

SUMMARY
Signaling by the cytokine transforming growth factor β (TGF-β) has been implicated in a multitude of biological functions; however, TGF-β signaling, particularly in the CNS, remains largely unexplored. ANK3 variants (encoding ankyrin-G) are associated with bipolar disorder, intellectual disability, and autism spectrum disorder, while mutations in USP9X, which encodes a deubiquitinase, are associated with X-linked intellectual disability and autism in humans. Here, we show that TGF-β signaling promotes Usp9X phosphorylation, which enhances its interaction with ankyrin-G and stabilizes ankyrin-G in spines, leading to spine enlargement. Using in situ proximity ligation combined with structured illumination superresolution microscopy, we characterize the postsynaptic spatial organization of phosphorylation-dependent regulation of Usp9X/ankyrin-G interactions in dendrites and its quantitative relationship with spine morphology and number. These data reveal a cytokine-mediated mechanism regulating protein stability in spines and suggest a role for deubiquitination and TGF-β signaling in neurodevelopmental disorder pathogenesis and treatment.
In Brief
Yoon et al. show that phosphorylation of a deubiquitinating enzyme by a cytokine enhances the stabilization of synaptic scaffolding protein during dendritic spine development, and its alterations result in deficient synaptic structural maintenance, with relevance for neurodevelopmental disorders.
Graphical Abstract
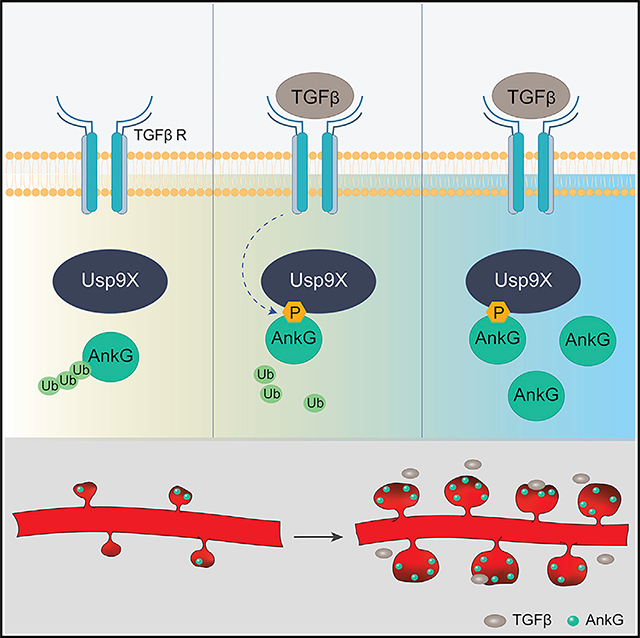
INTRODUCTION
Structural plasticity of spiny synapses, consisting of their formation, maturation, stabilization, remodeling, and elimination, underlies the development, function, and remodeling of brain circuits and contributes to behavior and cognition. Conversely, abnormalities of synaptic structural plasticity have emerged as cellular substrates in the pathogenesis of neurodevelopmental disorders (Forrest et al., 2018; Penzes et al., 2011). Accordingly, dendritic spine alterations have been observed in postmortem studies of patients with schizophrenia (SZ), bipolar disorder (BD), autism spectrum disorders (ASDs), and intellectual disability (ID) (Glantz and Lewis, 2000; Hutsler and Zhang, 2010; Irwin et al., 2000; Konopaske et al., 2014).
Consistent with this, recent genomic studies support a key role for genes encoding postsynaptic proteins in the pathogenesis of neurodevelopmental disorders (Gilman et al., 2011; Purcell et al., 2014). Among these, rare and common variants at the ANK3 gene locus were identified in patients with ASD (Sanders et al., 2012) and ID/attention-deficit/hyperactivity disorder (ADHD) (Iqbal et al., 2013) and are among the most strongly associated risk factors for BD in genome-wide association studies (GWASs) (Ferreira et al., 2008; Schulze et al., 2009; Stahl et al., 2019). ANK3 encodes ankyrin-G, which acts as a scaffold linking plasma membrane proteins to the actin/β-spectrin cytoskeleton (Bennett and Healy, 2008). Multiple isoforms of ankyrin-G (190/270/480 kDa) are expressed in the brain and share four conserved domains: an ankyrin repeat domain (ANKRD), a spectrin-binding domain, a death domain, and a regulatory domain (Smith and Penzes, 2018). The giant 270/480-kDa isoforms have well-characterized roles at the axon initial segment (AIS) and nodes of Ranvier (Bennett and Healy, 2008), and the 190-kDa form is enriched in dendrites and postsynaptic densities (PSDs) (Jordan et al., 2004) and regulates spine structure (Smith et al., 2014). Despite their importance, little is known about how the levels of ANKRD proteins, in particular at synapses, are regulated.
One of the major mechanisms controlling postsynaptic proteins is their regulated degradation by the ubiquitin-proteasome system (UPS), which also plays an important role in synaptic remodeling (van Roessel et al., 2004). Synapse remodeling is accompanied by changes in protein turnover, which occurs via regulated increases or decreases in protein degradation (Ehlers, 2003). Proteins are generally targeted for degradation by the UPS through the covalent addition of polyubiquitin chains to their lysine residues. Deubiquitinating enzymes (DUBs) oppose this process by removing ubiquitin from substrate proteins. Activation or inhibition of DUBs induces synaptic plasticity (Cartier et al., 2009). Recent studies support a role for dysregulation of the UPS pathway in SZ, BD (Bousman et al., 2010), ID (Hollstein et al., 2015; Johnson et al., 2020), and ASDs (Glessner et al., 2009), suggesting that protein turnover may play an important role in neurodevelopmental disorders. However, little is known about the synaptic targets of DUBs and about how deubiquitination of PSD proteins impacts spine plasticity and contributes to disease pathogenesis.
Transforming growth factor β (TGF-β) is a cytokine with important functions in the immune system but immune-independent roles have been reported (Massagué, 2012). TGF-β binds to TGF-β receptors I and II, which are Ser/Thr kinases that phosphorylate intracellular downstream substrates. TGF-β has been shown to play important roles in neuronal development and function (Heupel et al., 2008; Krieglstein et al., 2011). Abnormal TGF-β signaling has been implicated in cancer, heart disease, obesity and diabetes, but also in SZ and ASDs (Ashwood et al., 2008; Kasherman et al., 2020; Pietersen et al., 2014). However, the role of TGF-β in dendritic spine structural plasticity has not been explored and its interaction with neurodevelopmental and psychiatric disorder risk factors is not clearly defined.
