

Abstract
Mutations in histidyl-tRNA synthetase (HARS1), an enzyme that charges tRNA with the amino acid histidine in the cytoplasm, have only been associated to date with autosomal recessive Usher syndrome type III and autosomal dominant Charcot-Marie-Tooth disease type 2W. Using massive parallel sequencing, we identified bi-allelic HARS1 variants in a child (c.616G>T, p.Asp206Tyr and c.730delG, p.Val244Cysfs*6) and in two sisters (c.1393A>C, p.Ile465Leu and c.910_912dupTTG, p.Leu305dup), all characterized by a multi-system ataxic syndrome. All mutations are rare, segregate with the disease, and are predicted to have a significant effect on protein function. Functional studies helped to substantiate their disease-related roles. Indeed, yeast complementation assays showing that one out of two mutations in each patient is loss-of-function, and the reduction of mRNA and protein levels and enzymatic activity in patient’s skin-derived fibroblasts, together support the pathogenicity of the identified HARS1 variants in the patient phenotypes. Thus, our efforts expand the allelic and clinical spectrum of HARS1-related disease.
Keywords: hereditary ataxia, autosomal recessive, exome sequencing, multigene resequencing panel, yeast complementation assay, aminoacylation assay
Aminoacyl-tRNA synthetases are ubiquitously expressed and highly conserved enzymes that catalyze the conjugation of tRNA to cognate amino acids in the cytoplasm, in mitochondria, or in both locations (Antonellis & Green, 2008). Histidyl-tRNA synthetase (HARS1) is a homodimeric class IIa aminoacyl tRNA synthetase that charges tRNA with the amino acid histidine in the cytoplasm. Mutations in HARS (also referred to as HARS1, MIM #142810) were found to cause two different disorders through either autosomal dominant or autosomal recessive inheritance, a feature shared with only four other ARS enzymes (Meyer-Schuman & Antonellis, 2017). Puffenberger and collaborators initially identified three Old Order Amish patients who were homozygous for a missense mutation and who had Usher Syndrome type IIIB syndrome (USH3B, MIM #614504), an autosomal recessive disorder characterized by motor impairment, and progressive vision and hearing loss in childhood. Subsequently, several studies revealed monoallelic mutations in HARS1 as the cause of dominantly inherited Charcot-Marie-Tooth disease type 2W (CMT2W, MIM #616625), a motor and sensory peripheral neuropathy affecting both lower and upper limbs (Puffenberger et al., 2012; Vester et al., 2013; Safka Brozkova et al., 2015; Abbott et al., 2018; Royer-Bertrand et al., 2019). Herein, we describe a novel HARS1-related phenotype in three patients from two families harboring bi-allelic mutations in the histidyl-tRNA synthetase gene. This study was approved by the pediatric ethic committee of the Tuscany Region, Italy and cellular and DNA studies were performed with parental written informed consents.
Patient 1 is a 10-year-old boy born to healthy, unrelated parents after a complicated pregnancy with risk of placental abruption. At birth, he was admitted to neonatal intensive care unit because of hypoglycemia. Psychomotor development was delayed, and the boy could sit unassisted at age 9 months. Parents reported shuffling gait for a few minutes with head and upper limb tremor since age 1.5 years. He could walk with support at age 2 years mostly with a broad base and spastic gait. The patient was tested for cognitive delay at the age of 3 years and received a diagnosis of mild intellectual disability. He could speak words at age 2.5 years, but speech was dysarthric and sometimes difficult to understand. In spite of a normal brain MRI at that age, the patient presented gait ataxia with leg spasticity, dysarthria and dysmetria, and head and hand action tremor. At age 5, brain MRI showed T2-hyperintense signal in the upper cerebellar peduncles and subthalamic nuclei with slight enlargement of the inferior vermis interfolial spaces. At that time, the clinical phenotype had progressed with more marked cerebellar and pyramidal tract involvement with oculomotor apraxia, nystagmus, and presence of athetoid movements. Latest neurological examination at age 10 years showed a stable clinical and neuroimaging condition (Figure 1, upper panels) in an otherwise normal child (height/weight z-score of −1.15). An older sister, now age 14 years, is healthy. Scale for the assessment and rating of ataxia (SARA) score was 28/40, and spastic paraplegia rating scale (SPRS) score was 36/52 suggestive of severe neurological impairment.
