European Journal of Pharmaceutical Sciences, 2017, 99, 45–65. [DOI] [PubMed] [Google Scholar]
- 112.Yoshida T, Lai TC, Kwon GS and Sako K, Expert opinion on drug delivery, 2013, 10, 1497–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takemoto Y, Ajiro H, Asoh T.-a. and Akashi M, Chemistry of Materials, 2010, 22, 2923–2929. [Google Scholar]
- 114.Mc Gann MJ, Higginbotham CL, Geever LM and Nugent MJ, International journal of pharmaceutics, 2009, 372, 154–161. [DOI] [PubMed] [Google Scholar]
- 115.Vivek R, Babu VN, Thangam R, Subramanian K and Kannan S, Colloids and Surfaces B: Biointerfaces, 2013, 111, 117–123. [DOI] [PubMed] [Google Scholar]
- 116.Kim AR, Lee SL and Park SN, International journal of biological macromolecules, 2018, 118, 731–740. [DOI] [PubMed] [Google Scholar]
- 117.Fan X, Wang T and Miao W, Journal of Polymer Research, 2018, 25, 215. [Google Scholar]
- 118.Basak P and Adhikari B, Journal of Materials Science: Materials in Medicine, 2009, 20, 137. [DOI] [PubMed] [Google Scholar]
- 119.Zhang L, Wang L, Guo B and Ma PX, Carbohydrate Polymers, 2014, 103, 110–118. [DOI] [PubMed] [Google Scholar]
- 120.Bakker MH and Dankers PY, in Self-assembling Biomaterials, Elsevier, 2018, pp. 177–204. [Google Scholar]
- 121.Bastings MM, Koudstaal S, Kieltyka RE, Nakano Y, Pape A, Feyen DA, Van Slochteren FJ, Doevendans PA, Sluijter JP and Meijer E, Advanced healthcare materials, 2014, 3, 70–78. [DOI] [PubMed] [Google Scholar]
- 122.Dankers PY, van Luyn MJ, Huizinga-van der Vlag A, van Gemert GM, Petersen AH, Meijer E, Janssen HM, Bosman AW and Popa ER, Biomaterials, 2012, 33, 5144–5155. [DOI] [PubMed] [Google Scholar]
- 123.Fundueanu G, Mocanu G, Constantin M, Carpov A, Bulacovschi V, Esposito E and Nastruzzi C, International journal of pharmaceutics, 2001, 218, 13–25. [DOI] [PubMed] [Google Scholar]
- 124.Fundueanu G, Constantin M, Bucatariu S and Mocanu G, Journal of Nanoscience and Nanotechnology, 2017, 17, 4643–4648. [Google Scholar]
- 125.Racovita S, Lungan M, Bunia I, Popa M and Vasiliu S, Reactive and functional polymers, 2016, 105, 103–113. [Google Scholar]
- 126.Sun J and Zhou Z, Drug design, development and therapy, 2018, 12, 383. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 127.Pahuja P, Arora S and Pawar P, Expert Opinion on Drug Delivery, 2012, 9, 837–861. [DOI] [PubMed] [Google Scholar]
- 128.Park H, Woo EK and Lee KY, Journal of Controlled Release, 2014, 196, 146–153. [DOI] [PubMed] [Google Scholar]
- 129.Liu S, Oderinde O, Hussain I, Yao F and Fu G, Polymer, 2018, 144, 111–120. [Google Scholar]
- 130.Su J, Gels, 2018, 4, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Streuli C, Current opinion in cell biology, 1999, 11, 634–640. [DOI] [PubMed] [Google Scholar]
- 132.Chuang C-H, Lin R-Z, Melero-Martin JM and Chen Y-C, Artificial cells, nanomedicine, and biotechnology, 2018, 46, S434–S447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao X, Lang Q, Yildirimer L, Lin ZY, Cui W, Annabi N, Ng KW, Dokmeci MR, Ghaemmaghami AM and Khademhosseini A, Advanced healthcare materials, 2016, 5, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sheldon R, Schoevaart R and Van Langen L, Biocatalysis and Biotransformation, 2005, 23, 141–147. [Google Scholar]
- 135.Kunjukunju S, Roy A, Shekhar S and Kumta PN, International journal of biological macromolecules, 2018, 115, 176–184. [DOI] [PubMed] [Google Scholar]
- 136.Sokic S and Papavasiliou G, Acta biomaterialia, 2012, 8, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lutolf M, Lauer-Fields J, Schmoekel H, Metters AT, Weber F, Fields G and Hubbell JA, Proceedings of the National Academy of Sciences, 2003, 100, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ulijn RV, Bibi N, Jayawarna V, Thornton PD, Todd SJ, Mart RJ, Smith AM and Gough JE, Materials Today, 2007, 10, 40–48. [Google Scholar]