Here, we investigated the regulation of ankyrin-G protein stability at synapses. Previously, we found that the ID-associated DUB Usp9X interacts with ankyrin-G and regulates its stability, maintaining dendritic spines persistently (Yoon et al., 2020). In the current study, we show that the neuroprotective factor TGF-β initializes a signaling cascade causing Usp9X phosphorylation and subsequent stabilization of ankyrin-G in spines. Using structured illumination microscopy (SIM), we found that ankyrin-G and Usp9X localize to distinct nanostructures within spines, the number of which correlates to mushroom spine head size, while proximity ligation assay (PLA)/SIM confirmed that TGF-β increases ankyrin-G’s interaction with Usp9X. These data reveal that TGF-β can enhance stabilization of ankyrin-G in spines through the increase of Usp9X binding ability toward its target substrate.
RESULTS
TGF-β Signaling Pathway Enhances the Stabilization of Ankyrin-G
Usp9X has been shown to modulate the TGF-β signaling pathway by controlling the stability of Smad transcription factors (Stegeman et al., 2013). However, because TGF-β receptors, which are Ser/Thr kinases, can also act by phosphorylating other substrates, we hypothesized that TGF-β signaling may also directly regulate Usp9X function and its interaction with substrates for deubiquitination. We thus investigated the role of TGF-β in regulating the Usp9X/ankyrin-G interaction. Ankyrin-G was costained for either TGF-β receptor I or TGF-β receptor II to understand the expression patterns of TGF-β receptor in ankyrin-Gpositive neurons. TGF-β receptors I and II were localized to both the AIS and dendrites, and they overlapped strongly with ankyrin-G in dendrites (Figure 1A); both receptor proteins were present at significantly higher levels in the dendrites than the AIS (Figure 1B). Next, to determine whether TGF-β affected the expression of ankyrin-G and Usp9X, neurons were treated with TGF-β for 15 min, 30 min, 1 h, 3 h, and 6 h. As expected, TGF-β treatment resulted in a robust increase in Smad2 phosphorylation after 1 h, a known signaling component downstream of TGF-β (Figure 1C). Interestingly, ankyrin-G protein levels increased after TGF-β treatment, while Usp9X levels were not affected. Our previous studies have suggested that ankyrin-G plays an important role in spine development (Smith et al., 2014; Yoon et al., 2020). Therefore, we tested the impact of TGF-β on dendritic spine morphology in cultured cortical neurons. TGF-β treatment caused a significant increase in spine head size and density (Figures 1E and 1F). Taken together, the TGF-β-mediated increase in ankyrin-G may regulate spine development.
Figure 1. TGF-β Signaling Enhances the Expression Level of Ankyrin-G.

(A) Confocal image of a neuron immunostained for ankyrin-G and TGF-βRI or TGF-βRII in the axon initial segment (AIS) and dendrite. Scale bars, 20 μm (left); 5 μm (right).
(B) Comparing intensities in the AIS (white bars) and dendrite (black bars) (n = 6 for each group). *p < 0.05; ***p < 0.001 by two-tailed unpaired t test. All data are presented as mean ± SEM.
(C) Representative images of western blot. Time-dependent stimulation of expression of ankyrin-G after treatment with TGF-β (20 ng/mL) in cultured neurons. Phosphorylation of Smad2 (p-Smad2) was used for confirmation of TGF-β signaling activity. β-Actin was the internal loading control.
(D) Confocal images of neurons transfected with mCherry, and subsequently treated with vehicle (top) or TGF-β (20 ng/mL) (bottom) for 1 h. Scale bar, 2 μm.
(E and F) Spine head area (E) and density (F) with vehicle (plain pattern) or TGF-β (comb pattern) (n = 9 neurons for each group). *p < 0.05; ***p < 0.001 by two-tailed unpaired t test. All data are presented as mean ± SEM.
TGF-β Signaling Regulates Spine Morphology through the Interaction of Usp9X with Ankyrin-G
To validate TGF-β-mediated stabilization of ankyrin-G and the interaction of Usp9X and ankyrin-G within a cellular context, an in situ PLA was performed as previously reported (Yoon et al., 2020). The PLA method allows the interaction of cellular proteins to be spatially and quantitatively visualized. The PLA signal was significantly increased in both dendritic shafts and spine heads upon TGF-β treatment (Figures 2A and 2B).
Figure 2. TGF-β-Enhanced Interaction of Ankyrin-G and Usp9X Regulates Spine Morphogenesis.

(A) SIM image of mCherry-expressing neurons (red) with treatment of vehicle or TGF-β (20 ng/mL) for 1 h to detect the interaction between ankyrin-G and Usp9X by PLA (cyan). Scale bar, 10 μm, top. Magnified image of boxed spine and dendrite (bottom). Scale bar, 2 μm, bottom.
(B) The number of PLA puncta per 10 μm in neurons treated with vehicle or TGF-β. Bars also show the distribution of PLA puncta in spines (white) and dendrites (gray) (vehicle, n = 28 neurons; TGF-β, n = 33 neurons). **p < 0.01; ***p < 0.001 by two-tailed unpaired t test.
(C) Confocal images of neurons transfected with control RNAi (control) or shRNA against Usp9X (shUsp9X) or ankyrin-G (shAnkG) and subsequently treated with vehicle (top) or TGF-β (20 ng/mL) (bottom) for 1 h. Scale bar, 5 μm.
(D and E) Spine head area (D) and density (E) in control (black bars), shUsp9X (red bars), and shAnkG (blue bars) with vehicle (plain pattern) or TGF-β (comb pattern) (n = 20 neurons for each group). *p < 0.05; ***p < 0.001; †††p < 0.001 by one-way ANOVA followed by non-parametric statistical analysis. All data are presented as mean ± SEM.
As TGF-β signaling is a known regulator of synaptic development and function (Krieglstein et al., 2011), we hypothesized that it may regulate dendritic spine morphogenesis through Usp9X and its substrate ankyrin-G. We therefore applied TGF-β to cortical neurons after knockdown of ankyrin-G or Usp9X with short hairpin RNA (shRNA) or control neurons. TGF-β treatment of cultured cortical neurons resulted in increased spine area and density (Figures 2C–2E). Interestingly, the increase in spine area produced by TGF-β was prevented by knockdown of Usp9X (shUsp9X) or ankyrin-G (shAnkG). In addition, the observed alterations in spine density were prevented by shAnkG and attenuated by shUsp9X. These results strongly implicate Usp9X and ankyrin-G as downstream effectors of TGF-β receptor activation.
