

Abstract
Target engagement assays are crucial for establishing the mechanism-of-action of small molecules in living systems. Integral membrane transporters, due to their specialized biophysical properties and activity assays, can present a challenging protein class for assessing cellular engagement by small molecules. Here, we describe the chemical proteomic discovery of alpha-chloroacetamide (αCA) compounds that covalently modify cysteine-54 (C54) of the MPC2 subunit of the mitochondrial pyruvate carrier (MPC). We leverage this finding to create an alkyne-modified αCA, YY4-yne, that serves as a versatile cellular target engagement probe for MPC2 in click chemistry-enabled western blotting or global mass spectrometry-based proteomic experiments. Using YY4-yne, we demonstrate that UK-5099, an alpha-cyanocinnamate inhibitor of the MPC complex, first discovered more than 30 years ago, but still with a poorly defined mechanism-of-action, engages MPC2 with remarkable selectivity in human cells. These findings support a model where UK-5099 inhibits the MPC complex by binding to C54 of MPC2 in a covalent reversible manner that can be quantified in cells using the YY4-yne probe.
Keywords: Chemical proteomics, mitochondrial pyruvate complex, Target engagement, α-chloroacetamide, cysteine, reversible covalency
Graphical Abstract
We report the chemical proteomic discovery of covalent, irreversible ligands for the human mitochondrial pyruvate carrier (MPC) and demonstrate their utility as cellular engagement probes for this important integral membrane metabolite transporter.
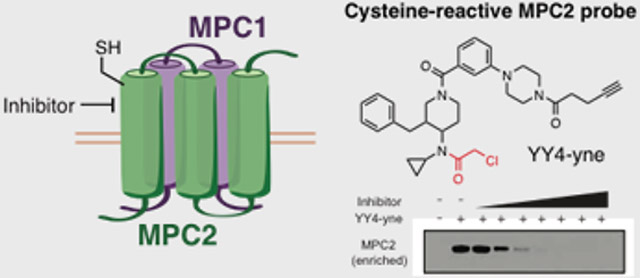
As part of a broader research program aimed at characterizing the protein targets of electrophilic compounds in primary human T cells[1] using a mass spectrometry (MS)-based chemical proteomic method that quantifies cysteine reactivity on a global scale[2], we discovered herein an alpha-chloroacetamide (αCA) YY4 (1; Figure 1A) that strongly engages cysteine-54 (C54) in subunit 2 of the mitochondrial pyruvate carrier (MPC2) (Figure 2A, B). This interaction was of interest because, to our knowledge, covalent, irreversible ligands have not been described for MPC2. Additional C54 of MPC2 was not sensitive to structurally related αCA compounds (e.g., EV-93 (3); Figures 1A and 2A, B and Supplementary Table 1) or broadly reactive electrophilic fragments (Figure S1)[1]. The restricted structure-activity relationship (SAR) displayed by C54 in MPC2 was suggestive of residence in a specific small molecule-binding pocket. Motivated by the important role of the MPC in human metabolism[3], combined with the dearth of assays available to evaluate endogenous MPC activity and pharmacological inhibition in cells[4], we set out to further investigate the YY4-MPC2 interaction.
Figure 1.

Chemical structures of compounds used in the study. A) Structures of αCAs YY4 (1) and corresponding alkyne probe YY4-yne (2), and inactive control compounds EV-93 (3). B) Structure of the MPC inhibitor UK-5099 (4).
Figure 2.

Cysteine reactivity profiles of primary human T cells treated with YY4 or EV-93 (10 μM, 3 h). A) Waterfall plots showing reactivity ratios, or R values (DMSO/compound), for cysteines in the particulate fraction of YY4- or EV-93-treated cells, as measured by MS-based proteomics. C54 of MPC2 is marked in both graphs. B) Representative MS1 chromatograms for the MPC2 tryptic peptide containing C54 (K.WGLVC*AGLADMAR.P) from YY4- or EV-93-treated primary human T cells. C) Anti-MPC2 Western blot showing HMW band shift and reduced signal intensity for MPC2 from human T cells treated with YY4, but not UK-5099 (10 μM of each compound, 3 h) or DMSO.
