

Abstract
One of the many ways by which bacteria control gene expression is through cis-acting regulatory mRNA elements called riboswitches. By specifically binding to small molecules or metabolites and pairing the binding event to an RNA structure change, riboswitches link a metabolic input to a transcriptional or translational output. For over a decade, isothermal titration calorimetry (ITC) has been used to investigate how riboswitches interact with small molecules. We present methods for assaying RNA-ligand interactions using ITC and analyzing resulting data to estimate thermodynamic parameters associated with binding.
Keywords: riboswitch, isothermal titration calorimetry, RNA folding, heat capacity
Introduction
In the course of investigating the functions of biological macromolecules, it is often desirable to measure binding interactions under tightly controlled conditions in vitro. Of the many possible experimental routes, isothermal titration calorimetry (ITC) is a label-free method for obtaining the binding event’s thermodynamic parameters, including association constant (Ka), binding enthalpy (ΔH), and stoichiometry (n). From these, the Gibbs free energy (ΔG) and binding entropy (ΔS) are also derived. The sample requirements for ITC are often much larger than other common approaches (e.g., fluorescence), and the throughput is low, with a single titration taking about 1–2 hours. To lessen the burden of these drawbacks, performing other experiments in parallel with ITC can in part mitigate sample and time loss and aid the interpretation of results. However, the advantage of using ITC is that one may observe in solution any interaction characterized by a relatively modest binding enthalpy without having to perturb the system by introducing a label and properly characterize and develop the assay detecting that label. Moreover, for the researcher seeking crystals and high-resolution structural information, preparing large amounts of RNA is usually not the rate-limiting step to experimental success.
For riboswitches, which are RNAs that regulate gene expression through ligand binding (Fig. 1), ITC has proven to be a robust method for analyzing RNA-ligand interactions [1,2]. Riboswitches function by specifically binding a metabolite, which promotes or stabilizes one riboswitch conformational state, often coupled to a change in mRNA transcript abundance or translation efficiency (reviewed in [3–6]). Although in vitro transcription or translation assays can directly measure the effects of a ligand binding on riboswitch modulation of gene expression, binding specificity is best dissected through direct binding assays. This is particularly true when the exact mechanism by which gene regulation is achieved is either unclear or complex [7,8,6]. The portion of the riboswitch sufficient for specific ligand binding is referred to as the aptamer domain, and is the most commonly chosen subject for ITC experiments.
Figure 1.
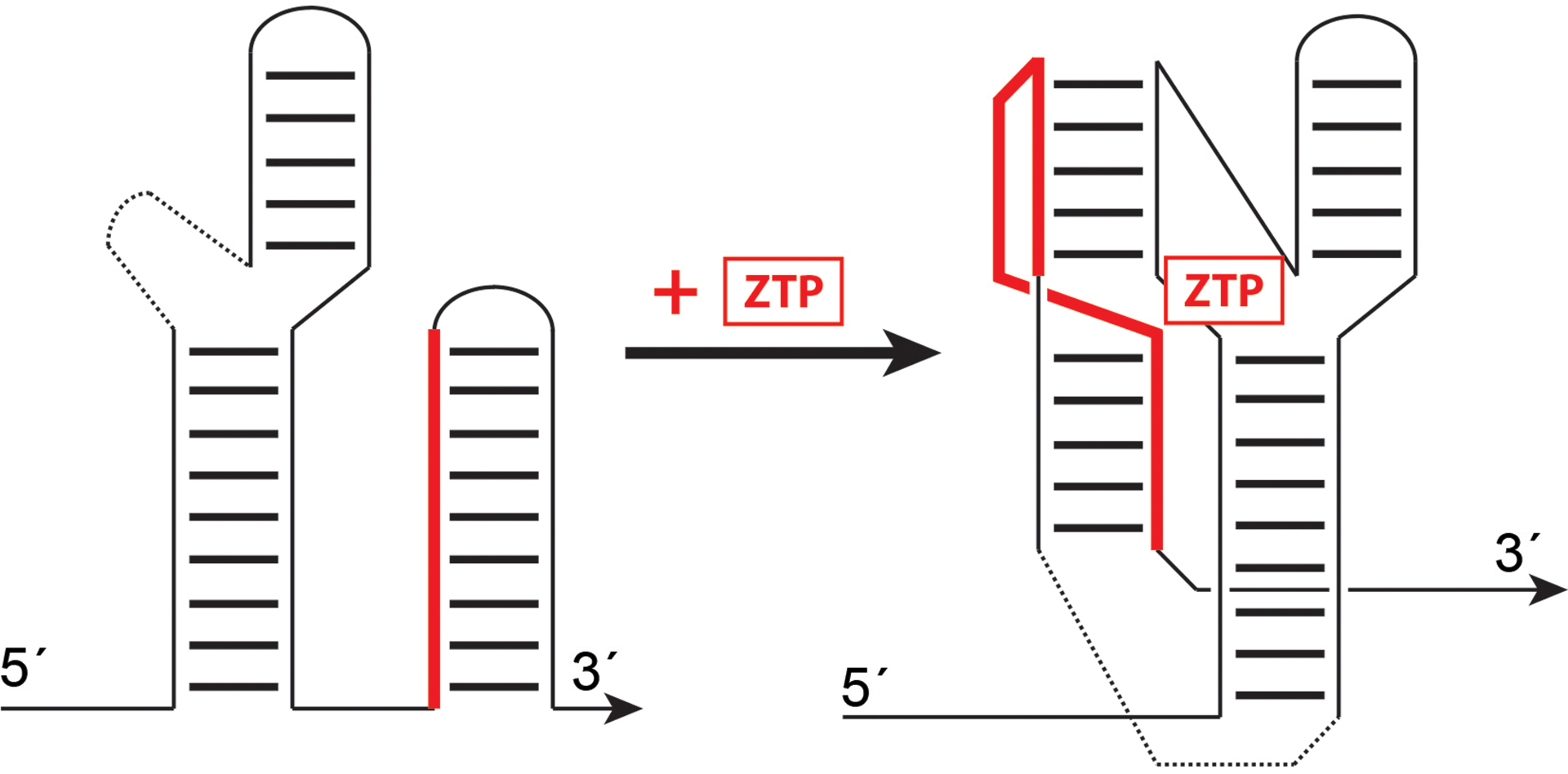
Found primarily in the 5´ untranslated regions of bacterial mRNAs, riboswitches alter gene expression in response to direct and specific small molecule binding. As exemplified by the ZTP riboswitch, the RNA exists in one conformational state in which a switch sequence (solid red) pairs to form a stable hairpin linked to transcription or translation regulation of a downstream 3´ open reading frame (not shown). In the presence of ZTP, an alternative pairing of the switch sequence is favored, resulting in an alternative secondary structure that binds ZTP and disfavors downstream hairpin formation.