Figure 1. Brain MRI scans.
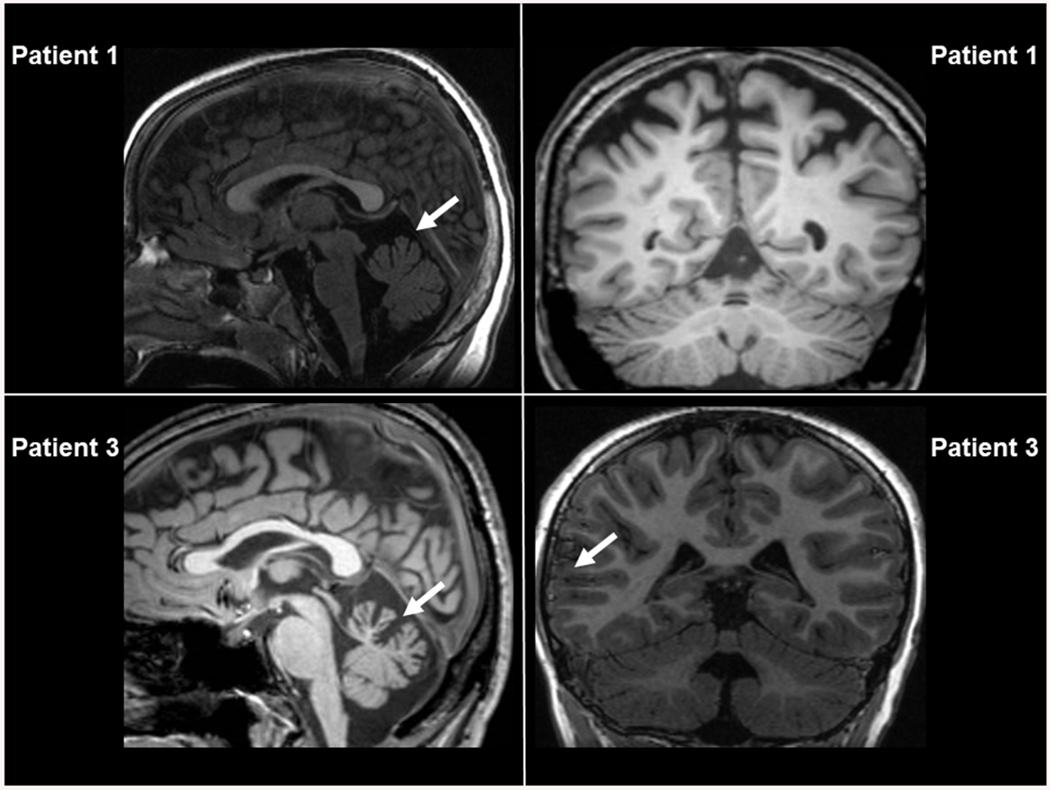
Sagittal and coronal sections taken from patients 1 and 3. Arrows indicates abnormalities detected (see text).
Patient 2 is a 41-year-old woman; her height is 154 cm, and her weight is 45 kg (z-score of −2.62). She presented in the neonatal period with weak sucking and a motor delay. She grew poorly and she could walk unaided at age 4 years with broad gait. There was a significant speech delay and severe intellectual disability. Hearing and visual functions were unremarkable. Her disease progressed slowly. At the age of 40 years neurological examination was significant for ataxic gait possible only with support, normal eye movements, hyponasal voice with dysarthria, absent nystagmus and dysmetria, decreased muscle tone, absent knee and ankle jerks, and severe distal weakness in the lower limbs. Physical examination showed scoliosis, pes planus with hammertoe, and ligamentous laxity. The patient also presented with urinary urgency. MRI showed microcephaly, and a slight enlargement of cerebellar interfolial spaces. Nerve conduction studies revealed slowed motor velocity and increased latency. SARA score was 20/40, and SPRS score was 22/52.