TGF-β-Dependent Phosphorylation of Usp9X Enhances Its Interaction with Ankyrin-G
TGF-β receptors are serine/threonine (S/T) kinases, and the TGF-β signaling pathway regulates the activity of a wide range of downstream S/T kinases, such as Smads, extracellular signal-regulated kinase, JUN N-terminal kinase, and mitogen-activated protein kinase (Akhurst and Hata, 2012). As Usp9X levels remained consistent following TGF-β stimulation, we hypothesized that post-translational modifications (PTMs) of Usp9X may be altered. We identified three S/T phosphorylation consensus sites (Serine 1,593/1,600/1,609) within the Usp9X peptidase domain (residues 1,555–1,958), and previous mass spectrometry studies have revealed that these sites are subject to PTM (Hornbeck et al., 2015). All three serine sites are predicted to lie distal to the catalytic triad and are likely exposed for PTM (Figure 3A). To confirm the interaction between the ANKRD of ankyrin-G and the peptidase domain of Usp9X, we conducted a pulldown assay. Nickel-bead-bound His-Usp9X1,547–1,962 showed pull-down of AnkG1–807 (Figure 3B). In order to assess whether these sites are subject to PTM following TGF-β stimulation, we carried out mutagenesis on the isolated peptidase domain of Usp9X (Usp9XWt) to create a phosphorylation-null triple mutant (Usp9XS3A). TGF-β treatment produced a robust increase in serine phosphorylation in Usp9XWt, but not in Usp9XS3A, suggesting that Usp9X is a downstream target of TGF-β receptor activation (Figure 3C). Interestingly, treatment with TGF-β enhanced the reciprocal interaction between AnkG1–807 and Usp9XWt, but this effect was not observed with Usp9XS3A (Figure 3C). To validate the interaction of Usp9X and ankyrin-G within a cellular context, we employed in situ PLA. This technique labels interacting proteins that fall within a defined, 16-nm distance as calculated from the number of nucleotides in the attached DNA arms (Trifilieff et al., 2011), a strong indicator of direct protein-protein interaction or complex formation. The PLA signal increased significantly after TGF-β treatment; however, this effect was absent when Usp9XS3A was employed (Figures 3D and 3E). As both 190-kDa and 480-kDa ankyrin-G isoforms share an ANKRD, we tested the interaction of Usp9X with the giant ankyrin-G (ankyrin-G 480). Consistent with the 190-kDa isoform, TGF-β-mediated Usp9X phosphorylation showed the enhancement in ankyrin-G 480 binding to Usp9X (Figure S1). Taken together, TGF-β-mediated phosphorylation of the peptidase domain of Usp9X enhances the interaction between ankyrin-G and Usp9X, an effect that may underlie the morphological effects on dendritic spine area, density, and axonal outgrowth.
Figure 3. TGF-β-Mediated Phosphorylation of Usp9X Enhances the Interaction with Ankyrin-G.

(A) Usp9X homology model (gray) was generated using crystal structure (PDB: 5WCH) as template. S1,593, S1,600, and S1,609 (orange) are positioned on a loop absent from crystal structure 5WCH. Energy minimization of this loop allowed an energetically favorable conformation to be predicted (blue) and suggests that all serine sites lie distal to the catalytic triad (magenta).
(B) Representative western blot of His-Usp9X1,547–1,962, expressed and purified from E. coli, was used to pull down HEK293T-expressed hemagglutinin (HA)-AnkG1–808 by nickel affinity purification. A GFP-transfected sample was used as a negative control.
(C) Representative western blot of HA-AnkG1–807 with co-expressing FLAG-Usp9X1,555–1,958 (FLAG-Usp9XWt) or FLAG-Usp9X1,555–1,958 (S1,593;1,600;1,609A) (FLAG-Usp9XS3A) construct from HEK293T cells. Immunoprecipitation (IP) of phosphoserine (pSer) and detection of phosphorylated Smad2 (p-Smad2) were performed to confirm TGF-β-mediated signaling.
(D) In situ PLA measurement of the interaction between HA-AnkG1–807 and FLAG-Usp9XWt or FLAG-Usp9XS3A in HEK293T cells. Scale bar, 5 μm.
(E) Quantification of PLA signal from (D) (n = 9–16 cells). HA-AnkG1–807 and FLAG-Usp9XWt (black bars) or FLAG-Usp9XS3A (red bars) with vehicle (plain pattern) or TGF-β (comb pattern). ***p < 0.001; †††p < 0.001 by two-way ANOVA followed by Bonferroni post-tests. All data are presented as mean ± SEM. TGF-β (−), vehicle; TGF-β (+), 20 ng/mL TGF-β for 1 h.
See also Figure S1.
TGF-β-Mediated Phosphorylation of Usp9X Regulates Spine Morphogenesis through Interaction with Ankyrin-G
Confocal images showed that PLA signals in the soma and dendrites after TGF-β treatment increased upon coexpression of ankyrin-G with Usp9XWt, but not with Usp9XS3A (Figures 4A and 4B). Similar experiments also showed ankyrin-G 480 and Usp9X interaction in the AIS (Figure S2). We then utilized SIM to investigate the spatial relationship between the sites of ankyrin-G-Usp9X interaction within spine heads and spine head size (Figure 4C). Likewise, the number of PLA puncta in dendritic shaft and spines increased in ankyrin-G with Usp9XWt with TGF-β treatment (Figure S3). Spine heads containing ankyrin-G were significantly larger than spine heads without ankyrin-G (Figure 4D), as previously reported (Smith et al., 2014). Interestingly, TGF-β treatment increased the ratio of ankyrin-G-containing spines coincident with an increase in PLA-positive spines (Figure 4E), and it promoted an increase in spine head size (Figure 4F). PLA signals were partially overlapping with and around ankyrin-G and Usp9X clusters and localized to the perisynaptic areas of spines (Figures 5A and 5B). To analyze the effects of TGF-β treatment on the correlation among the size of ankyrin-G, Usp9X, PLA nanodomains, and spine heads, equivalence tests were performed with confidence intervals. Unexpectedly, TGF-β treatment did not show any superior effects on correlations (Figures 5C–5F). Spine heads, which include ankyrin-G, Usp9X, and PLA nanodomains concurrently, showed the same phenotype whether TGF-β treated or not, suggesting that TGF-β treatment increases the number of ankyrin-G-containing spines and spine head size through upregulation of the interaction between Usp9X and ankyrin-G.
