

Abstract
T. gondii is an obligate intracellular parasite that establishes life-long infection in a wide range of hosts, including humans and rodents. To establish a chronic infection, pathogens often exploit the tradeoff between resistance mechanisms, which promote inflammation and kill microbes, and tolerance mechanisms, which mitigate inflammatory stress. Signaling through the type I IL-1 receptor (IL-1R) has recently been shown to control disease tolerance pathways in endotoxemia and salmonella infection. However, the role of the IL-1 axis in T. gondii infection is unclear. Here we show that IL-1R−/− mice can control T. gondii burden throughout infection. Compared to wildtype mice, IL-1R−/− mice have more severe liver and adipose tissue pathology during acute infection, consistent with a role in acute disease tolerance. Surprisingly, IL-1R−/− mice had better long-term survival than wildtype mice during chronic infection. This was due to the ability of IL-1R−/− mice to recover from cachexia, an immune-metabolic disease of muscle wasting that impairs fitness of wildtype mice. Together, our data indicate a role for IL-1R as a regulator of host homeostasis, and point to cachexia as a cost of long-term reliance on IL-1 mediated tolerance mechanisms.
Introduction
Toxoplasma gondii (T. gondii) is an obligate intracellular parasite that chronically infects a wide range of hosts. T. gondii is a natural rodent pathogen, but also infects humans. An estimated 30% of the global human population is infected (1). A host is infected by ingesting food contaminated with parasite cysts, which invade the small intestine. Over the first two weeks of infection, tachyzoites, the rapidly replicating form of the parasite, can be detected in almost every tissue (2). Infection elicits IL-12, IFN-γ, and IL-6 to promote a TH1-polarized adaptive immune response that is necessary to restrict systemic infection. However, intermediate hosts remain infected for life, harboring low levels of bradyzoite tissue cysts in the brain, muscle, and other tissues. A sustained immune response is essential to suppress parasite growth during chronic infection: chronically infected mice and humans present with elevated sera titers of T. gondii-specific antibodies and IFN-γ, and immune suppression triggers parasite recrudesce (3–5). However, the long-term consequences of sustained inflammation during T. gondii infection are not entirely clear.
Inflammasomes are cell-intrinsic innate immune signaling platforms linked to the release of IL-1 family cytokines and, often, inflammatory cell death. In T. gondii infection, mice deficient in the inflammasome sensors NLRP1 and NLRP3 and the effector caspases-1 and -11 have defects controlling parasite load (6, 7). These pathways release alarmins in the IL-1 family. However, there are conflicting reports regarding the role of IL-1α, IL-1β, and their receptor IL-1R1 during T. gondii infection. Elevated IL-1α and IL-1β have been observed in the sera (1 week post-T. gondii infection) and brain by 2–3 weeks post-infection (8, 9). Pretreating Swiss Webster mice with IL-1α and IL-1β protected them from a lethal dose of T. gondii (10). One study reported increased mortality in IL-1R-deficient (IL-1R−/−) mice following intraperitoneal (i.p.) T. gondii infection (11), while another reported no deficiencies in parasite restriction (12). A third study reported that orally infected IL-1R−/− mice had significantly improved survival, reduced intestinal pathology, and decreased parasite load compared to wildtype mice (13). Taken together, the role of the IL-1 axis on T. gondii burden and infection outcome is unclear.
We were interested in revisiting the requirement for IL-1R signaling in T. gondii infection based on an emerging role of the IL-1 axis in regulating disease tolerance during endotoxemia and bacterial infection (14–17). Disease tolerance programs are defined as shifts in homeostasis that protect the host from bystander damage caused by infection and inflammation without directly affecting pathogen load (18). In contrast, resistance programs promote host fitness by directly mediating pathogen clearance. Tolerance mechanisms are as important for survival as restriction mechanisms, but are far less well understood, particularly in T. gondii infection. We found that IL-1R was not necessary to control parasite burden during acute or chronic i.p. T. gondii infection. Consistent with a role in disease tolerance, IL-1R−/− mice had increased cell death in the liver and adipose tissue compared to wildtype mice during acute infection. Unexpectedly, infected IL-1R−/− mice had better long-term survival than wildtype mice in chronic infection. IL-1R−/- mice recovered from acute weight loss, hepatomegaly and muscle atrophy. In contrast, wildtype mice exhibited sustained cachexia, an immune-metabolic wasting disease that led to a poor survival outcome in chronic infection (8, 19–21). These data indicate that IL-1R signaling controls cytoprotective disease tolerance programs that benefit the host during acute T. gondii infection. However, in the context of chronic disease, IL-1R can drive cachexia and ultimately impair host fitness.
Materials and Methods
Mouse strains/husbandry
C57BL/6, IL-1R−/− (B6.129S7-Il1r1tm1Imx), mice were purchased from Jackson Laboratories and IL-1α−/− (22) and IL-1β−/− (23) mice were a gift from John Lukens at the University of Virginia. Mice were bred and housed in accordance with the University of Virginia Institutional Animal Care and Use Committee AAALAC and IACUC protocol #4107-12-18.
Infections and assessment of parasite burden
Me49 cysts were passaged in vivo as described (8). 10–14 week male mice were cross-housed on dirty bedding for 2 weeks to normalize microbiota then infected with 10 Me49 cysts by intraperitoneal infection. Parasite burden was determined by dolichos staining, counting at 10X magnification (cyst diameter measured in Fiji) (8); or by qPCR of T. gondii 529bp Repeat Element (RE) compared to mouse beta actin as described (24) and analyzed using the ΔCt method. Brain DNA was extracted with TRIzol Reagent (Invitrogen) and used at 30 ng DNA per qPCR reaction. Adipose DNA was isolated by NucleoSpin DNA Lipid Tissue kit (Machery-Nagel), and used at 10–30 ng DNA per qPCR reaction. Liver DNA was isolated as described (25), and used at 500 ng DNA per qPCR reaction. The following Taqman primer/probes were used: 529bp Repeat Element (RE) forward: 5’-CACAGAAGGGACAGAAGTCGAA-3’; reverse: 5’-CAGTCCTGATATCTCTCCTCCAAGA-3’; probe: 5’-CTACAGACGCGATGCC-3’ (IDT, (24)); mouse beta actin: Mm02619580_g (ThermoFisher Scientific).
Histology and Immunohistochemistry
Tissues were fixed overnight in 4% paraformaldehyde at 4°C and paraffin-embedded, sectioned, and stained with hematoxylin and eosin, Von Kossa Stain Kit (American Mastertech Scientific) or anti-cleaved caspase-3 (Cell Signaling Technology). H&E samples were scored for necrosis by a blinded pathologist using the following scale: 1) 1%-25%, 2) 26%-50%, 3) 51%-75%, 4) 76%-100% of the area affected. Inflammatory lesion diameter was measured with Fiji. Caspase-3 staining was determined using the color deconvolution Fiji plug-in (26) and thresholded on negative controls.
Immunofluorescence Microscopy
Following deparaffinization, slides were antigen-retrieved with boiling sodium citrate buffer (10 mM sodium citrate buffer + 0.05% Tween-20, pH 6.0), blocked and permeabilized for an hour (2% donkey serum, 2% goat serum, 0.1% Triton-X, 0.05% Tween-20), and stained overnight at 4°C with rabbit anti-mouse ASC (Santa Cruz, N-15), or rat anti-mouse CD45 (Biolegend, 30-F11) diluted 1:50 in blocking buffer. The next morning, following washing, samples were incubated for one hour at room temperature in secondary antibody diluted 1:200 in blocking buffer (donkey anti-rabbit AF594 (Jackson Immunoresearch, 711-585-152) or goat anti-rat IgG AF647 (Life Technologies, A21247)). Following washing, samples were incubated for 5 minutes in 10 μg/mL DAPI, washed 3 times in PBS, and mounted in Vectashield (Vector Laboratories). Click-iT™ Plus TUNEL Assay (Invitrogen) was used for TUNEL staining, following manufacturer’s instructions. Slides were imaged on a Zeiss LSM 880 confocal microscope (Carl Zeiss) and images were processed in Fiji.
Flow Cytometry
Liver and epigonadal visceral white adipose tissues were prepared for flow cytometry after cardiac perfusion with 10 mL HBSS (5 mM HEPES, 0.5 mM EDTA), as described (27). Following mechanical disruption, tissue was Liberase (Roche)-digested for 30 minutes at 37°C (shaking at 130 RPM) and filtered. Cells were separated on a 40% iodixanol density gradient by centrifugation at 1038 g for 25 minutes (no brake). Cells were blocked with anti-CD16/32, stained with Zombie Aqua (Biolegend) and antibodies (Biolegend): F4/80-FITC (BM8), Ly6C-PerCP/Cy5.5 (HK1.4), CD19-PE/Cy7 (6D5), CD11c-APC (N418), CD11b-AF700 (c1/70), I-A/I-E-APC/Cy7 (M5/114.15.2), CD45-PacBlue (30-F11), Ly6G-BV605 (1A8), CD3e-FITC (145–2C11), CD62L-PE-dazzle (MEL-14), CD8a-PerCP/Cy5.5 (53–6.7), PD-1-PE/Cy7 (29F.1A12), CD4-APC (GK1.5), CD44-BV605 (IM7). Beckman Coulter CytoFLEX and FlowJo were used to assess immune cell populations using the following gating strategy: Cells, singlets, live cells, CD4+ T cells (CD45+CD3e+CD4+), CD8+ T cells (CD45+CD3e+CD8+), B cells (CD45+CD19+), Kupffer cells (liver only, CD45+F480hiCD11blo), macrophages (CD45+F480intCD11bhiLy6clo), inflammatory monocytes (CD45+F480intCD11bhiLy6chi), dendritic cells (CD45+F480−CD11c+MHCII+), and neutrophils (CD45+F480−CD11b+ Ly6G+SSChi).