Patient 3 is the 37-year-old sister of patient 2; her height is 135 cm, and her weight is 41 kg (z-score of −2.85). She had a similar neurodevelopmental disorder to patient 2 and has used hearing aids since the age of 28 years because of conductive hearing deficits. At the age of 36 years neurological examination was significant for unsteady gait possible with support and steppage, absent tendon reflexes and indifferent plantar response. The patient presented with distal muscle weakness and lower limb hypotrophy. She had hyponasal speech without dysarthria, absent nystagmus and slight dysmetria at finger-to-nose test. Physical examination showed scoliosis, knee valgus, and pes planus with hammertoe. Brain MRI showed microcephaly, enlargement of the inferior vermis cistern and atrophy of left operculum (Figure 1, lower panels). Heart, visual, liver and kidney functions were normal. SARA score was 21/40, and SPRS score was 23/52. As for her sister, clinical scores indicate moderate-severe impairment of cerebellum and corticospinal tract neurons.
Exome sequencing (TruSightOne, Illumina, San Diego, California) was performed in patient 1, whilst a targeted multigene resequencing panel analysis encompassing 194 genes related to inherited ataxias (Nimblegen, Roche, Basel, Switzerland; gene list available upon request) was used for patients 2 and 3. Sequencing was carried out using a NextSeq500 Illumina platform (Illumina, San Diego, California) and a customized bioinformatics pipeline adopting the Ingenuity Variant Analysis (https://www.qiagenbioinformatics.com/products/ingenuity-variant-analysis/, Qiagen, Hilden, Germany) suite was employed as described before (D’Amore et al., 2018). Prior to Next Generation Sequencing (NGS) analysis, all patients had been tested for pathological trinucleotide repeat expansions at FRDA, SCA type 1, 2, 3, 6, 7, 17 and FXTAS loci. We found that patient 1 is compound heterozygous for two HARS1 variants (RefSeq: NM_002109.6; UniProt [https://www.uniprot.org]: P12081): one frameshift variant (c.730delG, p.Val244Cysfs*6) and one missense variant (c.616G>T, p.Asp206Tyr) (Supporting Information). We further found that patients 2 and 3 (who are sisters) harbored one missense variant (c.1393A>C, p.Ile465Leu) and one in-frame insertion variant (c.910_912dupTTG, p.Leu305dup) in compound heterozygosity (Supporting Information). All mutations were confirmed by Sanger sequencing and segregated with the disease phenotypes (Supporting Information). Patient 1 shares p.Val244Cysfs*6 with the father and healthy sister, whilst p.Asp206Tyr was inherited from the mother. Patients 2 and 3 share p.Leu305dup with their mother, and we therefore inferred that p.Ile465Leu was inherited from the father, since we could not genotype paternal DNA. Whilst the p.Ile465Leu has a very low frequency (MAF=0.000008) in gnomAD (https://gnomad.broadinstitute.org/), the other variants are absent in both gnomAD and our in-house multi-gene panel and WES cohort.
To predict the impact of the missense variants on HARS1 protein functions, we used an in silico pipeline comprehensive of nine algorithms which included SIFT (https://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), FATHMM-MKL (http://fathmm.biocompute.org.uk/fathmmMKL.htm), GERP, LRT (https://varsome.com/), Mutation Assessor (http://mutationassessor.org/r3/), Provean (http://provean.jcvi.org/index.php) and CADD (https://cadd.gs.washington.edu/). Eight out of nine of the abovementioned prediction tools predicted deleterious effects for p.Asp206Tyr and p.Ile465Leu (Supplementary Table 1). Of note, p.Asp206Tyr and p.Leu305dup are in the catalytic domain, which is crucial for the enzymatic activity, whereas p.Ile465Leu is located in the tRNA binding domain, which is important for recognizing the correct tRNA molecules (Supporting Information).