The MPC is an integral membrane transporter composed of two subunits, MPC1 and MPC2[5], that facilitates the transport of cytosolic pyruvate into the mitochondrial matrix and consequently plays a crucial role in cellular metabolism[6]. Small-molecule inhibitors have been described for the MPC[7], including an α-cyanocinnamate UK-5099 (4; Figure 1B) that is speculated to act by a covalent reversible mechanism[8] and has been used to study MPC involvement in pyruvate-related metabolic processes[9]. However, so far, monitoring MPC activity and inhibition in human cells has depended on technically intricate metabolic labeling assays or the introduction of exogenous MPC fusion proteins as BRET-based biosensors[4].
Recognizing that covalent ligands have been converted into versatile target engagement probes for other protein classes, including, for instance, hydrolases[10], kinases[11], glycosidases[12], and GTPases[13], we set out to investigate whether the YY4-MPC2 interaction could be similarly leveraged for developing a cellular engagement probe for the MPC complex. MPC2 is a small protein (14 kDa) with only a single cysteine – C54 (Figure S2) – the site of covalent modification by YY4 mapped in our chemical proteomic experiments. Given the small size of MPC2, we wondered whether covalent modification by YY4 would cause a band shift in the protein migration by SDS-PAGE. Consistent with this hypothesis, MPC2 migrated as a higher molecular weight (HMW) species in YY4-treated vs DMSO-treated human T cells, as detected by Western blotting with commercial anti-MPC2 antibodies (Figure 2C). Curiously, however, the signal intensity for MPC2 was also substantially lower in YY4-treated cells (Figure 2C). Neither the HMW shift nor reduction in signal intensity in MPC2 was observed in T cells treated with UK-5099 (Figure 2C).
The epitope for the commercial anti-MPC2 antibody is unclear, only stated as being a synthetic peptide corresponding to residues surrounding N33 (#46141, Cell Signaling Technology). We initially assumed that YY4 modification of C54 in MPC2 would not disrupt antibody recognition and alternatively considered that the reduction in MPC2 signal could reflect loss of MPC2 protein in cells treated with YY4. We tested this hypothesis by developing an alkyne analogue of YY4 – YY4-yne (2; Figure 1A) – suitable for direct labeling and enrichment of MPC2 from human T cells by conjugation to an azide-biotin tag[14] using copper-catalyzed azide alkyne cycloaddition (CuAAC) chemistry[15]. We performed time-course studies comparing MPC2 signals enriched from T cells treated with YY4-yne measured by MS-based proteomics (Figure 3A) or Western blotting (Figure 3B), which revealed a time-dependent increase and maintenance of YY4-yne-enriched MPC2 signals throughout the treatment period (Figure 3A, B), despite substantial reductions in total MPC2 signal observed by Western blotting in unenriched lysates from YY4-yne-treated cells (Figure 3B). These results indicate that modification of C54 by YY4-yne does not alter MPC2 content in cells, but rather impairs recognition of this protein by the commercial anti-MPC2 antibody.
Figure 3.

Characterization of MPC2 interactions with the YY4-yne probe. A) MPC2 signals in human T cells treated with YY4-yne (10 μM) or DMSO for the indicated times, after which YY4-yne-labeled proteins were enriched by CuAAC to an azide-biotin tag and analyzed by multiplexed (isobaric tandem mass tagging) MS-based proteomics. Data represent average values +/− SD for two replicate experiments. B) YY4-yne-coupled Western blotting of MPC2 from streptavidin-enriched fractions of T cells treated with YY4-yne (10 μM) or DMSO for the indicated times. C) YY4-yne-coupled Western blotting fro T cells treated with the indicated compounds (YY4, EV-93, and UK-5099; 10 μM, 3 h) or DMSO, followed by YY4-yne (10 μM, 2 h). Note that the stronger apparent Western blot signals for YY4-yne-labeled MPC2 in enriched samples likely reflect the larger amount of total cellular protein used as input for streptavidin enrichment experiments (see Supporting Information for more details).
