

Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating disorder of the CNS. Bile acids are cholesterol metabolites that can signal through receptors on cells throughout the body, including in the CNS and the immune system. Whether bile acid metabolism is abnormal in MS is unknown. Using global and targeted metabolomic profiling, we identified lower levels of circulating bile acid metabolites in multiple cohorts of adult and pediatric patients with MS compared with controls. In white matter lesions from MS brain tissue, we noted the presence of bile acid receptors on immune and glial cells. To mechanistically examine the implications of lower levels of bile acids in MS, we studied the in vitro effects of an endogenous bile acid, tauroursodeoxycholic acid (TUDCA), on astrocyte and microglial polarization. TUDCA prevented neurotoxic (A1) polarization of astrocytes and proinflammatory polarization of microglia in a dose-dependent manner. TUDCA supplementation in experimental autoimmune encephalomyelitis reduced the severity of disease through its effects on G protein–coupled bile acid receptor 1 (GPBAR1). We demonstrate that bile acid metabolism was altered in MS and that bile acid supplementation prevented polarization of astrocytes and microglia to neurotoxic phenotypes and ameliorated neuropathology in an animal model of MS. These findings identify dysregulated bile acid metabolism as a potential therapeutic target in MS.
Keywords: Metabolism, Neuroscience
Keywords: Multiple sclerosis, Neurodegeneration
Introduction
Multiple sclerosis (MS) is a chronic demyelinating disorder characterized by inflammation and neurodegeneration (1). Both genetic and environmental factors play a role in the etiopathogenesis of this disease (2–4). Metabolomics allows the measurement of the abundance of small molecules in biological matrices and provides information downstream of techniques such as genomics and proteomics (5). The circulating metabolome can be substantially affected by environmental exposures and gut microbiota and hence provides insight into these important factors that influence MS risk and pathogenesis (6). Several studies have demonstrated abnormalities in the metabolome in MS (7–9).
Bile acids are end-products of cholesterol metabolism and have multiple biological functions besides aiding lipid absorption in the gut (10). Primary bile acids are mostly produced in the liver, but other tissues are capable of producing bile acid precursors (oxysterols) through an alternative “acidic” pathway. Primary bile acids are modified (conjugated with glycine or taurine) and then stored in the gall bladder until they are secreted into the gut, where they are further modified through enzymes present in gut bacteria to produce secondary bile acids (Figure 1A and ref. 10). The majority of bile acids are reabsorbed and then undergo enterohepatic recirculation through the portal venous system, with a small fraction reaching the systemic circulation. Circulating bile acids can act on multiple receptors, both nuclear (farnesoid X receptor [FXR]) and cell surface (G protein–coupled bile acid receptor, [GPBAR1]), found on cells including those in the brain and the immune system (11, 12). An FXR agonist led to amelioration of disease in an animal model of MS — experimental autoimmune encephalomyelitis (EAE) — through antiinflammatory effects on myeloid cells (13, 14). GPBAR1 activation also produces antiinflammatory effects in innate immune cells and can affect glial cells (15, 16). Additionally, bile acid supplementation with tauroursodeoxycholic acid (TUDCA) and ursodeoxycholic acid (UDCA) has demonstrated neuroprotection in various models of neurodegeneration (17–19).
Figure 1. Bile acid metabolism is altered in MS.

(A) Overview of bile acid metabolism. (B) Heatmap of mean standardized bile acid metabolite concentrations derived from untargeted metabolomic profiling in the discovery cohort consisting of patients with RRMS (n = 56), patients with PMS (n = 52), and HCs (n = 50). Multiple bile acid metabolites in both primary and secondary bile acid metabolism pathways were lower in the MS groups compared with controls. Asterisks denote significant differences compared with HCs based on multivariate linear regression models adjusted for age, sex, and race (P < 0.05). (C) Box plots of pathway deregulation scores for primary bile acid metabolism in the discovery cohort demonstrated significant abnormality in the PMS group compared with controls. For all box plots, the center line indicates the median, the box indicates the 25th and 75th percentiles, the whiskers indicate 1.5 × IQR, and the dots indicate outliers. (D) Box plots of pathway deregulation scores for secondary bile acid metabolism in the discovery cohort show significant abnormality in the RRMS and PMS groups compared with the control group. P values for C and D were derived from multivariate linear regression models adjusted for age, sex, and race. (E) Heatmap of mean standardized bile acid metabolite concentrations derived from targeted metabolomic profiling in the validation cohort consisting of patients with RRMS (n = 50), patients with PMS (n = 125), and HCs (n = 75). Multiple bile acid metabolites in both primary and secondary bile acid metabolism pathways were lower in the MS groups compared with the control group. Asterisks denote significant differences as in B. (F) Box plots of pathway deregulation scores for primary bile acid metabolism in the validation cohort demonstrate significant abnormality in the PMS group compared with controls. (G) Box plots of pathway deregulation scores for secondary bile acid metabolism in the validation cohort show significant abnormality in the PMS group compared with controls. P values in F and G were derived as in C and D. (H) An increase in the DCA (sum of all DCA metabolites) to CA (sum of all CA metabolites) metabolite ratio was noted in the PMS group compared with the control group. The ratios of DCA to individual conjugated CA metabolites were also higher in the PMS group (I and J). (K) The ratio of the sum of UDCA and LCA metabolites to the sum of CDCA metabolites was higher in the PMS group compared with the control group. Data in H–K represent the mean ± SEM, and P values were determined using a linear regression model.
Although the role of altered bile acid metabolism has been extensively explored in metabolic diseases, only recently were abnormalities described in neurological disease (12, 20). In a large cohort of patients with Alzheimer’s disease (AD), abnormal circulating bile acid metabolite levels predicted worse outcomes and were hypothesized to be related to alterations of the gut microbiota (20). Gut dysbiosis has also been described in patients with MS who have an altered abundance of several bacterial species involved in bile acid metabolism (21–23). The brain contains 20% of the body’s cholesterol, and bile acid production is an important means of cholesterol clearance. In fact, the enzymes required for the alternative pathway of bile acid synthesis (leading to production of oxysterols) are expressed in the CNS. In EAE, neuroinflammation is associated with altered cholesterol metabolism in astrocytes (24), and abnormalities in circulating bile acid metabolites have been reported (25), providing a rationale for their evaluation in MS. Since bile acids have important effects on key inflammatory pathways and can be neuroprotective, we hypothesized that bile acid metabolism may play a role in modulating neuroinflammation in MS.