The ITC methods described herein are suited to studying interactions between small molecule ligands and their RNA binders, as exemplified by our recent work on the Bacillus subtilis cyclic diadenosine monophosphate (c-di-AMP) riboswitch [9] and Fusobacterium ulcerans ZTP riboswitch [10]. Thus, these ITC protocols are meant to inform and interpret other experimental findings in the course of structural studies. In these experiments, what is actually measured by ITC (i.e., the response) is the power required to maintain the same temperature between two adiabatic cells, one containing a sample and another containing a reference (e.g., water). Upon titration of one binding partner from a syringe into another in the sample cell, heat is released (exothermic) or absorbed (endothermic), which affects the amount of power (reported in μcal/sec) required to keep the sample cell at the same temperature as the reference cell. A more thorough description of calorimeters is described elsewhere [11]. The response power (Q) is related to the binding enthalpy ΔH by a number of instrumental parameters, including the volume of the cell, V, as described by:
where f(Ka, ML, MR) is the amount of RNA-ligand complex, which can be expressed as a function of the total ligand concentration (ML), total RNA concentration (MR), and Ka, as previously described and clearly derived for 1:1 binding [12]. By ITC, riboswitch-ligand interactions are generally characterized by binding enthalpies of approximately −40 to −20 kcal/mol at temperatures ranging from 15–37 °C [13], requiring 5–100 μM RNA in the sample cell, which is ~200 μL for the commonly used MicroCal iTC200 calorimeter.
Materials
2.1. Instrumentation
iTC200 (Microcal-Malvern) calorimeter, or equivalent device
C1000 Thermal Cycler (Biorad), equivalent thermal cycler, or heat blocks
NanoDrop, UV-Vis spectrophotometer (Thermo-Fisher Scientific), or equivalent device
Akta Purifier (GE Healthcare), or equivalent liquid chromatography system
2.2. Reagents
0.22 μm syringe-driven sterile filters (Millipore), 0.22 μm disposable bottle-top sterile filters (Corning), and 0.22 μm centrifugal filters (Millipore). All stocks, buffers, and RNA are filtered.
0.5 M HEPES (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) buffer stock, adjusted to pH 7.4 with KOH, filtered and stored at room temperature.
4 M KCl stock, stored at room temperature.
1 M MgCl2 stock, stored at room temperature.
10–100 mM stock of 5-aminoimidazole-4-carboxamide ribonucleotide monophosphate (ZMP) (Sigma), brought up in diethylpyrocarbonate (DEPC) treated water (see Note 1), and solubilized by addition of 5 M KOH.
In vitro transcribed RNA(s), purified and resuspended in DEPC-treated water (see Note 2)
ITC buffer: typically 25–50 mM HEPES, pH 7.4, 50–150 mM KCl, and 1–20 mM MgCl2, in ribonuclease (RNase) free water, conditions matching the RNA folding buffer. This buffer should be prepared as a 250–500 mL stock to be used throughout the experiment, sterile filtered, degassed, and stored at room temperature. It is extremely important to use the same exact preparation of ITC buffer throughout the experiment for preparing ligand and RNA stocks, washing the sample cell, etc.
2.3. Isothermal calorimetry experiments
Gastight ITC loading Hamilton syringe, 500 μL, ideally reserved solely for RNA experiments
RNase-free water, methanol, and detergent (i.e., Contrad 70) for washing and cleaning calorimeter cell and syringe between experiments.
Software: NITPIC [14] is used for the integration of thermograms and background subtraction, and SEDPHAT [15] is used for fitting, both of which are available as freeware. The SEDPHAT freeware companion program, GUSSI, can be used for preparation of publication-quality images. Alternatively, data can be fitted and plotted in Origin (OriginLab Corp.), PEAK-ITC (Malvern), or other commercially available software.