Despite the apparent perturbation of antibody recognition of MPC2 caused by YY4(-yne)-reactivity, sufficient signal intensity was still observed for streptavidin-enriched MPC2 in YY4-yne-coupled Western blotting to furnish a convenient method to measure the engagement of MPC2 by small molecule inhibitors in cells. Pre-treatment of human T cells, for instance, with YY4 or UK-5099, but not the inactive compound EV-93, blocked YY4-yne-enriched MPC2 protein (Figure 3C). We also note that UK-5099 blocked the reduction in total anti-MPC2 signal intensity observed in lysates of T cells treated with YY4-yne (Figure 3C, input). This result is consistent with the proposed covalent, reversible mechanism of action for UK-5099, which would not be expected to permanently modify MPC2 after denaturation of the protein prior to Western blotting. Finally, we found that similar results were obtained in vitro, where T cell lysates were pre-treated with compounds prior to exposure to YY4-yne, although the extent of engagement of MPC2 by YY4(-yne) appeared lower, as reflected in the total and enriched signals measured in standard or YY4-yne-coupled Western blotting, respectively (Figure S3). This result suggests that YY4(-yne) reacts better with MPC2 in cells, where the integrity of the MPC complex may be better maintained.
Despite the widespread use of UK-5099 as an MPC inhibitor, its precise mechanism of action and broader proteomic selectivity remain poorly understood. The latter item was of particular interest to us, as structurally related α-cyanoacrylamides have emerged as a class of covalent reversible ligands for diverse proteins[16]. We first assessed the relative cellular potencies of MPC2 engagement by UK-5099 and YY4 using YY4-yne-coupled Western blotting, revealing that both compounds exhibited sub-μM IC50 values (Figure 4A, B). We then evaluated the broader proteomic interactions profiles for UK-5099 and YY4 at concentrations >10X their respective cellular IC50 values for MPC2 engagement (10 μM of each compound). Following treatment with compounds for 3 h, human T cells were exposed to YY4-yne (10 μM, 1 h), lysed, conjugated to a biotin-azide tag by CuAAC, and biotinylated proteins enriched by streptavidin beads, digested on-bead with trypsin, and characterized by quantitative MS-based proteomics. These experiments confirmed that both UK-5099 and YY4, but not the inactive control compound EV-93 (10 μM), strongly engaged MPC2 in cells, as revealed by the blockade of YY4-yne enrichment of this protein (Figure 4C). UK-5099 further showed remarkable selectivity for MPC2 and did not block the enrichment of any other YY4-yne-reactive proteins in T cells (Figure 4C, y-axis, left plot). This selectivity profile contrasted with YY4, which decreased the enrichment of several additional YY4-yne-reactive proteins (Figure 4C, x-axis, left plot). MPC2 still showed an exceptional profile in YY4-treated cells, however, in that most of the other YY4-sensitive proteins were also blocked in their YY4-yne enrichment by EV-93 (Figure 4C, right plot). These data, taken together, indicate that UK-5099 shows excellent proteomic selectivity for MPC2, as readout through competitive profiling experiments with the YY4-yne cellular engagement probe.
Figure 4.

YY4-yne serves as a cellular target engagement probe for MPC2. A) YY4-yne-coupled Western blotting of human T cells treated with YY4 or UK-5099 (0.01–10 μM, 3 h) or DMSO followed by YY4-yne (10 μM, 2 h). B) IC50 values calculated from YY4-yne-coupled Western blotting. YY4: 0.02 μM (13–37 nM, 95% confidence interval (CI)); UK-5099: 0.88 μM (0.4–1.8 μM 95% CI). Data represent average values +/− SD for two independent experiments. C) Scatter plots showing competition ratios for proteins engaged by the indicated compounds, as measured by blockade of enrichment with the YY4-yne probe. Human T cells were treated with compounds (YY4, EV-93, and UK-5099; 10 μM, 3 h) or DMSO, followed by treatment with YY4-yne (10 μM, 1 h), after which YY4-yne-labeled proteins were enriched by CuAAC conjugation to an azide-biotin tag and analyzed by MS-based proteomics. Data are for one experiment representative of two replicate experiments.