In this study, we first identified altered bile acid metabolism in MS using both global and targeted metabolomic approaches in adult and pediatric patients with MS. We then examined the in vitro effects of a secondary bile acid (TUDCA) on microglial and astrocytic inflammatory polarization. Finally, we demonstrated that TUDCA supplementation ameliorated neuroinflammation in EAE through its effects on GPBAR1, suggesting that intervention in this dysregulated metabolic pathway may provide a new therapeutic strategy for MS.
Results
Bile acid metabolism is altered in adult-onset MS.
We performed global metabolomic profiling on plasma from a discovery cohort consisting of adult patients with relapsing-remitting MS (RRMS) (n = 56) , adult patients with progressive MS (PMS) (n = 51), and healthy controls (HCs) (n = 52) and identified 25 metabolites related to bile acid metabolism (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI129401DS1). The demographic characteristics are shown in Table 1. As expected, the patients with RRMS were younger and the patients with PMS were older than the HCs. Figure 1B depicts a heatmap of bile acid metabolite concentrations in the 3 groups. We observed lower levels of multiple primary bile acid metabolites in the PMS group than in the control group (Figure 1B and Supplemental Figure 1A). Comparison of pathway deregulation scores between groups, adjusting for age, sex, and race, revealed significantly higher scores (denoting greater abnormality) for primary bile acid metabolism in the PMS group (Figure 1C). Lower levels of multiple secondary bile acid metabolites were detected in both RRMS and PMS groups compared with the HC group (Figure 1B and Supplemental Figure 1B). Secondary bile acid metabolism pathway deregulation scores (adjusted for age, sex, and race) were higher for both MS groups compared with the control group (Figure 1D).
Table 1. Demographic characteristics of the discovery and validation cohorts.
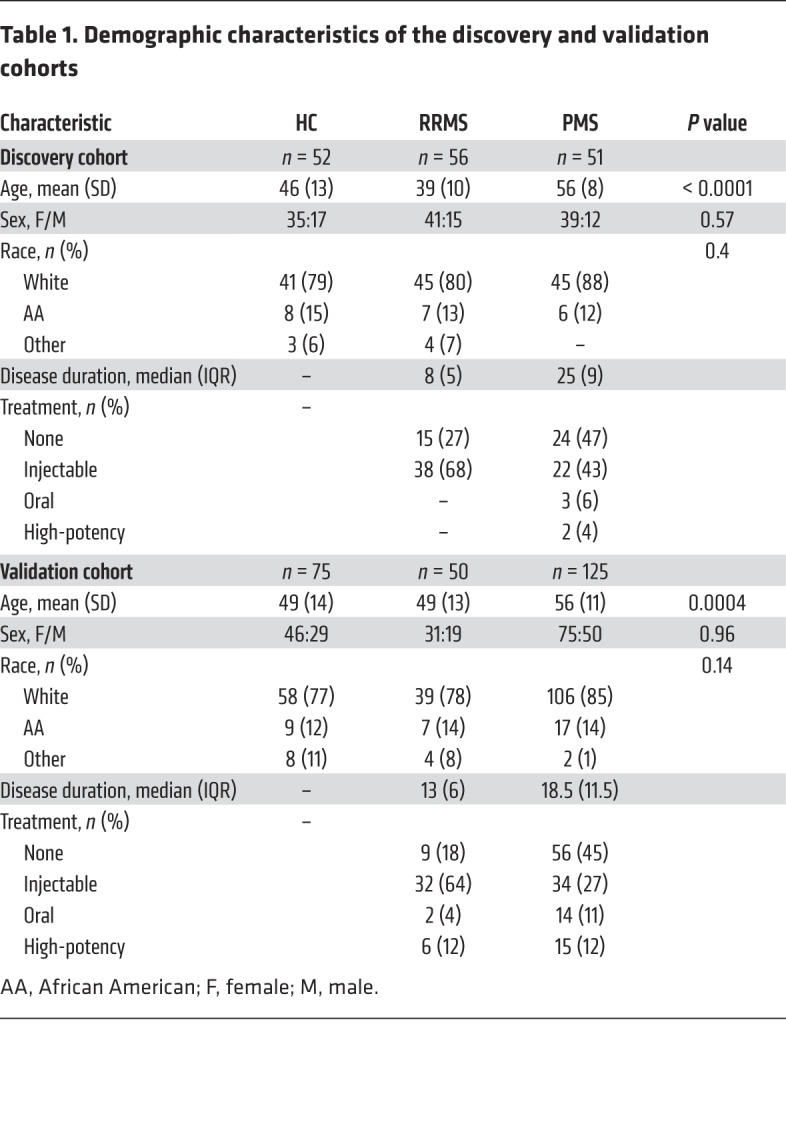
To extend our initial observations, we performed targeted metabolomic profiling of 15 bile acids (Supplemental Table 2) in plasma from a larger cohort of patients with MS (n = 175) and from controls (n = 75). The demographic characteristics are shown in Table 1. Again, the patients with PMS were older than the patients with RRMS and the HCs. Figure 1E shows a heatmap of concentrations of bile acid metabolites in the 3 groups. Lower levels of multiple primary bile acids were seen in the PMS group compared with levels in the control group (Figure 1E and Supplemental Figure 1C). Several of the bile acids that had lower concentrations overlapped with those identified in the global metabolomics analyses in the discovery cohort. We observed a reduction in multiple secondary bile acids in the PMS group compared with the control group (Figure 1E and Supplemental Figure 1D). We did not detect a reduction in secondary bile acids in the RRMS group compared with the control group (Figure 1E and Supplemental Figure 1D). A comparison of between-groups pathway deregulation (adjusting for age, sex, and race) revealed significantly higher scores (denoting greater abnormality) for both primary and secondary bile acid metabolism in the PMS group compared with scores for the control group (Figure 1, F and G).
We also assessed the effect of treatment on bile acid metabolism and noted no relationship between treatment status and pathway deregulation scores for either primary or secondary bile acid metabolism in univariate and multivariate analyses. Restricting our analysis to untreated patients with MS, we found similar results when comparing the MS groups with the HCs. In the discovery cohort, we noted higher pathway deregulation scores (greater abnormality) for primary bile acid metabolism in untreated patients with PMS compared with HCs (P = 0.01, Supplemental Figure 2A). We also noted higher pathway deregulation scores for secondary bile acid metabolism in both untreated RRMS and PMS patient groups compared with scores for the HC group (P = 0.02 and P = 0.001, respectively, Supplemental Figure 2B). In the second cohort, we noted higher pathway deregulation scores for primary bile acid metabolism in the untreated patients with PMS compared with scores for the HCs (P = 0.002, Supplemental Figure 2C). We also noted higher pathway deregulation scores for secondary bile acid metabolism in the untreated patients with PMS compared with the HC scores (P = 0.02, Supplemental Figure 2D).