Cytokine Measurements
Sera cytokines were measured by Luminex or ELISA (Invitrogen unless noted). Tissue homogenate cytokines were measured by ELISA: IFN-γ (88–7314); TNF-α (88–7324); IL-1α (88-5019-88); IL-6 (eBioscience 50-112-8863); IL-1β (88-7013-88); IL-10 (88-7105-86); IL-1Ra (R&D DY480) and are normalized to total protein per well.
Tissue Weights and Body Mass Composition
Tissues were harvested as previously described (8). Body composition was measured for each mouse prior to infection, and then at 2 and 9 weeks post-infection by using the EchoMRI™-100H Body Composition Analyzer, which uses Quantitative Magnetic Resonance to measure total lean and fat in unanesthetized mice.
Statistical Analysis
Two-tailed unpaired Student’s t-tests were performed with a confidence level of 95% and Holm-Sidak multiple comparisons correction (as noted) using GraphPad Prism 8. Data are presented as the mean ± SEM, *p< 0.05; **p< 0.01; ***p < 0.001, **** p< 0.0001.
Results
IL-1R signaling impairs host survival during chronic T. gondii infection independent of parasite burden.
Mice deficient in the IL-6 (28), TNF-α (29), and IFN-γ (30, 31) cytokine pathways succumb to acute or early chronic T. gondii infection due to a failure to restrict parasite growth. To test whether IL-1R signaling plays a similar role in parasite restriction, IL-1R-deficient (IL-1R−/−) or control C57BL/6 (WT) mice were intraperitoneally infected with 10 Me49 T. gondii cysts. Contrary to expectation, IL-1R−/− mice had significantly improved long-term survival compared to WT mice (Fig. 1A, 6-18 weeks post-infection). Improved survival was not due to better parasite clearance, as IL-1R−/− mice had a similar parasite load in the brain as WT mice at 9 weeks post-infection (Fig 1B–C). T. gondii cyst morphology and inflammatory infiltrate in the brain were similar in infected IL-1R−/− and WT mice by H&E staining (Supp. Fig. 1).
FIGURE 1.
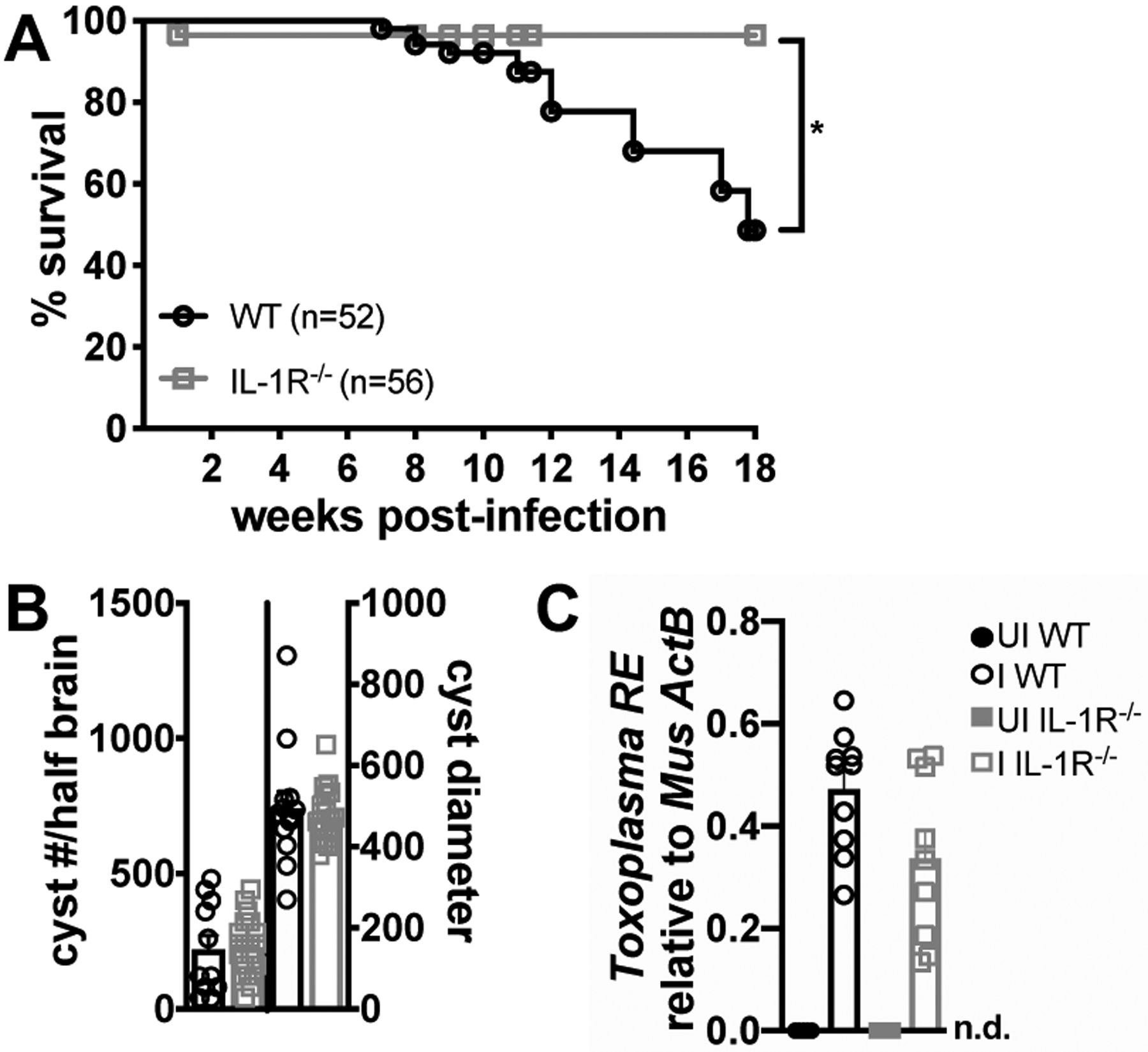
IL-1R−/− mice have improved long-term survival of T. gondii infection than wildtype mice independent of parasite burden. 10–14 week old male C57Bl/6 (WT) or IL-1R−/− mice were intraperitoneally infected with 10 Me49 Toxoplasma cysts. (A) Kaplan-Meier curves of infected C57BL/6 (WT) or IL-1R−/− mice (n = 52–55 mice per group, data pooled from twelve experiments). p-value is 0.0210 by Log-rank test. (B) Cyst burden and diameter in dolichos stained brain mash at 9 weeks post-infection (wpi) (n = 12–21 mice per group, data pooled from three experiments). (C) qPCR quantification of T. gondii 529-bp repeat element (T. gondii RE) gDNA relative to mouse β-actin in the brain at 9 wpi (n = 5–10 mice per group, data pooled from two experiments). n.d., not detectable. Data are shown as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, **** P < 0.0001 by unpaired Student’s T test with Holm-Sidak method to correct for multiple comparisons.
IL-1R signaling limits cell death in the adipose tissue and liver during acute infection without affecting parasite load, consistent with a role in disease tolerance.
Based on the observation that IL-1R−/− mice had better survival in chronic infection but similar parasite load, we reasoned that IL-1R signaling in acute infection could shape a fundamentally distinct immune environment with long-lasting effects on survival. Alternatively, sustained IL-1R signaling could prevent recovery in the chronic phase of infection. To assess the role of IL-1R signaling in acute infection, infected mice were euthanized at 2 weeks post-infection, when parasites are systemic and have infected most tissues in the mouse. We were surprised to observe numerous large lesions on the epigonadal visceral white adipose tissue (vWAT) of IL-1R−/− mice (Fig. 2A, arrows). Although lesions were sometimes observed in the vWAT of infected WT mice, they were noticeably smaller and less frequent. These lesions were consistent with a histopathological definition of fat necrosis, which is acellular areas devoid of inflammation and occasional calcification (32, 33), and is consistent with the fat necrosis phenotype observed in TLR11−/− mice 5 days post-infection with 20 Me49 cysts (34). Increased calcium deposition in IL-1R−/− vWAT was confirmed by Von Kossa stain (Fig. 2B, brown staining outlined, with pink nuclear counterstain). Necrotic lesions, defined as acellular regions surrounded by inflammatory infiltrate (Fig. 2C, outlined) were more frequent in IL-1R−/− vWAT than WT (Fig. 2D). By 9 weeks post-infection the lesions had largely resolved (Fig. 2A, 2C–D). T. gondii can directly induce tissue damage by lysing out of infected cells. T. gondii load was similar in WT and IL-1R−/− vWAT, indicating that the difference in necrosis was not due to parasite overgrowth in the IL-1R−/− mice (Fig. 2E).
FIGURE 2.