To determine the effect of each genotype on gene expression, RNA and protein samples were obtained from primary fibroblasts isolated from the three patients, and real-time qPCR and western blot analysis were performed. In patient 1, p.Asp206Tyr but not Val244Cysfs*6 was expressed at the cDNA level suggesting that the mRNA carrying the p.Val244Cysfs*6 was unstable or subjected to nonsense-mediated decay (Supporting information). In contrast, the missense and in-frame insertion variants in patients 2 and 3 were expressed at the mRNA level (Supporting information). Quantitative PCR and western blotting indicated a significant reduction of HARS1 mRNA (Figure 2A) and protein expression (Figure 2B), respectively, in patient skin fibroblasts.
Figure 2. Functional studies in skin fibroblasts and yeast.
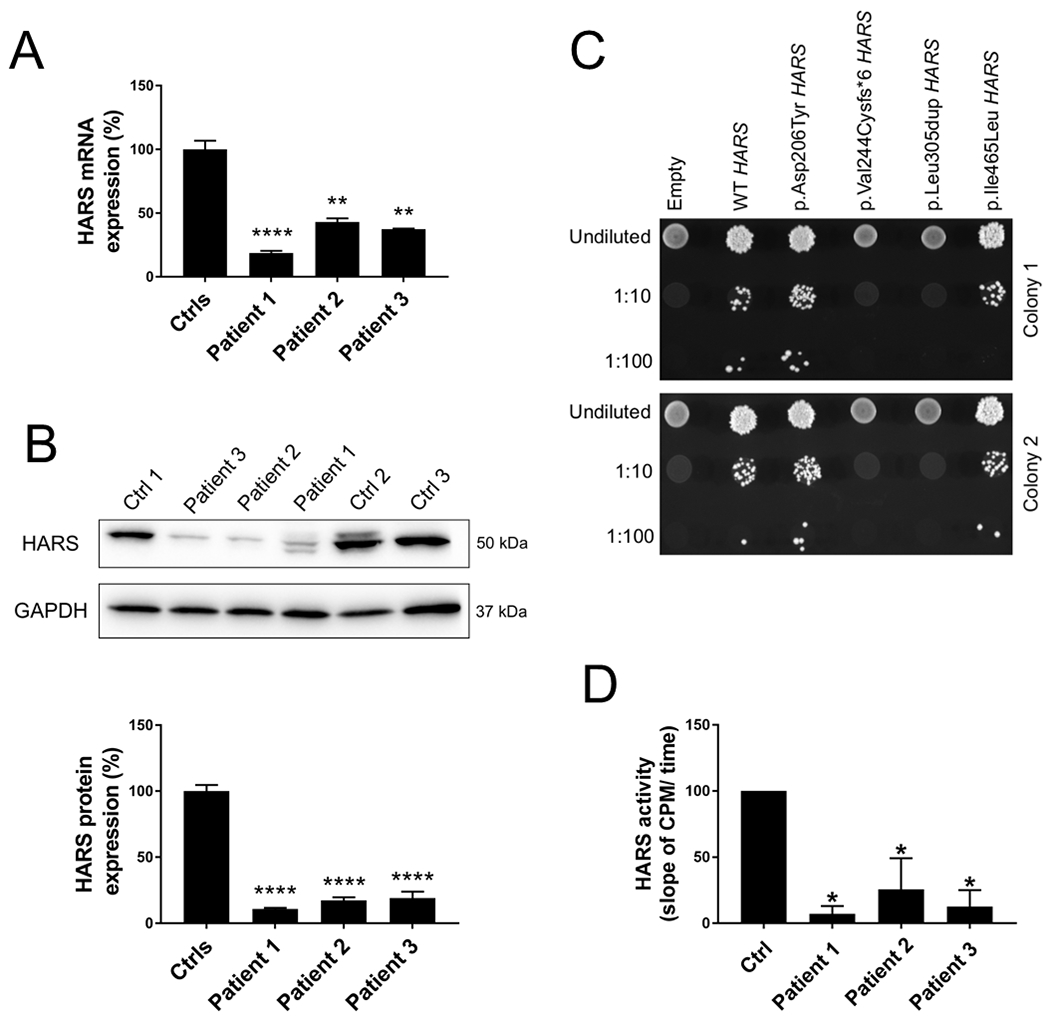
(A) Real time quantitative PCR showed a significant reduction of HARS1 mRNA level in all patients. Data are shown as mean ± SEM and were normalized to control fibroblasts. Ctrl (for controls) n=22; n=3 in each patient. One-way ANOVA was used for statistical analysis. ** indicates p<0.01, **** indicates p<0.0001. (B) Representative western blot (top) used for HARS1 protein detection. Quantification (bottom), showed a severe reduction of protein expression in all patients. Data are shown as mean ± SEM of three independent experiments (n= 9) and were normalized to control fibroblasts. One-way ANOVA was used for statistical analysis. **** indicates p<0.0001. (C) HARS1 variants display loss-of-function effects in yeast complementation assays. Yeast lacking endogenous HTS1 were transformed with a vector with no HARS1 insert (‘empty’), or with vectors containing wild-type or variant HARS1. The vector used in each experiment is indicated across the top. Cultures were plated undiluted or diluted (1:10 or 1:100) on media containing 5-FOA and grown at 30°C. Two independently transformed colonies were tested (upper and lower panels). (D) HARS1 aminoacylation activity as determined by the slope of counts per minute over time. Data shown as mean ± SEM of five independent experiments and were normalized to control fibroblasts. One-way ANOVA was used for statistical analysis. * p< 0.02.
