

Abstract
Metagenomic sequencing has greatly enhanced the discovery of viral genomic sequences; however, it remains challenging to identify the host(s) of these new viruses. We developed VirHostMatcher-Net, a flexible, network-based, Markov random field framework for predicting virus–prokaryote interactions using multiple, integrated features: CRISPR sequences and alignment-free similarity measures (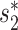 and WIsH). Evaluation of this method on a benchmark set of 1462 known virus–prokaryote pairs yielded host prediction accuracy of 59% and 86% at the genus and phylum levels, representing 16–27% and 6–10% improvement, respectively, over previous single-feature prediction approaches. We applied our host prediction tool to crAssphage, a human gut phage, and two metagenomic virus datasets: marine viruses and viral contigs recovered from globally distributed, diverse habitats. Host predictions were frequently consistent with those of previous studies, but more importantly, this new tool made many more confident predictions than previous tools, up to nearly 3-fold more (n > 27 000), greatly expanding the diversity of known virus–host interactions.
and WIsH). Evaluation of this method on a benchmark set of 1462 known virus–prokaryote pairs yielded host prediction accuracy of 59% and 86% at the genus and phylum levels, representing 16–27% and 6–10% improvement, respectively, over previous single-feature prediction approaches. We applied our host prediction tool to crAssphage, a human gut phage, and two metagenomic virus datasets: marine viruses and viral contigs recovered from globally distributed, diverse habitats. Host predictions were frequently consistent with those of previous studies, but more importantly, this new tool made many more confident predictions than previous tools, up to nearly 3-fold more (n > 27 000), greatly expanding the diversity of known virus–host interactions.
INTRODUCTION
Viruses are the most abundant and highly diverse biological entities on Earth (1,2). Viruses infect all domains of life, including archaea, bacteria and eukaryotes. For prokaryotic viruses, especially those that infect bacteria, there have been extensive studies about their diversity (3,4), functions (5–7) and impact on microbial communities through virus–host interactions (8–11). In particular, prokaryotic viruses can significantly impact human health (12–14) and the functioning of many ecosystems (15–17) such as marine and soil habitats. Therefore, characterizing virus–host interactions is a critical component to understanding how biological systems work. Viruses are traditionally studied using culture-based isolation techniques that provide direct identification of virus–host pairs (VHPs). Isolation approaches are, however, low throughput and limited to hosts that are cultivable. Compared to the predicted number of extant viruses, a relatively small number of viruses have been discovered via isolation-based approaches with current estimates indicating that 75–85% of viruses remain uncharacterized (11,18). With the advent of metagenomic sequencing technologies, genetic material from microbes including viruses, regardless of cultivability, can be sequenced. Metagenomic shotgun sequencing, especially the metagenomic sequencing of virus-like particles, has tremendously accelerated the discovery of previously unknown viruses. An example is crAss-like phages, a highly abundant family of ubiquitous human gut viruses, originally discovered from the cross-assembly of fecal viral metagenomic samples (19).
Identifying the hosts of viruses is important for understanding the impact of viruses on the host dynamics and thus host community diversity and function. Computational methods have been developed to infer the hosts of new viruses. Many bacteria and archaea possess CRISPR virus defense systems whereby the host incorporates some virus DNA fragments into its own genome forming interspaced short palindromic repeats (CRISPR) spacers. Therefore, shared CRISPR regions are direct evidence supporting virus–host interactions (16,19) and have been used for host prediction for viruses in previous studies (20,21). Genome alignment matches between virus and host genomes due to integrated prophages or horizontal gene transfer are another piece of strong evidence used in predicting the host of a virus (5,16). However, the above methods are limited by their low accessibility: it is estimated that CRISPRs are only present in ∼10% of sequenced bacterial genomes (22,23); many viruses infect hosts under a lytic mode without integration to the host genome; and many viruses do not extensively share host genes. Thus, CRISPRs and alignment-based approaches are not applicable for predicting many viral hosts.
Several investigators have utilized the fact that viruses are often more similar to their hosts, compared to non-host species, in terms of genome-wide signature, i.e. k-mer usage, because viruses and their hosts live in the same environment and viruses use the hosts’ replication mechanism for replication (11,20,24,25). This information has been used to predict the host of a virus as the one closest to the viral genome based on some similarity measures using k-mers. These methods in general have decent prediction accuracy, though the mechanism behind this phenomenon is not fully understood. One plausible explanation is that viruses tend to adopt the codon usage of their hosts in order to utilize the hosts’ translational machinery (26,27). The recently developed dissimilarity measure 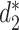 that subtracts expected k-mer frequency from the observed frequency achieves the highest reported host prediction accuracy among all current genomic signature-based measures, including commonly used Euclidean and Manhattan distances (24). Similarly, Galiez et al. (25) predicted the host of a virus to be the one for which results from a Markov chain model analysis had the highest likelihood score. The method has good prediction accuracy for short viral fragments. These genomic signature-based measures are often referred to as alignment-free sequence comparison measures. The high correlation between virus and host abundance profiles across different samples also serves as evidence for virus–host interaction (19), but its accuracy is not as high as the above methods (20). Edwards et al. (20) recently provided a comprehensive evaluation of several different computational approaches for virus–host predictions.
that subtracts expected k-mer frequency from the observed frequency achieves the highest reported host prediction accuracy among all current genomic signature-based measures, including commonly used Euclidean and Manhattan distances (24). Similarly, Galiez et al. (25) predicted the host of a virus to be the one for which results from a Markov chain model analysis had the highest likelihood score. The method has good prediction accuracy for short viral fragments. These genomic signature-based measures are often referred to as alignment-free sequence comparison measures. The high correlation between virus and host abundance profiles across different samples also serves as evidence for virus–host interaction (19), but its accuracy is not as high as the above methods (20). Edwards et al. (20) recently provided a comprehensive evaluation of several different computational approaches for virus–host predictions.
In addition to the methods using features defined between a pair of virus and host genomes, some researchers have used virus–virus similarity networks to infer the host of a query virus (28,29). The high similarity between viruses may indicate a common host or very close host relatedness. Network-based prediction models, whereby unknown entities are predicted based on the features of their neighbors in a network, have been successfully applied to many biological problems, including predicting protein functions using protein–protein interaction networks (30,31), inferring disease genes based on gene–gene networks (32–34) and predicting the target of new drugs using drug–drug, drug–target and target–target similarity networks (35). A few attempts have been made to exploit the possibility of predicting viral hosts based on virus–virus network information. Different principles such as gene homology (36,37), protein family (38) and genome similarity (28,39,40) were used to define the virus–virus relationships in networks. Villarroel et al. proposed HostPhinder (28), a method to predict the host of a virus by searching for the virus that shares the most k-mers from a database of viruses with known hosts. Zhang et al. (29) identified the important k-mer features of viruses infecting the same host genera, and built a classifier to predict whether or not a new virus belongs to the same group of viruses. One drawback of the network-based approach is that the performance can diminish if the query virus is highly divergent from the known viruses in the current network.
Though various methods have been proposed for predicting virus–host interactions, the highest accuracy is only 43% at the genus level using a single type of information. With the increasing number of viruses being discovered, there is a demand for a tool that is able to accurately and rapidly predict the hosts of viruses, incorporating all types of virus–host and virus–virus features. In this paper, we have developed a network-based integrated framework for predicting virus–prokaryote interactions based on multiple types of information: virus–virus similarity, virus–host alignment-free similarity, virus–host shared CRISPR spacers and virus–host alignment-based matches. To the best of our knowledge, this is the first time that multiple types of features are effectively integrated into a network to complement each other and enhance the prediction accuracy of virus–prokaryote interactions. This integrated method markedly improved the accuracies in predicting virus–prokaryote interactions for complete viral genomes from 43% to 59% at the genus level, and yielded 86% accuracy at the phylum level, the highest among all the existing methods. The prediction framework also had decent accuracy for shorter viral contigs even as short as 10 kb. We have used our method to infer the host of the first isolated strain of the crAssphage, 1811 marine viral genomes and >27 000 viral contigs from various environments. We have provided a user-friendly program, VirHostMatcher-Net, that uses this framework to predict virus–prokaryote interactions. Finally, VirHostMatcher-Net provides a flexible and expandable network-based framework for ongoing refinement of virus–prokaryote prediction methods.
MATERIALS AND METHODS
Datasets
All data generated or analyzed during this study are available from previously published studies (38,41,42) or are included in this paper and the Supplementary Data. We collected 2288 RefSeq viral genomes with known hosts at the genus level from NCBI as of 11 November 2019. Among them, 826 viruses have specific hosts (at strain level) and those were used for training. The training set includes 817 viruses that infect bacteria and 9 that infect archaea. The other 1462 viruses were used for validation. For simplicity of presentation, we will use ‘host’ to refer to ‘prokaryotic host’ throughout the rest of the paper. The hosts of the viruses from which the viruses were originally isolated were collected based on the key words ‘isolate_host=’ or ‘host=’ within each GenBank file. Furthermore, for a subset of 826 viral genomes, their hosts were reported at the strain, subspecies or serovar level, and only a single host genome was reported in the NCBI genome database for that particular strain, subspecies or serovar. We used the 826 viruses with known specific host genomes as the training set. The other viruses either have more than one specific host strains or have host taxonomic information only down to the genus or species level.
We applied our method to a set of 1811 marine virus genomes that were studied in (41). The dataset is available from ftp://ftp.genome.jp/pub/db/community/EVG2017. In addition, we predicted the hosts of 111 167 viral contigs that were assembled previously from various environmental metagenomic samples (38). Accession numbers of those viral contigs are available in Supplementary Table S19 of Paez-Espino et al. (38). The accession numbers for the novel VHPs predicted exclusively by our method can be found in Additional Files 7–9 in the Supplementary Data.
Outline of the model
We formulate the virus–host interactions using a Markov random field (MRF) model (30,42,43). Given a set of viruses {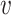 1,
1, 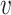 2, …,
2, …, 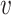 n} and a set of hosts {b1, b2, …, bm}, we define the set of VHPs and their interaction statuses,
n} and a set of hosts {b1, b2, …, bm}, we define the set of VHPs and their interaction statuses,
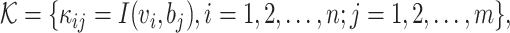 |
where I(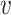 , b) = 1 if
, b) = 1 if 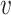 infects b and I(
infects b and I(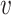 , b) = 0 otherwise. We construct a VHP network where nodes are VHPs and edge weights are the pairwise similarities between two VHPs.
, b) = 0 otherwise. We construct a VHP network where nodes are VHPs and edge weights are the pairwise similarities between two VHPs.
The interaction statuses of all VHPs depend on two essential components: (i) the likelihood of the interaction status of each individual VHP and (ii) the linkage between each VHP and all others. In the following sections, we first show how an MRF model can take the first component into consideration. Next, we introduce a similarity measure that describes the linkage between a pair of VHPs. Then, we define all other features that can be used to estimate the second component. Finally, we derive two models for host prediction given virus genomes and contigs, respectively. We emphasize that the MRF model described below is used to motivate our methods for predicting virus–host interactions. As for most practical problems, the assumptions of the models are most likely violated. The final prediction model is evaluated using an independent dataset of virus–host relationships.
An MRF approach for virus–host interactions
We model the likelihood of virus–host interaction statuses by considering two components: the fraction of interacting VHPs among all the VHPs and the similarity network among the VHPs. For the first component, we use a Bernoulli model that assumes the interaction statuses of VHPs are independent. For the second component, we use a network model based on the similarity network among the VHPs. The two components are integrated by multiplying the probabilities from both components. More specifically, the likelihood of an assignment 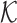 of the infection statuses for all the VHPs in the network is proportional to the likelihood of the assignments of the VHP nodes and the likelihood of the pairwise labels of VHPs given the network. Let π be the probability for a VHP to interact. Then, for each pair (
of the infection statuses for all the VHPs in the network is proportional to the likelihood of the assignments of the VHP nodes and the likelihood of the pairwise labels of VHPs given the network. Let π be the probability for a VHP to interact. Then, for each pair (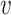 i, bj), the likelihood of the interaction status,
i, bj), the likelihood of the interaction status, 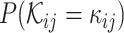 , can be expressed as
, can be expressed as 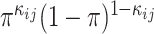 according to the Bernoulli model. By considering all VHPs and assuming their assignments are independent, the likelihood of an assignment of
according to the Bernoulli model. By considering all VHPs and assuming their assignments are independent, the likelihood of an assignment of  is equal to the product of the likelihood for all the VHPs, that is
is equal to the product of the likelihood for all the VHPs, that is
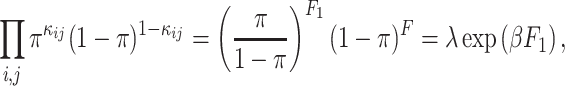 |
(1) |
where F1 = ∑κij,  is the size of
is the size of 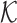 ,
, 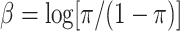 and λ = (1 − π)F.
and λ = (1 − π)F.