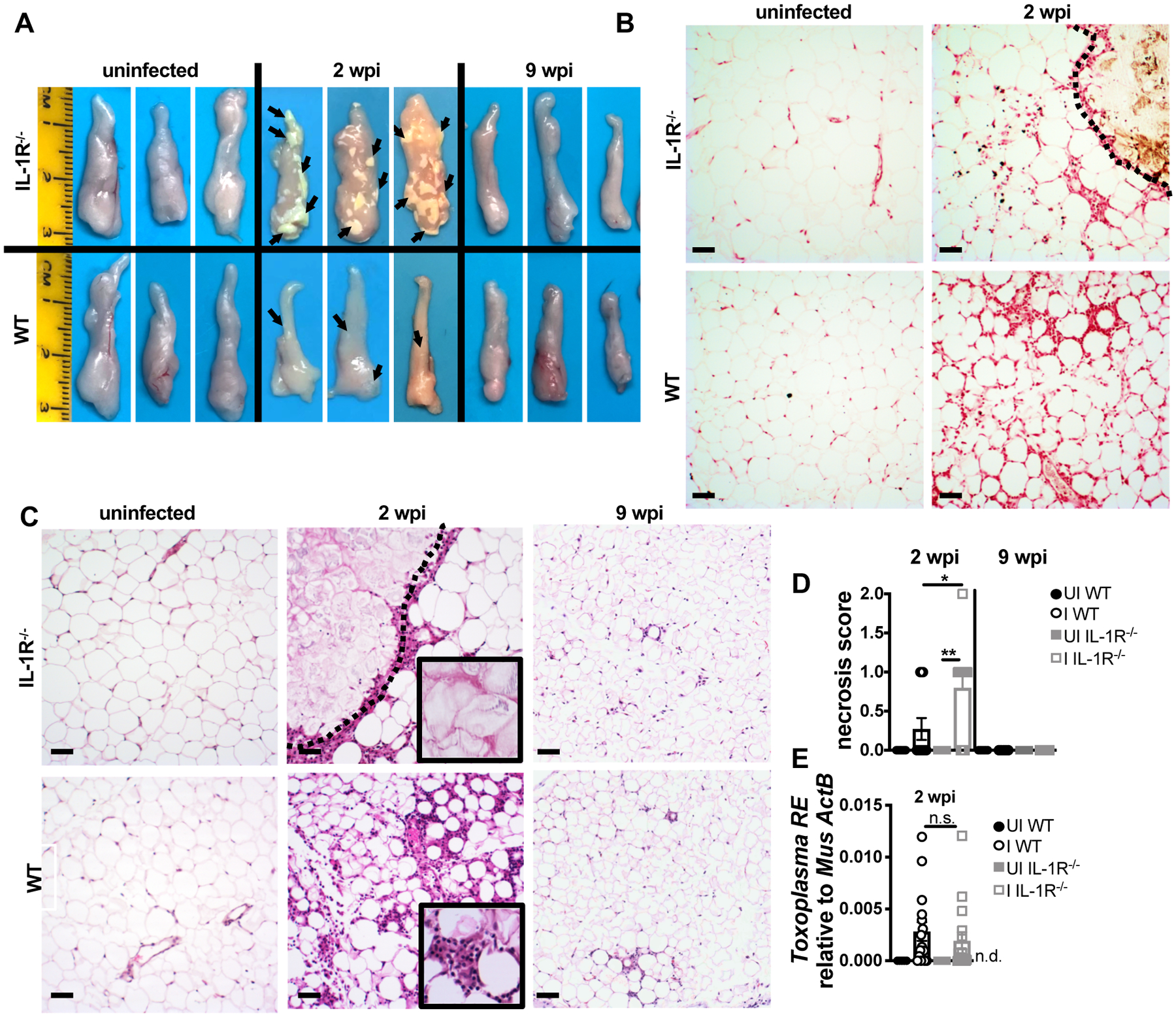
IL-1R signaling limits cell death in the adipose tissue during acute T. gondii infection. (A) Epididymal visceral white adipose tissue (vWAT) was harvested at 2 wpi (representative of five experiments). Representative images of vWAT sections stained with Von Kossa stain (brown, calcium; red, nuclei) (B) or hematoxylin/eosin (H&E, necrotic lesion outlined) (C). (D) H&E Necrosis scored by a blinded pathologist (n = 6–8 mice, pooled from two experiments). (E) qPCR quantification of T. gondii RE gDNA in the vWAT at 2 wpi, relative to mouse β-actin (n = 5–15 mice per group, pooled from four experiments). Scale bars = 50 μm. n.d., not detectable. Data are shown as the mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001, n.s., not significant by unpaired Student’s T test.
Consistent with previous observations, hepatomegaly was also observed in WT and IL-1R−/− mice at 2 weeks post-infection (35, 36). The livers of WT and IL-1R−/− mice had focal, perivascular immune infiltrate of similar size and frequency (Fig. 3A–B). However, IL-1R−/− livers had significantly more enzymatically active caspase-3 staining by immunohistochemistry than WT livers (Fig. 3C–D), an early measure of apoptotic cell death. Immune infiltrate was also observed at 9 weeks post-infection (Fig. 3A), however, this was morphologically consistent with extramedullary hematopoiesis and active caspase-3 staining was not observed (Fig. 3C). Similar to the vWAT, WT and IL-1R−/− livers had similar T. gondii burden (Fig. 3E), indicating that the increased cell death in IL-1R−/− livers was not due to increased parasite load at 2 weeks post-infection. Taken together, these data are consistent with the conclusion that IL-1R signaling is cytoprotective in the liver and vWAT during acute infection, implicating a role in disease tolerance in acute T. gondii rather than the expected role in T. gondii resistance.
FIGURE 3.
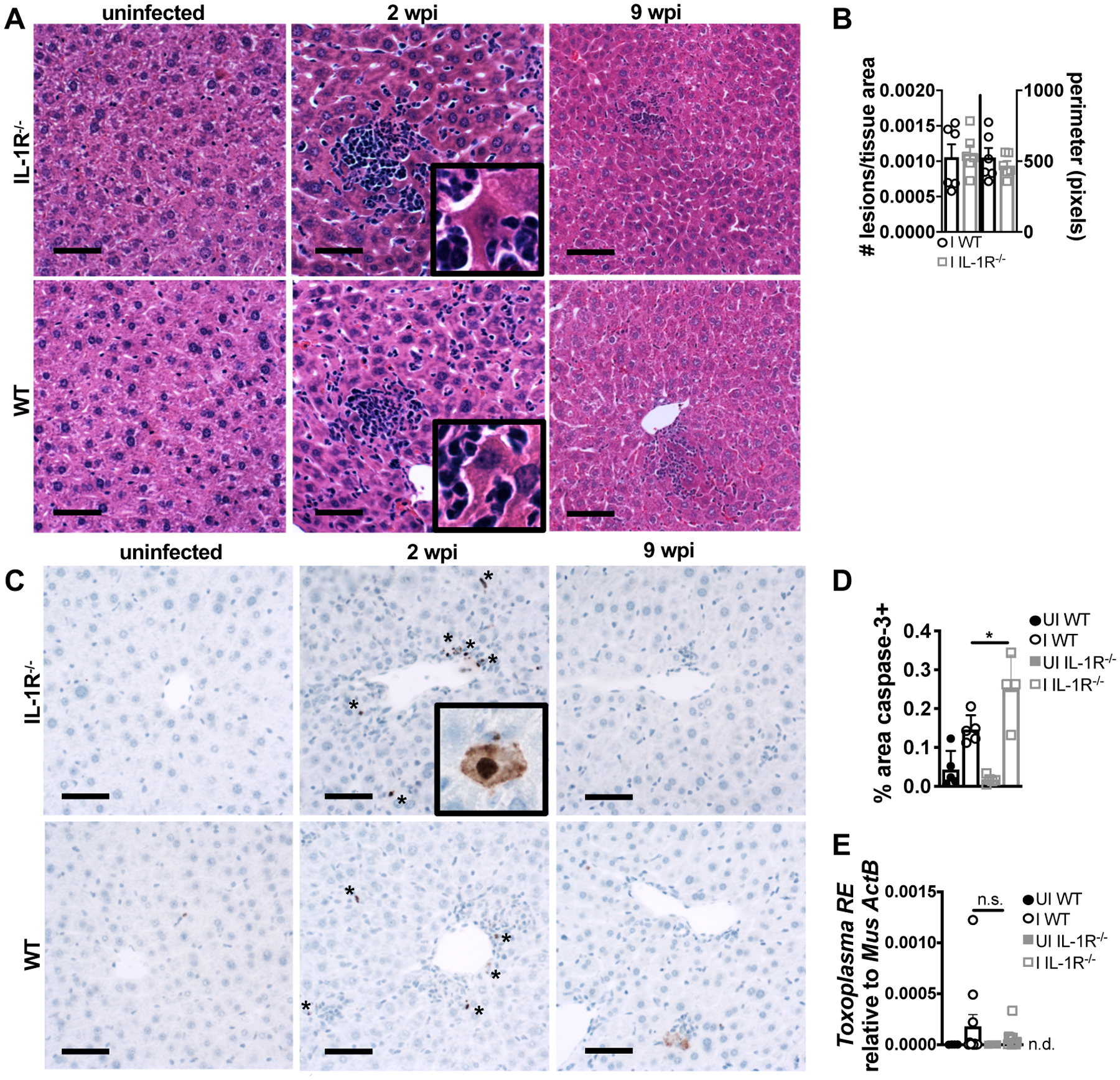
IL-1R signaling limits caspase-3 activation in the liver during acute T. gondii infection. At 2 wpi liver was harvested and sectioned for H&E staining (A). (B) Focal immune lesions were counted across slide scans of whole liver sections and lesion perimeter was measured (n = 6 mice per group, pooled from two experiments). (C) Immunohistochemistry for cleaved caspase-3 (brown stain, asterisks) quantified in (D) (n = 4–5 mice per group, representative of two experiments). (E) qPCR quantification of T. gondii RE gDNA in the liver at 2 wpi, relative to mouse β-actin (n = 4–11 mice per group, pooled from three experiments). Scale bars = 50 μm. n.d., not detectable. Data are shown as the mean ± SEM (except D, where they are SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001, n.s., not significant by unpaired Student’s T test.