In summary, we have described herein the first, to our knowledge, covalent, irreversible ligands for the MPC and demonstrated their utility as cellular engagement probes for this important metabolite transporter. Using the YY4-yne probe, we have provided evidence that UK-5099, a long-standing and putatively covalent reversible inhibitor of the MPC[8], shows excellent potency (IC50 ~ 0.02 μM) and proteomic selectivity in human cells. UK-5099 has been previously postulated to engage an (as of yet unidentified) cysteine on MPC1[8, 17], but our data suggest instead that this compound may bind to C54 of MPC2. We also note that we did not enrich MPC1 with YY4-yne, indicating that the αCA probes described herein only interact with the MPC2 subunit of the MPC. We acknowledge, however, that it remains possible UK-5099 inhibits the MPC by binding to MPC1, or at an MPC1-MPC2 interface, in a manner that indirectly blocks YY4-yne reactivity with MPC2. Possibly consistent with the binding site for UK-5099 involving both MPC1 and MPC2, MPC1 mutants have been identified that show resistance to UK-5099[5b] and MPC2 homomeric complexes are apparently insensitive to UK-5099[17], while, on the other hand, this compound has been shown to block radiolabeled thiazolidinedione binding to MPC2[7b].
We should also emphasize that, while using irreversible chemical probes to monitor the cellular engagement of proteins by reversible compounds is well-documented[18], these experiments require attention to the kinetics of irreversible probe reactivity, as reversible binding of competitor ligands can be underestimated, or even completely overlooked if incubation periods extend well-beyond the time required for complete probe reactivity with a protein target of interest in cells. Finally, our initial SAR data with αCA compounds support that C54 of MPC2 resides within a druggable pocket suitable for the future development of covalent, irreversible inhibitors of the MPC, which may offer advantages over covalent, reversible compounds like UK-5099 in terms of sustaining target engagement in increasingly complex biological systems (e.g., in vivo animal models). More generally, our findings highlight how broad cysteine reactivity profiling of electrophilic compounds can identify target engagement probes for monitoring the cellular activity and small molecule interactions of challenging protein classes such as integral membrane metabolite transporters.
Experimental Section
Experimental details and compound characterization.
Supplementary Material
Acknowledgements
This work was supported by NIH CA231991 (B.F.C.), a Clinical Translational Science Award (UL1 TR001114), a Damon-Runyon Cancer Research Foundation fellowship to X.Z. (DRG-2341-18), and a Life Sciences Research Foundation Fellowship to E.V.V.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Vinogradova EV, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Remillard D, Lum KM, Simon GM, Kemper EK, Lazear MR, Yin S, Blewett MM, Dix MM, Nguyen N, Shokhirev MN, Chin E, Lairson L, Forli S, Teijaro JR, Cravatt BF, bioRxiv 2019. doi: 10.1101/808113 [DOI] [Google Scholar]
- [2].a) Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF, Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Vanderperre B, Bender T, Kunji ER, Martinou JC, Curr Opin Cell Biol 2015, 33, 35–41; [DOI] [PubMed] [Google Scholar]; b) Bender T, Martinou JC, Biochim Biophys Acta 2016, 1863, 2436–2442. [DOI] [PubMed] [Google Scholar]
- [4].Compan V, Pierredon S, Vanderperre B, Krznar P, Marchiq I, Zamboni N, Pouyssegur J, Martinou JC, Mol Cell 2015, 59, 491–501. [DOI] [PubMed] [Google Scholar]