Next consider the relationship between two VHPs in the network. The probability of two similar VHPs having the same 0–1 status is higher than the probability of having different 0–1 assignments. Let 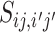 be the similarity between two VHPs (
be the similarity between two VHPs ( i, bj) and
i, bj) and 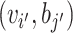 . Conditional on the similarity between two VHPs, we model the probability for them to be labeled as (1, 1), (1, 0) and (0, 0) by
. Conditional on the similarity between two VHPs, we model the probability for them to be labeled as (1, 1), (1, 0) and (0, 0) by 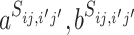 and
and 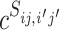 , respectively, where a, b and c are parameters. Mathematically, we can write the probability of (
, respectively, where a, b and c are parameters. Mathematically, we can write the probability of ( i, bj) labeled as κij and
i, bj) labeled as κij and 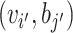 labeled as
labeled as 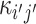 by
by
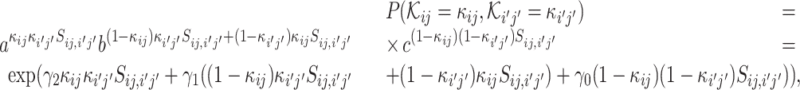 |
where γ2 = log(a), γ1 = log(b) and γ0 = log(c). We assume that the labeling of the VHP pairs is independent. Then, we can multiply the above equation over all the VHP pairs to obtain
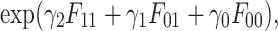 |
(2) |
where 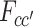 is defined as the sum of similarities among VHP pairs labeled as (c, c′), c, c′ = 0, 1, namely
is defined as the sum of similarities among VHP pairs labeled as (c, c′), c, c′ = 0, 1, namely
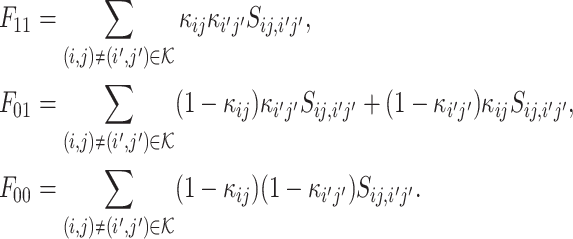 |
By multiplying Equations (1) and (2) and then normalizing to a probability distribution, we model the probability of the assignment conditional on the similarity network as
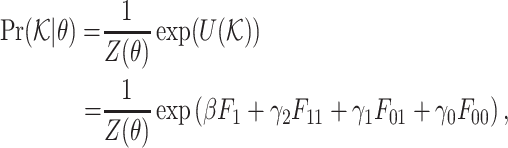 |
where θ = (β, γ2, γ1, γ0) are the parameters, 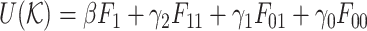 , and Z(θ) is the normalizing factor.
, and Z(θ) is the normalizing factor.
With this distribution function, for any 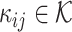 , we can calculate
, we can calculate
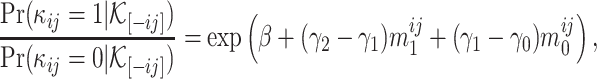 |
where
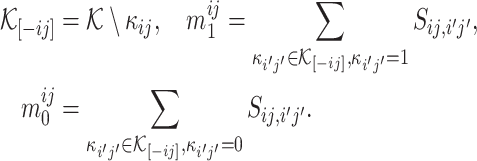 |
Then, the log odds of the probability 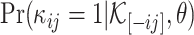 is
is
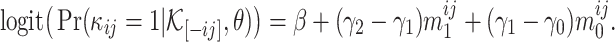 |
Denote γ+ = γ2 − γ1 and γ− = γ1 − γ0. We have
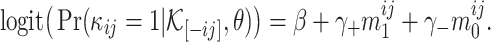 |
The similarity between two VHPs and the generalized probability model for a VHP to interact
The MRF network model is constructed based on the similarity between pairs of VHPs  . Various similarity measures between VHPs can be defined. In this study, we define the similarity between two VHPs as the similarity between the two viruses plus the similarity between the two hosts. To measure the similarity between two genomic sequences, we previously developed dissimilarity measures
. Various similarity measures between VHPs can be defined. In this study, we define the similarity between two VHPs as the similarity between the two viruses plus the similarity between the two hosts. To measure the similarity between two genomic sequences, we previously developed dissimilarity measures  and
and 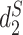 for alignment-free sequence comparison using k-mers as genomic signatures (44–47), and showed that the dissimilarity measures
for alignment-free sequence comparison using k-mers as genomic signatures (44–47), and showed that the dissimilarity measures 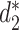 and
and 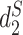 have high correlation with alignment-based distance measures (48). Since viruses are highly diverse and alignments of highly divergent sequences are challenging, alignment-free measures are more suitable for sequence comparison than the alignment-based methods. Furthermore, Ahlgren et al. (24) showed that
have high correlation with alignment-based distance measures (48). Since viruses are highly diverse and alignments of highly divergent sequences are challenging, alignment-free measures are more suitable for sequence comparison than the alignment-based methods. Furthermore, Ahlgren et al. (24) showed that 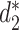 outperformed
outperformed 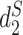 for the comparison of virus and bacterial sequences for the purpose of virus–host interaction prediction. Therefore, here we choose to use
for the comparison of virus and bacterial sequences for the purpose of virus–host interaction prediction. Therefore, here we choose to use 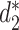 and transform it to
and transform it to 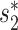 to measure the similarity between two sequences.
to measure the similarity between two sequences.
For each sequence, we represent it by the normalized k-mer frequency vector 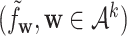 , where
, where  is the set of alphabets {A, C, G, T}, k is the length of k-mer and
is the set of alphabets {A, C, G, T}, k is the length of k-mer and
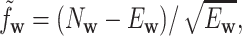 |
with  and
and  being the observed and expected numbers of occurrences of word
being the observed and expected numbers of occurrences of word  in the sequence. The expected count is calculated under a Markov chain model for the sequence as described below. Since it was shown in (24) that k = 6 and second-order Markov chain performed well in virus–host interaction prediction, we choose k = 6 and second-order Markov chain in this study. The similarity between two sequences,
in the sequence. The expected count is calculated under a Markov chain model for the sequence as described below. Since it was shown in (24) that k = 6 and second-order Markov chain performed well in virus–host interaction prediction, we choose k = 6 and second-order Markov chain in this study. The similarity between two sequences, 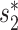 , is defined as the uncentered correlation between their corresponding normalized frequency vectors. That is,
, is defined as the uncentered correlation between their corresponding normalized frequency vectors. That is,
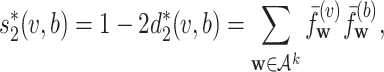 |
where 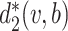 is the dissimilarity measure used in the previous studies, and
is the dissimilarity measure used in the previous studies, and 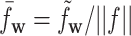 with ||f|| being the Euclid norm of the feature vector
with ||f|| being the Euclid norm of the feature vector 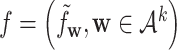 and the superscript indicates the virus
and the superscript indicates the virus 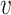 or bacterial b sequence. Thus, we define the similarity
or bacterial b sequence. Thus, we define the similarity
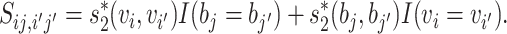 |
Plugging 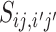 into the logit function, we have
into the logit function, we have
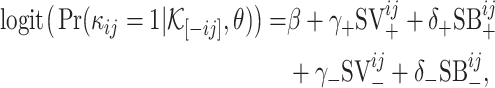 |
(3) |
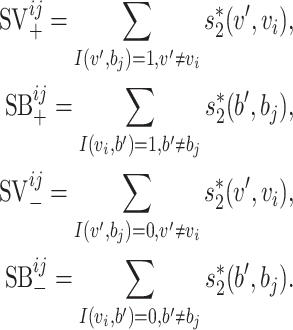 |
The above formulation takes into account both the similarity network between viruses and the similarity network between hosts. In our dataset, however, each virus has only one reported host. So, when we train the model using the current dataset, both 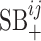 and
and 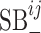 are set to zero. Then, the model reduces to
are set to zero. Then, the model reduces to
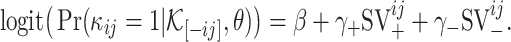 |
Though the terms 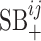 and
and 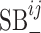 cannot be used given the current dataset, as more VHPs are collected in the training data, the host–host similarity network will contribute to the prediction model and the two-layer MRF network will be fully utilized based on Equation (3).
cannot be used given the current dataset, as more VHPs are collected in the training data, the host–host similarity network will contribute to the prediction model and the two-layer MRF network will be fully utilized based on Equation (3).
Incorporating similarity between virus and host for interaction prediction
The assumption that any VHP has the same probability π for interaction is not realistic. Different pairs of virus and host have different features that affect the probability of interaction. For example, the probability can be associated with the similarity between the virus and the host (24). Thus, the probability π is modeled specifically to each individual pair (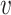 i, bj),
i, bj),
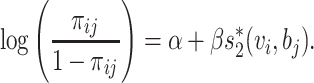 |
(4) |
Then, the logit model with the generalized probability can be written as
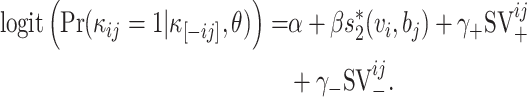 |
Therefore, the network-based MRF for predicting virus–host interaction is finally written as a logistic regression model where the predictors are the features of virus–virus similarity and virus–host similarity,
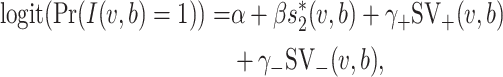 |
(5) |
where α is a constant and (β, γ+, γ−) measure the contributions of the features 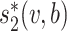 , SV+(
, SV+(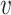 , b) and SV−(
, b) and SV−(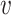 , b), respectively. We expect β and γ+ to be positive and γ− to be negative. However, we do not make these assumptions and let the data inform us the values of these parameters. To learn the parameters, we trained the model in a smaller training dataset, and predicted virus–host interactions in the network of all viruses and hosts. Since the scales of SV+(
, b), respectively. We expect β and γ+ to be positive and γ− to be negative. However, we do not make these assumptions and let the data inform us the values of these parameters. To learn the parameters, we trained the model in a smaller training dataset, and predicted virus–host interactions in the network of all viruses and hosts. Since the scales of SV+(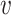 , b) and SV−(
, b) and SV−(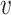 , b) are proportional to the size of the dataset, in practice we used the normalized variables, that is
, b) are proportional to the size of the dataset, in practice we used the normalized variables, that is
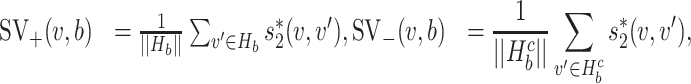 |
where Hb = { ′|I(
′|I( ′, b) = 1,
′, b) = 1, 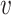 ′ ≠
′ ≠ 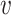 },
}, 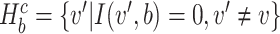 and ||·|| is the size of the set. When Hb or
and ||·|| is the size of the set. When Hb or 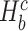 is an empty set, the value of SV+(
is an empty set, the value of SV+(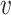 , b) or SV−(
, b) or SV−(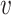 , b) is set to zero.
, b) is set to zero.
To achieve the best performance, in addition to the similarity score 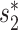 , we integrate other types of features, including the CRISPR score and the alignment score between the virus
, we integrate other types of features, including the CRISPR score and the alignment score between the virus 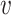 and host b into the framework.
and host b into the framework.
Sharing of CRISPR spacers between the virus and the host
The CRISPR systems play an important role as an adaptive and heritable immune system for prokaryotes. They help the host fight against the invasion of specific viruses by inserting small fragments of viral genomes (typically 21–72 bp) as spacers into a CRISPR locus. The spacers are transcribed and are used as a guide by a Cas complex to target the degradation of the corresponding viral DNA (49).
Given a host genome, the CRISPR locus can be computationally located and thus the spacers can be extracted. In our study, we used the CRISPR Recognition Tool (50) to find spacers. The spacers in a host genome (if available) were aligned to a viral genome by blastn (51) and alignment with E-value <1 was recorded. This threshold was chosen the same as the one used in a previous study (20). Since a lower E-value between a spacer and a virus genome indicates high similarity between them, we use −log(E-value) to measure the strength of association between the spacer and the virus genome. It is possible that a host genome may contain multiple spacers and the strongest association between these spacers and the virus genome indicates the strength of association between the host and the virus. Therefore, for each pair of virus and host, we define the score SCRISPR(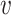 , b) as the largest value of −log(E-value). If there is no match between a virus and host, a score of zero is assigned. Details of the programs and parameters used in this analysis are given in the Supplementary Data.
, b) as the largest value of −log(E-value). If there is no match between a virus and host, a score of zero is assigned. Details of the programs and parameters used in this analysis are given in the Supplementary Data.
With the CRISPR information, we modify the model of πij in Equation (4) to
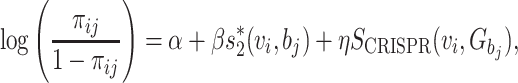 |
and our logistic regression model in Equation (5) to
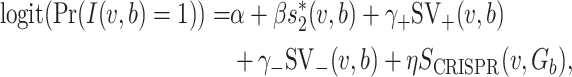 |
(6) |
where Gb is the set of hosts that belong to the same genus as host b, and
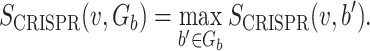 |
Due to the limited availability of CRISPR information in the training data, as shown in Figure 2, we group hosts by genus for the CRISPR feature.
Figure 2.

Distributions of the different feature values among 826 interacting and non-interacting VHPs. The positive set consists of 826 known infecting VHPs (positive set) and the same number of randomly selected virus and host pairs were used as the non-interacting, negative set. (A) Box plots of similarity defined by 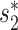 (v, b). (B) Box plots of the log-likelihood scores given by WIsH. (C) Box plots of SV+(v, b) scores. (D) Box plots of the SV−(v, b) scores. (E) Box plots of BLAST scores. (F) Box plots of the CRISPR scores. For all figures, the horizontal bar displays the median; boxes display the first and third quartiles; whiskers depict minimum and maximum values; and points depict outliers beyond the whiskers.
(v, b). (B) Box plots of the log-likelihood scores given by WIsH. (C) Box plots of SV+(v, b) scores. (D) Box plots of the SV−(v, b) scores. (E) Box plots of BLAST scores. (F) Box plots of the CRISPR scores. For all figures, the horizontal bar displays the median; boxes display the first and third quartiles; whiskers depict minimum and maximum values; and points depict outliers beyond the whiskers.
The fraction of virus genome aligned to the host genome
Viruses and their hosts frequently exchange genetic material and viruses play important roles in horizontal gene transfer. Therefore, similar regions in virus and host genomes can provide a strong evidence for linking a virus into its potential host. On the one hand, phages, especially the temperate phages, are able to integrate their own genomes to the hosts. On the other hand, phages can obtain genetic material from their hosts. If a genetic element brings an evolutionary advantage to the virus, the borrowed genetic segment will be preserved in the viral genome (20). One example is cyanophages, phages that infect cyanobacteria. Many cyanophages acquire and express host photosystem genes that are thought to bolster photosynthetic energy during infection (52).