Methods
3.1. RNA refolding and sample application
The quality of prepared RNA is paramount to experimental success (see Note 2). Initial experimental goals should include monitoring the RNA before and after the titration to observe signs of cleavage due to RNase contamination and other changes in oligomeric state, including dimerization and aggregation.
RNA is refolded in ITC buffer (e.g., for the F. ulcerans ZTP riboswitch, 50 mM HEPES-KOH, pH 7.4, 150 mM KCl, and 10 mM MgCl2), which has been chosen to match empirically determined RNA refolding conditions (see Note 3).
RNA folding reactions are brought up to 300 μL with ITC buffer prior to loading into the calorimeter cell. To completely fill the 200 μL iTC200 sample cell without introducing bubbles, ~280 μL of the RNA sample is injected via a Hamilton syringe. The remaining ~20 μL of RNA solution are kept for post-ITC analysis to be used as a pre-titration control (see Note 4).
3.2. ITC experiments
3.2.1. Choice of cell and syringe solutions and concentrations
It is critical to choose the appropriate concentrations of RNA and ligand for performing a titration, which is sensitive to the choice of the parameter c [11], described by:
where MR is the total concentration of RNA in the cell and Kd is the apparent dissociation constant. If c is too small (c < 0.1), essentially no transition will be observed, and if c is too large (c > 1000), as for tight binding, an extremely steep transition will be observed, preventing calculation of Ka (see Note 5). Thus, a prior knowledge of Ka (to the nearest log) is desirable, as c should ideally be within 10–100. For measuring the interaction between riboswitches and small molecule ligands, the experimental setup is to inject ligand from the syringe into the cell containing riboswitch RNA. This arrangement minimizes the highest concentration of RNA necessary to perform the experiment, as it is easier technically to have the ligand at a higher concentration in the syringe, ML, and avoids aggregation at higher RNA concentrations. As a starting point, ML is chosen to be 10*MC, which results in a ~2:1 ratio of ligand:RNA by the end of the titration, assuming 40 μL ligand injected into a 200 μL cell for this instrument. Prior to experimental injections, a small (0.4 μL) initial volume is first injected and later discarded during analysis. All subsequent injections are fixed to be the same size, typically 1 to 2 μL. For the F. ulcerans ZTP riboswitch, which binds in a 1:1 stoichiometry to ZMP with a Kd of ~0.5 μM, MR is chosen to be 20 μM (i.e., c = 40), and ML is chosen to be 200 μM. These are also the chosen concentrations for riboswitch mutants, but titrations require higher cell and syringe concentrations for weaker binders. The n observed may be slightly less than unity, which indicates that a fraction of the RNA is misfolded (Fig. 2) or that the concentration is inaccurate. For the F. ulcerans ZTP riboswitch, size exclusion chromatography (SEC) revealed a large fraction of RNA present as dimer and higher oligomerization states [10]. In this case, post-titration SEC was useful in determining the source of the discrepancy by revealing RNA species eluting at larger elution volumes from the gel filtration column [10]. Purification of RNA by SEC virtually eliminated the incompetent fraction, as observed by a shift in the x-value of the titration inflection point to ~1:1 molar ratio (Fig. 2).