Figure 4. TGF-β-Mediated Phosphorylation of Usp9X in Dendrites Is Required for the Development of Spines.

(A) Confocal images of primary cortical neurons for detection of interaction between the whole construct of HA-AnkG and FLAG-Usp9XWt or FLAG-Usp9XS3A with PLA. Scale bar, 20 μm.
(B) Bar graph of PLA signal with vehicle (plain pattern) or TGF-β (comb pattern) treatment in HA-AnkG and FLAG-Usp9XWt (black bars) or FLAG-Usp9XS3A (red bars) expressing cells (n = 16 neurons for each group). ***p < 0.001; †††p < 0.001 by two-way ANOVA followed by Bonferroni post-tests. All data are presented as mean ± SEM.
(C) In situ PLA/SIM images of dendritic regions in neurons from (A). Scale bar, 5 μm.
(D–F) Spine head size in the presence or absence of ankyrin-G (D) and with vehicle (plain pattern) or TGF-β (comb pattern) treatment (F); theratio of spines expression HA-AnkG and PLA signal (E) (n = 16 neurons for each group). ***p < 0.001; †††p < 0.001 by two-tailed unpaired t test (D) or one-way ANOVA followed by non-parametric statistical analysis (F). All data are presented as mean ± SEM.
See also Figures S2 and S3.
Figure 5. TGF-β-Mediated Phosphorylation of Usp9X Regulates Spine Morphogenesis through Interaction with Ankyrin-G.

(A and B) SIM images of a spine (A) from FLAG-Usp9XWt in Figure 4C. Lower panels show ratiometric images and colocalization (white). Scale bar, 0.5 μm. 3D reconstruction (B) of a PLA-SIM image of a spine from TGF-β (+). Scale bar, 1 μm.
(C–F) Correlation plots of the area of FLAG-Usp9X versus HA-AnkG nanodomains (C) and the area of FLAG-Usp9X (D) or HA-AnkG (E) nanodomains versus the area of PLA nanodomains and spine head area versus the area of PLA nanodomains (F) from the HA-AnkG + FLAG-Usp9XWt + TGF-β(−) group or the HA-AnkG + FLAG-Usp9XWt + TGF-β(+) group. Two-tailed non-parametric Spearman correlation was performed, and the 90% confidence interval (CI) was considered for equivalence tests. TGF-β (−): vehicle; TGF-β (+): 20 ng/mL TGF-β for 1 h.
Phosphorylation of Usp9X Is Required for TGF-β-Dependent Spine Enlargement
To better understand the role of TGF-β and phosphorylation of the Usp9X peptidase domain in regulating spine head size and spine density, we replaced endogenous Usp9X with the wild-type or S3A Usp9X peptidase domain and assessed the effect of TGF-β treatment on spines. We transfected neurons with either control or shUsp9X and co-transfected with GFP-tagged Usp9XWt or Usp9XS3A. Overexpression of Usp9XS3A significantly decreased spine head size and spine density in control shRNA-transfected neurons (Figure 6A). In addition, overexpression of Usp9XWt rescued the effect of shUsp9X on spine head size and spine density; however, overexpression of Usp9XS3A could not rescue the spine phenotype. The effect of TGF-β was prevented by overexpression of Usp9XS3A (Figures 6B and 6C). These data suggest that phosphorylation of the peptidase domain of Usp9X is important for regulating spine head size and generating new spines in response to TGF-β.
Figure 6. Replacement of RNAi-Resistant GFP-Usp9XWt, but Not GFP-Usp9XS3A, Induces TGF-β-Dependent Spine Enlargement.

(A) Confocal images of neurons transfected with control RNAi (control) or shUsp9X and GFP-Usp9XWt or GFP-Usp9XS3A (green) and subsequently treated with vehicle or TGF-β (20 ng/mL) for 1 h in a dendritic region outlined by tRFP (red) expression. Scale bar, 5 μm.
(B and C) Spine head area (B) and density (C) in control or shUsp9X and GFP-Usp9XWt (black bars) or GFP-Usp9XS3A (red bars) with vehicle (plain pattern) or TGF-β (comb pattern) (n = 12–18 neurons for each group). Vehicle versus TGF-β: *p < 0.05, ***p < 0.001; GFP-Usp9XWt versus GFP-Usp9XS3A: ††p < 0.01, †††p < 0.001 by one-way ANOVA followed by non-parametric statistical analysis. TGF-β (−), vehicle; TGF-β (+), 20 ng/mL TGF-β for 1 h. All data are presented as mean ± SEM.
DISCUSSION
Based on these results, we propose that TGF-β induces spine growth by enhancing the interaction between Usp9X and ankyrin-G, stabilizing it through deubiquitination in dendritic spine heads. Using SIM, we mapped the subsynaptic localization and TGF-β-regulated interaction of ankyrin-G and Usp9X and assessed their relationship to spine morphology. We identify TGF-β-dependent phosphorylation of Usp9X as an upstream regulatory mechanism of ankyrin-G and spine stability.
We show that TGF-β regulates the Usp9X-ankyrin-G module and spine morphology. Usp9X has been shown to mediate TGF-β signaling by deubiquitinating Smad4 proteins, thereby modulating their transcription factor properties in the nucleus (Dupont et al., 2009). However, here, we report a novel signaling pathway involving TGF-β and Usp9X; Usp9X is a direct downstream target of TGF-β-dependent phosphorylation, which is required for its interaction with the important postsynaptic protein ankyrin-G.
TGF-β signaling has been extensively implicated in neuronal functions, including hippocampal development (Stegeman et al., 2013), axonal specification (Yi et al., 2010), developmental synaptic refinement (Bialas and Stevens, 2013), and dendritic growth and excitatory-inhibitory synaptic balance in dopaminergic neurons (Luo et al., 2016), as well as hippocampal synaptic plasticity and memory (Caraci et al., 2015) and reversal learning (Luo et al., 2016). Loss of TGF-β signaling induces improper CNS development, including neuronal degeneration and deficits in both glutamatergic and GABAergic synapses (Heupel et al., 2008). However, TGF-β has not been implicated in dendritic spine plasticity.