Similar to the method in (20), we used blastn to find similarities between each pair of virus and host genomes. For each VHP, their similarity, Sblastn(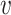 , b), is defined as the fraction of the virus genome that can be mapped to the host genome. Only matches with percent identity >
, b), is defined as the fraction of the virus genome that can be mapped to the host genome. Only matches with percent identity >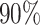 are used for prediction. Note that different parts of the virus genome can be matched to different positions on the host genome and all contribute to the coverage percentage. We used the same parameter settings as in (20) for our analysis. Details of the program and parameters used in this analysis are given in the Supplementary Data.
are used for prediction. Note that different parts of the virus genome can be matched to different positions on the host genome and all contribute to the coverage percentage. We used the same parameter settings as in (20) for our analysis. Details of the program and parameters used in this analysis are given in the Supplementary Data.
Finally, with the CRISPR feature and the alignment-based similarity, we have the following model:
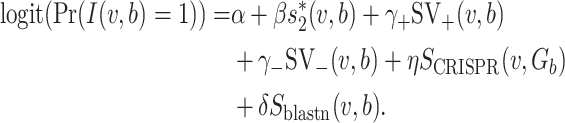 |
(7) |
Incorporation of WIsH score for predicting hosts of virus contigs
In many metagenomic studies, the whole genome of a virus may not be available. Instead, only parts of the virus genome referred to as contigs that were assembled from shotgun reads are known. Several algorithms such as VirFinder, VirSorter, etc. (53–58) can be used to decide whether the contigs come from virus genomes. Our objective is to predict the hosts for full virus genomes as well as viral contigs.
Galiez et al. (25) recently developed a program, WIsH, to predict the hosts of viral contigs and showed that WIsH outperforms 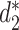 for predicting the hosts of viral contigs as short as 5 kb. WIsH trains a homogeneous Markov chain model for each host genome, and calculates the likelihood of a viral contig based on each Markov chain model. Instead of using
for predicting the hosts of viral contigs as short as 5 kb. WIsH trains a homogeneous Markov chain model for each host genome, and calculates the likelihood of a viral contig based on each Markov chain model. Instead of using 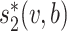 as a feature, we hereby replace it with the log-likelihood of viral contig
as a feature, we hereby replace it with the log-likelihood of viral contig 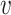 fitting to the Markov chain model of bacteria b, SWIsH(
fitting to the Markov chain model of bacteria b, SWIsH(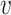 , b). WIsH (25) scores were computed using WIsH 1.0 with the default parameters. Then, the model for predicting the host b of viral contig
, b). WIsH (25) scores were computed using WIsH 1.0 with the default parameters. Then, the model for predicting the host b of viral contig 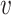 becomes
becomes
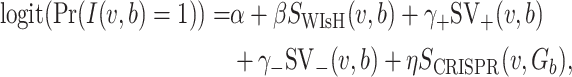 |
(8) |
corresponding to Equation (6), and
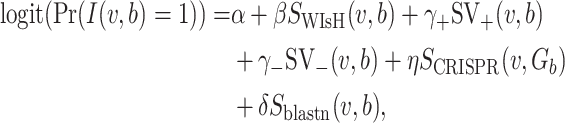 |
(9) |
corresponding to Equation (7).
Note that both SV+(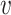 , b) and SV−(
, b) and SV−(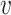 , b) are still computed by
, b) are still computed by 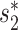 , since WIsH is not able to depict the similarities between viral contigs.
, since WIsH is not able to depict the similarities between viral contigs.
Model training and evaluation
Among the 2288 viruses obtained from NCBI, we used the set of 826 viruses whose exact host genome sequences were known and the set of their corresponding 185 hosts as the positive training set. We randomly select 826 pairs of virus–host within the 826 viruses and 185 hosts as negative training data. To alleviate potential false negative interactions, we required that the selected host for each virus is not in the same phylum level as the true host. We then learned the model based on the training data for the various models. We repeated the selection of negative training sets for 100 times. For real applications and the software, we set the coefficients by averaging over 100 times of the training procedure to reduce randomness.
It is possible that the selected 826 non-interacting pairs may contain some positive yet unknown interaction pairs, which may influence the training and test results. We recognized this possibility while assuming the fraction of such pairs is relatively low since the virus–host interaction is specific so that the overall fraction of virus–host interacting pairs among all the pairs is very small. The additional requirement that the host in a negative VHP comes from a different phylum level further mitigates this potential problem.
The trained models were then used to predict the hosts of the remaining 1462 viruses against 62 493 candidate prokaryotic hosts. For each virus, we estimated its probability of infecting any hosts, and the one with the highest probability was predicted as its host. For a taxonomic group 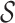 at an upper taxonomic level containing a set of hosts, we define the prediction score between
at an upper taxonomic level containing a set of hosts, we define the prediction score between 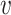 and
and 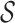 as the maximum probability between
as the maximum probability between 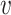 and all hosts in
and all hosts in 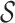 , that is
, that is
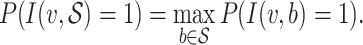 |
We predict the host group of the virus 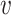 by the one having the highest prediction score
by the one having the highest prediction score 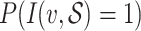 . In case of ties, we first checked the number of hosts having the highest probability in each group and chose the one with the largest number of hosts having the highest probability. Further, if there were more than one taxon with the same number of bacteria having the highest probability, all taxa were reported.
. In case of ties, we first checked the number of hosts having the highest probability in each group and chose the one with the largest number of hosts having the highest probability. Further, if there were more than one taxon with the same number of bacteria having the highest probability, all taxa were reported.
We then compared the predicted host taxonomic groups with the true taxonomic group of every virus at several taxonomic levels: genus, family, order, class and phylum. At a particular taxonomic level 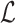 , let
, let 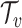 be the set of predicted groups and
be the set of predicted groups and 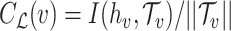 , where
, where 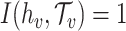 if the true host of
if the true host of 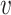 , h
, h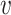 , belongs to the set of the predicted host groups
, belongs to the set of the predicted host groups 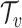 , and
, and 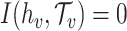 , otherwise. The prediction accuracy for a certain taxonomic level is defined as
, otherwise. The prediction accuracy for a certain taxonomic level is defined as
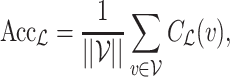 |
where 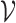 is the set of viruses for prediction.
is the set of viruses for prediction.
Clustering of viral contigs
To examine the relatedness of viral contigs for novel host predictions, proteins encoded on viral contigs were predicted by Prodigal 2.6.3 (with default parameters). BLASTp 2.6.0 was then used to search for similar proteins shared between viral contigs. The percentage of genes shared between two contigs was defined as the number of pairs of homologous proteins between the two contigs divided by the average number of proteins of the two contigs.
Consideration of virus–host co-abundance in host prediction
In order to investigate whether co-abundance can help the prediction of virus–host interactions, we incorporated this feature to the model in a smaller dataset to evaluate its contribution. The dataset included a subset of 2695 prokaryotic reference genomes and 1403 viruses (see below). A total of 148 stool metagenomic samples from the Human Microbiome Project (HMP) (59) and 103 metagenomes from the Tara Ocean (filter size 0.22–3 μm) (60) were collected. We used centrifuge (56) (centrifuge-1.0.3-beta) to compute the abundance of virus and bacteria genomes in each of the metagenomes, resulting in an abundance profile of a 251-dimensional vector for every virus and host genome. The co-abundance feature Sco-abundance(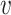 , b) was defined by the Pearson correlation between the abundance profiles for the pair of virus and bacterium. We then modified the integrated model to
, b) was defined by the Pearson correlation between the abundance profiles for the pair of virus and bacterium. We then modified the integrated model to
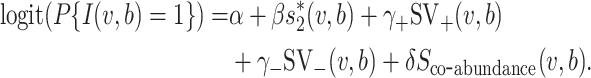 |
We compared the performance of this model with that of the model in Equation (5). Both models were trained based on a subset of 308 viruses and 50 hosts, including 308 pairs of true interacting pairs and 308 randomly chosen negative pairs. After both models were trained, we predicted the hosts of 1095 viruses. The results are shown in Additional File 14 in the Supplementary Data. The co-abundance feature itself had weak prediction ability and adding it to the model did not help prediction. Therefore, we did not consider it as a feature in the final model presented in the main text.
Alternative methods
Support vector machines (SVMs) and random forests (RFs) are among the most popular machine learning tools for classification (61). In this study, for each pair of virus and host, we considered two network-based features introduced by the MRF framework, SV+ and SV−, and three additional features: 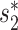 , Sblastn and SCRISPR. We learned the SVM and RF models based on the five features using the same training data by 5-fold cross-validation. The learned models were then evaluated on the validation set. Additional File 16 in the Supplementary Data shows SVMs and RFs do not perform as well as our integrated MRF-based approach. The details are given in the Supplementary Data.
, Sblastn and SCRISPR. We learned the SVM and RF models based on the five features using the same training data by 5-fold cross-validation. The learned models were then evaluated on the validation set. Additional File 16 in the Supplementary Data shows SVMs and RFs do not perform as well as our integrated MRF-based approach. The details are given in the Supplementary Data.
Software
We developed a computational tool, VirHostMatcher-Net, implementing our network-based integrated method for virus–host predictions. The software is publicly available at https://github.com/WeiliWw/VirHostMatcher-Net. The tool supports parallel computing and has the option of choosing the type of query viruses (complete genomes or contigs). It also provides the option of specifying a customized subset of candidate hosts for prediction. The tool provides informative outputs including all the feature scores of the query viruses against all candidate hosts, and a summarized table listing top predictions for each virus with their feature scores, score percentiles and accuracy. The score percentile of a VHP is defined as the percentile of this score among all scores between that virus and all the candidate hosts. A large percentile suggests high relevance of the feature score. The percentile of SV−, the only feature with a negative coefficient, is reversed to be consistent with other feature score percentiles. The percentile information helps to better understand how relevant each feature score is for a particular prediction. We also provide ‘accuracy’ that gives the fraction of correct predictions when VHPs with prediction scores above the particular threshold are declared as interacting.
RESULTS
A novel network-based integrated framework for predicting virus–host interaction
We collected from NCBI the genomes of a set of known virus–host interaction pairs, S+, and generated a set of random VHPs that most likely do not interact, S−, as the training data for this study. Our objective was to develop a machine learning approach to predict the probability of interaction between a query VHP (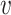 , b), denoted as P(I(
, b), denoted as P(I(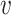 , b) = 1), where I(
, b) = 1), where I(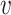 , b) denotes the interaction status of a virus
, b) denotes the interaction status of a virus 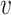 and a host b with value 1 indicating interaction and 0 indicating no interaction. In order to achieve the best performance, we comprehensively considered various factors that contribute to the interaction of a VHP (
and a host b with value 1 indicating interaction and 0 indicating no interaction. In order to achieve the best performance, we comprehensively considered various factors that contribute to the interaction of a VHP (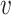 , b). First, if a virus is genetically close to viruses infecting a particular host, this virus is highly likely to infect the same host (28,29). On the other hand, if a virus infects a host, the virus should be genetically distant from the viruses that do not infect the host. Second, the similarity among hosts indicates the possibility of infection by the same virus (62,63). If a potential host belongs to the same taxon as the known host of the virus, then that host is likely to be infected by the virus. Third, the similarity between VHPs in terms of genomic signatures reflects the likelihood of interaction (24). If a virus genome is similar to a host genome in terms of the alignment-free k-mer usage pattern, the pair is predicted to have a high probability of interacting. Finally, the existence of virus–host shared CRISPR spacers and the alignment-based matches (i.e. BLASTn) is strong evidence of interaction.
, b). First, if a virus is genetically close to viruses infecting a particular host, this virus is highly likely to infect the same host (28,29). On the other hand, if a virus infects a host, the virus should be genetically distant from the viruses that do not infect the host. Second, the similarity among hosts indicates the possibility of infection by the same virus (62,63). If a potential host belongs to the same taxon as the known host of the virus, then that host is likely to be infected by the virus. Third, the similarity between VHPs in terms of genomic signatures reflects the likelihood of interaction (24). If a virus genome is similar to a host genome in terms of the alignment-free k-mer usage pattern, the pair is predicted to have a high probability of interacting. Finally, the existence of virus–host shared CRISPR spacers and the alignment-based matches (i.e. BLASTn) is strong evidence of interaction.
Altogether, virus–virus similarity, host–host similarity and virus–host similarity can be integrated to form a two-layer network connecting viruses and hosts. Thus, we constructed a VHP network where nodes are VHPs and edge weights are the pairwise similarities between VHPs. We developed an integrated network-based MRF approach that systematically and comprehensively integrates various types of features to predict interacting VHPs. The probability of a given VHP to be interactive is based on the characteristics of this VHP itself, and the connectivity between this VHP and its neighbor VHPs in the network. Intuitively, the characteristics of a VHP itself include alignment-free score, the fraction of alignment-based matches and the existence of shared CRISPR spacers. The connectivity between this VHP and other VHPs is defined based on the genome similarity between the virus and other viruses infecting the same host. The outline of the framework is demonstrated in Figure 1. The details of the models for this framework can be found in the ‘Materials and Methods’ section.
Figure 1.