Figure 2.
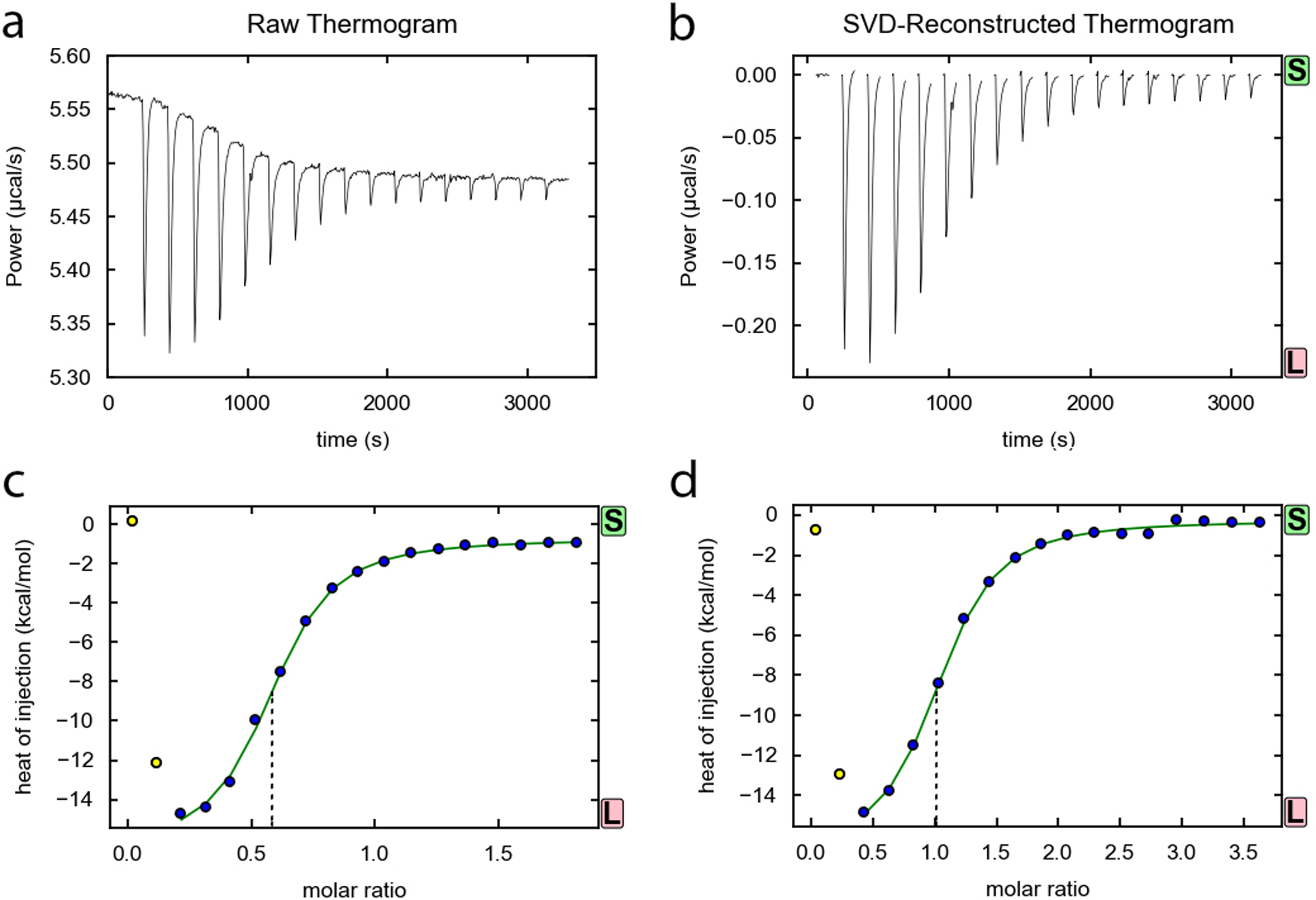
Example titration of ZTP riboswitch with ZMP. (a) Raw data prior to baseline correction and (b) data after baseline correction for a binding isotherm of 20 μM Fusobacterium ulcerans ZTP riboswitch titrated with 200 μM ZMP at 37 °C, as corrected by and viewed in NITPIC [14]. (c) Fit of binding isotherm prior to SEC purification and (d) of a separate titration after SEC purification of the peak corresponding to the monomeric RNA. Note the shift in the x-value of the inflection point (indicated by dashed lines) from (c) to (d) indicating the change in the fraction competent for binding. For (c), the incompetent fraction was ~45%. The first two points in each titration were excluded.
3.2.2. Varying temperature, Mg2+ concentration, and buffer
The goal is to measure the heats of binding evolved from the interaction between the RNA and ligand, but in principle, other heats may occur during titration. For example, the sources of the artifacts include buffer mismatch between the syringe and the cell, salt binding, or protonation/deprotonation. For this reason, the exact same buffer must be use in the ITC cell and syringe. Controls include titrating ligand into buffer and buffer into RNA, which may be subtracted from the actual titration in SEDPHAT prior to fitting data. Although these controls are often very small heats, similar to the heats observed from titrating water into water, occasionally they indicate the presence of contaminants (e.g., dimethyl sulfoxide or salt) in the ligand solution (Fig. 3).
Figure 3.
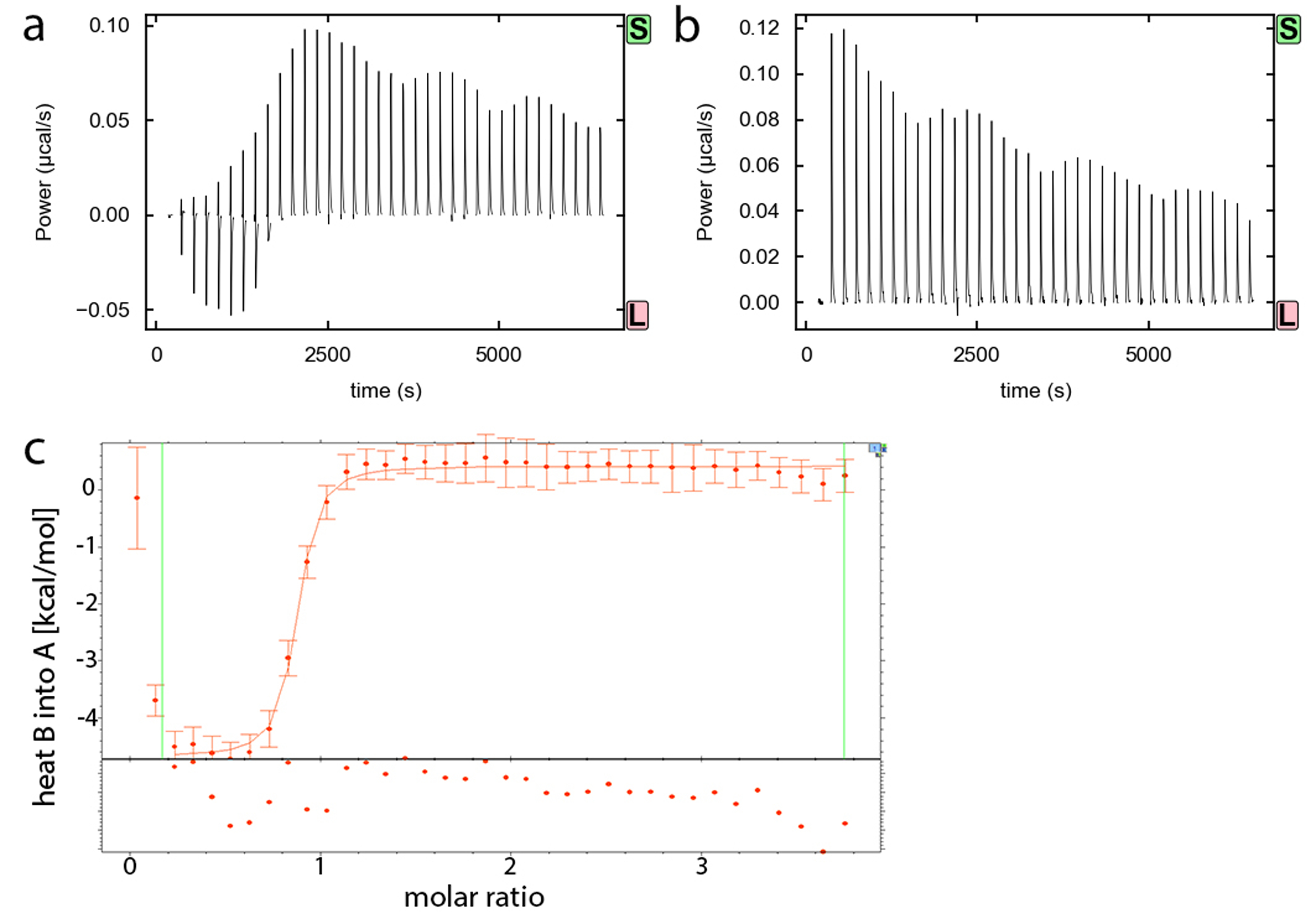
Example titration requiring background subtraction due to ligand heats of dilution. (a) Binding isotherm of Bacillus subtilis c-di-AMP riboswitch A10G mutant (20 μM) titrated with 400 μM c-di-AMP. (b) Binding isotherm of ITC buffer titrated with 400 μM c-di-AMP. (c) Background subtracted titration fit as viewed in SEDPHAT [15]. Vertical lines indicate the data range chosen for fitting, excluding the first two data points, and points below the axis are residuals.
For a new RNA, initial titrations should begin at 25 °C and 10 mM MgCl2. The first series of experiments should include titrations performed at other temperatures (e.g., 15–37 °C) and MgCl2 concentrations (0–20 mM MgCl2). For riboswitches, it is not uncommon to observe a dependence on MgCl2, typically reflecting the involvement of tertiary interactions or ligand binding.
Blank titrations of buffer into RNA and ligand into buffer are performed for background subtraction, if necessary.
3.2.3. Choice of other experimental parameters