To model metabolism of bile acids in the gut, we studied the ratio of secondary bile acid metabolites to their primary bile acid precursors, since secondary bile acid production requires multiple reactions catalyzed by enzymes present in the gut microbiota (20, 26). Targeted metabolomics showed a significant increase in the ratio of deoxycholic acid (DCA) metabolites (DCA, glycodeoxycholic acid [GDCA], taurodeoxycholic acid [TDCA]) to cholic acid (CA) metabolites (CA, glycocholic acid [GCA], taurocholic acid [TCA]) in the PMS group, with a trend toward a higher ratio in the RRMS group (Figure 1H). Individual ratios of DCA to either glycine- or taurine-conjugated (G/T) forms of CA were also elevated in the PMS group (Figure 1, I and J) but not the RRMS group. A similar increase was seen in the ratio of lithocholic acid (LCA) and UDCA metabolites to chenodeoxycholic acid (CDCA) metabolites in the PMS but not the RRMS group compared with controls (Figure 1K). We observed no difference in the ratios of circulating conjugated to unconjugated primary or secondary bile acids between the groups (G/T-CDCA/CDCA, G/T-CA/CA, G/T-DCA/DCA, G/T-LCA/LCA).
Overall, the data from both metabolomic approaches in the adult cohorts revealed alterations in bile acid metabolism in patients with MS, with greater abnormality noted in the PMS group.
Bile acid metabolism is altered in pediatric-onset MS.
We performed global metabolomic profiling in a cohort of patients with pediatric-onset MS and age-, race-, and sex-matched controls drawn from the UCSF Pediatric MS Center. The subjects’ demographic characteristics are shown in Table 2. We detected lower levels of several primary and secondary bile acid metabolites in the pediatric MS patients compared with levels in controls (Figure 2, A–C, and Supplemental Figure 3). Several of the metabolites that were measured in this cohort and were differentially altered between groups overlapped with those in the adult cohorts (Supplemental Table 5). Comparison of pathway deregulation scores for primary bile acid metabolism revealed a greater abnormality in the pediatric MS group than in the control group (Figure 2D), whereas no significant difference was noted in pathway deregulation scores for secondary bile acid metabolism (Figure 2E).
Table 2. Demographic characteristics of the pediatric cohort.

Figure 2. Bile acid metabolism is altered in pediatric-onset MS.

(A–C) Box plots of individual bile acid metabolites in the pediatric-onset MS and control groups (n = 31 each), demonstrating lower metabolite levels in the MS group. (D) Box plots of pathway deregulation scores for primary bile acid metabolism in patients with pediatric-onset MS and controls, demonstrating significant abnormality in the metabolic pathway in the pediatric MS group. (E) Box plots of pathway deregulation scores for secondary bile acid metabolism in patients with pediatric-onset MS and controls. (F) An increase in the ratio of DCA (sum of all DCA metabolites) to CA (sum of all CA metabolites) metabolites was noted in the MS group. For all box plots, the center line indicates the median, the box indicates the 25th and 75th percentiles, the whiskers indicate 1.5 × IQR, and the dots represent outliers. The P values for A–E were derived from multivariate linear regression models adjusted for age, sex, and race. In F, error bars represent the SEM, and the P value was determined using a 2-tailed, unpaired Student’s t test.
We detected a significant increase in the ratio of DCA/CA metabolites, similar to that seen in adult patients with MS (Figure 2F). In this cohort of patients with pediatric-onset MS from an independent site, we identified bile acid metabolism abnormalities similar to those noted in adult patients with MS.
Bile acid receptors are expressed in white matter lesions from patients with MS.
After establishing altered bile acid metabolism in both adult and pediatric patients with MS, we enquired whether the key receptors associated with bile acid signaling are present in MS brain lesions. Although a previous study demonstrated the presence of FXR in inflammatory cells in active lesions from patients with RRMS (13), the expression of bile acid receptors has not, to our knowledge, been studied in lesions from patients with PMS.
In white matter lesions (WMLs) from PMS autopsy tissue (Figure 3A), FXR+ cells were present in the center (Figure 3, A and B, red box, and Figure 3D) and at the edge (Figure 3, A and B, blue boxes, and Figure 3F) of lesions. The expression of FXR paralleled the distribution and nuclear localization within macrophages (CD68+ cells) as reported previously (Figure 3, C–F, and ref. 13). Compared with control brain white matter, MS normal-appearing white matter (NAWM), mixed active/inactive lesions, and active lesions had a higher number of FXR+ cells (Supplemental Figure 4, A–E).
Figure 3. The FXR, a nuclear bile acid receptor, is detected in demyelinating lesions in PMS brains.

(A) Immunohistochemistry for PLP identified a WML in an MS brain. (B) Numerous FXR+ cells were detected within the lesion. (C) CD68 staining showed numerous CD68+ macrophages within the center of the lesion (red boxes from A and B), consistent with an active lesion. (D) Numerous FXR+ cells were detected within the center of the lesion, corresponding to the CD68+ macrophages. (E) Several CD68+ cells were also noted at the edge of the lesion (blue boxes from A and B). (F) Numerous FXR+ cells were also observed within the edge of the lesion shown in E. Scale bars: 500 μm (A and B); 100 μm (C–F).
Staining of active and mixed active/inactive WMLs also revealed the presence of GPBAR1 (Figure 4A), a cell-surface bile acid receptor, on cells within these lesions. We noted that expression of GPBAR1 appeared to be greatest in the active lesion and was less pronounced in mixed active/inactive lesions, with the lowest expression noted in control white matter (Figure 4, B–D, and Supplemental Figure 4, F–J). The increased expression of GPBAR1 in demyelinated lesions was also confirmed using quantitative PCR (qPCR), which demonstrated increased expression of GPBAR1 in WMLs compared with expression levels in NAWM (Figure 4M).
Figure 4. The cell-surface bile acid receptor GPBAR1 is detected in demyelinating lesions in PMS brains.