To determine the functional consequences of each allele in vivo, yeast complementation assays were employed using established methods (Vester et al., 2013; Oprescu, Beg, Griffin & Antonellis, 2017). Complementation studies were performed using a previously validated haploid yeast strain with the endogenous HARS1 orthologue (HTS1) deleted and viability maintained via a maintenance vector harboring HTS1 and URA3 (Vester et al., 2013; Abbott et al., 2018). Each variant was modeled in human HARS1 and then transformed into the yeast strain described above. Growth was evaluated on 5-Fluoroorotic acid (5-FOA), which is toxic to strains expressing URA3 and selects for loss of the maintenance vector (Boeke, Lacroute, & Fink, 1984). These analyses revealed that: (1) wild-type human HARS1 supports yeast cell growth, while a vector with no HARS1 insert does not; (2) p.Asp206Tyr and p.Ile465Leu HARS1 allow growth consistent with wild-type HARS1 in this assay; and (3) p.Val244Cysfs*6 and p.Leu305dup do not support any yeast growth, consistent with these alleles having a complete loss-of-function effect (Figure 2C). When measured in skin fibroblasts, HARS1 aminoacylation activity, tested by incubating whole cell extracts with [14C] histidine, tRNAHis, and ATP using established methods (Zhang et al., 2014), showed a dramatic reduction of tRNA charging in all patients (Figure 2D).
Taking together our findings, we report three patients in two unrelated families carrying bi-allelic HARS1 variants present with novel clinical features. All three patients presented with microcephaly, mild-to severe intellectual disability, skeletal deformities, and ataxic broad base gait with clinical features affecting the cerebellar and pyramidal tract system; none of these phenotypes have been previously associated with HARS1 variants. Further, choreo-athetoid movements in patient 1 and dystonic postures in the two sisters characterize this as an atypical ataxia-related recessive phenotype. Consistent with previously reported HARS1 variants, hearing loss was found in patient 3 and peripheral motor nerve involvement ―the hallmark of CMT2W― occurred in patient 2. No peripheral nerve conduction study was performed in patient 3, in which tendon reflexes were absent, associated with hypotrophy and weakness, suggesting a motor peripheral neuropathy. In contrast, retinal dysfunction, typical of HARS1-related Usher Syndrome IIIB, was not found in any patient.