A typical titration will begin with a 0.4 μL injection followed by 17 injections of 2 μL each. Substantially more injections than this (i.e., >25) can lead to larger experimental errors, as the signal to noise ratio will decrease for smaller heats. The minimum absolute heat of the second injection should be larger than +/−3 μcal, as recommended by Malvern. If the measured heat is smaller, either loading concentrations should be increased, or number of injections decreased. As few as 10 injections can be used to estimate binding reactions with smaller heats [16].
The time between injections is initially chosen to be 180 s, which in most cases should give enough time to re-equilibrate the power baseline. However, the baseline should be inspected and spacing adjusted when appropriate. Insufficient intervals of stable baseline between injections will complicate thermogram integration and affect the final data. To measure binding of cyclic diguanosine monophosphate (c-di-GMP) to the c-di-GMP riboswitch, binding was notably slower in the presence of kanamycin B, and 20 min spacing was required [17].
The reference power should be set depending on the expected heat of binding. For the typically exothermic heats of binding for riboswitches, for which exceeding the instrumental power range is not a concern, reference power is set to 5–6 μcal/sec. For endothermic binding, a smaller reference power should be used (e.g., 1 μcal/sec). Exothermic reactions with large heats of binding might require increasing the reference power.
Maintaining a clean calorimeter
Between runs, the cell should be flushed with ~50 mL water, followed by ITC buffer. The cell should be filled with water when not in use. The syringe should be flushed with water and methanol to dry.
For deeper cleaning of either the cell or the syringe, a detergent like Contrad 70 (10% solution) should be used, followed by flushing with ample amounts of water.
The reference cell (containing degassed ultrapure water) should be changed regularly. Before performing titrations with samples, we routinely perform test titrations of water into water or 5 M CaCl2 into 0.4 M EDTA in 10 mM MES buffer, pH 5.6.
3.3. Data analysis
The output data file is opened in NITPIC and baseline corrected using the EXECUTE command. The preliminary fit performed by NITPIC (log Ka, n, ΔH) provides starting estimates for further refinement by SEDPHAT. If background subtraction or outlier removal is required, or binding is not 1:1, the NITPIC fit will be poor. In SEDPHAT, background subtraction should yield a binding isotherm from which the x-value of the inflection point of a single transition can be determined by eye (Fig. 3).
In SEDPHAT, binding isotherms can be fit to a variety of models that include simple 1:1 binding (A+B), more complex models (e.g., A+B+B, A+B+C), and global fits to several sets of data, such as titrations at varying T to determine heat capacity (ΔCp) (Fig. 4). For 1:1 binding, in which sample A in the syringe is titrated into sample B in the cell, the “A+B” model is employed, fitting Ka, ΔH, and the incompetent fraction of A (incfA) (see Note 6). For low c, incfA is fixed to 0.