When IL-1α and IL-1β protein levels were assessed in the tissue we found that IL-1β was elevated in the vWAT (Fig. 4A). IL-1α and IL-1β were both significantly elevated in the liver of WT and IL-1R deficient mice. Other published disease tolerance models have reported significant differences in tissue pathology and function regulated independently of inflammatory cytokine levels or immune infiltrate (16, 37, 38). Consistent with these models, TNF-α and IL-6 were similar in the vWAT and liver of IL-1R−/− and WT mice at 2 weeks post-infection (Fig. 4A–B). IFN-γ levels were not elevated in the vWAT, but were elevated in the liver, where IL-1R−/− livers had a slight but significantly elevated level compared to WT. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) is an indicator of late stage apoptosis as well as inflammatory, programmed cell death pathways that release IL-1α and IL-1β, however, these processes can be difficult to detect in vivo due to immune clearance of damaged cells. A low but significant elevation of TUNEL positive cells were detected in the vWAT (Supp. Fig. 2A) and livers (Supp. Fig. 2B) of infected WT and IL-1R−/− vWAT. ASC ‘specks,’ which form downstream of some inflammasomes, were also observed in the vWAT of infected mice (Supp. Fig 2C); and, very infrequently in liver sections (less than one per 10 fields of view at 40x, data not shown).
FIGURE 4.
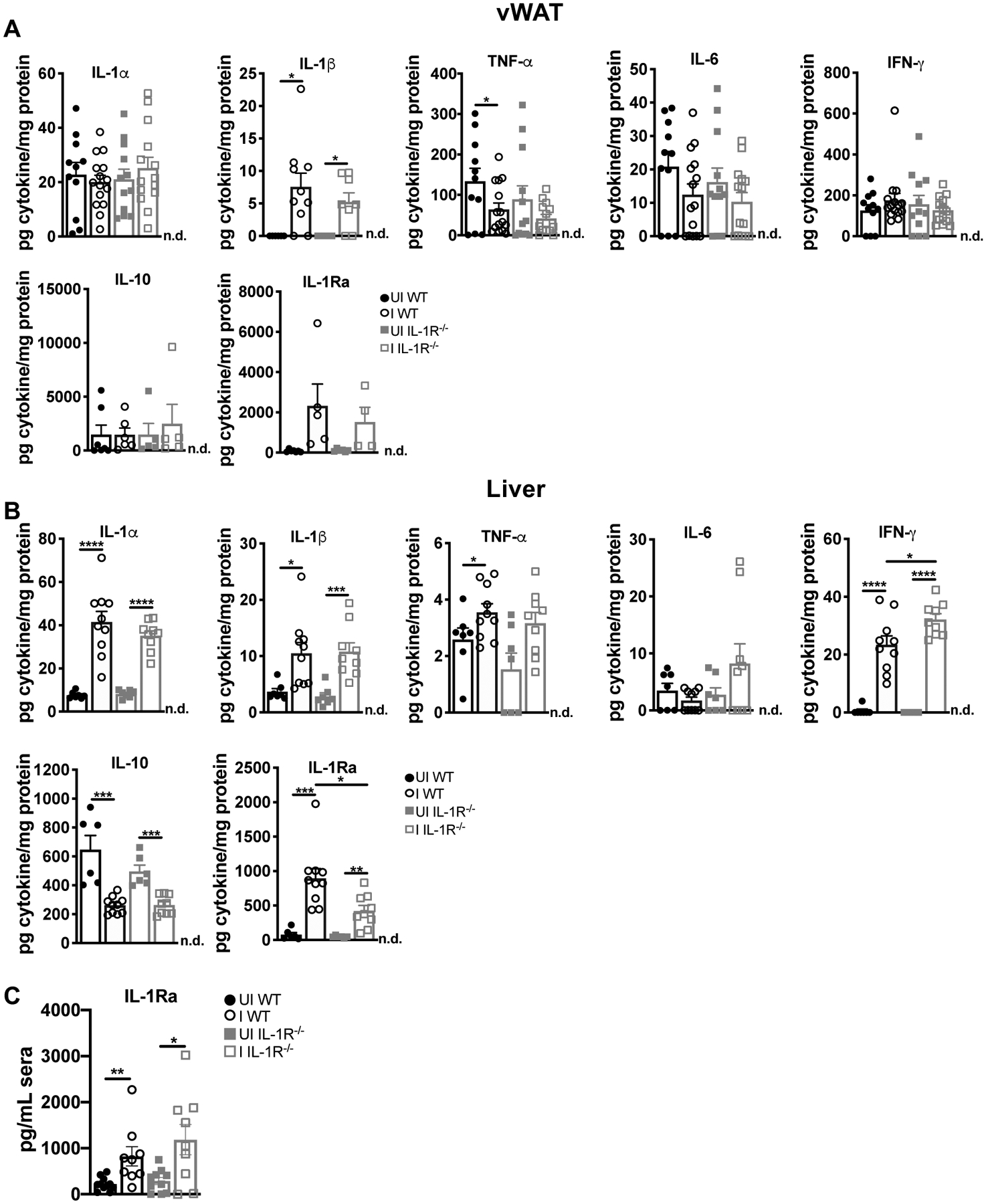
IL-1R ligands are elevated in the liver and vWAT at 2 weeks post-infection. Cytokine concentration in tissue lysate were measured by ELISA in (A) vWAT (n = 5–15 mice per group, pooled from three experiments. IL-1β, IL-10, and IL-1Ra are from two experiments) or (B) liver (n = 7–10 mice per group, pooled from two experiments). n.d., not detectable. Data are shown as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, ****P<0.0001, n.s., not significant by unpaired Student’s T test.
IL-10 is a regulatory cytokine necessary to control lethal immunopathology during T. gondii infection (36, 39, 40). Levels of IL-10 were not elevated in the vWAT of infected mice relative to uninfected controls, and were lower in infected WT and IL-1R−/− mice relative to uninfected animals, suggesting that the cytoprotective effect of IL-1R in WT tissues was not mediated by IL-10. IL-1R antagonist (IL-1Ra) is a competitive inhibitor of IL-1R signaling that is transcriptionally regulated by IL-1R signaling among other NF-κB inducers (41). IL-1Ra was higher in the vWAT of infected mice relative to uninfected animals, although this was not significant. IL-1Ra was significantly higher in the liver (Fig 4B) and sera (Fig 4C) of infected WT and IL-1R−/− mice: however, there was significantly more IL-1Ra in the WT livers compared to IL-1R−/− mice, consistent with positive regulation of IL-1Ra by IL-1R signaling.
To determine if the increased pathology in IL-1R−/− was associated with a greater magnitude or altered distribution of immune infiltrating cells, mice were perfused with HBSS, and the liver and adipose tissues were dissociated for analysis by flow cytometry. Infected WT and IL-1R−/− mice had a similar overall number of infiltrating CD45+ immune cells (Supp. Fig. 3A) and a similar distribution of infiltrating immune cell types in the adipose tissue and liver (Supp. Fig. 3B–C). Minimal contamination from blood-derived cells was confirmed by i.v. injecting anti-CD45-PacBlue five minutes prior to perfusion and staining with CD45-BV711 (data not shown) (42). While it is possible that more subtle difference in immune cell populations exist, these data indicate that the enhanced cell death in the IL-1R−/− mice was not simply due to a greater magnitude of inflammatory infiltrate, innate cytokine production, or altered parasite burden. These data also refute our initial hypothesis that chronic survival of IL-1R−/− is established during the acute phase of T. gondii infection.
IL-1R signaling during T. gondii infection drives chronic cachexia.