- 139.Seliktar D, Zisch AH, Lutolf MP, Wrana JL and Hubbell JA, Journal of Biomedical Materials Research Part A, 2004, 68A, 704–716. [DOI] [PubMed] [Google Scholar]
- 140.Liu Y, He W, Zhang Z and Lee B, Gels, 2018, 4, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thoniyot P, Tan MJ, Karim AA, Young DJ and Loh XJ, Advanced Science, 2015, 2, 1400010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bian L, Zhai DY, Tous E, Rai R, Mauck RL and Burdick JA, Biomaterials, 2011, 32, 6425–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Eivazzadeh-Keihan R, Chenab KK, Taheri-Ledari R, Mosafer J, Hashemi SM, Mokhtarzadeh A, Maleki A and Hamblin MR, Materials Science and Engineering: C, 2020, 107, 110267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yan J, Miao Y, Tan H, Zhou T, Ling Z, Chen Y, Xing X and Hu X, Materials Science and Engineering: C, 2016, 63, 274–284. [DOI] [PubMed] [Google Scholar]
- 145.Zhou M, Lozano N, Wychowaniec JK, Hodgkinson T, Richardson SM, Kostarelos K and Hoyland JA, Acta biomaterialia, 2019, 96, 271–280. [DOI] [PubMed] [Google Scholar]
- 146.Blaeser A, Million N, Campos DFD, Gamrad L, Köpf M, Rehbock C, Nachev M, Sures B, Barcikowski S and Fischer H, Nano Research, 2016, 9, 3407–3427. [Google Scholar]
- 147.Hans ML and Lowman AM, Current Opinion in Solid State and Materials Science, 2002, 6, 319–327. [Google Scholar]
- 148.Satarkar NS, Biswal D and Hilt JZ, Soft Matter, 2010, 6, 2364–2371. [Google Scholar]
- 149.Liang R, Wei M, Evans DG and Duan X, Chemical Communications, 2014, 50, 14071–14081. [DOI] [PubMed] [Google Scholar]
- 150.Kuttappan S, Mathew D, Jo J.-i., Tanaka R, Menon D, Ishimoto T, Nakano T, Nair SV, Nair MB and Tabata Y, Acta Biomaterialia, 2018, 78, 36–47. [DOI] [PubMed] [Google Scholar]
- 151.Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK and Roy S, The American journal of pathology, 2015, 185, 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Matson JB and Stupp SI, Chemical communications, 2012, 48, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li I-C, Moore AN and Hartgerink JD, Biomacromolecules, 2016, 17, 2087–2095. [DOI] [PubMed] [Google Scholar]
- 154.Wickremasinghe NC, Kumar VA and Hartgerink JD, Biomacromolecules, 2014, 15, 3587–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Truong VX, Ablett MP, Richardson SM, Hoyland JA and Dove AP, Journal of the American Chemical Society, 2015, 137, 1618–1622. [DOI] [PubMed] [Google Scholar]
- 156.Luo Y and Shoichet MS, Nature materials, 2004, 3, 249. [DOI] [PubMed] [Google Scholar]
- 157.Wosnick JH and Shoichet MS, Chemistry of Materials, 2007, 20, 55–60. [Google Scholar]
- 158.Aizawa Y, Wylie R and Shoichet M, Advanced materials, 2010, 22, 4831–4835. [DOI] [PubMed] [Google Scholar]
- 159.Webber MJ, Tongers J, Renault M-A, Roncalli JG, Losordo DW and Stupp SI, Acta biomaterialia, 2010, 6, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stendahl JC, Rao MS, Guler MO and Stupp SI, Advanced Functional Materials, 2006, 16, 499–508. [Google Scholar]
- 161.Lee O-S, Stupp SI and Schatz GC, Journal of the American Chemical Society, 2011, 133, 3677–3683. [DOI] [PubMed] [Google Scholar]