For global fits to determine ΔCp, for example, binding isotherms from several temperatures (e.g., 20, 25, 31, and 37 °C) are opened simultaneously. All data files are typically fit by four global parameters—Ka and ΔH values at the reference temperature of 25 °C, incfA, and ΔCp. The apparent Ka(T) and ΔH(T) for each experiment are linked to reference temperature values by their ΔCp dependence [18,19]. It is beneficial to perform titrations for the global analysis using the same sample stock solutions. This will allow using additional constraints in the fit to limit the number of fitting parameters. For example, the incompetent fraction incfA for samples prepared by dilutions from the same stock can usually be treated as a global parameter, identical for all data sets. For global fits, the importance of each isotherm is weighted by its noise (as viewed in EXPERIMENTAL PARAMETERS in SEDPHAT) to prevent over-fitting of noisy isotherms (Fig. 4).
Figure 4.
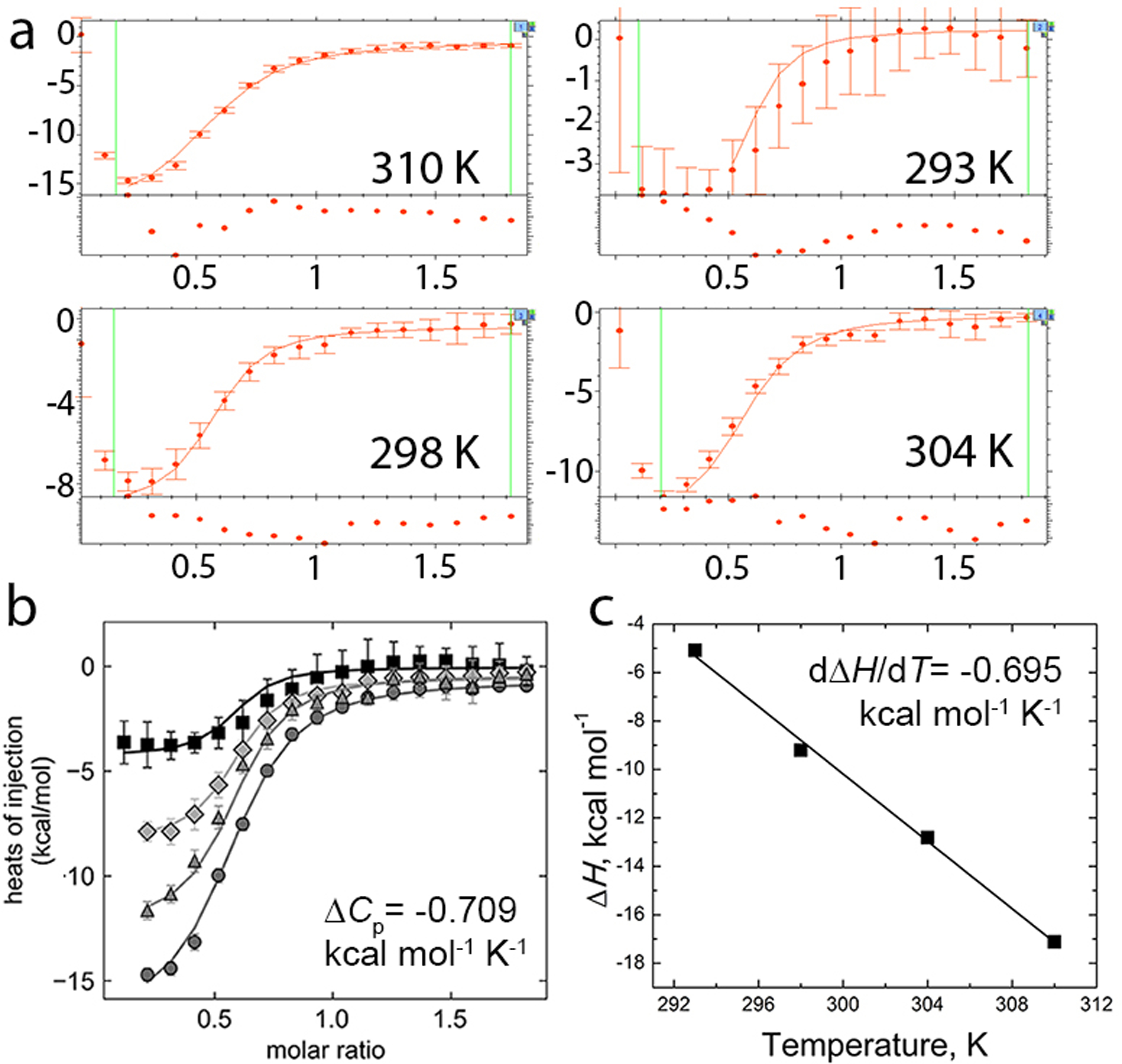
Representative global fit to calculate ΔCp. (a) SEDPHAT global fit of ΔCp, Ka, ΔH, and incompetent fraction of RNA from binding isotherms of four temperatures. All binding isotherms contained 20 μM F. ulcerans ZTP riboswitch titrated with 200 μM ZMP. Note the higher noise for the 293 K binding isotherm. (b) The same data and fits as plotted in GUSSI with 293 K data indicated by squares, 298 K data indicated by diamonds, 304 K data indicated by triangles, and 310 K data indicated by circles. Also shown is the ΔCp obtained from the SEDPHAT global fit of Ka at 298 K, ΔH at 298 K, ΔCp, and the incompetent fraction of RNA. (c) Linear fit of ΔH versus T from binding isotherms in (a) except that ΔH, Ka, and incompetent fractions at each temperature were fit locally. Over the measured temperatures, the relationship between ΔH and T is linear, and the slope (dΔH/dT) is ΔCp.