Based on our observation that IL-1R is cytoprotective during acute infection but negatively impacts long-term survival, we turned our attention to the role of IL-1R signaling during the chronic phase of disease. We, and others, have shown that infection with T. gondii can trigger sustained weight loss, inflammation, and muscle atrophy, consistent with the clinical definition of cachexia (8, 20, 43). Cachexia is an immune-metabolic disease of muscle wasting that predicts and directly contributes to mortality in a wide range of chronic diseases (19). IL-1, TNF-α, IL-6, and IFN-γ comprise a cytokine signature conserved across etiologies of cachexia. We hypothesized that sustained IL-1 signaling during chronic T. gondii infection could promote cachexia, explaining the poor long-term survival of wildtype mice compared to IL-1R knockouts. To examine cachexia, we first measured body mass. Infected IL-1R−/− and WT mice both lost approximately 10% of their body mass by 1–4 weeks post infection (Fig. 5A), including fat and lean mass (Fig 5B). However, IL-1R−/− mice regained weight between 5 and 11 weeks post-infection (Fig 5A–B) whereas WT mice remained wasted. Infected IL-1R−/− and WT mice were anorexic at 1–2 weeks post-infection; however, both groups recovered eating during chronic infection (Fig. 5C). Infected IL-1R−/− mice lost significantly more muscle mass than WT mice by 2 weeks post-infection, but significantly regained muscle mass by 9 weeks post-infection. In contrast, WT mice continued to lose muscle mass from 2 to 9 weeks post-infection (Fig. 5D, Quad). Consistent with liver damage observed at acute infection, both WT and IL-1R−/− mice had hepatomegaly 2 weeks post-infection, although compared to uninfected mice, WT livers atrophied by 9 weeks post-infection whereas IL-1R−/− livers returned to normal weight (Fig. 5D, liver). Consistent with clinical observations that adipose tissue levels do not always correlate with cachexia severity, WT and IL-1R−/− mice regained subcutaneous adipose tissue, but the vWAT remained significantly wasted in both genotypes at 9 weeks post-infection (Fig. 5D).
FIGURE 5.
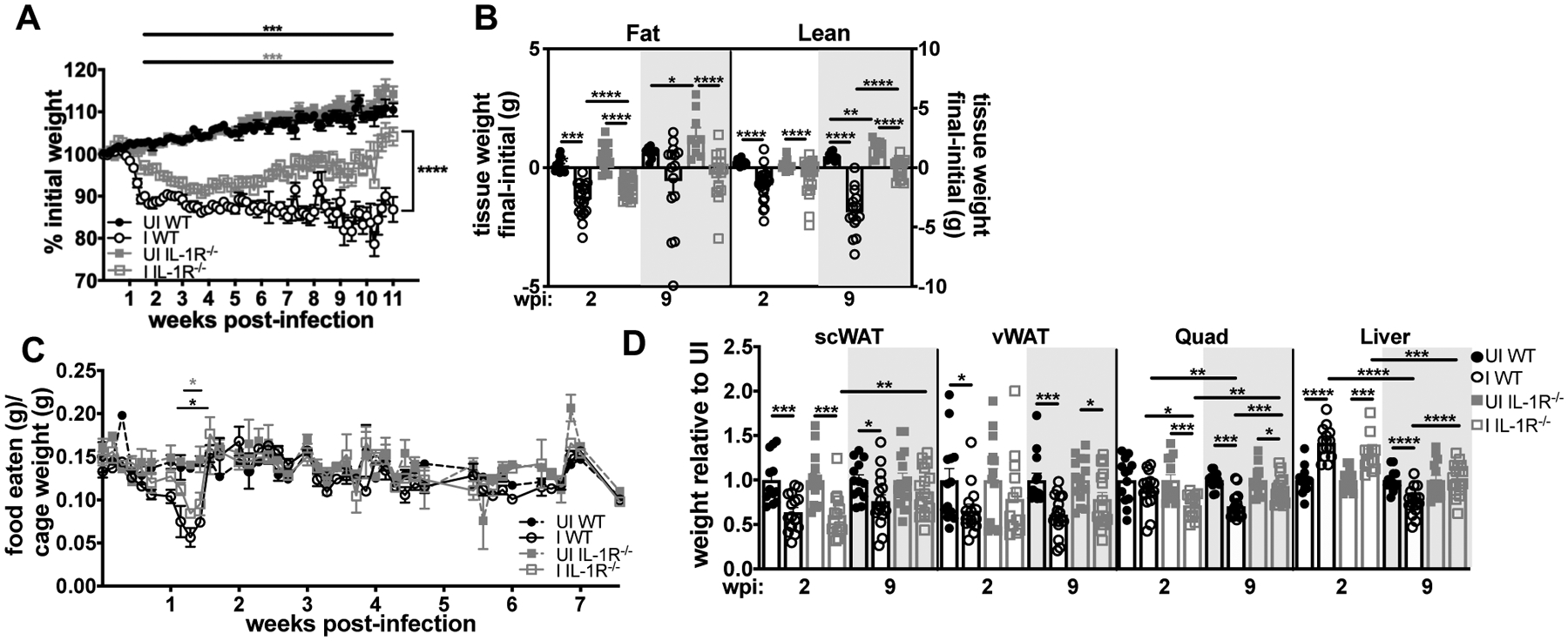
Acute weight loss is IL-1-independent, but IL-1R−/− mice are protected from chronic Toxoplasma-induced cachexia. (A) Weight curves showing percent of initial weight from 0–11 wpi (n = 34–48 mice, pooled from seven experiments). Horizontal bars denote the span of significant differences between uninfected and infected wildtype (black asterisks) or IL-1R−/− mice (gray asterisks). (B) Echo MRI quantification of fat (left) and lean (right) mass at 2 or 9 wpi (n = 9–22 mice, pooled from two to four experiments). (C) Mouse chow was weighed daily, and food intake per cage normalized to total body weight was determined (n = 2–4 cages per group, pooled from two experiments). Asterisks indicate significant differences between UI and I WT (black) or UI and I IL-1R−/− (gray). (D) Inguinal subcutaneous white adipose tissue (scWAT), vWAT, quadriceps (Quad), and liver at 2 wpi or 9 wpi (n = 12–18 mice per group, pooled from three experiments). Data are shown as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, ****P<0.0001, n.s., not significant by unpaired Student’s T test with Holm-Sidak method to correct for multiple comparisons.
We next assessed levels of cachexia-associated cytokines in WT and IL-1R−/− mice at 9 weeks post- infection (19, 44). TNF-α, IL-6, and IFN-γ were significantly elevated in WT and IL-1R−/− serum relative to uninfected controls (Fig. 6A). Although this was expected based on the essential role these cytokines play in restricting parasite growth (28, 31, 45), these data also suggest that elevated serum TNF-α (previously named cachectin), IL-6, and IFN-γ are not sufficient to sustain cachexia during T. gondii infection in the absence of IL-1R signaling. IL-1α and IL-1RA were also significantly elevated in sera of chronically infected, cachectic WT mice (Fig 6A). When cytokine levels were assessed in wildtype tissues, IL-1α was sustained in the liver of infected WT mice and brain lysates had increased IL-1α, IL-1β, and IL-1RA protein levels relative to uninfected mice (Fig. 6B). Although IL-1 expression is not a direct measure of release, these data suggest that the liver and brain may be sites of sustained IL-1R signaling in chronic infection. Although IL-1α and IL-1β were not elevated in muscle lysate, IL-1Ra was significantly increased, suggesting that skeletal muscle may be sensitive to IL-1R signaling due to circulating IL-1α and/or IL-1α or IL-1β levels in muscle microenvironments that are below the threshold of detection in whole tissue lysate.
FIGURE 6.