(A) Immunohistochemistry for PLP and CD68 identified an active and a mixed active/inactive MS WML, with GPBAR1+ cells detected within both of these lesions. Comparison of GPBAR1 staining (red) in an active lesion (B), a mixed active/inactive lesion (C), and control brain (D) revealed increased staining in MS lesions compared with control white matter. Double immunostaining for PLP (green) and GPBAR1 (red) shows the presence of GPBAR1+ cells (E) and vessels (F) in areas of demyelination. (G–I) Double immunostaining using GFAP (green) and GPBAR1 (red) demonstrates GPBAR1 staining in GFAP+ astrocytes in MS lesions. (J–L) Double immunostaining with CD68 (green) and GPBAR1 (red) demonstrates GPBAR1 staining in CD68+ macrophages in MS lesions. (M) Comparison of PLP and GPBAR1 gene expression in MS WML and NAWM revealed decreased PLP expression within the lesions with increased expression of GPBAR1, similar to findings noted on immunohistochemistry (n = 4 in each group). Scale bars: 200 μm (A); 100 μm (B–F); 20 μm (G–I). *P < 0.05, by 2-tailed Mann-Whitney U test. Data in M represent the mean ± SEM.
Within WMLs, GPBAR1 appeared to be expressed on cells with morphology resembling that of astrocytes (Figure 4E) as well as in vascular structures (Figure 4F). Double immunostaining confirmed the presence of GPBAR1 staining in cells positive for glial fibrillary acidic protein (GFAP), confirming that astrocytes in MS lesions expressed cell-surface bile acid receptors (Figure 4, G–I). Additionally, we noted the presence of CD68+ cells expressing GPBAR1, confirming that macrophages/myeloid cells in MS lesions also expressed this receptor (Figure 4, J–L).
The presence of bile acid receptors on immune and glial cells in chronic MS lesions raised the possibility that these metabolites could play a role in modulating neuroinflammation.
An endogenous bile acid blocks neurotoxic polarization of astrocytes and proinflammatory polarization of microglia.
Since bile acid receptors are present on astrocytes and inflammatory cells in MS tissue, we investigated the effects of a bile acid, TUDCA, on these cell populations in vitro. We chose TUDCA, since it is currently being tested in multiple human trials for various neurological disorders including MS (ClinicalTrials.gov: NCT03423121) (19).
We examined the effects of TUDCA on murine astrocytes that were treated with IL-1α, TNF-α, and C1q to polarize them to a neurotoxic (A1) phenotype (27). We tested the effect of 2 doses of TUDCA and an agonist of GPBAR1 (INT-777) under these polarizing conditions. TUDCA treatment blocked the upregulation of A1-specific gene transcripts in a dose-dependent manner (Figure 5A). Interestingly, we found that INT-777 partially recapitulated the effects of TUDCA on astrocyte polarization. No difference in cell viability between the various treatment conditions was observed (Supplemental Figure 5, A and B).
Figure 5. Endogenous bile acid blocks the neurotoxic polarization of astrocytes and proinflammatory polarization of microglia.

(A) Astrocytes were isolated from neonatal mouse brains using ACSA2 bead selection and were polarized to an inflammatory phenotype using a cocktail of IL-1α, TNF-α, and C1q for 24 hours in the presence or absence of TUDCA or the GPBAR1 agonist INT-777. qPCR of genes associated with various astrocyte phenotypes revealed that expression of multiple A1-specific genes was significantly downregulated in the 70-μM TUDCA condition compared with vehicle. Downregulation of several A1-specific genes with INT-777 treatment was also noted, but to a lesser degree than with 70-μM TUDCA . Data were derived from 3 independent experiments with 6 biological replicates. Groups were compared using a Kruskal-Wallis test with Dunn’s multiple comparisons test, and asterisks represent P < 0.05. (B) Schema showing concentration of ACM from A0 (nonpolarized), A1 plus vehicle, and A1 plus 70-μM TUDCA conditions and testing of the effects on viability of murine oligodendrocytes over a 24-hour period. (C) Oligodendrocyte (OL) cell death was reduced with 70-μM TUDCA ACM compared with vehicle (normalized to cell death from A0 ACM). Data were derived from 3 independent biological replicates and represent the mean ± SEM. ****P < 0.001, by unpaired, 2-tailed Student’s t test. (D) Representative images reveal oligodendrocyte death. ACM from the 2 conditions showed greater cell death (green) in the vehicle-treated condition than in the 70-μM TUDCA–treated condition. Scale bars: 200 μm. (E) Microglia were isolated from neonatal mouse brains using CD11b bead selection and polarized to an inflammatory M1 phenotype by treatment with LPS and IFN-γ in the presence or absence of differing doses of TUDCA. RNA was then isolated to determine the expression of the NOS2 gene, which is a marker of M1 polarization, and showed reduced expression of the gene under TUDCA-treated conditions compared with vehicle. Gene expression levels were also compared for IL1A (F) and TNFA (G), which are factors produced by microglia critical for A1 polarization, under the different treatment conditions. Expression of IL1A was significantly reduced, with a similar trend for TNFA, under the 70-μM TUDCA condition compared with vehicle. Data in E–G were derived from 4 independent experiments with 6 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.005, by 1-way ANOVA with Dunnett’s test for multiple comparisons. For the box plots in E–G, the center line indicates the median, the boxes indicate the 25th and 75th percentiles, the whiskers indicate 1.5 × IQR, and the dots indicate outliers.
We also examined the effect of astrocyte-conditioned media (ACM) obtained under these different conditions on oligodendrocyte viability (Figure 5B). Since ACM from A1 astrocytes can be toxic to neurons and oligodendrocytes, we tested the effects of ACM obtained under conditions of nonpolarization (A0), A1 polarization (vehicle), and A1 polarization in the presence of TUDCA (70 μM) on the viability of murine oligodendrocytes. As expected, ACM from TUDCA-treated astrocytes caused significantly less oligodendrocyte cell death than did vehicle conditions, in keeping with the changes noted in gene expression (Figure 5, C and D).
We polarized murine microglia to a proinflammatory phenotype by treating them with IFN-γ and LPS, with or without TUDCA present. We noted a reduction in the expression of the NOS2 gene (Figure 5E), a marker for M1 polarization, and a reduction in IL1A and TNFA transcription in the presence of TUDCA (Figure 5, F and G). Since these molecules have been implicated in the generation of neurotoxic reactive astrocytes, reduced production from microglia would be expected to be beneficial in neuroinflammatory/neurodegenerative disorders (27). The presence of TUDCA in microglial cultures had no adverse effect on cell viability (Supplemental Figure 5, C and D).