Notes
Note 1. DEPC is extremely toxic and should be handled with gloves and proper protective clothing in a fume hood or well-ventilated area. We prepare DEPC-treated water by thoroughly mixing 4 L water with 1 mL of DEPC, followed by autoclaving for 40 min at 121 °C to eliminate remaining DEPC.
Note 2. Purification of RNA requires careful handling to prevent contamination with RNase present in the environment, especially on human fingers [20]. It is best to avoid using glassware, pipette tips, or Eppendorf tubes of unknown provenance. Most proteins purified to apparent homogeneity, including T7 RNA polymerase, will still contain some RNase. RNA prepared for ITC follows the transcription protocol of Milligan et al [21], updated by introducing self-cleaving ribozymes to give homogeneous 5´ ends [22] and 2´-methoxy modified DNA primers to give homogeneous 3´ ends [23,24]. After purification through denaturing polyacrylamide gel electrophoresis (PAGE) containing 8 M urea, RNA is eluted from gel slices using an Elutrap (GE Healthcare), concentrated in Amicon spin concentrators, washed with 1 M KCl, washed several times with DEPC-treated water, and filtered with 0.22 μm spin filters. A small aliquot of the RNA is diluted ten times (or more) for determining the concentration using the extinction coefficient at 260 nm and for judging post-preparation purity by denaturing PAGE.
Note 3. Optimizing RNA refolding conditions is often the very first series of experiments performed with an RNA molecule of interest. The goal is to assay heterogeneity (via native gel electrophoresis, analytical size exclusion chromatography, light scattering, etc.) over a range of folding conditions to find conditions for which the RNA is a single folded species. Before this achieved, however, ITC is useful for comparing Ka and n between samples, with the best behaved samples having an expected n of 1. As a starting point, RNA is heated and cooled in solutions in which the concentrations of monovalent ions (50 or 150 mM KCl or NaCl) and divalent ions (0, 1, or 10 mM MgCl2) are varied to sparsely search folding condition space. The temperatures for refolding are also varied, starting with “snap cooling” (95 °C for 2 min, then place on ice), slow cooling (95 °C for 2 min, slowly cool to 25 °C), and step cooling (e.g., 95 °C for 2 min, 60 °C for 2 min, add MgCl2, then place on ice). At high temperature, MgCl2 will cleave RNA, so MgCl2 is added after cooling, and the RNA+Mg2+ mixture is incubated at 37 °C for 15 min. For steps involving slow cooling, thermal cyclers are used to control cooling rate.
Note 4. After the titration has completed, RNA samples may be pooled and repurified (i.e., by denaturing gel electrophoresis). Alternatively, the post-titration sample should be saturated with ligand, as judged by the titration results, so the sample is perfectly suited to be used in other experiments comparing ligand-bound and ligand-free states, or concentrated and used for crystallization screens, as described previously [25].
Note 5. For titrations with low c (e.g., c = 1), Ka may be approximated by fixing n to a value determined previously [26], or by studying the temperature dependence of low c titrations [27]. Although this will introduce error if the actual n is different from the fixed n, it allows determining the Ka for weakly binding RNAs for which increasing the RNA and ligand concentrations is problematic due to sample requirements or aggregation. In cases in which the Kd for a ZTP riboswitch mutant to ZMP is relatively weak—for example, the A34U and A34G mutants bound with Kd of 30–50 μM [10]—twice as many injections at half volume are performed at higher ligand concentration to ensure that the final ligand concentration is ~4 times higher than the Kd. For measurements with the glutamine riboswitch, which binds with a ~250 μM Kd, sample cell concentrations of up to 0.3–0.5 mM RNA (~20 mg/mL) were required (T. Numata and A. Ferré-D’Amaré, unpublished results). In this case, concentrated RNA samples were washed extensively with ITC buffer to prevent buffer mismatch.
Note 6. The appearance of the SEDPHAT windows can be simplified to show only the ITC-specific options by selecting the ITCsy mode from the HELP menu. It is important, especially when fitting includes a large number of parameters, to ensure that the global χ2 minimum has been reached. This can be tested by refitting the data with different fitting algorithms (i.e. interchanging the Marquardt–Levenberg and Simplex from the OPTIONS/FITTING OPTIONS Menu) and confirming that the global χ2 is not decreasing. The error intervals of final fitting parameters can be calculated by accessing SEDPHAT statistical functions (CONFIDENCE INTERVAL SEARCH WITH THE PROJECTION METHOD option from the STATISTICS menu).
Acknowledgments
We thank S. Bachas, M. Chen, N. Demeshkina, C. Fagan, T. Numata, Lj. Sjekloca, and R. Trachman III for helpful discussions. This work was partially supported by the intramural program of the NHLBI, NIH, and by a Lenfant Biomedical Fellowship to C.P.J.