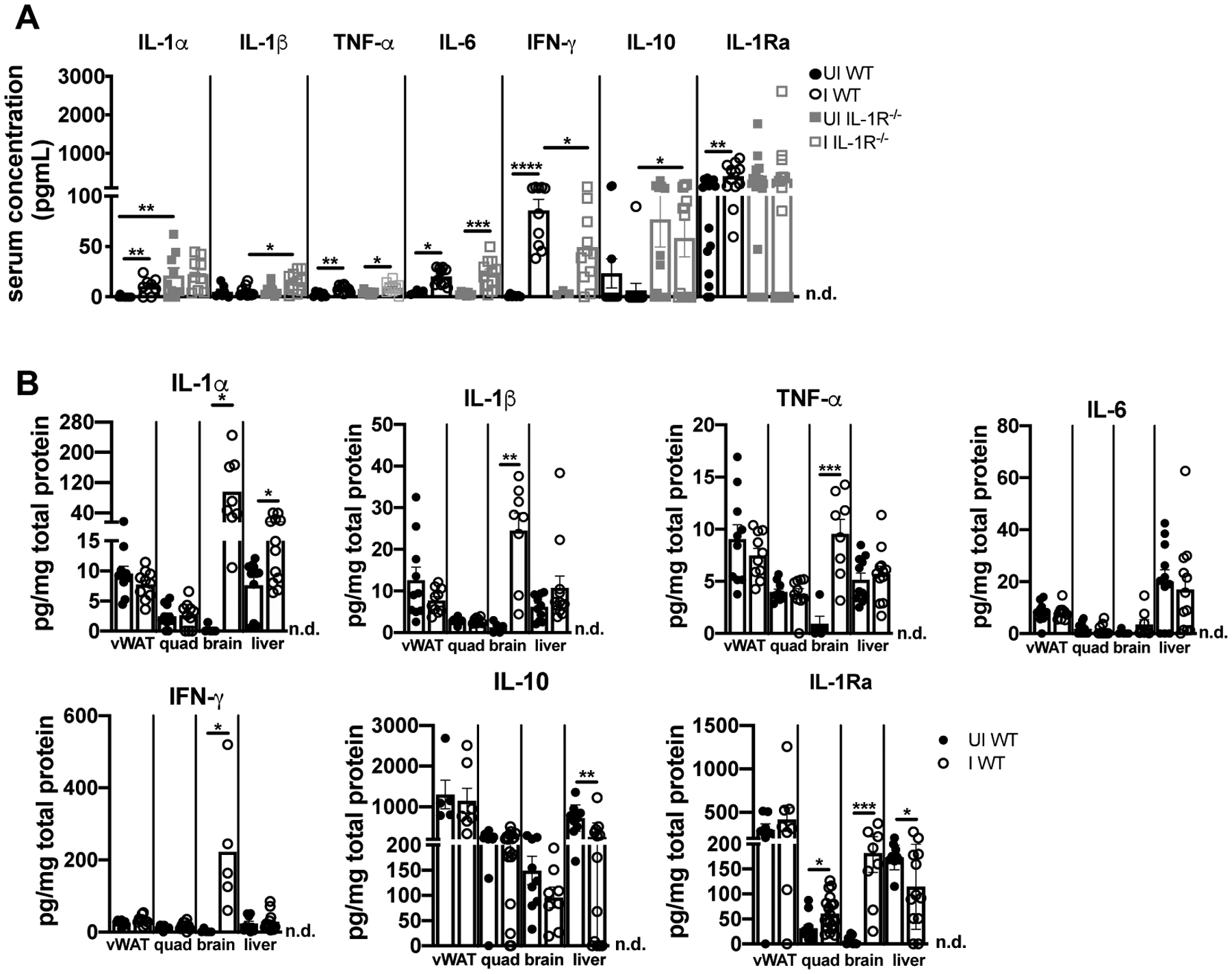
IL-1α remains chronically elevated in the circulation, liver, and brain of cachectic mice. (A) Serum cytokines at 9 wpi measured by Luminex (n = 3–10 mice per group, pooled from two experiments) or ELISA (IL-1α, IL-10, and IL-1Ra n = 9–17 mice per group, pooled from two to three experiments). (B) Cytokines in tissue lysates were determined by ELISA (n = 5–19 mice per group, pooled from three experiments. Brain data pooled from two experiments). n.d., not detectable. Data are shown as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, ****P<0.0001, by unpaired Student’s T test with Holm-Sidak method to correct for multiple comparisons.
To determine if IL-1α, IL-1β, or both cytokines were necessary for chronic T. gondii-induced cachexia, IL-1α−/− or IL-1β−/− mice were infected and weights were monitored for up to 16 wpi. Mice deficient in either IL-1α−/− or IL-1β−/− had improved survival compared to wild type animals (Supp. Fig. 4A). Both IL-1α−/− and IL-1β−/− animals and were partially protected from weight loss by 12 weeks post-infection (Supp. Fig. 4B). (Note that death of the most severely cachectic infected wildtype mice from 12–16 weeks post infection increased the mean body mass of this group and a loss of the statistically significant difference between infected IL-1α−/− and IL-1β−/− groups). However, neither the IL-1α−/− or IL-1β−/− groups fully regained body mass relative to uninfected animals by 16 weeks post infection. Similar to infected wildtype mice, the muscle and liver of infected IL-1β−/− mice were significantly wasted relative to uninfected controls (Supp. Fig 4C–D). IL-1α−/− mice had larger quadriceps muscles than IL-1β−/− mice, and the muscle and liver mass trended larger in IL-1α−/− mice relative to WT, however, this was not significant (Supp. Fig 4C–D). These data, in conjunction sustained tissue cytokine levels (Figure 6), suggest that IL-1α and IL-1β both likely contribute to chronic cachexia during T. gondii infection. Together, our data demonstrate that the IL-1R axis plays a dual role in disease progression during T. gondii infection: IL-1R signaling limits tissue damage during acute infection; however, cachexia is a long-term cost of IL-1R signaling during the chronic phase of infection.
Discussion
Our data indicate that IL-1R plays a distinct role in the innate inflammatory response during T. gondii infection. In contrast to better-studied effectors IL-6, TNF-α, and IFN-γ, which are necessary for T. gondii resistance (28, 31, 45), IL-1R signaling protects mice from acute liver and adipose tissue pathology without affecting parasite load. This result is consistent with the definition of a disease tolerance mechanism, which is a tissue-intrinsic pathway that enables tissue function during immunological stress without directly affecting pathogen burden (46). However, in the long-run, an intact IL-1R signaling axis negatively impacts host survival during chronic infection by driving cachexia.
Our studies suggest that IL-1α and IL-1β may both contribute to acute weight loss and cachexia during T. gondii infection. Release of functional IL-1β requires cleavage by the effector casapase-1 downstream of inflammasome activation (47), whereas bioactive IL-1α can be released downstream of a broader range of cell death and damage events. Unlike IL-1R−/− mice, mice deficient in the inflammasome adaptor ASC, sensors NLRP1 and NLRP3, or effector caspases-1 and/or caspase-11 cannot control T. gondii replication and have decreased survival in acute or early chronic infection (6, 7, 48, 49). Interpreting these data in the context of our results suggest that inflammasome activation restricts T. gondii growth through an IL-1R independent mechanism. Interestingly, pyroptotic cell death has not been observed in T. gondii infected mouse macrophage or human monocytes (6, 11, 50, 51). However, it is possible that other cell types undergo pyroptosis in response to T. gondii infection, or that there is crosstalk between the inflammasome and other pathways that restrict the parasite in vivo, like iNOS (52) and/or or interferon-regulated GTPase pathways (53, 54). Alternatively, the inflammasome also mediates IL-18 release, which has been shown to be critical for parasite restriction (7), particularly in the absence of TLR11 (48). In mice, TLR11 recognizes T. gondii profilin to signal through MyD88 and NF-kB, which initiates transcription of pro-IL-1 (in addition to other effector functions that contribute to a protective Th1 immune response). Notably, TLR11−/− mice also develop fat necrotic plaques that are visible by eye similar to what we observed in IL-1R−/− mice during acute infection, suggesting a potential mechanism of IL-1R axis activation in the vWAT during acute infection (55).
Our study is not the first to investigate the role of IL-1R signaling in T. gondii infection; however, results, at least at the outset, seem to conflict. A 1998 study showed that IL-1α or IL-1β administration one day before infection protected Swiss Webster mice from an LD100 dose of type III T. gondii (10). Although parasite load was not measured, all experimental groups had comparable T. gondii specific IgG and evidence of brain infection. In 2007 La Rosa and colleagues reported that IL-1R antagonist treatment did not affect parasite burden and stated that IL-1R−/− mice are no more susceptible to T. gondii infection than wildtype controls (12). By contrast, Gorfu et al found that mice deficient in IL-1R or the IL-18 axis were more susceptible than WT mice to i.p. infection with 10,000 type II 76K T. gondii tachyzoites, and susceptibility correlated with increased parasite load by luciferase assay (11). These data do not necessarily conflict with our results, in fact we might expect that a failure in tolerance biology would result in increased host death at an LD50, as used here, compared to our milder infection conditions. It is also important to note that although tolerance biology does not directly target pathogen growth, signal load can be indirectly impacted over time.
In a study by Villeret et al. IL-1R-deficient mice orally infected with type II T. gondii (76K) had more severe acute gut pathology, increased neutrophil and myeloid cell recruitment, and elevated pro-inflammatory cytokines compared to wildtype mice (13). Several important differences between this model and our study may explain the pathological effects of IL-1R in the gut and the protective effect of IL-1R signaling we observed in the liver and fat. First, during oral infection, the small intestine barrier is compromised, leading to TLR recognition of commensal microbiota, so it is unclear whether changes in immune infiltration are due to a lack of IL-1R signaling directly or an indirect effect of increased interaction with commensals (56–58). A not mutually exclusive alternative is that the small intestine has a different tolerance threshold for inflammation, distinct IL-1R-responsive cell types, or overall levels IL-1α and/or IL-1β released than the fat and liver. Importantly, our conclusion agrees with the Villeret study in IL-1R signaling was not necessary to restrict T. gondii and, overall, the loss of IL-1 promoted host survival during T. gondii infection.
Future experiments will be necessary to determine if the cytoprotective effect IL-1R observed in the liver and adipose tissue is cell-intrinsic or indirect. IL-1β pretreatment has been shown to protect primary murine hepatocytes from Fas-ligand mediated death (59), and prevent hepatocyte death in vivo after treatment with a Fas activating antibody (60) or TNF-α (61). Ishibe et al. found that IL-1RA deficient mice (which have increased IL-1R signaling) are protected from acetaminophen-induced liver injury, and pretreatment with IL-1α was sufficient to significantly attenuate liver pathology (62). Together, these studies indicate that IL-1R signaling can directly promote survival in liver parenchymal cells, however, less is known in adipose tissue.
From a clinical perspective, promoting tolerance biology has become a topic of therapeutic interest as a strategy to mitigate the negative aspects of inflammation without increasing the risk of infection. While this may be a beneficial treatment strategy to treat resolving infections or acute inflammatory events, an extension of the tolerance/resistance paradigm is that disease tolerance programs must come at a cost to the organism, otherwise they would be selected for homeostatic use. Consistent with this theoretical framework, our data indicate that IL-1R signaling is cytoprotective during acute infection, but ultimately promotes cachexia. Precisely how IL-1R signaling promotes cachexia remains to be determined; however, there is evidence to suggest central nervous system dependent and independent mechanisms could be at play.