We find that TGF-β signaling induces phosphorylation of Usp9X within the peptidase domain, strengthening its interaction with ankyrin-G. Phosphorylation of key interacting partners in response to extracellular signaling may fine-tune ankyrin-G function at synapses. Interestingly, alteration of TGF-β signaling has been reported in SZ and ASD patients (Ashwood et al., 2008; Kasherman et al., 2020; Pietersen et al., 2014). Moreover, mutations (D1606E and P1613T) within the peptidase domain in close proximity to serine 1,600/1,609, have been identified in patients with mild developmental delay and speech delay (Johnson et al., 2020).
Our SIM imaging reveals, for the first time, the nanoscale spatial organization of the regulation of a deubiquitination pathway in spines by an extracellular signal. SIM has been instrumental in showing that ankyrin-G is localized to perisynaptic regions and spine necks (Smith et al., 2014). Here the combination of PLA and SIM provides the unprecedented ability to examine phosphorylation-dependent protein-protein interactions in situ at the nanoscale level. In particular, we show that the phosphorylation-dependent interaction of ankyrin-G with Usp9X occurs primarily in a region of the spine head facing the interior of the spine and not in the neck. In addition, SIM and PLA/SIM allowed accurate quantitative analysis of the relationships between the size of TGF-β-regulated deubiquitinase-substrate interaction nanodomains and spine morphological parameters.
We previously found that the absence of Usp9X in mice drastically reduces the levels of multiple ANKRD proteins, including Shank2, Shank3, and ankyrin-B, indicating that Usp9X is likely the major DUB regulating their stability during this developmental period (Yoon et al., 2020). Taken together with the present data, this suggests a mechanism of convergence between multiple neurodevelopmental risk factors, whereby one paracrine signaling molecule with important roles in brain development and disease regulates the interaction of two other important risk factors independently implicated in neurodevelopmental disorders.
Multiple studies have shown that TGF-β and its related family members have neuroprotective actions in several models of brain injury and degeneration (Krieglstein et al., 2011), suggesting potentially therapeutic synaptotrophic actions. For example, TGF-β protects synapses against Aβ oligomers in an Alzheimer’s disease model (Diniz et al., 2017); it protects neurons in Huntington’s disease (Hughes et al., 1999), Parkinson’s disease (Gonzalez-Aparicio et al., 2010), and ischemia (Wu et al., 1999) and promotes nigrostriatal dopaminergic neuron survival (Krieglstein et al., 1995). By enhancing the efficiency of Usp9X-mediated deubiquitination toward its substrates, leading to stabilization of ankyrin-G and perhaps other ANKRD proteins, TGF-β could be developed into a therapeutic strategy to recover synaptic deficits in neurodevelopmental disorders.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Penzes (p-penzes@northwestern.edu).
Materials Availability
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Dissociated cultures of primary cortical neurons were prepared from P0 C57BL/6J (The Jackson Laboratory) pups. All procedures were approved by Northwestern University’s Animal Care and Use Committee and were in compliance with the National Institutes of Health standards. Brains were dissected in ice-cold Leibowitz’s L-15 media with penicillin/streptomycin, and cortical tissue isolated, digested with 0.25% trypsin-EDTA solution at 37°C for 10 min, and mechanically dissociated in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1.4 mM L-glutamine, and 6.0 g/L glucose. Cortical neurons were plated at 320,000 cells per 18 × 18 mm coverslip precoated with 50 μg/ml poly-D-lysine (Sigma) and 2 μg/ml laminin (Sigma). Neuronal cultures were maintained at 37°C in 5% CO2 in Neurobasal media supplemented with B27 and GlutaMax-1 and penicillin/streptomycin. Neurons were transfected at day in vitro (DIV) 21 with Lipofectamine 2000 (Invitrogen). 4 μg total DNA and Lipofectamine 2000 were diluted in DMEM + HEPES (10 mM), mixed thoroughly together, and incubated for 20–30 minutes at 37°C before adding to cultured cells. Following transfection, neurons were supplanted in antibiotic-containing feeding media containing half conditioned and half fresh media, and allowed to express constructs for 4 days.
METHOD DETAILS
Plasmid construction
3XHA-AnkG (EX-Mm25668-M06) and 3XFlag-Usp9X (EX-Mm24322-M12) were purchased from GeneCopoeia. GFP-AnkG 480 was generously given by Dr. Vann Bennett (Duke University School of Medicine) and the construct was cloned into pReceiver-M06 (GeneCopoeia). shRNA constructs were purchased from Origene, in the pGFP-C-RS vector with a turboGFP element or the pRFP-C-RS vector with a turboRFP element to enable identification of transfected cells. The N-terminal fragment of 3XHA-AnkG (amino acids 1–807) and the peptidase domain fragment of 3XFlag-Usp9X (amino acids 1555–1958; 3XFlag-Usp9X1555−1958) were generated as described previously (Yoon et al., 2020). The 3XFlag-Usp9X1555−1958 triple serine mutant (S1593A, S1600A, S1609A; S3A) was generated using a QuickChange Site-Directed Mutagenesis Kit (Agilent) as per the manufacturer’s instructions. All constructs were verified by sequencing (Genewiz).
SIM imaging
Imaging and reconstruction parameters were empirically determined with the assistance of the expertise in the Nikon Imaging Center at Northwestern University. Acquisition was set to 10MHz, 14 bit with EM gain and no binning. Auto exposure was kept between 100–300ms and the EM gain multiplier restrained below 300. Conversion gain was held at 1x. Laser power was adjusted to keep look-up tables (LUTs) within the first quarter of the scale (< 4000). Reconstruction parameters (0.96, 1.19, and 0.17) were kept consistent across experiments and imaging sessions. Resolution of images was validated with full-width half maximum (FWHM) measurements of a small structure within the image. Three-dimensional reconstructions of dendritic spines were created in the Nikon Elements Software by merging of the channels.
Immunocytochemistry
Cells were fixed for 10 min in 4% formaldehyde in phosphate-buffer saline (PBS) at 4°C. After 3 PBS washes of 5 min each, fixed neurons were permeabilized and blocked simultaneously in PBS containing 1% bovine serum albumin (BSA) and 0.3% Triton X-100 for 1 h at room temperature. Primary antibodies were added in PBS containing 1% BSA and 0.3% Triton X-100 overnight at 4°C, followed by 3 × 10 min washes in PBS. Secondary antibodies were incubated for 1 h at room temperature, also in 1% BSA and 0.3% Triton X-100 in PBS. Three further washes (5 min each) were performed before coverslips were mounted using Fluorescent Mounting Medium (Dako).