References
- 1.Liberman JA, Bogue JT, Jenkins JL, Salim M, Wedekind JE (2014) ITC analysis of ligand binding to preQ(1) riboswitches. Methods in enzymology 549:435–450. doi: 10.1016/B978-0-12-801122-5.00018-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert SD, Batey RT (2009) Monitoring RNA-ligand interactions using isothermal titration calorimetry. Methods in molecular biology 540:97–114. doi: 10.1007/978-1-59745-558-9_8 [DOI] [PubMed] [Google Scholar]
- 3.Jones CP, Ferré-D’Amaré AR (2015) RNA quaternary structure and global symmetry. Trends in biochemical sciences 40 (4):211–220. doi: 10.1016/j.tibs.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serganov A, Nudler E (2013) A decade of riboswitches. Cell 152 (1–2):17–24. doi: 10.1016/j.cell.2012.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batey RT (2012) Structure and mechanism of purine-binding riboswitches. Quarterly reviews of biophysics 45 (3):345–381. doi: 10.1017/S0033583512000078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones CP, Ferre-D’Amare AR (2017) Long-Range Interactions in Riboswitch Control of Gene Expression. Annual review of biophysics. doi: 10.1146/annurev-biophys-070816-034042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chauvier A, Picard-Jean F, Berger-Dancause JC, Bastet L, Naghdi MR, Dube A, Turcotte P, Perreault J, Lafontaine DA (2017) Transcriptional pausing at the translation start site operates as a critical checkpoint for riboswitch regulation. Nature communications 8:13892. doi: 10.1038/ncomms13892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caron MP, Bastet L, Lussier A, Simoneau-Roy M, Masse E, Lafontaine DA (2012) Dual-acting riboswitch control of translation initiation and mRNA decay. Proceedings of the National Academy of Sciences of the United States of America 109 (50):E3444–3453. doi: 10.1073/pnas.1214024109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones CP, Ferré-D’Amaré AR (2014) Crystal structure of a c-di-AMP riboswitch reveals an internally pseudo-dimeric RNA. The EMBO journal. doi: 10.15252/embj.201489209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones CP, Ferré-D’Amaré AR (2015) Recognition of the bacterial alarmone ZMP through long-distance association of two RNA subdomains. Nature structural & molecular biology 22 (9):679–685. doi: 10.1038/nsmb.3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiseman T, Williston S, Brandts JF, Lin LN (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Analytical biochemistry 179 (1):131–137 [DOI] [PubMed] [Google Scholar]
- 12.Indyk L, Fisher HF (1998) Theoretical aspects of isothermal titration calorimetry. Methods in enzymology 295:350–364 [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Jones CP, Ferré-D’Amaré AR (2014) Global analysis of riboswitches by small-angle X-ray scattering and calorimetry. Biochimica et biophysica acta. doi: 10.1016/j.bbagrm.2014.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, Schuck P (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Analytical chemistry 84 (11):5066–5073. doi: 10.1021/ac3007522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Piszczek G, Schuck P (2015) SEDPHAT--a platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76:137–148. doi: 10.1016/j.ymeth.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tellinghuisen J (2005) Optimizing experimental parameters in isothermal titration calorimetry. The journal of physical chemistry B 109 (42):20027–20035. doi: 10.1021/jp053550y [DOI] [PubMed] [Google Scholar]
- 17.Baird NJ, Inglese J, Ferré-D’Amaré AR (2015) Rapid RNA-ligand interaction analysis through high-information content conformational and stability landscapes. Nature communications 6:8898. doi: 10.1038/ncomms9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson AD, Murphy KP (1997) Protein Structure and the Energetics of Protein Stability. Chemical reviews 97 (5):1251–1268 [DOI] [PubMed] [Google Scholar]
- 19.Tan A, Tanner JJ, Henzl MT (2008) Energetics of OCP1-OCP2 complex formation. Biophysical chemistry 134 (1–2):64–71. doi: 10.1016/j.bpc.2008.01.005 [DOI] [PubMed] [Google Scholar]
- 20.Holley RW, Apgar J, Merrill SH (1961) Evidence for the liberation of a nuclease from human fingers. The Journal of biological chemistry 236:PC42–43 [PubMed] [Google Scholar]
- 21.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic acids research 15 (21):8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferré-D’Amaré AR, Doudna JA (1996) Use of cis- and trans-ribozymes to remove 5’ and 3’ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic acids research 24 (5):977–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao C, Zheng M, Rudisser S (1999) A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. Rna 5 (9):1268–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helmling C, Keyhani S, Sochor F, Furtig B, Hengesbach M, Schwalbe H (2015) Rapid NMR screening of RNA secondary structure and binding. Journal of biomolecular NMR 63 (1):67–76. doi: 10.1007/s10858-015-9967-y [DOI] [PubMed] [Google Scholar]
- 25.Da Veiga C, Mezher J, Dumas P, Ennifar E (2016) Isothermal Titration Calorimetry: Assisted Crystallization of RNA-Ligand Complexes. Methods in molecular biology 1320:127–143. doi: 10.1007/978-1-4939-2763-0_9 [DOI] [PubMed] [Google Scholar]
- 26.Turnbull WB, Daranas AH (2003) On the value of c: can low affinity systems be studied by isothermal titration calorimetry? Journal of the American Chemical Society 125 (48):14859–14866. doi: 10.1021/ja036166s [DOI] [PubMed] [Google Scholar]
- 27.Tellinghuisen J (2008) Isothermal titration calorimetry at very low c. Analytical biochemistry 373 (2):395–397. doi: 10.1016/j.ab.2007.08.039 [DOI] [PubMed] [Google Scholar]