IL-1R signaling in the central nervous system has been shown to be important for controlling appetite and eating behavior (63). However, in our model, anorexia is not IL-1R dependent or sustained. Both i.p. administration of IL-1α and intracerebroventricular infusion of IL-1β in rats has been demonstrated to induce anorexia-independent muscle loss (64, 65). Mice that received tumor xenografts engineered to express IL-1α had more severe wasting than mice that received control tumor xenografts (independent of food intake), and blockade of local IL-1R signaling by intra-tumoral administration of IL-1Ra significantly less fat and muscle wasting in the C-26 murine model of colon cancer (66, 67). The IL-1R axis has also been shown to regulate glucose and lipid metabolism (68, 69) Similar to metabolic shifts associated with cachexia, mice deficient in IL-1Ra are smaller, have impaired fat storage, abnormal lipid metabolism, and elevated resting baseline energy expenditure (69–71). Of note, the majority of these studies were performed before a modern clinical definition of cachexia was established, indicating that it may be time to revisit a role for IL-1 in current models of cachexia.
T. gondii infection has several unique advantages as a tool to study the inflammatory mediators in cachexia. First, the kinetics of T. gondii cachexia more closely resemble clinical disease than current surgical or tumor cachexia models, which are lethal after only several days of weight loss (72, 73). This study shows genetic rescue of cachexia after peak weight loss has occurred, which is significant because it indicates that the inflammatory networks responsible for maintaining cachexia are distinct from those that initiate disease. Second, the parameter of pathogen burden can be used to distinguish between immune effectors primarily designed to promote inflammation (resistance biology) versus those targeting tissue homoeostasis (tolerance biology). Targeting resistance effectors should reduce damage-to-self, but may not correct the tolerance-induced homeostatic shifts that occur in response to the inflammatory environment. Consistent with this model, blocking the cachexia-associated cytokine TNF-α (a pathogen resistance signaling molecule) has repeatedly failed to treat clinical cachexia (31). Recently, however, a first-in-class monoclonal antibody targeting IL-1α (MABp1) was shown to improve body mass in a phase 1 dose escalation trial for metastatic cancers (74). While further studies in disease-matched cachectic and weight stable patients are necessary, these findings suggest that dual therapies targeting both tolerance and resistance pathways may be a more efficacious way to restore homeostasis during chronic diseases like cachexia. Given the conserved role IL-1 plays across etiologies of clinical cachexia, we anticipate that mechanisms uncovered in T. gondii cachexia may have important implications for cachexia in general.
Supplementary Material
Key Points.
IL-1R signaling is not necessary to control acute or chronic T. gondii burden
IL-1R signaling limits cell death in liver and adipose tissue at acute infection
IL-1R prevents recovery from acute weight loss and drives chronic cachexia
Acknowledgements
We thank S. Batista and Dr. T. Harris for sharing the initial observation that IL-1R−/− mice regain weight after T. gondii infection and for critical feedback. We thank Dr. J. V. Cross for evaluation of the manuscript, Dr. K. Tung for histopathology advice, and M. Gonzalez for assistance with flow cytometry. The authors have no competing financial interest.
This work was supported by NIH K22 AI116727 (S.E.E.)
References
- 1.Flegr J, Prandota J, Sovičková M, and Israili ZH. 2014. Toxoplasmosis--a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS One 9: e90203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Cristina M, Marocco D, Galizi R, Proietti C, Spaccapelo R, and Crisanti A. 2008. Temporal and spatial distribution of Toxoplasma gondii differentiation into Bradyzoites and tissue cyst formation in vivo. Infect. Immun 76: 3491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pernas L, Ramirez R, Holmes TH, Montoya JG, and Boothroyd JC. 2014. Immune profiling of pregnant toxoplasma-infected US and Colombia patients reveals surprising impacts of infection on peripheral blood cytokines. J. Infect. Dis 210: 923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luft BJ, and Remington JS. 1992. Toxoplasmic Encephalitis in AIDS. Clin. Infect. Dis 15: 211–222. [DOI] [PubMed] [Google Scholar]
- 5.Khurana S, and Batra N. 2016. Toxoplasmosis in organ transplant recipients: Evaluation, implication, and prevention. Trop. Parasitol 6: 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ewald SE, Chavarria-Smith J, and Boothroyd JC. 2014. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun 82: 460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, Masters S, Sher A, Leppla SH, Moayeri M, Saeij JPJ, and Grigg ME. 2014. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatter JA, Kouche YM, Melchor SJ, Ng K, Bouley DM, Boothroyd JC, and Ewald SE. 2018. Toxoplasma gondii infection triggers chronic cachexia and sustained commensal dysbiosis in mice. PLoS One 13: e0204895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter CA, Abrams JS, Beaman MH, and Remington JS. 1993. Cytokine mRNA in the central nervous system of SCID mice infected with Toxoplasma gondii: Importance of T-cell-independent regulation of resistance to T. gondii,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang HR, Grau GE, and Pechère JC. 1990. Role of TNF and IL-1 in infections with Toxoplasma gondii. Immunology 69: 33–7. [PMC free article] [PubMed] [Google Scholar]
- 11.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, Masters S, Sher A, Leppla SH, Moayeri M, Saeij JPJJ, and Grigg ME. 2014. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, and Turka LA. 2008. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A 105: 3855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villeret B, Brault L, Couturier-Maillard A, Robinet P, Vasseur V, Secher T, Dimier-Poisson I, Jacobs M, Zheng S-G, Quesniaux VF, and Ryffel B. 2013. Blockade of IL-1R signaling diminishes Paneth cell depletion and Toxoplasma gondii induced ileitis in mice. Am. J. Clin. Exp. Immunol 2: 107–16. [PMC free article] [PubMed] [Google Scholar]
- 14.Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I, Aychek T, Dinarello CA, Apte RN, and Voronov E. 2014. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 63: 598–609. [DOI] [PubMed] [Google Scholar]
- 15.Alves-Rosa F, Vulcano M, Beigier-Bompadre M, Fernández G, Palermo M, and Isturiz MA. 2002. Interleukin-1beta induces in vivo tolerance to lipopolysaccharide in mice. Clin. Exp. Immunol 128: 221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rao S, Schieber AMP, O’Connor CP, Leblanc M, Michel D, and Ayres JS. 2017. Pathogen-Mediated Inhibition of Anorexia Promotes Host Survival and Transmission. Cell 168: 503–516.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamin JT, Moore DJ, Bennett C, van der Meer R, Royce A, Loveland R, and Wynn JL. 2018. Cutting Edge: IL-1α and Not IL-1β Drives IL-1R1–Dependent Neonatal Murine Sepsis Lethality. J. Immunol 201: 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melchor SJ, and Ewald SE. 2019. Disease tolerance in Toxoplasma infection. Front. Cell. Infect. Microbiol 9: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R, and Anker SD. 2008. Cachexia: A new definition. Clin. Nutr 27: 793–799. [DOI] [PubMed] [Google Scholar]
- 20.Arsenijevic D, Girardier L, Seydoux J, Pechere JC, Garcia I, Lucas R, Chang HR, and Dulloo AG. 1998. Metabolic-cytokine responses to a second immunological challenge with LPS in mice with T. gondii infection. Am. J. Physiol 274: E439–45. [DOI] [PubMed] [Google Scholar]
- 21.Jin RM, Blair SJ, Warunek J, Heffner RR, Blader IJ, Wohlfert EA, Reid R, Blader IJ, Wohlfert EA, Jin RM, Blair SJ, Warunek J, Heffner RR, Blader IJ, and Wohlfert EA. 2017. Regulatory T Cells Promote Myositis and Muscle Damage in Toxoplasma gondii Infection. J. Immunol 198: 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuki T, Nakae S, Sudo K, Horai R, and Iwakura Y. 2006. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int. Immunol 18: 399–407. [DOI] [PubMed] [Google Scholar]
- 23.Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Ferguson TA, and Chaplin DD. 1996. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J. Exp. Med 183: 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasper DC, Sadeghi K, Prusa A-R, Reischer GH, Kratochwill K, Förster-Waldl E, Gerstl N, Hayde M, Pollak A, and Herkner KR. 2009. Quantitative real-time polymerase chain reaction for the accurate detection of Toxoplasma gondii in amniotic fluid. Diagn. Microbiol. Infect. Dis 63: 10–15. [DOI] [PubMed] [Google Scholar]
- 25.Pearson H, and Stirling D. 2003. DNA Extraction from Tissue In PCR Protocols Humana Press, New Jersey: 33–34. [DOI] [PubMed] [Google Scholar]
- 26.Ruifrok AC, and Johnston DA. 2001. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol 23: 291–9. [PubMed] [Google Scholar]
- 27.Mohar I, Brempelis KJ, Murray SA, Ebrahimkhani MR, and Crispe IN. 2015. Isolation of Non-parenchymal Cells from the Mouse Liver. In Humana Press, New York, NY: 3–17. [DOI] [PubMed] [Google Scholar]
- 28.Jebbari H, Roberts CW, Ferguson DJ, Bluethmann H, and Alexander J. 1998. A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunol. 20: 231–9. [DOI] [PubMed] [Google Scholar]