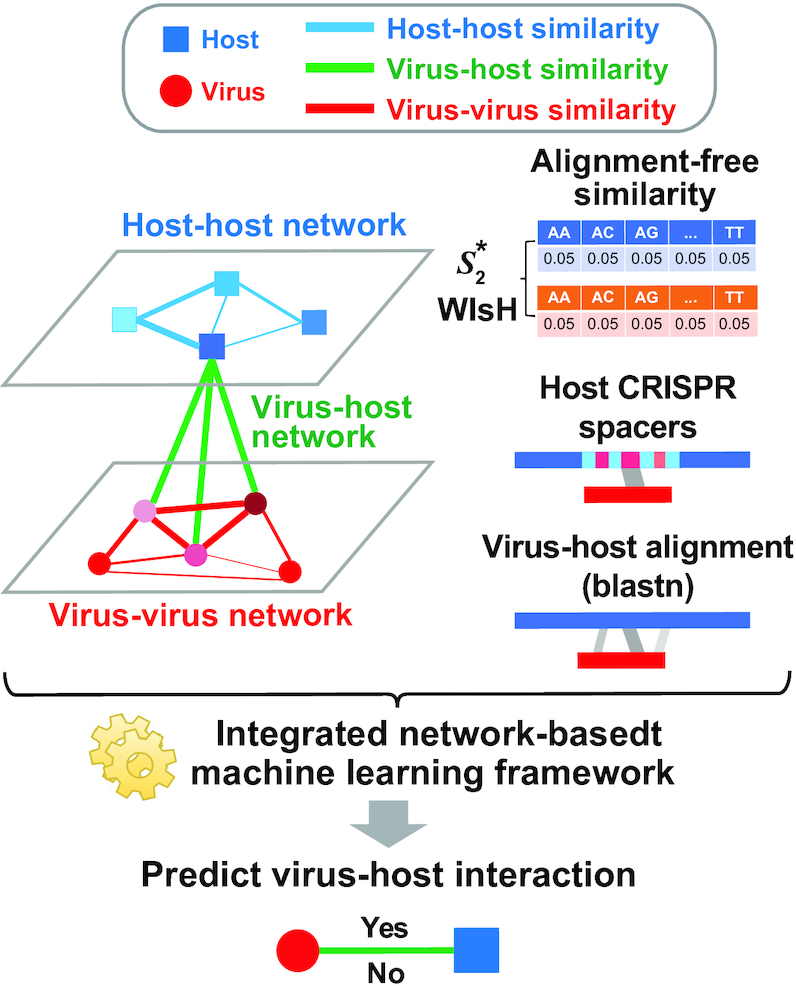
Overview of the network prediction framework. A novel two-layer network is constructed for representing virus–virus, host–host and virus–host similarities. Viruses (red circles) are connected based on sequence similarity (red edges). Similarly, hosts (blue squares) are connected based on sequence similarity (blue edges). The thickness of the edges indicates the degree of similarity. The interaction between a virus and host pair (green edges) can be predicted using multiple types of features: (i) the similarity between the virus and other viruses infecting the host; (ii) the similarity between the host and other hosts infected by the virus; (iii) the alignment-free sequence similarity between the virus and the host based on k-mer frequencies; (iv) the existence of shared CRISPR spacers between the virus and the host; and (v) alignment-based sequence matches between the virus and the host. Finally, a network-based machine learning model is used to integrate all different types of features and to predict the likelihood of the interaction of a VHP.
Feature scores are significantly different between positive and negative VHPs
We incorporated multiple types of features that contribute to the prediction of virus–host interactions. To assess the discriminatory power of each feature, we compared the distributions of the feature values between the virus–host interacting pairs and the non-interacting pairs. A set of 826 known virus–host interacting pairs was used as the positive set, and a set of the same number of randomly selected VHPs was used as the negative set. See the ‘Materials and Methods’ section for details of the data collection and the simulation of negative pairs. We used a one-sided t-statistic to test whether the feature values in the positive set are significantly higher or lower than the ones in the negative set.
First, the alignment-free similarity score 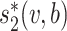 was used to measure the similarity between virus and host pairs, where
was used to measure the similarity between virus and host pairs, where 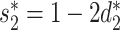 and the k-mer-based dissimilarity score
and the k-mer-based dissimilarity score 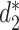 is defined in our previous work (24). The measure
is defined in our previous work (24). The measure 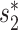 has an advantage over other classical similarity measures because of its precise correction of background noise, and has shown superior accuracy for predicting virus–host interactions (24). See the ‘Materials and Methods’ section for the definition of
has an advantage over other classical similarity measures because of its precise correction of background noise, and has shown superior accuracy for predicting virus–host interactions (24). See the ‘Materials and Methods’ section for the definition of 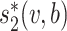 . The
. The 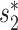 score had significantly higher values (P-value <2.2e−16, one-sided t-test) for positive VHPs than the negative pairs (Figure 2A). The mean
score had significantly higher values (P-value <2.2e−16, one-sided t-test) for positive VHPs than the negative pairs (Figure 2A). The mean 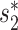 similarity score between positive pairs was 0.52, while the mean
similarity score between positive pairs was 0.52, while the mean 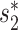 similarity between negative pairs was 0.24.
similarity between negative pairs was 0.24.
The WIsH score, proposed by Galiez et al. (25), is another alignment-free similarity measure for a VHP. It uses a log-likelihood score of a Markov chain model to measure similarity between viruses and hosts. We computed the WIsH scores for both positive and negative VHPs, and found that the WIsH scores for positive virus–host interacting pairs were significantly higher than those for the negative VHPs (P-value = 1e−10; Figure 2B). In fact, we observed that the WIsH and 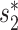 scores were highly correlated (Pearson correlation coefficient ρ = 0.85, P-value <2.2e−16). We predicted a VHP as interacting if one of the similarity measures,
scores were highly correlated (Pearson correlation coefficient ρ = 0.85, P-value <2.2e−16). We predicted a VHP as interacting if one of the similarity measures, 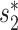 or WIsH, was above a threshold and, by changing the threshold, the corresponding receiver operating characteristic curve was plotted. The area under the receiver operating characteristic curve, which measures the discriminative ability between positive and negative pairs, was 0.91 for
or WIsH, was above a threshold and, by changing the threshold, the corresponding receiver operating characteristic curve was plotted. The area under the receiver operating characteristic curve, which measures the discriminative ability between positive and negative pairs, was 0.91 for 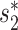 and 0.86 for WIsH (Additional File 1 in the Supplementary Data). Though the distinguishing power using WIsH was lower than that of
and 0.86 for WIsH (Additional File 1 in the Supplementary Data). Though the distinguishing power using WIsH was lower than that of 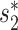 using complete genomes, WIsH was previously shown to be more effective than
using complete genomes, WIsH was previously shown to be more effective than 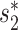 when predicting hosts of partial viral genomes (25). Therefore, we decided to use
when predicting hosts of partial viral genomes (25). Therefore, we decided to use 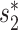 to measure virus–host alignment-free similarity when the length of viral sequence is close to the size of a complete genome, and to use WIsH to measure the virus–host similarity for short contigs.
to measure virus–host alignment-free similarity when the length of viral sequence is close to the size of a complete genome, and to use WIsH to measure the virus–host similarity for short contigs.
Second, for a given VHP (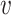 , b), we defined the similarity between a virus
, b), we defined the similarity between a virus 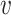 and other viruses infecting the host b, denoted as SV+(
and other viruses infecting the host b, denoted as SV+(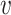 , b), and likewise, the similarity between virus
, b), and likewise, the similarity between virus 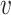 and other viruses not infecting the host b, denoted as SV−(
and other viruses not infecting the host b, denoted as SV−(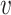 , b). See the ‘Materials and Methods’ section for the details of their definitions. We hypothesized that, for a true interacting VHP (
, b). See the ‘Materials and Methods’ section for the details of their definitions. We hypothesized that, for a true interacting VHP ( , b), other viruses that infect the same host b should exhibit high similarity to the virus
, b), other viruses that infect the same host b should exhibit high similarity to the virus  , resulting in a high SV+(
, resulting in a high SV+( , b). At the same time, other viruses not infecting the host b should have low similarity to the virus
, b). At the same time, other viruses not infecting the host b should have low similarity to the virus  , resulting in a low SV−(
, resulting in a low SV−(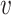 , b). For a non-interacting VHP, the above trend of SV+(
, b). For a non-interacting VHP, the above trend of SV+(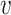 , b) and SV−(
, b) and SV−(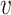 , b) should be opposite. Consistent with our hypothesis, SV+(
, b) should be opposite. Consistent with our hypothesis, SV+(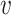 , b) scores were significantly higher for positive VHPs than negative pairs, and vice versa for SV−(
, b) scores were significantly higher for positive VHPs than negative pairs, and vice versa for SV−(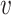 , b) scores (both P-values <2.2e−16; Figure 2C and D).
, b) scores (both P-values <2.2e−16; Figure 2C and D).
Third, we included information from CRISPR matches and alignment-based genome similarity between viruses and hosts. The CRISPR score was defined as the highest alignment score between the predicted CRISPR spacers in a host and a viral genome, and the alignment-based matching score was defined as the fraction of virus genome that significantly matches the host genome using blastn (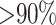 identity; see the ‘Materials and Methods’ section). Thus, for simplicity, we refer to the alignment-based matching score as the BLAST score. Both CRISPR and BLAST scores were significantly higher for the true interacting VHPs than the non-interacting pairs with P-values of 0.0001 and <2.2e−16 for one-sided t-tests, respectively. Figure 2E and F also shows the limited frequency of CRISPR and BLAST matches between viruses and hosts.
identity; see the ‘Materials and Methods’ section). Thus, for simplicity, we refer to the alignment-based matching score as the BLAST score. Both CRISPR and BLAST scores were significantly higher for the true interacting VHPs than the non-interacting pairs with P-values of 0.0001 and <2.2e−16 for one-sided t-tests, respectively. Figure 2E and F also shows the limited frequency of CRISPR and BLAST matches between viruses and hosts.
Integrated approach markedly increases host prediction accuracy
We integrated the multiple types of features proposed previously to predict virus–host interactions using a general framework of MRF, where the nodes were VHPs and edges were the similarities between the VHPs. We investigated the prediction accuracies of the newly developed integrated models in Equations (6) and (7) (see the ‘Materials and Methods’ section), and compared the accuracies with those using the individual features. The model in Equation (6) incorporates the network features including virus–virus similarities SV+ and SV−, the virus–host similarity 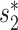 and the CRISPR score. The model in Equation (7) combines features in Equation (6) plus the BLAST scores. For each of the integrated models, we learned the parameters using the 826 positive and the same number of negative VHPs, and then tested the trained model on the remaining 1462 viruses for which their true hosts are known against 62 493 candidate hosts.
and the CRISPR score. The model in Equation (7) combines features in Equation (6) plus the BLAST scores. For each of the integrated models, we learned the parameters using the 826 positive and the same number of negative VHPs, and then tested the trained model on the remaining 1462 viruses for which their true hosts are known against 62 493 candidate hosts.
We assessed the prediction accuracies of the trained models using an independent set of 1462 viruses at different taxonomic levels, including genus, family, order, class and phylum. For each virus, we computed the prediction scores between this virus and all candidate hosts (n = 62 493) using the trained models, and predicted the host as the one having the highest prediction score. The prediction accuracy was calculated as the percentage of viruses whose predicted hosts had the same taxonomy as their respective known hosts. Host prediction accuracies were markedly higher for the integrated approach using network features and CRISPR scores than using 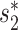 or CRISPR scores alone (Figure 3). For example, at the genus level, prediction accuracy was 31% and 43% when using
or CRISPR scores alone (Figure 3). For example, at the genus level, prediction accuracy was 31% and 43% when using 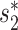 and CRISPR, respectively. Combining network similarity features together with CRISPR score (Equation 6) increased prediction accuracy to 59%, or a 1.4-fold increase.
and CRISPR, respectively. Combining network similarity features together with CRISPR score (Equation 6) increased prediction accuracy to 59%, or a 1.4-fold increase.
Figure 3.
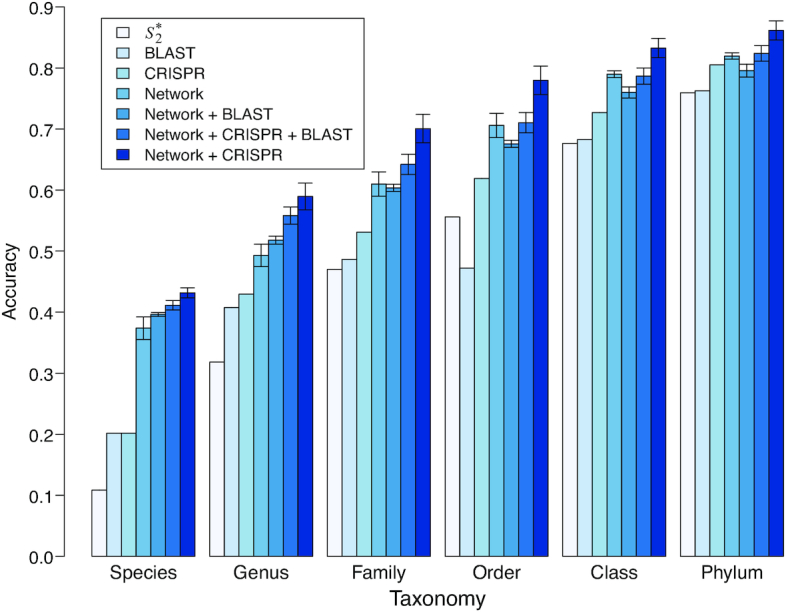
Prediction accuracies of the different approaches for 1462 viruses. Prediction accuracies for 1462 viral genomes whose true hosts are known against 62 493 candidate hosts, binned by taxonomic level. The first three bars show results using individual features of 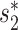 (v, b), CRISPR score or alignment-based similarity score (blastn), respectively. The remaining bars show results with integrated network models, trained using 826 positive and the same number of negative VHPs as in Figure 2. In order, these are the model in Equation (5) that incorporates the network-based features SV+(v, b) and SV−(v, b), alignment-free virus–host similarity
(v, b), CRISPR score or alignment-based similarity score (blastn), respectively. The remaining bars show results with integrated network models, trained using 826 positive and the same number of negative VHPs as in Figure 2. In order, these are the model in Equation (5) that incorporates the network-based features SV+(v, b) and SV−(v, b), alignment-free virus–host similarity 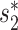 (v, b), in addition to the blastn scores (‘Network + BLAST’), the model in Equation (7) (‘Network + CRISPR + BLAST’), and the model in Equation (6) (‘Network + CRISPR’). Error bars for the network-based results depict 95% confidence intervals using 100 replicates of negative training sets (random VHPs).
(v, b), in addition to the blastn scores (‘Network + BLAST’), the model in Equation (7) (‘Network + CRISPR + BLAST’), and the model in Equation (6) (‘Network + CRISPR’). Error bars for the network-based results depict 95% confidence intervals using 100 replicates of negative training sets (random VHPs).