Several pieces of evidence support the pathogenicity of the identified HARS1 variants. First, similar clinical features are present in two unrelated kindred. These patients all presented with an overlapping, recessive multi-system syndrome with evidence of a progressive neurodevelopmental disorder affecting the cerebellum and corticomotor tract and associated with intellectual disability. Second, the nature of the identified alleles combined with in silico predictions are consistent with pathogenicity. Third, our combined functional studies demonstrate that each allele affects HARS1 function: (i) qPCR and western blotting revealed a significant reduction of HARS1 mRNA and protein expression; (ii) studies in yeast showed that p.Val244Cysfs*6 and p.Leu305dup cause a loss-of-function effect; (iii) aminoacylation assays using patient cells indicated a strong and significant reduction of tRNA charging. Importantly, each of these assays have been shown to accurately predict the functional consequences of pathogenic ARS alleles (Oprescu et al., 2017). The fact that all of our patients harbor at least one mutation that is not a null allele is not surprising, considering the crucial role of ARSs enzymes that requires a residual activity for viability. In sum, based on these genetic, phenotypic, and functional data, the patients we describe here expand the clinical manifestations of HARS1 variants and enlarge the set of aminoacyl-tRNA synthetases associated with ataxia and movement disorders.
Pathogenic variants in ARS-encoding loci have been implicated in a large number of genetic diseases embracing a wide spectrum of different phenotypes, causing impairments in different tissues (Meyer-Schuman & Antonellis, 2017, Royer-Bertrand et al., 2019). Both loss-of-function and gain-of-function mechanisms have been proposed for dominant ARS-associated diseases while loss-of-function effects are clearly the culprit in the recessive phenotypes (Wallen & Antonellis, 2013). Impairment in the central nervous system in ARS-associated diseases could be caused by decreased aminoacylation only in neurons, cells with high energy and protein synthesis demands, and not in other tissues (Royer-Bertrand et al., 2019) ) but it is also possible that specific alleles affect residual ARS activity only in exclusive brain regions (Fuchs et al., 2019). Further experimentation is required to test these hypotheses.
In conclusion, combined clinical, genetic, in silico, in vitro, and in vivo analyses corroborate the hypothesis that the bi-allelic mutations in HARS1 described here are the cause of a previously unreported ataxia-related recessive phenotype in our patients. Whilst the mechanisms that link impaired HARS1 activity to a specific brain involvement during development remain unclear, our patients broaden the clinical and allelic heterogeneity of HARS1-related disease.
Supplementary Material
Supplementary Table. In silico prediction of HARS1 missense mutations.
ACKNOWLEDGEMENTS
D.G. is the recipient of a PhD fellowship in the PhD program in Neuroscience, University of Florence. M.E.K. is supported by the NIH Medical Scientist Training Program Training Grant (GM007863), the NIH Cellular and Molecular Biology Training Grant (GM007315), and an NIH National Research Service Award (F31) from the National Institute of Neurological Disorders and Stroke (NS113515). R.M. is supported by the Michigan Pre-doctoral Training in Genetics Program (GM007544) and an NIH National Research Service Award (F31) from the National Institute of Neurological Disorders and Stroke (NS108510). A.A. is supported by a grant from the National Institute of General Medical Sciences (GM118647). F.M.S. is supported by the Italian Ministry of Health-Ricerca Finalizzata RF-2016-02361610 and MIT-OMICS, and the E-RARE-3 Joint Transnational Call grant PREPARE (MoH; project 3398).
Funding information:
University of Florence, Grant/Award Number: PhD fellowship in the PhD program in Neuroscience (D.G.); NIH Medical Scientist Training Program, Grant/Award Number: GM007863 (M.E.K.); NIH Cellular and Molecular Biology, Grant/Award Number: GM007315 (M.E.K.); National Institute of Neurological Disorders and Stroke, Grant/Award Number: NIH National Research Service Award (F31) NS113515 (M.E.K.); Michigan Pre-doctoral Training in Genetics Program, Grant/Award Number: GM007544 (R.M.); National Institute of Neurological Disorders and Stroke, Grant/Award Number: NIH National Research Service Award (F31) NS108510 (R.M.); National Institute of General Medical Sciences, Grant/Award Number: GM118647 (A.A.); Italian Ministry of Health-Ricerca Finalizzata, Grant/Award Numbers: RF-2016-02361610 and MIT-OMICS (F.M.S.); E-RARE-3 Joint Transnational Call grant PREPARE, Grant/Award Numbers: MoH, project 3398 (F.M.S.).