- 29.Schlüter D, Kwok L-Y, Lütjen S, Soltek S, Hoffmann S, Körner H, and Deckert M. 2003. Both lymphotoxin-alpha and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J. Immunol 170: 6172–82. [DOI] [PubMed] [Google Scholar]
- 30.Deckert-Schlüter M, Rang A, Weiner D, Huang S, Wiestler OD, Hof H, and Schlüter D. 1996. Interferon-gamma receptor-deficiency renders mice highly susceptible to toxoplasmosis by decreased macrophage activation. Lab. Invest 75: 827–41. [PubMed] [Google Scholar]
- 31.Suzuki Y, Orellana MA, Schreiber RD, and Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240: 516–8. [DOI] [PubMed] [Google Scholar]
- 32.Kiryu H, Rikihisa W, and Furue M. 2000. Encapsulated fat necrosis - A clinicopathological study of 8 cases and a literature review. J. Cutan. Pathol 27: 19–23. [DOI] [PubMed] [Google Scholar]
- 33.Misumi I, Starmer J, Uchimura T, Beck MA, Magnuson T, and Whitmire JK. 2019. Obesity Expands a Distinct Population of T Cells in Adipose Tissue and Increases Vulnerability to Infection. Cell Rep. 27: 514–524.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yarovinsky F, Hieny S, and Sher A. 2008. Recognition of Toxoplasma gondii by TLR11 Prevents Parasite-Induced Immunopathology. J. Immunol 181: 8478–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atmaca HTK, Gazya CAN, Canpolat SL, and Kul OU. 2013. Hepatic stellate cells increase in Toxoplasma gondii infection in mice. Parasit. Vectors 6: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R, Müller W, Trinchieri G, and Sher A. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol 157: 798–805. [PubMed] [Google Scholar]
- 37.Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, Zhang C, Ring AM, Young LH, and Medzhitov R. 2019. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 178: 1231–1244.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot J-D, Booth CJ, and Medzhitov R. 2016. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 166: 1512–1525.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neyer LE, Grunig G, Fort M, Remington JS, Rennick D, and Hunter CA. 1997. Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect. Immun 65: 1675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson EH, Wille-Reece U, Dzierszinski F, and Hunter CA. 2005. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J. Neuroimmunol 165: 63–74. [DOI] [PubMed] [Google Scholar]
- 41.La E, and Fischer SM. 2001. Transcriptional regulation of intracellular IL-1 receptor antagonist gene by IL-1 alpha in primary mouse keratinocytes. J. Immunol 166: 6149–55. [DOI] [PubMed] [Google Scholar]
- 42.Gibbings SL, and V Jakubzick C. 2018. Isolation and Characterization of Mononuclear Phagocytes in the Mouse Lung and Lymph Nodes. Methods Mol. Biol 1809: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, Wang W, Zhang S, Iwakura Y, Meng G, Fu YX, Hou B, and Tang H. 2018. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell. Mol. Immunol 15: 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matthys P, and Billiau A. 1997. Cytokines and cachexia. Nutrition 13: 763–770. [DOI] [PubMed] [Google Scholar]
- 45.Johnson LL 1992. A protective role for endogenous tumor necrosis factor in Toxoplasma gondii infection. Infect. Immun 60: 1979–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medzhitov R, Schneider DS, and Soares MP. 2012. Disease Tolerance as a Defense Strategy. Science (80-.) 335: 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinon F, Burns K, and Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10: 417–26. [DOI] [PubMed] [Google Scholar]
- 48.López-Yglesias AH, Camanzo E, Martin AT, Araujo AM, and Yarovinsky F. 2019. TLR11-independent inflammasome activation is critical for CD4+ T cell-derived IFN-γ production and host resistance to Toxoplasma gondii. PLOS Pathog. 15: e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coutermarsh-Ott SL, Doran JT, Campbell C, Williams TM, Lindsay DS, and Allen IC. 2016. Caspase-11 Modulates Inflammation and Attenuates Toxoplasma gondii Pathogenesis. Mediators Inflamm. 2016: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, and Frickel E. 2019. Human GBP 1 is a microbe‐specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L, and Lodoen MB. 2019. Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin D-independent release of IL-1β during infection of primary human monocytes. PLOS Pathog. 15: e1007923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan IA, Schwartzman JD, Matsuura T, and Kasper LH. 1997. A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proc. Natl. Acad. Sci 94: 13955–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halonen SK, Taylor GA, and Weiss LM. 2001. Gamma interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect. Immun 69: 5573–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao YO, Khaminets A, Hunn JP, and Howard JC. 2009. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 5: e1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yarovinsky F, Hieny S, and Sher A. 2008. Recognition of Toxoplasma gondii by TLR11 prevents parasite-induced immunopathology. J. Immunol 181: 8478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molloy MJ, Grainger JR, Bouladoux N, Hand TW, Koo LY, Naik S, Quinones M, Dzutsev AK, Gao J-L, Trinchieri G, Murphy PM, and Belkaid Y. 2013. Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe 14: 318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benson A, Pifer R, Behrendt CL, V Hooper L, and Yarovinsky F. 2009. Gut Commensal Bacteria Direct a Protective Immune Response against Toxoplasma gondii. Cell Host Microbe 6: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liesenfeld O 2002. Oral Infection of C57BL/6 Mice with Toxoplasma gondii: A New Model of Inflammatory Bowel Disease? J. Infect. Dis 185: S96–S101. [DOI] [PubMed] [Google Scholar]
- 59.Lutz A, Sanwald J, Thomas M, Feuer R, Sawodny O, Ederer M, Borner C, Humar M, and Merfort I. 2014. Interleukin-1β Enhances FasL-Induced Caspase-3/-7 Activity without Increasing Apoptosis in Primary Mouse Hepatocytes. PLoS One 9: e115603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takehara T, Hayashi N, Tatsumi T, Kanto T, Mita E, Sasaki Y, Kasahara A, and Hori M. 1999. Interleukin 1β protects mice from Fas-mediated hepatocyte apoptosis and death. Gastroenterology 117: 661–668. [DOI] [PubMed] [Google Scholar]
- 61.Bohlinger I, Leist M, Barsig J, Uhlig T, Tiegs G, and Wendel A. 1995. Interleukin-1 and nitric oxide protect against tumor necrosis factor α-induced liver injury through distinct pathways. Hepatology 22: 1829–1837. [PubMed] [Google Scholar]
- 62.Ishibe T, Kimura A, Ishida Y, Takayasu T, Hayashi T, Tsuneyama K, Matsushima K, Sakata I, Mukaida N, and Kondo T. 2009. Reduced acetaminophen-induced liver injury in mice by genetic disruption of IL-1 receptor antagonist. Lab. Investig 89: 68–79. [DOI] [PubMed] [Google Scholar]
- 63.Layé S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, and Parnet P. 2000. Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am. J. Physiol. Integr. Comp. Physiol 279: R93–R98. [DOI] [PubMed] [Google Scholar]
- 64.Fong Y, Moldawer LL, Marano M, Wei H, Barber A, Manogue K, Tracey KJ, Kuo G, Fischman DA, and Cerami A. 1989. Cachectin/TNF or IL-1 alpha induces cachexia with redistribution of body proteins. Am. J. Physiol 256: R659–65. [DOI] [PubMed] [Google Scholar]
- 65.Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, Levasseur PR, Graham K, Khan S, Damaraju S, Colmers WF, Baracos VE, and Marks DL. 2011. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med 208: 2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strassmann G, Masui Y, Chizzonite R, and Fong M. 1993. Mechanisms of experimental cancer cachexia. Local involvement of IL-1 in colon-26 tumor. J. Immunol 150: 2341–5. [PubMed] [Google Scholar]
- 67.Kumar S, Kishimoto H, Chua HL, Badve S, Miller KD, Bigsby RM, and Nakshatri H. 2003. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am. J. Pathol 163: 2531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burke SJ, Batdorf HM, Burk DH, Martin TM, Mendoza T, Stadler K, Alami W, Karlstad MD, Robson MJ, Blakely RD, Mynatt RL, and Collier JJ. 2018. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet β-cell de-differentiation. Mol. Metab 14: 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuki T, Horai R, Sudo K, and Iwakura Y. 2003. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J. Exp. Med 198: 877–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Somm E, Henrichot E, Pernin A, Juge-Aubry CE, Muzzin P, Dayer J-M, Nicklin MJH, and Meier CA. 2005. Decreased fat mass in interleukin-1 receptor antagonist-deficient mice: impact on adipogenesis, food intake, and energy expenditure. Diabetes 54: 3503–9. [DOI] [PubMed] [Google Scholar]
- 71.Hirsch E, Irikura VM, Paul SM, and Hirsh D. 1996. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. U. S. A 93: 11008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deboer MD 2009. Animal models of anorexia and cachexia. Expert Opin. Drug Discov 4: 1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ballarò R, Costelli P, and Penna F. 2016. Animal models for cancer cachexia. Curr. Opin. Support. Palliat. Care 10: 281–287. [DOI] [PubMed] [Google Scholar]
- 74.Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS, Piha-Paul S, Wheler JJ, Fu S, Tsimberidou AM, Stecher M, Mohanty P, Simard J, and Kurzrock R. 2014. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 15: 656–666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.