Confocal microscopy
Confocal images of immunostained neurons were obtained with a Nikon C2+ confocal microscope. Images of neurons were taken using the 63x oil-immersion objective (NA = 1.4) as a z series of 8–10 images, averaged 2 times, taken at 0.4 μm intervals, with 1024×1024 pixel resolution. Detector gain and offset were adjusted in the channel of cell fill (turboRFP or turboGFP or mCherry) to include all spines and enhance edge detection.
Image analysis from confocal microscopy and SIM
Colocalization highlighter images (ankyrin-G, Usp9X, TGFβRI, and TGFβRII) and Manders’ colocalization coefficients were determined in ImageJ with the use of the MBF set of plugins. ImageJ recalculates the pixel values of images between 0–65535 for 16-bit images and the ‘Auto’ option in the Brightness/Contrast was chosen to apply to the optimal contrast of images. To outline a region of interest (ROI) along dendrites or cell bodies, images were auto-thresholded (threshold = (average background + average objects)/2) and converted into binary images. Total immunofluorescence intensity for each cluster was measured automatically. For dendritic spine quantification, two-dimensional, background-subtracted maximum projection reconstructions of images for dendritic spine morphometric analysis (area), and quantification of spine linear density (# of spines/10 μm dendritic length) were performed using ImageJ software. A threshold was applied to the maximum projection images to include all detectable spines, and regions along dendrites containing dendritic spines were manually traced to enclose spines but not dendritic shafts or other structures. Dendritic spine “objects,” restricted to objects with areas greater than 0.1 μm2, were automatically detected, and the area of each spine head was measured. Two dendritic branches (approximately 100 μm) of each neuron were analyzed. Only spines on secondary and tertiary apical dendrites were measured to reduce variability. Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters. To analyze the number of PLA puncta, ImageJ examined the image of 32.38 × 32.38 μm area taken by SIM. All images transferred to an 8-bit format and applied a threshold option as a range 0 and 30. Next, thresholded images processed convert to mask and watershed with a binary option. Finally, an option for “analyze particles” was selected to count the number of PLA puncta automatically. Counted puncta were separately recorded inside and outside of transfected neurons and divided by analyzed area (punta number / μm2).
Coimmunoprecipitation assays
Coimmunoprecipitation assays were performed as described in Yoon et al. (2020) with modifications. For assays of overexpressed AnkG and Usp9X constructs, HEK293T cells were transfected with 3XHA-AnkG1−807 and 3XFlag-Usp9XWt or 3XFlag-Usp9XS3A using PEI transfection reagent (Sigma) in 100-mm dishes. Cells were treated in lysis buffer (20 mM Tris pH 7.5, 1% Triton X-100, 150 mM NaCl, 1 mM EGTA, 1mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) with protease inhibitor cocktail), and equal protein aliquots of cell lysates (700 μg) were incubated with 3 μg of antibody overnight at 4°C followed by the addition of 30 μl of Sepharose A/G slurry for 4 h at 4°C. The complexes were washed three times with washing buffer. Samples were then denatured (5 min at 95°C). The immunoprecipitated proteins were separated with 10% SDS-PAGE and detected by western blotting.
Pulldown assay
Recombinant protein was expressed in E. Coli, and purified by nickel affinity as previously described (Yoon et al., 2020). Protein was assessed to be > 95% pure by Coomassie stain, and protein was snap-frozen in liquid nitrogen for storage. HEK293T cells were transfected with GFP or AnkG1−808, and expressed for 3 days before lysis in PBS + 0.5% Triton X-100. Cells were scraped down, and incubated 1 hour before centrifugation (5,000 × g, 5 mins) to remove insoluble material. Nickel beads were prewashed in lysis buffer and incubated with lysis buffer or saturated with recombinant Usp9X (0.8 mg on 50 ul beads), before dilution of Triton X-100 (2.5x dilution in PBS + 30 mM imidazole, final = 12.5 mM imidazole, 0.2% Triton X-100). Lysates were split into each condition and incubated with nickel beads for 2 hours. Beads were pelleted (1000 × g, 1 min) and washed with PBS + 30 mM imidazole for 30 minutes two times. Final elution was performed using PBS + 2 M NaCl and brief vortexing. Samples were loaded on a 4%–20% SDS gel and ECL was performed.
Proximity ligation assay (PLA)
Cortical neurons plated on coverslips were transfected with 3XHA-AnkG and 3XFlag-Usp9XWt or 3XFlag-Usp9XS3A at DIV 14. At 3 days post-transfection, the transfected cells were treated with 20 ng/ml of TGFβ (eBioscience) for 1 h before fixation. HEK293T cells were transfected with 3XHA-AnkG1−807 and 3XFlag-Usp9XS3A using PEI transfection reagent. At 24 h post-transfection, the transfected cells were treated with 20 ng/ml of TGFβ (eBioscience; 14–8342-62) for 1 h before fixation. All procedures of PLA were performed according to the manufacturer’s instructions (Sigma). To confirm the transfected cells, mouse 488 conjugated-Flag antibody and rabbit 568 conjugated-HA antibody were used. For negative control, in situ PLA probe anti-mouse MINUS was replaced by anti-goat MINUS. To detect PLA signal, far red reagent (Sigma, DUO92013) was used.