Thus, TUDCA, an endogenous bile acid, blocked the in vitro polarization of astrocytes to an A1 neurotoxic phenotype and the polarization of microglia to a proinflammatory phenotype.
TUDCA supplementation ameliorates EAE through effects on GPBAR1.
Since multiple circulating bile acid levels were lower in patients with MS than in HCs and TUDCA prevented the proinflammatory polarization of microglia and astrocytes, we next tested the effect of TUDCA supplementation in EAE, an animal model of MS.
EAE was induced by active immunization with MOG35–55 peptide, and mice were randomized to receive either TUDCA (500 mg/kg, orally) or vehicle when they reached a disease severity score of 1 (Figure 6A). Treatment was continued until day 28 after immunization, and we compared the behavioral scores between the 2 groups and the pathological measures of demyelination and immune cell infiltration in the spinal cord.
Figure 6. TUDCA supplementation ameliorates EAE.

(A) Eight- to 9-week-old female C57/BL6 mice were subcutaneously immunized with MOG35–55 and CFA in addition to intraperitoneal injection of pertussis toxin (PTX) on day 0 and day 2. Mice were then monitored for signs of EAE and, at disease onset, were randomized to oral gavage with either TUDCA or vehicle until day 28 after immunization. (B) Behavioral scores demonstrate that TUDCA treatment resulted in reduced severity of EAE disease. Data were derived from a representative experiment of 1 of 3 independent experiments and represent the mean ± SEM. *P < 0.05, by Mann-Whitney U test. (C and D) Representative images of Black-gold staining of spinal cords of mice with EAE treated with either TUDCA or vehicle show reduced demyelination with TUDCA treatment. Quantification is shown in the plot in D. (E and F) Immunostaining for infiltrating myeloid cells (Mac-2+) demonstrated reduced infiltration in the TUDCA-treated group compared with the control. Quantification is shown in the plot in F. (G and H) Staining for GFAP showed reduced astrocytosis in the TUDCA-treated group compared with the control. This is quantified in the plot in H. (I and J) Staining for Iba-1 and iNOS shows a decrease in INOS+Iba-1+ cells (M1 macrophages/microglia) in the TUDCA-treated group compared with the control. Quantification is shown in the plot in J. (K and L) Images show a reduced number of PSMβ8+GFAP+ cells (neurotoxic A1 astrocytes) in the TUDCA-treated group compared with the control. This is quantified in the plot in L. Scale bars: 200 μm (E and G); 50 μm (I and K). *P < 0.05 and **P < 0.005, by unpaired, 2-tailed Student’s t test (D, F, H, J, and L). Data represent the mean ± SEM (D, F, H, J, and L).
We noted a reduction in the severity of EAE based on behavioral scores (Figure 6B) as well as pathological measures — reduced demyelination (Figure 6, C and D), reduced infiltration of myeloid cells (Figure 6, E and F), and reduced astrocytosis (Figure 6, G and H).
GPBAR1 was detected on both astrocytes (GFAP+) and microglia/macrophages (Mac-2+) in the spinal cords of mice with EAE (Supplemental Figure 6). We evaluated the presence of proinflammatory (M1) microglia/macrophages using staining for inducible NO synthase (iNOS) and Iba-1 and noted a reduction in the proportion of iNOS+Iba-1+ cells (Figure 6, I and J). We also evaluated A1 astrocytes in EAE spinal cord tissue using staining for proteasome subunit β type-8 (PSMβ8) and GFAP, since PSMβ8 was previously noted to be a marker of neurotoxic (A1) astrocytes (27). We detected a reduction in PSMβ8+GFAP+ cells with TUDCA treatment (Figure 6, K and L), suggesting a reduction in A1 astrocytes. These data are consistent with the in vitro data regarding the effects of TUDCA on both microglial and astrocytic polarization.
Since CD4+ T cells are key players in the pathogenesis of the EAE model, we tested the effects of TUDCA on CD4+ T cell proliferation and cytokine production and noted no direct antiinflammatory effects of TUDCA on CD4+ T cells (Supplemental Figure 8). We also examined the effect of TUDCA treatment on infiltration of T cells into the CNS, as well as its effect on T cell phenotype and cytokine production at a late-peak time point in mice with EAE. These mice had received treatment with TUDCA or vehicle for 1 week before collection of CNS tissue for evaluation of the inflammatory infiltrate. We saw no difference between the 2 groups in T cell infiltration or phenotype (Supplemental Figure 9). These findings suggest that TUDCA treatment does not mediate its effects in EAE through alteration of T cell function.
Bile acids can act on a variety of receptors, and TUDCA also has non–receptor-mediated effects on various cell types (28, 29). To further examine the mechanism of action of TUDCA in neuroinflammation, we induced EAE in WT and GPBAR1-KO mice and randomized them to treatment with either TUDCA or vehicle at the onset of clinical disease. We found that TUDCA treatment again reduced EAE severity in WT mice (Figure 7A), but no therapeutic effect was noted in the GPBAR1-KO mice (Figure 7B).
Figure 7. Therapeutic effect of TUDCA supplementation in EAE is dependent on GPBAR1.

Nine- to 11-week-old female WT or GPBAR1-KO C57/BL6 mice were subcutaneously immunized with MOG35–55 and CFA in addition to intraperitoneal injection of PTX on day 0 and day 2. Mice were then monitored for signs of EAE, and at disease onset, the mice were randomized to oral gavage with either 500 mg/kg TUDCA or vehicle until day 28 after immunization. (A) Behavioral scores showed that TUDCA treatment resulted in reduced severity of EAE in WT mice. (B) No significant difference was noted in EAE severity between the 2 treatment groups in GPBAR1-KO mice. Individual data points are shown along with the mean ± SEM. Data were derived from 2 independent experiments. ***P < 0.001, by Mann-Whitney U test.
These data suggest that TUDCA supplementation reduced innate immune cell infiltration and astrocytosis, resulting in amelioration of neuroinflammation through the effects of TUDCA on GPBAR1.
Discussion
In this study, we demonstrated that bile acid metabolism was altered in both adult and pediatric-onset MS, with lower levels of multiple circulating bile acid metabolites in the MS groups compared with levels in the HCs. We also demonstrated the presence of 2 key bile acid receptors — FXR and GPBAR1 — on glial and immune cells in MS brain tissue. We then identified the direct antiinflammatory effects of a secondary bile acid, TUDCA, on astrocytes and microglia in vitro and showed that supplementation with TUDCA ameliorated disease in a mouse model of MS through its effects on GPBAR1.