Alignment-based BLAST scores alone had a prediction accuracy of 41%, comparable to that based on CRISPR scores. However, incorporating BLAST into the network model in Equation (5) or Equation (6) does not yield a better performance than the model in Equation (6) (Figure 3). Therefore, the model in Equation (6) that incorporates the network features, virus–host similarity 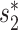 and CRISPR had the highest accuracy and was used in the subsequent host prediction applications. For the higher levels of taxonomy like family, order, class and phylum, the network-based integrated framework also achieved large improvements over the prediction accuracy of individual features, yielding 70%, 78%, 83% and 86% prediction accuracy, respectively. At the species level, the prediction accuracy is 43%. The estimated coefficients and the corresponding P-values of the features are shown in Table 1. All the coefficients had the expected signs that were consistent with the observations in Figure 2, and the statistical significance P-values for the coefficients were all <0.05.
and CRISPR had the highest accuracy and was used in the subsequent host prediction applications. For the higher levels of taxonomy like family, order, class and phylum, the network-based integrated framework also achieved large improvements over the prediction accuracy of individual features, yielding 70%, 78%, 83% and 86% prediction accuracy, respectively. At the species level, the prediction accuracy is 43%. The estimated coefficients and the corresponding P-values of the features are shown in Table 1. All the coefficients had the expected signs that were consistent with the observations in Figure 2, and the statistical significance P-values for the coefficients were all <0.05.
Table 1.
The estimated coefficients and corresponding P-values for host prediction features
| Model |
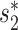
|
S WIsH | SV+ | SV− | S CRISPR | |
|---|---|---|---|---|---|---|
| Complete genomes | Coeff. | 16.41 | 4.44 | −27.38 | 0.13 | |
| using Equation (6)a | P-value | <2e−16 | <2e−16 | <2e−16 | 0.0002 | |
| Short contigs | Coeff. | 25.96 | 6.46 | −15.29 | 0.19 | |
| using Equation (8)b | P-value | <2e−16 | <2e−16 | <2e−16 | 0.0069 |
aResults for complete viral genomes using the network-based integrated model in Equation (6).
bResults for short viral contigs using the model in Equation (8).
‘Coeff.’ = coefficient. Since different negative training sets yielded slightly different estimated coefficients of the features, we show one example here.
Integrated approach improves host prediction accuracy of short viral sequences
Viral contigs assembled from metagenomic data often represent partial viral genomes. We tested an integrated model in Equation (8) that uses WIsH scores instead of 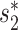 for measuring the alignment-free similarity between viruses and hosts. We evaluated the accuracy of the model for predicting the hosts of viral contigs at various lengths, and investigated the effect of viral sequence length on the prediction accuracy. To evaluate the performance of host prediction for short viral contigs, we randomly subsampled fragments of different lengths (1, 2, 5, 10 and 20 kb) from each of the 1462 viral genomes. For a given viral genome and a fixed contig length, we randomly chose a segment of fixed length uniformly from the genome. If the fixed length was longer than the size of the complete genome, we took the entire genome. This procedure was repeated 10 times for each contig length. We then computed all the features of the contigs using the same procedure as for the complete viral genome analyses, with the only difference being that
for measuring the alignment-free similarity between viruses and hosts. We evaluated the accuracy of the model for predicting the hosts of viral contigs at various lengths, and investigated the effect of viral sequence length on the prediction accuracy. To evaluate the performance of host prediction for short viral contigs, we randomly subsampled fragments of different lengths (1, 2, 5, 10 and 20 kb) from each of the 1462 viral genomes. For a given viral genome and a fixed contig length, we randomly chose a segment of fixed length uniformly from the genome. If the fixed length was longer than the size of the complete genome, we took the entire genome. This procedure was repeated 10 times for each contig length. We then computed all the features of the contigs using the same procedure as for the complete viral genome analyses, with the only difference being that 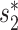 similarity was replaced with the WIsH score (25). The model was trained with the same set of 826 virus–host positive pairs and the same number of negative pairs using the same scheme as before by replacing
similarity was replaced with the WIsH score (25). The model was trained with the same set of 826 virus–host positive pairs and the same number of negative pairs using the same scheme as before by replacing 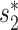 with the WIsH likelihood score. With the trained model, we predicted the hosts for all subsampled contigs. The results for different models on viral contigs of length 5 kb are shown in Figure 4. With WIsH score alone, the prediction accuracy at the genus level was 35%. Adding the network features SV+ and SV− improved the accuracy to 48%. Similar to the results for predicting complete viral genomes, the model in Equation (8) performed best (Figure 4). For viral contigs of length 5 kb, the model has 53% prediction accuracy at the genus level and 85% at the phylum level.
with the WIsH likelihood score. With the trained model, we predicted the hosts for all subsampled contigs. The results for different models on viral contigs of length 5 kb are shown in Figure 4. With WIsH score alone, the prediction accuracy at the genus level was 35%. Adding the network features SV+ and SV− improved the accuracy to 48%. Similar to the results for predicting complete viral genomes, the model in Equation (8) performed best (Figure 4). For viral contigs of length 5 kb, the model has 53% prediction accuracy at the genus level and 85% at the phylum level.
Figure 4.
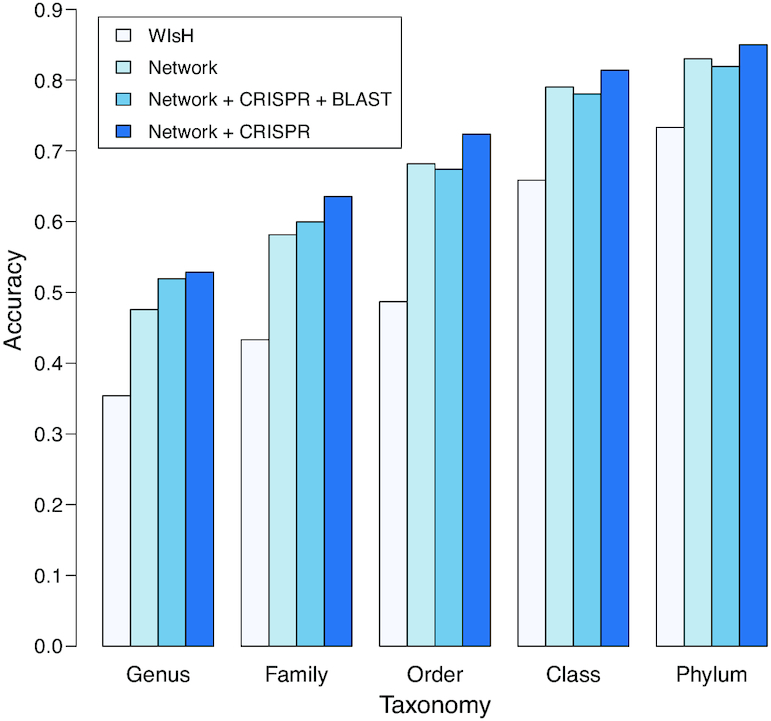
Prediction accuracies of the different approaches for viral contigs of length 5 kb. Prediction accuracies for viral contigs of length 5 kb, binned by taxonomic level. The first bar shows results using WIsH method alone, as in (25). The remaining bars show results with integrated network models, similar to Figure 3. All bars are calculated based on the average accuracies for 10 different sets of viral contigs.
The average prediction accuracies for each contig length are shown in Figure 5. Our model (solid lines) achieved a large improvement compared to the results of WIsH alone (dashed lines). For example, when the contig length was 20 kb, the prediction accuracy using our model was ∼19–26% higher than that of WIsH at the genus, family and order levels. As expected, the prediction accuracy of our model (solid lines) increases with contig lengths. For instance, at the genus level, the accuracy increased from 42% for 1 kb long contigs, 48% for 2 kb, to 53% for 5 kb, to 55% for 10 kb and to 57% for 20 kb (Figure 5). Given the results, we provide our framework with two models for host prediction: one for complete or nearly complete viral genomes using the model in Equation (6), and one for short viral contigs using the model in Equation (8).
Figure 5.

Prediction accuracies for contigs subsampled at various lengths from the 1462 virus genomes. Mean accuracies are shown at different taxonomic levels using WIsH scores only (dashed lines) or the integrated model in Equation (8) (solid line) that uses WIsH scores in place of 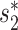 scores.
scores.
Thresholding on the prediction score further improves accuracy
In many situations, investigators are interested in making sure the predicted hosts are as accurate as possible, i.e. the predictions have high precision or low false discovery rate. Therefore, we investigated how the accuracy changes by thresholding on the predicted probability of interaction P(I(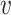 , b) = 1). In the above analysis, we predicted the host of every virus as the one with the highest score. However, sometimes the highest score was relatively low. For example, as shown in Figure 6, the highest prediction score among the 62 493 hosts for some viruses in the complete genome test set was as low as 0.31. Low scores may occur, for example, when the true host is not in the database of potential hosts. In order to improve the prediction accuracy, we can set a threshold such that host predictions are only made if the score is above that threshold. For instance, when a threshold was set at 0.95, there was an improvement of prediction accuracy at all taxonomic levels. Specifically at the genus level, accuracy was improved by
, b) = 1). In the above analysis, we predicted the host of every virus as the one with the highest score. However, sometimes the highest score was relatively low. For example, as shown in Figure 6, the highest prediction score among the 62 493 hosts for some viruses in the complete genome test set was as low as 0.31. Low scores may occur, for example, when the true host is not in the database of potential hosts. In order to improve the prediction accuracy, we can set a threshold such that host predictions are only made if the score is above that threshold. For instance, when a threshold was set at 0.95, there was an improvement of prediction accuracy at all taxonomic levels. Specifically at the genus level, accuracy was improved by 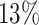 , from
, from 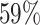 to
to 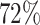 ; at the phylum level, accuracy was improved by 4%, from 86% to 90%.
; at the phylum level, accuracy was improved by 4%, from 86% to 90%.
Figure 6.
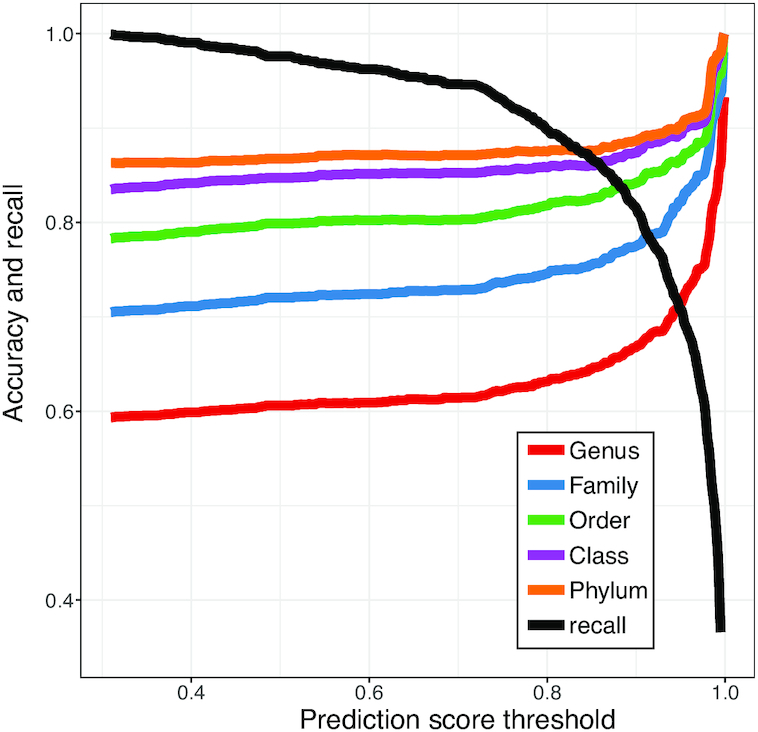
Improvement in host prediction by thresholding on the prediction score. By applying a given threshold, predictions were made only when the prediction score is above the threshold. Predictions were made using the whole genomes of 1462 viruses whose true hosts are known among 62 493 hosts as in Figure 3. The proportion of viruses that can be predicted (recall rate) decreases as the prediction accuracy at all levels increases.
Prediction accuracy varies for different viral families
Viruses from three major families, Siphoviridae, Myoviridae and Podoviridae, are highly represented in our evaluation dataset (42%, 24% and 18%, respectively). Previous host predictions with 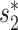 showed notable differences in prediction accuracy among these families (24). Therefore, we examined prediction accuracies using our model (Figure 7). We found that the Siphoviridae family of viruses in our dataset had generally higher prediction accuracy than other families of viruses, achieving 72% accuracy compared with the average accuracy of 59% for all types of viruses, consistent with previous results using the
showed notable differences in prediction accuracy among these families (24). Therefore, we examined prediction accuracies using our model (Figure 7). We found that the Siphoviridae family of viruses in our dataset had generally higher prediction accuracy than other families of viruses, achieving 72% accuracy compared with the average accuracy of 59% for all types of viruses, consistent with previous results using the 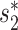 scores alone (24). The prediction accuracies for the different virus families with various thresholds on the prediction score are shown in Additional File 2 in the Supplementary Data. We also noticed that the top prediction scores for the Siphoviridae family of viruses are significantly higher than those for the other two families (Kolmogorov–Smirnov test, P-value <1e−15). The above observations may be explained by the fact that (i) Siphoviridae is the most abundant viral family in the training data (75%, n = 618) and (ii) siphoviruses typically have relatively narrow host ranges and podoviruses and myoviruses often have broader host ranges (64–66), though recent studies suggest that current isolation techniques may result in the under-representation of broad host range viruses and that the true host range of viruses is hard to define (62,67).
scores alone (24). The prediction accuracies for the different virus families with various thresholds on the prediction score are shown in Additional File 2 in the Supplementary Data. We also noticed that the top prediction scores for the Siphoviridae family of viruses are significantly higher than those for the other two families (Kolmogorov–Smirnov test, P-value <1e−15). The above observations may be explained by the fact that (i) Siphoviridae is the most abundant viral family in the training data (75%, n = 618) and (ii) siphoviruses typically have relatively narrow host ranges and podoviruses and myoviruses often have broader host ranges (64–66), though recent studies suggest that current isolation techniques may result in the under-representation of broad host range viruses and that the true host range of viruses is hard to define (62,67).
Figure 7.