Homology modeling
Human Usp9X (amino acids 1555–1975) was aligned to crystal structure (PDB: 5WCH) using Maestro multiple sequence viewer (Schrödinger, LLC), as described previously (Johnson et al., 2020). A homology model was generated using Maestro Bioluminate (Schrödinger, LLC) using energy minimization to model flexible loops absent from 5WCH. Images were prepared using PyMol (Schrödinger, LLC). > 97% amino acids are within accepted torsion angles.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed with GraphPad Prism 8. Two-sample comparisons were performed using two-tailed unpaired Student’s t test or two-tailed non-parametric Spearman correlation, and multiple comparisons were made using one-way ANOVA followed by non-parametric statistical analysis or two-way ANOVA followed by a Bonferroni test. Statistical details are given in each figure legend. Bar graphs are displayed as mean ± SEM. P values < 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-AnkG | NeuroMab Facility | Cat#75-146; RRID: AB_10673030 |
| Goat polyclonal anti-AnkG | Santa Cruz Biotechnology | Cat#sc-31778; RRID: AB_2289736 |
| Rabbit polyclonal anti-Usp9X | Abcam | Cat#ab19879; RRID: AB_470300 |
| Mouse monoclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Mouse monoclonal anti-HA (clone HA-7) | Sigma-Aldrich | Cat#H3663; RRID: AB_262051 |
| Rabbit polyclonal anti-HA | Enzo Life Sciences | Cat#ADI-MSA-106; RRID: AB_10615792 |
| Rabbit polyclonal anti-HA | Abcam | Cat#ab20084; RRID: AB_445319 |
| Mouse monoclonal anti-His | Millipore | Cat#05-949; RRID: AB_492660 |
| Mouse monoclonal anti-β-actin (clone AC-74) | Sigma-Aldrich | Cat#A2228; RRID: AB_476697 |
| Rabbit polyclonal anti-TGFβRI | Abcam | Cat#ab31013; RRID: AB_778352 |
| Rabbit polyclonal anti-TGFβRII | Abcam | Cat#ab186838; RRID: AB_2728775 |
| Rabbit monoclonal anti-p-Smad2 (Ser465/467) | Cell signaling Technology | Cat#3101; RRID: AB_331673 |
| Mouse monoclonal anti-Smad2 | Abcam | Cat#ab119907; RRID: AB_10899446 |
| Rabbit polyclonal anti-phosphoserine | Abcam | Cat#ab9332; RRID: AB_307184 |
| Mouse monoclonal anti-Flag-488 (clone M2) | Abcam | Cat#ab117505; RRID: AB_10972518 |
| Rabbit polyclonal anti-HA-568 | Synaptic System | Cat#245 003C3; RRID: AB_2619954 |
| Chicken polyclonal anti-mCherry | Abcam | Cat#ab205402; RRID: AB_2722769 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970; RRID: AB_300798 |
| Rabbit polyclonal anti-GFP | Thermo Fisher Scientific | Cat#A10260; RRID: AB_2534022 |
| Goat anti-chicken 488 | Thermo Fisher Scientific | Cat#A-11039; RRID: AB_2534096 |
| Goat anti-chicken 568 | Thermo Fisher Scientific | Cat#A-11041; RRID: AB_2534098 |
| Donkey anti-rabbit 488 | Thermo Fisher Scientific | Cat#A-21206; RRID: AB_2535792 |
| Goat anti-mouse 568 | Thermo Fisher Scientific | Cat#A-11031; RRID: AB_144696 |
| Bacterial and Virus Strains | ||
| E. coli HST08 | TaKaRa | Cat#636766 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse TGFβ1 | eBioscience | Cat#14-8342-62 |
| Critical Commercial Assays | ||
| Duolink® In Situ PLA® Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat#DUO92004 |
| Duolink® In Situ PLA® Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat#DUO92002 |
| Duolink® In Situ PLA® Probe Anti-Goat MINUS | Sigma-Aldrich | Cat#DUO92006 |
| Duolink® In Situ Detection Reagents FarRed | Sigma-Aldrich | Cat#DUO92013 |
| In-Fusion HD Cloning Kit | Clontech | Cat#638910 |
| QuickChange Site-Directed Mutagenesis Kit | Agilent Technologies | Cat#200516 |
| Experimental Models: Cell Lines | ||
| HEK293T/17 cells | ATCC | Cat#CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | #000664 |
| Oligonucleotides | ||
| AnkG480 F: TGA AGG AAT TCG GTA CCA TGG CTC ATG CCG CCT CCC AGTTA | This paper | N/A |
| AnkG480 R: GCG GCC GCA CTC GAG CTA GTG GGC TTT CTT CTC CAC ATT CCG | This paper | N/A |
| Recombinant DNA | ||
| pEZ-3XHA-AnkG | GeneCopoeia | Cat# EX-Mm25668-M06 |
| pEZ-3XFlag-Usp9X | GeneCopoeia | Cat#EX-Mm24322-M12 |
| pRFP-C-RS Control | Origene | Cat#TR30015 |
| pRFP-C-RS shUsp9X | Origene | Cat#TF510959 |
| pGFP-C-RS shAnkG | Smith et al., 2014 | https://www.cell.com/neuron/abstract/S0896-6273(14)00908-8 |
| pmCherry-C1 | Clontech | Cat#632524 |
| pEZ-3XHA-AnkG1–807 | Yoon et al., 2020 | HA-AnkG1–807 |
| pEZ-3XFlag-Usp9X1555–1958 | Yoon et al., 2020 | Flag-Usp9XWt |
| pEGFP-Usp9X1555–1958 | Yoon et al., 2020 | GFP-Usp9XWt |
| pEZ-3XFlag-Usp9X1555–1958 S1593;1600;1609A | Yoon et al., 2020 | Flag-Usp9XS3A |
| pEGFP-Usp9X1555–1958 S1593;1600;1609A | Yoon et al., 2020 | GFP-Usp9XS3A |
| pMCSG53-6XHis-Usp9X1547–1962 | Yoon et al., 2020 | His-Usp9X1547–1962 |
| pEZ-3XHA-AnkG480 | This paper | HA-AnkG480 |
| Software and Algorithms | ||
| FIJI (ImageJ) | NIH | https://fiji.sc/ |
| NIS-Elements v4.51.00 | Nikon | https://www.nikon.com/products/microscope-solutions/support/index.htm |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Maestro multiple sequence viewer | Schrödinger | https://www.schrodinger.com/ |
| Maestro Bioluminate | Schrödinger | https://www.schrodinger.com/ |
| PyMOL V1.8.2.0 | Schrödinger | https://pymol.org/2/ |
| Image Lab 3.0 | Bio-Rad Laboratories | https://www.bio-rad.com/en-us/product/image-lab-software |
Highlights.
Usp9X regulates ankyrin-G stability downstream of TGF-β to maintain spines
TGF-β-induced phosphorylation of Usp9X increases its interaction with ankyrin-G
In situ PLA/SIM reveal the spatial regulation of Usp9X/ankyrin-G interactions in spines
TGF-β is a potential therapeutic lead to correct synaptic deficits
ACKNOWLEDGMENTS
This work was supported by NIH grant R01-MH107182 to P.P. We thank the NU Nikon Center for Advanced Microscopy for use of the N-SIM instrument. Special thanks to Dr. Marc Forrest for proofreading and Ahreum Han for a graphical abstract.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107685.