Previous studies have demonstrated that bile acids have antiinflammatory effects on myeloid cells, including the prevention of M1 polarization and blockade of the NLRP3 inflammasome pathway (16). The balance of pro- and antiinflammatory microglial polarization is important for controlling the extent of damage and recovery in animal models of MS (30, 31). Recent studies have demonstrated that cues from microglia influence the astrocytic phenotype and affect neuroinflammation (27, 32). We confirmed that TUDCA not only prevented proinflammatory polarization of microglia, but also reduced the production of factors implicated in the polarization of astrocytes to an A1 phenotype. This is important, given recent data showing that A1 astrocytes are toxic to neurons and oligodendrocytes and that their number is increased in MS brains (27).
Besides the potential indirect effect through prevention of microglial polarization, we also found that TUDCA can act directly on astrocytes and block their polarization to an A1 phenotype. We noted that TUDCA treatment decreased upregulation of A1-specific genes in a dose-dependent manner. We also showed that the change in gene expression resulted in a functional change, that of reduced toxicity of the ACM to oligodendrocytes. This has important implications, since this subset of astrocytes could play a critical role in neurodegeneration in disorders such as Parkinson’s disease (PD), AD, and MS (27). To the best of our knowledge, this is the first example of an endogenous metabolite that directly blocks the polarization of astrocytes to a toxic phenotype. Since we noted that astrocytes in both humans and mice express GPBAR1, we tested whether the effects of TUDCA on astrocyte polarization could be replicated using the GPBAR1 agonist INT-777. We identified partial replication of the effects of TUDCA with INT-777, suggesting that this beneficial effect may be partially mediated through GPBAR1. A previous study has also reported that GPBAR1 may mediate the antiinflammatory effects of TUDCA on microglial cells (16). Thus, although our data suggest that GPBAR1 may be important for the effects of bile acids on astrocyte polarization, the precise mechanism requires further study.
The finding of altered serum levels of bile acids in MS is not entirely unexpected, given that gut dysbiosis has been reported in patients with MS in several studies (22, 33–35). Since modification of bile acids by the gut microbiota is a key step in their metabolism, the alteration in abundance of bacterial species that are involved in these processes could potentially explain altered circulating levels of bile acid metabolites in MS. Indeed, bacterial species including Clostridium and Parabacteroides, which have been noted to be reduced in abundance in MS, are important players in bile acid metabolism and have been linked to alteration of circulating levels of certain bile acids (23, 26, 36). The change in ratios of secondary to primary bile acids in both synthesis pathways also points to the potential role of gut dysbiosis as a key factor driving this change. The observation that these changes were more marked in progressive MS parallels the trend noted in AD, in which the change in bile acid profiles was more marked in AD than in mild cognitive impairment (20). This potentially suggests a relationship between greater abnormality in bile acid metabolism and neurodegeneration.
As the makeup of the gut microbiota is linked to an individual’s genome, circulating metabolite levels could also be driven by specific genetic factors (36). In a recent study, vitamin D receptor (VDR) polymorphisms were demonstrated to alter Parabacteroides abundance and ultimately impact the level of specific bile acids (36). Similarly, other MS-related genetic risk alleles could alter the gut microbiota and ultimately lead to metabolic changes such as those noted in our study. For example, ZFP36L1 (a gene identified in several MS GWAS studies) was recently demonstrated to modulate bile acid biosynthesis (37).
Besides altered gut microbial bile acid metabolism, altered bile acid profiles could result from abnormal endogenous production. Since cholesterol is the precursor of bile acids, alterations in cholesterol metabolism could potentially underlie changes in bile acid abundance. Cholesterol metabolism is intricately linked to inflammation and to T cell and innate immune cell function (38). TLR activation can also modify cholesterol metabolism by impairing the transport of cholesterol out of innate immune cells (39). Since cholesterol is a major brain component, alterations in brain cholesterol metabolism could also modify the circulating bile acid profile in patients with MS. Inflammation can alter cholesterol metabolism in the CNS, and this was the major abnormal pathway in a recent study evaluating spinal cord astrocyte gene expression profiles in EAE (24). In addition, the Western diet is associated with changes in peripheral and brain bile acid profiles and alteration of cholesterol metabolism in the brain (40). These data suggest that inflammation can alter endogenous cholesterol metabolism in the CNS and the periphery and could be a factor underlying the altered bile acid levels in the circulation of patients with MS.
We examined the distribution of 2 key bile acid receptors in MS brains. FXR is a nuclear bile acid receptor present in MS WMLs from patients with RRMS, primarily in immune cells (41). We found that in addition to active lesions, FXR expression can also be found in chronic MS lesions. In addition, we also identified the presence of GPBAR1 on astrocytes, macrophages, and vascular structures in MS lesions. GPBAR1 has been previously demonstrated to mediate several key antiinflammatory effects of bile acid ligands in immune cells including inhibition of the NLR family pyrin domain–containing 3 (NLRP3) inflammasome (12). The expression of GPBAR1 on astrocytes and macrophages raises the possibility that the circulating bile acid metabolites could potentially alter neuroinflammation through effects on these cells. In animal models, GPBAR1 expressed on the endothelium helped regulate chemotaxis of immune cells (42), and the presence of this receptor on vessels in the MS brain raises the possibility that it could help regulate entry of immune cells into the CNS parenchyma. Overall, the presence of both nuclear and surface receptors in MS brains at levels greater than those in control brain tissue reinforces the relevance of altered circulating bile acid levels in MS.
Previous studies have demonstrated that bile acid levels are reduced in chronic EAE, providing a strong rationale for testing bile acid supplementation in this animal model (25). Obeticholic acid, a synthetic bile acid and an agonist specific for FXR, led to amelioration of EAE, though the effects on astrocytes and microglia were not examined (14). Previous work also established that FXR+ cells are present in the CNS in EAE and that other FXR agonists can lead to amelioration of the disease (13). However, despite the expression of FXR in glial cells, a previous study demonstrated that an FXR agonist did not result in antiinflammatory effects in astrocytes or microglia (43). We demonstrated that astrocytes and myeloid cells in the CNS of EAE mice also expressed GPBAR1, thus providing additional support for evaluating the in vivo effects of TUDCA supplementation in this model. Indeed, recent studies suggest that TUDCA may have a potent effect on GPBAR1 rather than on FXR and hence is likely to differ from obeticholic acid in its mechanism of action that was previously tested in EAE. In addition to identifying a beneficial effect of TUDCA supplementation on EAE severity, we also noted reduced numbers of iNOS-expressing microglia/macrophages (M1 phenotype) and PSMβ8-expressing astrocytes (A1 phenotype). This provided in vivo corroboration of the antiinflammatory effects of bile acid supplementation that we noted initially in our in vitro experiments. We also investigated the mechanism of the beneficial effects of bile acid supplementation in the EAE model using GPBAR1-KO mice and showed a lack of therapeutic effect of TUDCA in these mice compared with in WT animals, suggesting that GPBAR1 activation is a critical mediator of the treatment effect of TUDCA. Future studies will help to identify the specific GPBAR1-expressing cell populations — peripheral myeloid cells, microglia, or astrocytes — that mediate this effect.