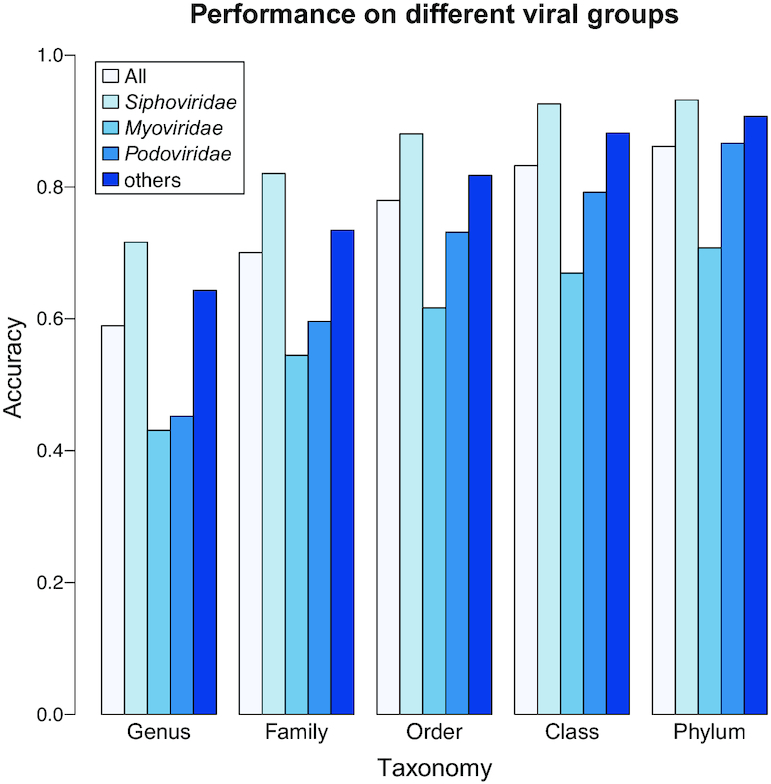
Differences in prediction accuracy across viral families. Prediction accuracies for different virus families within the order Caudovirales: Siphoviridae, Myoviridae and Podoviridae. For comparison, accuracies are shown for all viruses (‘all’) and for viruses outside of the Caudovirales or for which their virus families were not listed in the GenBank files (‘other’). Predictions were made using whole viral genomes with no thresholding.
To investigate whether the high host prediction accuracy for siphoviruses is due to their high abundance in the training set, we trained a new model only on podoviruses (n = 76) and myoviruses (n = 113), and tested the model on siphoviruses in the validation set (n = 607). Comparing the performance of this model with the model trained with the full training set, we found the difference in prediction accuracy is <1% for each taxonomic level, from the species to the phylum (Additional File 4 in the Supplementary Data). To further investigate the sensitivity of the model to the training data, we similarly trained a model excluding a certain group of viruses from the training set and evaluated the host prediction accuracy for that group of viruses in the validation set. The same procedure was conducted for several groups, including the other two major virus families (Myoviridae and Podoviridae) and groups of viruses infecting the common host taxonomic groups (Escherichia coli, Proteobacteria, Actinobacteria and Firmicutes). The overall decrease in host prediction accuracy for the excluded groups of viruses is on average 2.6%. The detailed results are provided in Additional File 4 in the Supplementary Data.
Prediction of the host of crAss-like phage ΦcrAss001
CrAssphage was first discovered through the cross-assembly of human fecal metagenomes and was originally published as an individual genome that is referred to as prototypical crAssphage (p-crAssphage) (19). Though crAssphage is ubiquitous in human gut samples and comprises up to 90% of the sequencing reads in some fecal viral metagenomes (19), little is known about the biological significance and the hosts of crAssphage, due to the difficulty of culturing crAssphage and the high divergence between crAssphage and known viruses. Different methods have been used to predict the hosts of crAssphage. Dutilh et al. (19) predicted its host as the phylum Bacteroidetes using the co-occurrence profile between crAssphage and 404 potential human gut bacteria hosts across 151 human gut metagenomes from the HMP. Ahlgren et al. (24) compared the alignment-free similarity between crAssphage and the potential hosts, and the genera Bacteroides, Coprobacillus and Fusobacterium were found to have significantly high similarity to crAssphage.
Recently, Shkoporov et al. (42) isolated a particular strain of crAssphage, ΦcrAss001, by enriching viral fraction gut samples on a collection of 54 bacteria strains from the human gut. They subsequently showed that ΦcrAss001 specifically infects only one of the 14 strains of Bacteroides tested, Bacteroides intestinalis 919/174. We first predicted the host of ΦcrAss001 using 22 species of the bacteria used to enrich ΦcrAss001 (and whose genomes are available) that span 4 phyla and 14 genera (Additional File 3 in the Supplementary Data). A B. intestinalis strain had the highest prediction score of 0.962, congruent with the experimental results of Shkoporov et al. (42). Alignment-based scores such as CRISPR and BLAST were all 0 and did not contribute to the prediction. The main contribution comes from the alignment-free similarity score 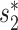 of 0.5 and the CRISPR signal. We then applied the integrated approach to predict the host of ΦcrAss001 using the large database of 62 493 host genomes and found that all of the top 25 predictions belong to the Bacteroidetes phylum, including 23 belonging to the genus Prevotella. ΦcrAss001 was classified as a genus VI crAssphage (42). Guerin et al. (68) previously hypothesized that genus VI crAss-like phages infect Prevotella based on the observation that these two genera of virus and host were both enriched in malnourished and healthy Malawian infants. Our host prediction of ΦcrAss001 is therefore consistent with this hypothesis.
of 0.5 and the CRISPR signal. We then applied the integrated approach to predict the host of ΦcrAss001 using the large database of 62 493 host genomes and found that all of the top 25 predictions belong to the Bacteroidetes phylum, including 23 belonging to the genus Prevotella. ΦcrAss001 was classified as a genus VI crAssphage (42). Guerin et al. (68) previously hypothesized that genus VI crAss-like phages infect Prevotella based on the observation that these two genera of virus and host were both enriched in malnourished and healthy Malawian infants. Our host prediction of ΦcrAss001 is therefore consistent with this hypothesis.
Host prediction for marine environmental viral genomes
Metagenomic sequencing has provided access to a broad range of viral genomes and has played an important role in studying uncharacterized marine viral genetic materials. Nishimura et al. (41) compiled a set of 1811 marine environmental viral genomes (EVGs) including those newly assembled from the Tara Ocean (6) and Osaka Bay viromes and previously reported EVGs (69–71). They predicted putative hosts of the EVGs based on the gene-based similarity between the EVGs and the cultured viral genomes with known hosts. In particular, they compiled another set of cultured viral genomes as a reference (RVGs) and created a proteomic tree for all EVGs and RVGs by the all-against-all distance matrix calculated from tBLASTx. They first assigned hosts by directly comparing the proteomic similarity between the EVGs and RVGs resulting in host assignment for 29 EVGs. They then constructed genus-level genomic operational taxonomic units (gOTUs) according to the proteomic tree. Based on the identification and phylogenetic analysis of various functional genes in EVGs and their closeness to related RVGs in the proteomic tree, they predicted the hosts of gOTUs at different host taxonomic levels (phylum to genus). In total, they predicted the hosts for 564 EVGs.
We used our integrated model in Equation (6) to predict the hosts for the 1811 EVGs using a set of 4034 marine bacteria as host candidates. We set a cutoff of 0.95 on the prediction score to ensure 90% prediction accuracy at the phylum level (Figure 6). With this cutoff, our model was able to make host predictions for 676 EVGs, among which 233 EVGs also had phylum-level host predictions by Nishimura et al. (see Additional File 6 in the Supplementary Data for the prediction results). Compared with the predictions of Nishimura et al., our method had consistent predictions for 172 (74%) out of the EVGs at the phylum level and 156 (77%) out of the 203 EVGs at the class level (only 203 EVGs have predictions by our method and Nishimura et al.). In particular, our predictions were consistent with the previous predictions for the entire group of 16 cyanobacteria viruses. For a group of viruses that Nishimura et al. predicted to infect Proteobacteria, our predictions agree with theirs in 24 out of 39 cases at the phylum level. For another group of 158 viruses that were previously predicted as Flavobacteriaceae (within the phylum Bacteroidetes) phages, our predictions were consistent with theirs for 127 viruses at the family level. Note that the inconsistency between our predictions and Nishimura et al. may due to the different choices of features used for prediction. Predictions of Nishimura et al. are based on the similarity between virus genomes, while our method uses not only the similarity between viruses, but also the CRISPR scores between virus and host genomes, which are direct evidence for interactions. In addition, our method was able to predict more hosts at lower taxonomic levels compared with the previous method. We had all 233 EVGs predicted at the order level or lower host taxonomic levels, a 9% increase in the number of EVGs that the previous method was able to predict.
For the 443 viruses whose hosts were not predicted previously and only predicted by our method, their predicted hosts include 4 phyla and 22 genera. In particular, we discovered 11 viruses infecting 8 novel host genera that are absent from the dataset of 2288 isolate virus genomes (Additional File 6 in the Supplementary Data).
Host prediction for metagenomic viral contigs from various habitats
Paez-Espino et al. (38) analyzed over 3000 geographically diverse metagenomic samples and identified 125 842 putative metagenomic viral contigs of median length 11 kb, revealing the extended viral genetic diversity in various environments (38). In the original prediction, the metagenomic viral contigs and other 2536 isolated contigs were first clustered into viral groups or singletons. They predicted the hosts of the viral contigs using a series of analyses including projecting the isolate viral host information onto viral groups, matching viral contigs to a database of 3.5 million CRISPR spacers found in prokaryotic genomes and identifying tRNA sequences in corresponding hosts. The analysis predicted hosts for 9992 (7.7%) viral contigs. To evaluate our integrated approach for host prediction, we first used our method in Equation (8) to predict the hosts of those putative metagenomic viral contigs. We then compared our predictions with those of Paez-Espino et al. by concentrating on 5105 metagenomic contigs whose previously predicted host families were present in our host database and having a prediction score above 0.95. Our predictions were consistent with the vast majority of the original predictions, having 96% consistency at the phylum level (Table 2). Our predictions matched the previous predictions at an even higher rate (97% at the phylum level) for 62.7% of viruses whose hosts were previously inferred based on direct evidence of CRISPR spacer matches or tRNA matches to the hosts. For viruses whose hosts were inferred indirectly based on the hosts of other viruses in the same viral groups, our predicted hosts had 93% consistency with those based on the previous method at the phylum level. Thus, the inconsistent predictions mostly occurred for the viruses whose hosts were previously inferred based on viral group membership. For those viruses with inconsistent predictions, 88% of our predictions had significant network scores (>95% percentile), 86% had significant WIsH scores and 43% had significant CRISPR scores.
Table 2.
Proportions of congruent predictions for viral contigs between our method and those in Paez-Espino et al. (38)
| Genus | Family | Order | Class | Phylum | |
|---|---|---|---|---|---|
| Overalla | 82% | 86% | 90% | 90% | 96% |
| Extensive predictions onlyb | 75% | 78% | 82% | 82% | 93% |
| Excluding extensive predictionsc | 86% | 91% | 95% | 95% | 97% |
aCalculated based on all 5105 metagenomic viral contigs.
bCalculated based on 3203 metagenomic viral contigs whose predictions were previously inferred indirectly from group membership instead of direct evidence.
cCalculated based on 1902 metagenomic viral contigs whose previous predictions were inferred directly by CRISPR spacer matches or tRNA matches.
We then predicted the hosts for the remaining available contigs that were not predicted in Paez-Espino et al. (n = 101 343; note not all of the contigs from Paez-Espino et al. are accessible at IMG/VR). Viruses were parsed by the type of sample from which they were obtained (human-associated, marine and all other environments/sample types) and predictions were made against collections of host genomes corresponding to the sample type (human-related genomes, n = 9097; marine genomes, n = 4034; or all 62 493 host genomes, respectively). This resulted in 7653, 12 014 and 8013 viral contigs with prediction scores above 0.95 (Additional Files 7–9 in the Supplementary Data) or 27 680 viral contigs in sum. In combination with viral contigs with overlapping predictions by Paez-Espino et al., we were able to make confident host predictions for 27% of all the remaining viral contigs, representing 2.7-fold more host predictions than previously by Paez-Espino et al.
We analyzed more specifically the predicted hosts for contigs with length ≥10 kb and for which ≥90% of their genes belong to known viral protein families (a criterion used in the original paper). There were 545 contigs from human-associated samples that met the above criterion, and we restricted our host predictions to 9097 human-associated bacterial genomes. In total, 173 human-associated viral contigs were successfully predicted by our method with a score above 0.95 (Additional File 10 in the Supplementary Data). The predicted hosts of these 173 viral contigs belonged to 12 host genera. In particular, we discovered 24 viral contigs predicted to infect four host genera that have no known infecting viruses. To study the virus diversity within those hosts, we clustered the 24 viral contigs based on their percentage of shared genes using the UPGMA hierarchical clustering method (Additional File 11 in the Supplementary Data). Some viruses infecting the same host genus were found in the same habitat. For example, all three viruses predicted to infect Prevotella were found in human tongue dorsum; the two viruses predicted to infect Neisseria were found in human supragingival plaque. On the other hand, the 18 viruses predicted to infect Veillonella were found in human tongue dorsum, throat and saliva, probably indicating a higher viral diversity in this host genus. Meanwhile, the large cluster of 10 viruses of host genus Veillonella was from different samples in multiple studies (as assessed by contig IDs, WUGC, Baylor and LANL representing different studies), indicating those VHPs were common across individuals.