Footnotes
ACCESSION NUMBERS
All mutations described can be found in the ClinVar genetic repository (https://www.ncbi.nlm.nih.gov/clinvar/) (Submission ID: SUB6272751, release is hold until published).
CONFLICT OF INTERESTS
The authors declare no conflict of interest
DATA SHARING STATEMENT
Data are available on request due to privacy/ethical restrictions
Supporting information. Additional supporting information include detailed materials and methods of quantitative real-time PCR, western blotting, yeast complementation assays and aminoacylation assay, supplemental figures showing HARS1 variants confirmations, cDNA mutation analysis and segregation study, localization and phylogenetic conservation, and supplemental references.
REFERENCES
- Abbott JA, Meyer-Schuman R, Lupo V, Feely S, Mademan I, Oprescu SN, … Francklyn C (2018). Substrate interaction defects in histidyl-tRNA synthetase linked to dominant axonal peripheral neuropathy. Human Mutation, 39(3), 415–432. 10.1002/humu.23380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A, & Green ED (2008). The Role of Aminoacyl-tRNA Synthetases in Genetic Diseases. Annual Review of Genomics and Human Genetics, 9(1), 87–107. 10.1146/annurev.genom.9.081307.164204 [DOI] [PubMed] [Google Scholar]
- Boeke JD, Lacroute F, & Fink GR (1984). A positive selection for mutants lacking orotidine-5’-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Molecular & General Genetics, 197(2), 345–346. 10.1007/bf00330984 [DOI] [PubMed] [Google Scholar]
- D’Amore A, Tessa A, Casali C, Dotti M, Filla A, Silvestri G, … Santorelli F (2018). Next Generation Molecular Diagnosis of Hereditary Spastic Paraplegias: An Italian Cross-Sectional Study Patients and Study Design. Frontiers in Neurology, 9(981), 1–13. 10.3389/fneur.2018.00981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, & Liu X (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human Molecular Genetics, 24(8), 2125–2137. 10.1093/hmg/ddu733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SA, Schene IF, Kok G, Jansen JM, Nikkels PGJ, van Gassen KLI, … van Hasselt PM (2019). Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genetics in Medicine, 21(2), 319–330. 10.1038/s41436-018-0048-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Schuman R, & Antonellis A (2017). Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Human Molecular Genetics, 26(R2), R114–R127. 10.1093/hmg/ddx231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprescu SN, Griffin LB, Beg AA, & Antonellis A (2017). Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods, 113, 139–151. 10.1016/j.ymeth.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, … Strauss KA (2012). Genetic Mapping and Exome Sequencing Identify Variants Associated with Five Novel Diseases. PLoS One, 7(1), e28936 10.1371/journal.pone.0028936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer-Bertrand B, Tsouni P, Mullen P, Campos Xavier B, Mittaz Crettol L, Lobrinus AJ, … Tran C (2019). Peripheral neuropathy and cognitive impairment associated with a novel monoallelic HARS1 variant. Annals of Clinical and Translational Neurology, 6(6), 1072–1080. 10.1002/acn3.791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safka Brozkova D, Deconinck T, Beth Griffin L, Ferbert A, Haberlova J, Mazanec R, … Baets J (2015). Loss of function mutations in HARS1 cause a spectrum of inherited peripheral neuropathies. Brain, 138(8), 2161–2172. 10.1093/brain/awv158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester A, Velez-Ruiz G, McLaughlin HM, Nisc Comparative Sequencing Program, Lupski JR, Talbot K, … Antonellis A (2013). A Loss-of-Function Variant in the Human Histidyl-tRNA Synthetase (HARS1) Gene is Neurotoxic In Vivo. Human Mutation, 34(1), 191–199. 10.1002/humu.22210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallen RC, & Antonellis A (2013). To Charge or Not to Charge: Mechanistic Insights into Neuropathy-Associated tRNA Synthetase Mutations. Current Opinion in Genetics & Development, 23(3), 302–309. 10.1016/j.gde.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, … Nabbout R (2014). Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. American Journal of Human Genetics, 94(4), 547–558. 10.1016/j.ajhg.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table. In silico prediction of HARS1 missense mutations.