Our study has several strengths. We demonstrated abnormal bile acid metabolism in 2 cohorts from different centers and in different age groups. In addition to evaluating this finding in humans, we also studied the effects of bile acids in vitro and in an animal model of MS. The limitations of the current study include the cross-sectional nature of the metabolomics analyses, which precluded an assessment of how bile acids change over time in MS and whether they are affected by specific MS disease–modifying therapies. Fewer bile acid metabolites were measured in the targeted cohort compared with the untargeted panel, i.e., not all metabolites were present in both data sets for direct comparison. However, we did note that the majority of metabolites had consistent changes across all the cohorts. In the adult cohorts, there were fewer controls that matched the age of the patients with PMS, however, a large metabolomics study that evaluated the effects of aging on the circulating metabolome did not identify any of the bile acids as being significantly associated with age (44). We did not have complete data regarding the feeding state at the time of the blood draw, and hence this was not controlled for. This is a limitation, since bile acid levels in the circulation change depending on the fasting state (45, 46). The likelihood that a difference in feeding status was driving the difference between the controls and the patients with MS is low, given the replication in cohorts of different ages and in multiple locations. Fasting status was also unlikely to influence the ratio of DCA/CA metabolites that was noted to be altered in both adult and pediatric MS patients. It is also important to point out that circulating bile acids are only a small proportion of the bile acid pool, therefore these levels may not necessarily reflect the overall bile acid pool status. Finally, human samples were obtained after disease onset, which precludes the evaluation of causality.
Future studies will help further define the relationship between bile acid levels and MS disease course. Additionally, ongoing clinical trials of supplementation with TUDCA in MS (ClinicalTrials.gov: NCT03423121) are likely to provide us with information regarding the effects of this intervention on circulating bile acid levels and ultimately on MS activity and disease course. These steps will potentially enable the results of this study to be translated to the clinic.
The results of this study expand our understanding of how alterations in the metabolome may affect aspects of MS disease pathophysiology and potentially impact processes that mediate neurodegeneration in multiple neurological diseases. We also provide a paradigm for future studies to identify additional metabolic pathways that could be targeted to modify the course of MS.
Methods
Patient cohorts.
We recruited adult patients with MS and HCs from the Johns Hopkins MS Center. Participants underwent phlebotomy for blood collection in addition to collection of clinical and demographic data. Blood was processed using our standard protocol and plasma stored at –80°C until the time of metabolomics analyses. This study included 2 adult cohorts: a discovery cohort consisting of 56 patients with RRMS, 51 patients with PMS, and 52 HCs who underwent global metabolomics analysis, and a validation cohort that consisted of 50 patients with RRMS, 125 patients with PMS, and 75 HCs who underwent targeted metabolomics analysis. Pediatric MS patients and HCs (n = 31 each) were recruited from the UCSF Pediatric MS Clinic. Participants underwent phlebotomy, and blood was processed and serum stored at –80°C until the time of metabolomics analyses (47).
Global metabolomics analyses.
Global metabolomics analyses were performed at Metabolon Inc. (Durham, North Carolina, USA) as previously described (7). In brief, recovery standards were added before the extraction process for quality control (QC). To remove protein and recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking (GenoGrinder 2000, Glen Mills) for 2 minutes, followed by centrifugation. The resulting extract was divided into 5 fractions: analysis by ultra-high-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) (positive ionization); UPLC-MS/MS (negative ionization); UPLC-MS/MS polar platform (negative ionization); gas chromatography–mass spectrometry; and 1 aliquot reserved for backup. Metabolite identification was performed by automated comparison of the ion features in the study samples with a reference library of standard metabolites. Quantification of peaks was performed using the AUC. Raw values for the area counts for each metabolite were normalized (correcting for variation resulting from instrument inter-day tuning differences) by the median value for each run day. The lists of metabolites detected in adult and pediatric cohorts are provided in Supplemental Tables 1 and 4.
Targeted metabolomics assay.
The targeted bile acid panel measured all of the major human primary and secondary bile acids and their respective glycine and taurine conjugates, which are listed in Supplemental Table 2. Bile acid concentrations were analyzed by LC-MS/MS. The individual analyte quantitation ranges (based on the analysis of 50 μL plasma) are also listed in Supplemental Table 2.
Calibration samples were prepared at 8 different concentration levels by spiking a PBS/BSA solution with corresponding calibration spiking solutions. Calibration samples, study samples, and QC samples were spiked with a solution of labeled internal standards and subjected to protein precipitation with an organic solvent (acidified MeOH). Following centrifugation, an aliquot of the organic supernatant was evaporated to dryness in a gentle stream of nitrogen. The dried extracts were reconstituted and injected onto an Agilent 1290/Sciex QTrap 6500 LC-MS/MS or Agilent Infinity II/Sciex TripleQuad 6500+ LC-MS/MS system equipped with a C18 reverse-phase HPLC column. The mass spectrometer was operated in negative mode using electrospray ionization. The peak area of each bile acid parent (pseudo-MRM mode) or product ion was measured against the peak area of the respective internal standard parent (pseudo-MRM mode) or product ion. Quantitation was performed using a weighted linear least squares regression analysis generated from fortified calibration standards prepared immediately before each run.
Single-level, pooled QC samples were used with most bile acid concentrations at the endogenous level. Given the low abundance of TUDCA, TCA, TLCA, GLCA, and LCA in this lot, the QCs were up-spiked to typically observed levels (standard C to D range). Because of the multiplexing of 15 analytes, only a single concentration level of QC samples was used. Raw data were collected and processed using AB SCIEX Analyst software, version 1.6.3. Data reduction was performed using Microsoft Excel 2013.