Similarly, we applied our method to a set of 558 marine viral contigs that were not predicted by Paez-Espino et al. using the same criteria as above. Prediction was restricted to the set of 4034 marine hosts defined previously by Ahlgren et al. Our model predicted hosts for 160 viral contigs using a score threshold of 0.95. The predicted hosts belonged to four host genera (Additional File 12 in the Supplementary Data). In particular, the newly identified VHPs expanded the universe of known Cellulophaga viral diversity, a nascent marine heterotrophic model system. Previously, Holmfeldt et al. (72), by sequencing 31 viral isolates, demonstrated the existence of several viral genera associated with this marine group. Here, we found additional 102 viral contigs that putatively infect Cellulophaga. Using the same gene-based method for hierarchical clustering as in (72), the newly discovered 102 viruses clustered into multiple groups, including one having 31 contigs (group A) and one having 17 contigs (group B), which are separate from the group containing the 31 known isolates (Additional File 13 in the Supplementary Data). Overall, we identified at least three novel genera with each having >10 viral contigs, representing a sizable increase from the previously known diversity. Genera were defined, for consistency as in Holmfeldt et al., as pairs of genomes sharing >40% of their gene content. Those new virus groups were found in multiple locations such as the Delaware Coast, Pacific Ocean and North Sea, indicating their ubiquity and potential impacts on communities of Cellulophaga, an important degrader of complex organic matter. In addition, our method predicted 49 viruses as cyanobacterial phages (cyanophages) infecting Prochlorococcus, a group of globally abundant marine cyanobacteria (73). We independently confirmed that 33 of these are actually cyanophages based on significant nucleotide or protein similarity to cyanophage isolate genomes (≥70% nucleotide identity for ≥10% of the contig or ≥50% of proteins on the contig shared ≥40% identity to cyanophage proteins). The remaining 16 contigs thus represent potentially novel lineages that have no significant nucleotide similarity to known cyanophage isolates. This showcases both the diversity of virus–host interactions and the power of our method to capture groups with relatively few known representatives.
Computational cost
For a set of 1500 complete viral genomes, the prediction requires no more than 16 GB of memory for host predictions. However, due to the implementation of WIsH score, it requires up to 100 GB for the same size of query viral contigs. In practice, we recommend analyzing the viral contigs in smaller groups at a time if the memory is a major constrain. Using an eight-core E5-2640v3 CPU, the analysis takes <1 h for 1500 complete genomes and <4 h for the same size of viral contigs.
DISCUSSION
The interactions between virus and prokaryotic hosts play important roles in human health and ecosystems. Millions of new viruses have been identified using high-throughput metagenomic sequencing technologies, but little is known about their biological functions and the prokaryotic hosts with which they interact. We developed a network-based integrated framework for predicting the hosts of prokaryotic viruses. The new method provides a sizable improvement on prediction accuracy compared with previous methods by integrating multiple measures for informing host prediction. Based on the evaluation of the methods using a large benchmark dataset containing 1462 viruses and 62 493 hosts, the method achieves 59% and 86% prediction accuracy at the genus and phylum levels, respectively, yielding 16% and 6% improvements at the genus and phylum levels compared to the highest accuracy achieved by previous single methods.
The novel two-layer network of virus–virus, host–host and virus–host genomic similarity lays the foundation for this method. The employment of a two-layer network is inspired by underlying biological phenomena. First, it is observed that genetically similar viruses tend to infect closely related hosts (62,63). So, the host of a new virus can be partly inferred based on the similarity to related viruses with known hosts. Similarly, the host of new viruses could potentially be inferred through similarity of hosts. Second, because viruses depend on the cellular machinery of their host to replicate, viruses often share highly similar patterns in codon usage or short nucleotide words with their hosts. The host of a new virus can be predicted using nucleotide word similarity between the virus and candidate hosts (11,20,24). Thus, the two-layer network model is a natural formulation of the biological relationships described above. Despite the fact that the viruses in our current database only have one reported host for each virus such that host–host network connections cannot be incorporated into the prediction model, the novel two-layer network can be fully implemented in the future as multiple hosts of viruses are revealed.
Multiple types of features, including shared sequences between host CRISPR spacers and viral genomes and virus–host BLAST matches, combined with the network-based features, were tested in the integrated framework for host prediction. The CRISPR and BLAST features are based on the biological process that some viruses and their hosts share a portion of their genomes due to CRISPR defense systems, horizontal gene transfer or prophage integration. Although these features have been investigated individually in previous studies (20,24,25,36), this is the first time that multiple types of features have been integrated into a unified framework for virus–host prediction. We interestingly found that addition of the BLAST feature did not significantly improve over the model that included CRISPR and k-mer frequency similarity, possibly because BLAST information is incorporated in informative CRISPR matching feature results. In the future, more sophisticated and sensitive approaches, beyond simple BLAST searches, could be developed for identifying genes shared between hosts and their phage via horizontal gene transfer. Our results show that the integrated method combining multiple features achieves a higher prediction accuracy than use of individual types of information.
Our model also markedly improved the host prediction accuracy on shorter viral fragments at all taxonomic levels when compared to WIsH (25), a recently developed probabilistic method for predicting hosts of viral contigs. Our method was able to obtain 57%, 55% and 53% prediction accuracies at the genus level for 20, 10 and 5 kb sequence lengths, respectively. The prediction accuracies for 20, 10 and 5 kb contigs were all above 84% at the phylum level.
Setting a minimum threshold for making predictions led to a notable improvement in accuracy. We also investigated the host prediction accuracy for different groups of viruses. Specifically, our observations indicate that viruses in the Siphoviridae family have higher prediction accuracy than the other Caudovirales families, consistent with the fact that siphoviruses tend to have a narrower range of target hosts (65,66). Likewise, restricting the possible hosts from all available prokaryotic genomes to a focused set of relevant microbes can help improve prediction accuracy, as was the case of predicting hosts of human-associated viruses using the 9097 human-related host genomes and predicting marine viruses using 4034 marine host genomes.
Our model was trained on a selected set of known VHPs, mostly represented by well-studied virus–host systems (e.g. E. coli viruses). Therefore, it was important to assess the sensitivity of our approach to the sets of viruses used for model training. The model was tested by excluding several groups of viruses, either by virus family (Myoviridae, Podoviridae, Siphoviridae) or by the taxonomic group of hosts they infect and then assessing accuracies for predicting the hosts of those groups of viruses (Additional File 4 in the Supplementary Data). Prediction accuracies were largely similar when using models trained with all available or the restricted sets of viruses and hosts, strongly supporting that our integrated approach can be extended to make predictions on novel groups of viruses. Indeed, in the applications above, we make confident new predictions for viruses for which their predicted host taxa are not represented in the training dataset. We conjecture that the applicability of this approach to novel viruses reflects that the features used and their underlying molecular processes are common across viral groups. In particular, CRISPR defense systems have been found across many prokaryotic phyla, and more importantly, the mechanisms and thus the molecular signals underlying the CRISPR defense systems are conserved.
We utilized our model to predict the host of a new strain of crAss-like phages, ΦcrAss001. Until the recent isolation of ΦcrAss001, the host of crAss-like phages was unknown, but surmised to be Bacteroidetes based on bioinformatic analyses (19). It was recently isolated and was found to infect B. intestinalis among a set of 54 strains belonging to 22 bacterial species (42). Our computational prediction for the host of ΦcrAss001 against the 22 species for which genomes were available is consistent with the culture-based results. When we predicted its host against the 62 493 candidate genomes, the genus Prevotella within the Bacteroidetes phylum was the top predicted host. Although this genus is different from the experimentally determined host, the prediction of Prevotella is consistent with the hypothesis of Guerin et al. (68) that genus VI crAss-like phages, to which crAss001 belongs, infect Prevotella.
We also applied our method to predict hosts for viruses in two large-scale metagenomic datasets, one focusing on marine viral genomes such as those discovered in Tara Oceans, and the other including viral contigs in over 3000 geographically diverse metagenomic samples including marine and HMP samples. Our predictions had high consistency with previous predictions made using simpler methods such as CRISPR or tRNA matches or gene-based similarity to known reference viruses. More importantly, our method greatly increased the number of viruses for which predictions could be made, nearly 3-fold more viruses than by Paez-Espino et al. These predictions were made using a minimum score threshold of 0.95, with a false discovery rate of <10% for nearly complete genomes and contigs of length >10 kb at the phylum level. The newly predicted VHPs revealed viruses for hosts without known infecting viruses, and also expanded the diversity of viruses for hosts with known isolate viruses, showcasing the usefulness of our method in expanding knowledge of hosts in both ways.
A major advantage of our network-based integrated framework is that it can be easily extended to incorporate more meaningful features that can better inform virus–host interactions in the future. Virus–host co-abundance profiles have been shown to provide some evidence of virus–host interactions (74,75), but Edwards et al. (20) suggested that its performance on host prediction was relatively poor compared to other measures such as CRISPR and sequence homology. Coenen et al. (76) also showed that virus–host correlations are poor predictors of virus–host interactions. Our preliminary analysis of incorporating such co-abundance data as a feature likewise showed the model did not benefit from adding the co-abundance feature (see Additional File 14 in the Supplementary Data). In general, co-abundance can be a misleading feature because virus–host interactions may not always yield positive or negative correlations depending on the complexity of virus lifestyles (e.g. lytic versus lysogenic) (77). In fact, we noticed that the feature coefficient for co-abundance when incorporated into the model was not statistically significant, indicating that the co-abundance cannot consistently be a useful predictor. Moreover, virus–host interactions are dynamic with delays and fluctuate over time, while metagenomic sampling only captures the community at a single time point. Also, the interactions can be nonlinear because of the complicated many-to-many virus–host networks (76). Likewise, non-specific hosts and viruses can exhibit spurious correlations due to the computational bias in terms of the compositional data where the abundance vector is constrained to a constant sum. Similarly, hosts may be incorrectly predicted to infect certain viruses because their hosts coincidentally share similar niches and dynamics. Significant co-abundance between a virus and a host nonetheless is consistent with and can support in some cases the discovery of a true virus–host interaction, but co-abundance evidence alone should be taken with caution. Although we do not exclude the possibility that co-abundance could be useful under certain environments or for certain types of viruses, it is not likely that a simple co-abundance measure based on non-time series samples can well describe the virus–host dynamics in general. More sophisticated model-based approaches that utilize virus and host abundances for host prediction could in theory be incorporated in our model in the future.
If other promising predictive virus–host features are discovered in the future, these can easily be incorporated into our framework. As noted above, inclusion of the BLAST feature did not significantly improve the prediction model. Simple nucleotide BLAST results, however, may not be best suited for detection of genes shared between cross-infecting viruses and their hosts. The discovery of auxiliary metabolic genes (AMGs) in viruses has emerged as a valuable means to connect viruses to their hosts (16,78–80). Protein-based homology searches or phylogenetic-based detection of AMGs may be more informative means for host prediction, and further development and incorporation of an improved AMG matching feature in our model framework could further improve host prediction.
Sequence-based and alignment-based measures such as CRISPR and BLAST scores generally have limited availability, but can provide solid evidence for virus–host interactions when such signals are present. On the other hand, alignment-free 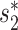 similarity can be computed for any VHPs, but may not always perform as well as CRISPR and BLAST. We compared the prediction accuracies for
similarity can be computed for any VHPs, but may not always perform as well as CRISPR and BLAST. We compared the prediction accuracies for 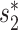 score and BLAST score when the hosts belonging to the true host genus of the viruses are removed from the candidates. The result showed that when the specific hosts were removed, the prediction accuracy for BLAST at the family level decreased markedly to 0.20, while the accuracy for
score and BLAST score when the hosts belonging to the true host genus of the viruses are removed from the candidates. The result showed that when the specific hosts were removed, the prediction accuracy for BLAST at the family level decreased markedly to 0.20, while the accuracy for 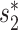 was 0.32 (Additional File 15 in the Supplementary Data). Therefore, alignment-based methods depend heavily on the existence of the true host in the database, and they can perform much worse than the alignment-free methods for predicting hosts of new viruses when the true host genus is not in the host candidate set. These results highlight again how the integrated framework combining both alignment-based and alignment-free features helps to complement the two types of methods and improve the overall prediction accuracy.
was 0.32 (Additional File 15 in the Supplementary Data). Therefore, alignment-based methods depend heavily on the existence of the true host in the database, and they can perform much worse than the alignment-free methods for predicting hosts of new viruses when the true host genus is not in the host candidate set. These results highlight again how the integrated framework combining both alignment-based and alignment-free features helps to complement the two types of methods and improve the overall prediction accuracy.
Although the new model makes sizable improvements over existing methods for both complete viral genomes and viral contigs at different taxonomic levels, the prediction accuracy at the genus level is still 59% for complete genomes and 55% for 10 kb contigs. It is expected that with an increased dataset of hosts and virus–host interactions for training our models, the prediction accuracy of our method will further increase. Our host dataset will be gradually updated to include more newly discovered VHPs for training and testing. However, we note that prediction accuracy at the phylum level is already very high (∼90%). Since there are many prokaryotic phyla (>75%) for which their viruses have yet to be identified, our tool is promising to greatly expand characterization of novel groups of viruses.
In summary, our novel network-based integrated approach demonstrates how integration of multiple features informative of virus–host interactions significantly improves host prediction than any single feature. Application of our method to a few datasets of metagenomically assembled contigs demonstrates the strong prediction ability of the model—yielding predictions largely congruent with previous methods but more importantly generating many more host predictions and identifying novel virus–host interactions than previous approaches. This approach will be valuable for identifying the putative hosts of newly discovered viral genomes, particularly for the flood of new viral metagenomic data currently being generated. The flexible nature of our prediction framework also has the potential to be updated as new computational theories and biological understanding in virus–host interactions become available.
DATA AVAILABILITY
Accession numbers for viral contigs in the real data studies can be found in the ‘Datasets’ section. All other relevant data for training and testing the model and code are available at https://github.com/WeiliWw/VirHostMatcher-Net.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Michael S. Waterman at the University of Southern California (USC) for helpful discussions. Dr Yang Lu and Dr Mengge Zhang at USC participated in the discussions and some of the calculations at the beginning of the project.
Notes
Present address: Jie Ren, Google Inc., Mountain View, CA, USA.
Contributor Information
Weili Wang, Quantitative and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
Jie Ren, Quantitative and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
Kujin Tang, Quantitative and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
Emily Dart, Biology Department, Clark University, Worcester, MA 01610, USA.