- 162.Kang MK, Colombo JS, D’Souza RN and Hartgerink JD, Biomacromolecules, 2014, 15, 2004–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Galler KM, Aulisa L, Regan KR, D’Souza RN and Hartgerink JD, Journal of the American Chemical Society, 2010, 132, 3217–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kumar VA, Taylor NL, Shi S, Wang BK, Jalan AA, Kang MK, Wickremasinghe NC and Hartgerink JD, ACS nano, 2015, 9, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kumar VA, Liu Q, Wickremasinghe NC, Shi S, Cornwright TT, Deng Y, Azares A, Moore AN, Acevedo-Jake AM and Agudo NR, Biomaterials, 2016, 98, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hachim D, Whittaker TE, Kim H and Stevens MM, Journal of Controlled Release, 2019, 313, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schultz GS and Wysocki A, Wound repair and regeneration, 2009, 17, 153–162. [DOI] [PubMed] [Google Scholar]
- 168.Şalaru DL, Arsenescu-Georgescu C, Chatzikyrkou C, Karagiannis J, Fischer A and Mertens PR, Nephrology Dialysis Transplantation, 2016, 31, 1781–1787. [DOI] [PubMed] [Google Scholar]
- 169.Higashiyama S, Abraham JA, Miller J, Fiddes JC and Klagsbrun M, Science, 1991, 251, 936–939. [DOI] [PubMed] [Google Scholar]
- 170.Bragg JC, Kweon H, Jo Y, Lee KG and Lin C-C, RSC advances, 2016, 6, 114353–114360. [Google Scholar]
- 171.Li Q, Niu Y, Diao H, Wang L, Chen X, Wang Y, Dong L and Wang C, Biomaterials, 2017, 148, 54–68. [DOI] [PubMed] [Google Scholar]
- 172.Li Q, Guo G, Meng F, Wang HH, Niu Y, Zhang Q, Zhang J, Wang Y, Dong L and Wang C, ACS Macro Letters, 2016, 5, 617–621. [DOI] [PubMed] [Google Scholar]
- 173.Jeon O, Lee K and Alsberg E, Small, 2018, 14, 1800579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Battig MR, Huang Y, Chen N and Wang Y, Biomaterials, 2014, 35, 8040–8048. [DOI] [PubMed] [Google Scholar]
- 175.Ellington AD and Szostak JW, nature, 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 176.Tuerk C and Gold L, science, 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 177.Abune L, Zhao N, Lai J, Peterson B, Szczesny S and Wang Y, ACS Biomaterials Science & Engineering, 2019, 5, 2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Brudno Y, Desai RM, Kwee BJ, Joshi NS, Aizenberg M and Mooney DJ, ChemMedChem, 2015, 10, 617–620. [DOI] [PubMed] [Google Scholar]
- 179.Mejia Oneto JM, Khan I, Seebald L and Royzen M, ACS Central Science, 2016, 2, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nguyen LH, Gao M, Lin J, Wu W, Wang J and Chew SY, Scientific reports, 2017, 7, 42212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gonzalez-Fernandez T, Rathan S, Hobbs C, Pitacco P, Freeman F, Cunniffe G, Dunne N, McCarthy H, Nicolosi V and O’Brien F, Journal of Controlled Release, 2019, 301, 13–27. [DOI] [PubMed] [Google Scholar]
- 182.McMillan A, Nguyen MK, Gonzalez-Fernandez T, Ge P, Yu X, Murphy WL, Kelly DJ and Alsberg E, Biomaterials, 2018, 161, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang S, Greenfield MA, Mata A, Palmer LC, Bitton R, Mantei JR, Aparicio C, De La Cruz MO and Stupp SI, Nature materials, 2010, 9, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Guvendiren M and Burdick JA, Nature communications, 2012, 3, 792. [DOI] [PubMed] [Google Scholar]
- 185.Mabry KM, Lawrence RL and Anseth KS, Biomaterials, 2015, 49, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Joseph CA, McCarthy CW, Tyo AG, Hubbard KR, Fisher HC, Altscheffel JA, He W, Pinnaratip R, Liu Y and Lee BP, ACS biomaterials science & engineering, 2018, 5, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wei Z and Gerecht S, Biomaterials, 2018, 185, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sani ES, Kheirkhah A, Rana D, Sun Z, Foulsham W, Sheikhi A, Khademhosseini A, Dana R and Annabi N, Science advances, 2019, 5, eaav1281. [DOI] [PMC free article] [PubMed] [Google Scholar]