- [5].a) Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC, Science 2012, 337, 93–96; [DOI] [PubMed] [Google Scholar]; b) Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J, Science 2012, 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gray LR, Tompkins SC, Taylor EB, Cell Mol Life Sci 2014, 71, 2577–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Halestrap AP, Biochem J 1975, 148, 85–96; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, Gracheva E, Korshunova Y, Trusgnich M, Karr R, Wiley SE, Divakaruni AS, Murphy AN, Vigueira PA, Finck BN, Kletzien RF, PLoS One 2013, 8, e61551; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN, Proc Natl Acad Sci U S A 2013, 110, 5422–5427; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Du J, Cleghorn WM, Contreras L, Lindsay K, Rountree AM, Chertov AO, Turner SJ, Sahaboglu A, Linton J, Sadilek M, Satrustegui J, Sweet IR, Paquet-Durand F, Hurley JB, J Biol Chem 2013, 288, 36129–36140; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hildyard JC, Ammala C, Dukes ID, Thomson SA, Halestrap AP, Biochim Biophys Acta 2005, 1707, 221–230. [DOI] [PubMed] [Google Scholar]
- [8].Halestrap AP, Biochem J 1976, 156, 181–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM, Mol Cell 2014, 56, 425–435; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, Olson KA, Miao R, Waller TC, Delker D, Kanth P, Jiang L, DeBerardinis RJ, Bronner MP, Li DY, Cox JE, Christofk HR, Lowry WE, Thummel CS, Rutter J, Nat Cell Biol 2017, 19, 1027–1036; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, Lee J, Wolfgang MJ, Patti GJ, Finck BN, Pearce EL, Bhattacharya D, Immunity 2016, 45, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Li W, Blankman JL, Cravatt BF, J Am Chem Soc 2007, 129, 9594–9595; [DOI] [PubMed] [Google Scholar]; b) Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF, Nat Chem Biol 2011, 7, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW, Biochemistry 2007, 46, 350–358; [DOI] [PubMed] [Google Scholar]; b) Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ, Proc Natl Acad Sci U S A 2010, 107, 13075–13080; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, Roberts LR, Schnute ME, Wang C, Hulce JJ, Wei B, Whiteley LO, Hayward MM, Cravatt BF, Nat Chem Biol 2014, 10, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu L, Armstrong Z, Schroder SP, de Boer C, Artola M, Aerts JM, Overkleeft HS, Davies GJ, Curr Opin Chem Biol 2019, 53, 25–36. [DOI] [PubMed] [Google Scholar]
- [13].Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y, Cancer Discov 2016, 6, 316–329. [DOI] [PubMed] [Google Scholar]
- [14].Speers AE, Cravatt BF, Chem Biol 2004, 11, 535–546. [DOI] [PubMed] [Google Scholar]
- [15].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew Chem Int Ed Engl 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- [16].a) Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J, J Am Chem Soc 2013, 135, 5298–5301; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J, Nat Chem Biol 2015, 11, 525–531; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Senkane K, Vinogradova EV, Suciu RM, Crowley VM, Zaro BW, Bradshaw JM, Brameld KA, Cravatt BF, Angew Chem Int Ed Engl 2019, 58, 11385–11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nagampalli RSK, Quesnay JEN, Adamoski D, Islam Z, Birch J, Sebinelli HG, Girard R, Ascencao CFR, Fala AM, Pauletti BA, Consonni SR, de Oliveira JF, Silva ACT, Franchini KG, Leme AFP, Silber AM, Ciancaglini P, Moraes I, Dias SMG, Ambrosio ALB, Sci Rep 2018, 8, 3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Adibekian A, Martin BR, Chang JW, Hsu KL, Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder PS, Rosen H, Cravatt BF, J Am Chem Soc 2012, 134, 10345–10348; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ogasawara D, Ichu TA, Vartabedian VF, Benthuysen J, Jing H, Reed A, Ulanovskaya OA, Hulce JJ, Roberts A, Brown S, Rosen H, Teijaro JR, Cravatt BF, Nat Chem Biol 2018, 14, 1099–1108; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Leung D, Hardouin C, Boger DL, Cravatt BF, Nat Biotechnol 2003, 21, 687–691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.