Julio Cesar Ignacio-Espinoza, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Jed A Fuhrman, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Jonathan Braun, Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
Fengzhu Sun, Quantitative and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
Nathan A Ahlgren, Biology Department, Clark University, Worcester, MA 01610, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institutes of Health [R01GM120624, 1R01GM131407, R01DK085691, P01DK046763]; National Science Foundation [DMS-1518001]; Gordon and Betty Moore Foundation Marine Microbiology Initiative [GBMF3779]; Simons Foundation Collaboration on Computational Biogeochemical Modeling of Marine Ecosystems/CBIOMES [549943 to JAF]; USC Provost Fellowship (in part) [to J.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Breitbart M., Rohwer F.. Here a virus, there a virus, everywhere the same virus?. Trends Microbiol. 2005; 13:278–284. [DOI] [PubMed] [Google Scholar]
- 2. Breitbart M., Salamon P., Andresen B., Mahaffy J.M., Segall A.M., Mead D., Azam F., Rohwer F.. Genomic analysis of uncultured marine viral communities. Proc. Natl Acad. Sci. U.S.A. 2002; 99:14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fierer N., Breitbart M., Nulton J., Salamon P., Lozupone C., Jones R., Robeson M., Edwards R.A., Felts B., Rayhawk S. et al.. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microb. 2007; 73:7059–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hurwitz B.L., Sullivan M.B.. The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One. 2013; 8:e57355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waller A.S., Yamada T., Kristensen D.M., Kultima J.R., Sunagawa S., Koonin E.V., Bork P.. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014; 8:1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brum J.R., Ignacio-Espinoza J.C., Roux S., Doulcier G., Acinas S.G., Alberti A., Chaffron S., Cruaud C., De Vargas C., Gasol J.M. et al.. Patterns and ecological drivers of ocean viral communities. Science. 2015; 348:1261498. [DOI] [PubMed] [Google Scholar]
- 7. Minot S., Sinha R., Chen J., Li H., Keilbaugh S.A., Wu G.D., Lewis J.D., Bushman F.D.. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011; 21:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mirzaei M.K., Maurice C.F.. Ménage à trois in the human gut: interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017; 15:397–408. [DOI] [PubMed] [Google Scholar]
- 9. Hannigan G.D., Duhaime M.B., Koutra D., Schloss P.D.. Biogeography and environmental conditions shape bacteriophage–bacteria networks across the human microbiome. PLoS Comput. Biol. 2018; 14:e1006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gómez P., Buckling A.. Bacteria–phage antagonistic coevolution in soil. Science. 2011; 332:106–109. [DOI] [PubMed] [Google Scholar]
- 11. Roux S., Hallam S.J., Woyke T., Sullivan M.B.. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife. 2015; 4:e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Norman J.M., Handley S.A., Baldridge M.T., Droit L., Liu C.Y., Keller B.C., Kambal A., Monaco C.L., Zhao G., Fleshner P. et al.. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015; 160:447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reyes A., Blanton L.V., Cao S., Zhao G., Manary M., Trehan I., Smith M.I., Wang D., Virgin H.W., Rohwer F. et al.. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl Acad. Sci. U.S.A. 2015; 112:11941–11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mills S., Shanahan F., Stanton C., Hill C., Coffey A., Ross R.P.. Movers and shakers: influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes. 2013; 4:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Srinivasiah S., Bhavsar J., Thapar K., Liles M., Schoenfeld T., Wommack K.E.. Phages across the biosphere: contrasts of viruses in soil and aquatic environments. Res. Microbiol. 2008; 159:349–357. [DOI] [PubMed] [Google Scholar]
- 16. Roux S., Brum J.R., Dutilh B.E., Sunagawa S., Duhaime M.B., Loy A., Poulos B.T., Solonenko N., Lara E., Poulain J. et al.. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature. 2016; 537:689–693. [DOI] [PubMed] [Google Scholar]
- 17. Rohwer F., Prangishvili D., Lindell D.. Roles of viruses in the environment. Environ. Microbiol. 2009; 11:2771–2774. [DOI] [PubMed] [Google Scholar]
- 18. Cann A.J., Fandrich S.E., Heaphy S.. Analysis of the virus population present in equine faeces indicates the presence of hundreds of uncharacterized virus genomes. Virus Genes. 2005; 30:151–156. [DOI] [PubMed] [Google Scholar]
- 19. Dutilh B.E., Cassman N., McNair K., Sanchez S.E., Silva G.G., Boling L., Barr J.J., Speth D.R., Seguritan V., Aziz R.K. et al.. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014; 5:4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edwards R.A., McNair K., Faust K., Raes J., Dutilh B.E.. Computational approaches to predict bacteriophage–host relationships. FEMS Microbiol. Rev. 2016; 40:258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J., Gao Y., Zhao F.. Phage–bacteria interaction network in human oral microbiome. Environ. Microbiol. 2016; 18:2143–2158. [DOI] [PubMed] [Google Scholar]
- 22. Burstein D., Sun C.L., Brown C.T., Sharon I., Anantharaman K., Probst A.J., Thomas B.C., Banfield J.F.. Major bacterial lineages are essentially devoid of CRISPR–Cas viral defence systems. Nat. Commun. 2016; 7:10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grissa I., Vergnaud G., Pourcel C.. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007; 8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahlgren N.A., Ren J., Lu Y.Y., Fuhrman J.A., Sun F.. Alignment-free d2* oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017; 45:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galiez C., Siebert M., Enault F., Vincent J., Söding J.. WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics. 2017; 33:3113–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carbone A. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 2008; 66:210–223. [DOI] [PubMed] [Google Scholar]
- 27. Gouy M., Gautier C.. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982; 10:7055–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Villarroel J., Kleinheinz K.A., Jurtz V.I., Zschach H., Lund O., Nielsen M., Larsen M.V.. HostPhinder: a phage host prediction tool. Viruses. 2016; 8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang M., Yang L., Ren J., Ahlgren N.A., Fuhrman J.A., Sun F.. Prediction of virus–host infectious association by supervised learning methods. BMC Bioinformatics. 2017; 18:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng M., Zhang K., Mehta S., Chen T., Sun F.. Prediction of protein function using protein–protein interaction data. J. Comput. Biol. 2003; 10:947–960. [DOI] [PubMed] [Google Scholar]
- 31. Jansen R., Yu H., Greenbaum D., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M.. A Bayesian networks approach for predicting protein–protein interactions from genomic data. Science. 2003; 302:449–453. [DOI] [PubMed] [Google Scholar]
- 32. Jiang R., Gan M., He P.. Constructing a gene semantic similarity network for the inference of disease genes. BMC Syst. Biol. 2011; 5:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Freudenberg J., Propping P.. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002; 18:S110–S115. [DOI] [PubMed] [Google Scholar]
- 34. Zhang W., Sun F., Jiang R.. Integrating multiple protein–protein interaction networks to prioritize disease genes: a Bayesian regression approach. BMC Bioinformatics. 2011; 12:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng F., Liu C., Jiang J., Lu W., Li W., Liu G., Zhou W., Huang J., Tang Y.. Prediction of drug–target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012; 8:e1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shapiro J.W., Putonti C.. Gene Co-occurrence Networks Reflect Bacteriophage Ecology and Evolution. mBio. 2018; 9:e01870-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lima-Mendez G., Van Helden J., Toussaint A., Leplae R.. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008; 25:762–777. [DOI] [PubMed] [Google Scholar]
- 38. Paez-Espino D., Eloe-Fadrosh E.A., Pavlopoulos G.A., Thomas A.D., Huntemann M., Mikhailova N., Rubin E., Ivanova N.N., Kyrpides N.C.. Uncovering Earth’s virome. Nature. 2016; 536:425–430. [DOI] [PubMed] [Google Scholar]
- 39. Wu G.A., Jun S.-R., Sims G.E., Kim S.-H.. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. Proc. Natl Acad. Sci. U.S.A. 2009; 106:12826–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Q., Jun S.-R., Leuze M., Ussery D., Nookaew I.. Viral phylogenomics using an alignment-free method: a three-step approach to determine optimal length of k-mer. Sci. Rep. 2017; 7:40712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishimura Y., Watai H., Honda T., Mihara T., Omae K., Roux S., Blanc-Mathieu R., Yamamoto K., Hingamp P., Sako Y. et al.. Environmental viral genomes shed new light on virus–host interactions in the ocean. mSphere. 2017; 2:e00359-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shkoporov A.N., Khokhlova E.V., Fitzgerald C.B., Stockdale S.R., Draper L.A., Ross R.P., Hill C.. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018; 9:4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li S.Z. Markov random field models in computer vision. European Conference on Computer Vision. 1994; Berlin: Springer; 361–370. [Google Scholar]
- 44. Song K., Ren J., Zhai Z., Liu X., Deng M., Sun F.. Alignment-free sequence comparison based on next-generation sequencing reads. J. Comput. Biol. 2013; 20:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song K., Ren J., Reinert G., Deng M., Waterman M.S., Sun F.. New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing. Brief. Bioinform. 2014; 15:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wan L., Reinert G., Sun F., Waterman M.S.. Alignment-free sequence comparison (II): theoretical power of comparison statistics. J. Comput. Biol. 2010; 17:1467–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reinert G., Chew D., Sun F., Waterman M.S.. Alignment-free sequence comparison (I): statistics and power. J. Comput. Biol. 2009; 16:1615–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ren J., Song K., Deng M., Reinert G., Cannon C.H., Sun F.. Inference of Markovian properties of molecular sequences from NGS data and applications to comparative genomics. Bioinformatics. 2015; 32:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horvath P., Barrangou R.. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010; 327:167–170. [DOI] [PubMed] [Google Scholar]
- 50. Bland C., Ramsey T.L., Sabree F., Lowe M., Brown K., Kyrpides N.C., Hugenholtz P.. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007; 8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 52. Sharon I., Battchikova N., Aro E.-M., Giglione C., Meinnel T., Glaser F., Pinter R.Y., Breitbart M., Rohwer F., Béjà O.. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J. 2011; 5:1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F.. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017; 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roux S., Enault F., Hurwitz B.L., Sullivan M.B.. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roux S., Faubladier M., Mahul A., Paulhe N., Bernard A., Debroas D., Enault F.. Metavir: a web server dedicated to virome analysis. Bioinformatics. 2011; 27:3074–3075. [DOI] [PubMed] [Google Scholar]
- 56. Kim D., Song L., Breitwieser F.P., Salzberg S.L.. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016; 26:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 58. Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N.. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015; 12:902–903. [DOI] [PubMed] [Google Scholar]
- 59. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012; 486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sunagawa S., Coelho L.P., Chaffron S., Kultima J.R., Labadie K., Salazar G., Djahanschiri B., Zeller G., Mende D.R., Alberti A. et al.. Structure and function of the global ocean microbiome. Science. 2015; 348:1261359. [DOI] [PubMed] [Google Scholar]
- 61. Hastie T., Tibshirani R., Friedman J.. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. 2009; Berlin: Springer. [Google Scholar]
- 62. Flores C.O., Meyer J.R., Valverde S., Farr L., Weitz J.S.. Statistical structure of host–phage interactions. Proc. Natl Acad. Sci. U.S.A. 2011; 108:E288–E297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Flores C.O., Valverde S., Weitz J.S.. Multi-scale structure and geographic drivers of cross-infection within marine bacteria and phages. ISME J. 2013; 7:520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sullivan M.B., Waterbury J.B., Chisholm S.W.. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003; 424:1047–1051. [DOI] [PubMed] [Google Scholar]
- 65. Wichels A., Biel S.S., Gelderblom H.R., Brinkhoff T., Muyzer G., Schütt C.. Bacteriophage diversity in the North Sea. Appl. Environ. Microb. 1998; 64:4128–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chibani-Chennoufi S., Bruttin A., Dillmann M.-L., Brüssow H.. Phage–host interaction: an ecological perspective. J. Bacteriol. 2004; 186:3677–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ross A., Ward S., Hyman P.. More is better: selecting for broad host range bacteriophages. Front. Microbiol. 2016; 7:1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guerin E., Shkoporov A., Stockdale S.R., Clooney A.G., Ryan F.J., Sutton T.D., Draper L.A., Gonzalez-Tortuero E., Ross R.P., Hill C.. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe. 2018; 24:653–664. [DOI] [PubMed] [Google Scholar]
- 69. Labonté J.M., Swan B.K., Poulos B., Luo H., Koren S., Hallam S.J., Sullivan M.B., Woyke T., Wommack K.E., Stepanauskas R.. Single-cell genomics-based analysis of virus–host interactions in marine surface bacterioplankton. ISME J. 2015; 9:2386–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bellas C.M., Anesio A.M., Barker G.. Analysis of virus genomes from glacial environments reveals novel virus groups with unusual host interactions. Front. Microbiol. 2015; 6:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mizuno C.M., Rodriguez-Valera F., Kimes N.E., Ghai R.. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013; 9:e1003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Holmfeldt K., Solonenko N., Shah M., Corrier K., Riemann L., VerBerkmoes N.C., Sullivan M.B.. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl Acad. Sci. U.S.A. 2013; 110:12798–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Flombaum P., Gallegos J.L., Gordillo R.A., Rincón J., Zabala L.L., Jiao N., Karl D.M., Li W.K., Lomas M.W., Veneziano D. et al.. Present and future global distributions of the marine cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl Acad. Sci. U.S.A. 2013; 110:9824–9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stern A., Mick E., Tirosh I., Sagy O., Sorek R.. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012; 22:1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Coutinho F.H., Silveira C.B., Gregoracci G.B., Thompson C.C., Edwards R.A., Brussaard C.P., Dutilh B.E., Thompson F.L.. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017; 8:15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Coenen A.R., Weitz J.S.. Limitations of correlation-based inference in complex virus–microbe communities. mSystems. 2018; 3:e00084-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weitz J.S., Beckett S.J., Brum J.R., Cael B., Dushoff J.. Lysis, lysogeny and virus–microbe ratios. Nature. 2017; 549:E1. [DOI] [PubMed] [Google Scholar]
- 78. Roux S., Hawley A.K., Beltran M.T., Scofield M., Schwientek P., Stepanauskas R., Woyke T., Hallam S.J., Sullivan M.B.. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife. 2014; 3:e03125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ahlgren N.A., Fuchsman C.A., Rocap G., Fuhrman J.A.. Discovery of several novel, widespread, and ecologically distinct marine Thaumarchaeota viruses that encode amoC nitrification genes. ISME J. 2019; 13:618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Anantharaman K., Duhaime M.B., Breier J.A., Wendt K.A., Toner B.M., Dick G.J.. Sulfur oxidation genes in diverse deep-sea viruses. Science. 2014; 344:757–760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers for viral contigs in the real data studies can be found in the ‘Datasets’ section. All other relevant data for training and testing the model and code are available at https://github.com/WeiliWw/VirHostMatcher-Net.