REFERENCES
- Akhurst RJ, and Hata A (2012). Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen RL, Croen LA, Ozonoff S, Pessah IN, and Van de Water J (2008). Decreased transforming growth factor beta1 in autism: a potential link between immune dysregulation and impairment in clinical behavioral outcomes. J. Neuroimmunol. 204, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, and Healy J (2008). Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol. Med. 14, 28–36. [DOI] [PubMed] [Google Scholar]
- Bialas AR, and Stevens B (2013). TGF-b signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bousman CA, Chana G, Glatt SJ, Chandler SD, Lucero GR, Tatro E, May T, Lohr JB, Kremen WS, Tsuang MT, and Everall IP (2010). Preliminary evidence of ubiquitin proteasome system dysregulation in schizophrenia and bipolar disorder: convergent pathway analysis findings from two independent samples. Am. J. Med. Genet. B. Neuropsychiatr. Genet 153B, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraci F, Gulisano W, Guida CA, Impellizzeri AA, Drago F, Puzzo D, and Palmeri A (2015). A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci. Rep. 5, 11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier AE, Djakovic SN, Salehi A, Wilson SM, Masliah E, and Patrick GN (2009). Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 29, 7857–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Matias I, Morgado J, Bérgamo Araujo AP, Melo HM, Seixas da Silva GS, Alves-Leon SV, de Souza JM, Ferreira ST, et al. (2017). Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer’s disease model. J. Neurosci. 37, 6797–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, et al. (2009). FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136, 123–135. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. ; Wellcome Trust Case Control Consortium (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Parnell E, and Penzes P (2018). Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 19, 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, and Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Aparicio R, Flores JA, and Fernandez-Espejo E (2010). Antiparkinsonian trophic action of glial cell line-derived neurotrophic factor and transforming growth factor β1 is enhanced after co-infusion in rats. Exp. Neurol. 226, 136–147. [DOI] [PubMed] [Google Scholar]
- Heupel K, Sargsyan V, Plomp JJ, Rickmann M, Varoqueaux F, Zhang W, and Krieglstein K (2008). Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev. 3, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein R, Parry DA, Nalbach L, Logan CV, Strom TM, Hartill VL, Carr IM, Korenke GC, Uppal S, Ahmed M, et al. (2015). HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J. Med. Genet. 52, 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PE, Alexi T, Williams CE, Clark RG, and Gluckman PD (1999). Administration of recombinant human Activin-A has powerful neurotrophic effects on select striatal phenotypes in the quinolinic acid lesion model of Huntington’s disease. Neuroscience 92, 197–209. [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, and Zhang H (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA, Roca LT, Vulto-van Silfhout AT, Nijhof B, Kramer JM, et al. (2013). Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum. Mol. Genet. 22, 1960–1970. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, and Greenough WT (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044. [DOI] [PubMed] [Google Scholar]
- Johnson BV, Kumar R, Oishi S, Alexander S, Kasherman M, Vega MS, Ivancevic A, Gardner A, Domingo D, Corbett M, et al. (2020). Partial loss of USP9X function leads to a male neurodevelopmental and behavioral disorder converging on transforming growth factor beta signaling. Biol. Psychiatry 87, 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, and Neubert TA (2004). Identification and verification of novel rodent postsynaptic density proteins. Mol. Cell. Proteomics 3, 857–871. [DOI] [PubMed] [Google Scholar]
- Kasherman MA, Premarathne S, Burne THJ, Wood SA, and Piper M (2020). The ubiquitin system: a regulatory hub for intellectual disability and autism spectrum disorder. Mol. Neurobiol. 57, 2179–2193. [DOI] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, and Benes FM (2014). Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Fischer WH, and Unsicker K (1995). TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 14, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein K, Zheng F, Unsicker K, and Alzheimer C (2011). More than being protective: functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci. 34, 421–429. [DOI] [PubMed] [Google Scholar]
- Luo SX, Timbang L, Kim JI, Shang Y, Sandoval K, Tang AA, Whistler JL, Ding JB, and Huang EJ (2016). TGF-β signaling in dopaminergic neurons regulates dendritic growth, excitatory-inhibitory synaptic balance, and reversal learning. Cell Rep. 17, 3233–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, and Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietersen CY, Mauney SA, Kim SS, Lim MP, Rooney RJ, Goldstein JM, Petryshen TL, Seidman LJ, Shenton ME, McCarley RW, et al. (2014). Molecular profiles of pyramidal neurons in the superior temporal cortex in schizophrenia. J. Neurogenet. 28, 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, Detera-Wadleigh SD, Akula N, Gupta A, Kassem L, Steele J, Pearl J, Strohmaier J, Breuer R, Schwarz M, et al. ; NIMH Genetics Initiative Bipolar Disorder Consortium (2009). Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol. Psychiatry 14, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, and Penzes P (2018). Ankyrins: roles in synaptic biology and pathology. Mol. Cell. Neurosci. 91, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Kopeikina KJ, Fawcett-Patel JM, Leaderbrand K, Gao R, Schürmann B, Myczek K, Radulovic J, Swanson GT, and Penzes P (2014). Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 84, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, Mattheisen M, Wang Y, Coleman JRI, Gaspar HA, et al. ; eQTLGen Consortium; BIOS Consortium; Bipolar Disorder Working Group of the Psychiatric Genomics Consortium (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman S, Jolly LA, Premarathne S, Gecz J, Richards LJ, Mackay-Sim A, and Wood SA (2013). Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFβ-mediated axonogenesis. PLoS ONE 8, e68287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Rives ML, Urizar E, Piskorowski RA, Vishwasrao HD, Castrillon J, Schmauss C, Slättman M, Gullberg M, and Javitch JA (2011). Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotechniques 51, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roessel P, Elliott DA, Robinson IM, Prokop A, and Brand AH (2004). Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 119, 707–718. [DOI] [PubMed] [Google Scholar]
- Wu DD, Lai M, Hughes PE, Sirimanne E, Gluckman PD, and Williams CE (1999). Expression of the activin axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 835, 369–378. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Barnes AP, Hand R, Polleux F, and Ehlers MD (2010). TGF-βeta signaling specifies axons during brain development. Cell 142, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Parnell E, Kasherman M, Forrest MP, Myczek K, Premarathne S, Sanchez Vega MC, Piper M, Burne THJ, Jolly LA, et al. (2020). Usp9X controls ankyrin-repeat domain protein homeostasis during dendritic spine development. Neuron 105, 506–521.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon request.