Sample analysis was carried out in the 96-well plate format containing 2 calibration curves and 6 QC samples (per plate) to monitor method performance. Four sample batches were prepared and analyzed. Following analysis, 4 samples were found to be above the limit of quantitation (ALOQ) and therefore reanalyzed at a 5-fold dilution. Precision was evaluated using the corresponding QC replicates in each sample run. Intra-run and inter-run precision (percentage of the coefficient of variation) of all analytes met the acceptance criteria. A summary of the absolute concentrations of bile acid metabolites measured by this technique is provided in Supplemental Table 3.
Immunohistochemistry of MS autopsy tissue.
All brains were collected as part of the tissue procurement program approved by the Cleveland Clinic IRB. MS brain tissues were characterized for demyelination by immunostaining 30-μm fixed-tissue sections for proteolipid protein (PLP) using previously described protocols (48–50). Briefly, fixed blocks of the tissue were cut on a sliding microtome, microwaved in 10 mM citric acid buffer (pH 6.0), and immunostained using the avidin-biotin complex procedure with DAB and rat anti-PLP (1:250, gift from Wendy Macklin, University of Colorado, Denver, Colorado, USA), mouse anti-FXR (1:500, Perseus Proteomics), mouse anti-CD68 (1:250, M0876, Dako), rabbit anti-GPBAR1 (1:250, Thermo Fisher Scientific), and anti-GFAP (1:250, MilliporeSigma) as described previously (Supplemental Table 7). Adjacent sections were used for double labeling (PLP-GPBAR1, CD68-GPBAR1, and GFAP-GPBAR1) along with the following respective secondary antibodies: biotinylated donkey anti–rat IgG, donkey anti–mouse IgG, and donkey anti–rabbit IgG (1:500, Vector Laboratories), and Alexa Fluor 488 donkey anti–rat IgG, Alexa Fluor 488 donkey anti–mouse IgG, and Alexa Fluor 594 donkey anti–rabbit IgG (1:500, Invitrogen, Thermo Fisher Scientific). Immunofluorescence-labeled sections were imaged using a Zeiss AX10 (Imager Z2) confocal microscope, and DAB-stained sections were imaged using a Leica DM5500 upright microscope (Leica Microsystems). PLP-GPBAR1, CD68-GPBAR1, and GFAP-GPBAR1 images were collected from the corresponding sections. The resultant images were processed using the Fiji version (http://fiji.sc) of ImageJ (NIH) free image processing software (http://rsbweb.nih.gov/ij), as described previously (48–50). Lesions evaluated from each MS patient were classified on the basis of Kuhlmann et al. (51), and details on the demographics of the patients with MS and the controls and the types of lesions evaluated are included in Supplemental Table 8.
qPCR method for MS autopsy tissue.
Total RNA was isolated from MS brain (demyelinated lesions [WMLs] and surrounding NAWM) using QIAGEN miRNA isolation kits according to the manufacturer’s instructions. Total RNA was reverse-transcribed to cDNA using SuperScript VILO cDNA Synthesis Kits (catalog 11754050, Applied Biosystems) as recommended. The expression of reported genes was checked using TaqMan Gene expression assays (Thermo Fisher Scientific) and GAPDH as endogenous controls in the reaction. Each sample was run in triplicate. ΔCt values were used to determine relative expression changes (2–ΔΔCt) and presented as fold change (FC). The following TaqMan probes were used: PLP- Hs00166914_m1, GAPDH- Hs03929097_g1, and GPBAR1- Hs01937849_m1. The demographics of the MS patients from whom the autopsy tissues were obtained are provided in Supplemental Table 9.
Murine microglia primary cultures.
Microglia were isolated from brains of P3 to P5 mouse pups (strain 556, C57/BL6N mice, Charles River Laboratories). After dissecting off the meninges, whole brains underwent mechanical and enzymatic dissociation using a Neural Tissue Dissociation Kit containing papain (Miltenyi Biotec). Microglia were then positively selected using CD11b magnetic beads (Miltenyi Biotec) and seeded onto poly-l-lysine–coated 6-well plates at a density of 3 × 105 cells/mL in DMEM/F12 containing 10% FBS, 1% penicillin/streptomycin, and 1% GlutaMAX (Gibco, Thermo Fisher Scientific). Microglial purity was confirmed by flow cytometry (CD11b+CD45loACSA2–A2B5–O4–). After 24 hours, cells were left unstimulated or stimulated with a combination of LPS (100 ng/mL) and recombinant murine IFN-γ (25 ng/mL, PeproTech). Concurrently, cells were treated with either vehicle, 20 μM TUDCA, or 70 μM TUDCA. After 18 hours, supernatants were collected, and cells were lysed and prepared for qPCR analysis.
Murine astrocyte primary cultures.
Astrocytes were isolated from brains of P3–P5 mouse pups (as above). Brains were enzymatically dissociated similarly to microglia (see above). Cells were Fc blocked with anti-CD16 and anti-CD32 antibodies, positively selected using ACSA-2 magnetic beads (Miltenyi Biotec), and then seeded onto poly-l-lysine–coated 6-well plates at a density of 2.5 × 105 cells/mL. Astrocytes were cultured in serum-free media containing 50% DMEM, 50% neurobasal, 1% penicillin/streptomycin, 1% GlutaMAX, 1 mM sodium pyruvate, N-acetyl cysteine (5 μg/mL), and 1× Sato, and supplemented with heparin-binding EGF (HB-EGF) (5 ng/mL, PeproTech) as previously described (27). Astroglial purity was confirmed by flow cytometry (ACSA2+CD11b–CD45–A2B5–O4–). Astrocytes were grown for approximately 7 days until cultures reached a desired density, with media changes every 2 days. Cells were then left unstimulated or stimulated to generate A1 polarized astrocytes by adding recombinant rat Il-1α (3 ng/mL, PeproTech), recombinant human TNF-α (30 ng/mL, MilliporeSigma), and C1q (400 ng/mL, MyBioSource). Concurrently, cells were treated with either vehicle, 20 μM TUDCA, 70 μM TUDCA, or 30 μM INT-777. After 24 hours, supernatants were collected, and cells were lysed and prepared for qPCR analysis.
Cell viability assay.
Cells were seeded onto 96-well plates (50,000 microglia or astrocytes/well) and grown for 7 days (astrocytes) or 24 hours (microglia). To assess the toxicity of bile acid treatment or other treatments, dead cells were stained with Cytotox Green or Cytotox Red Reagent (IncuCyte) following the manufacturer’s instructions. Plates were monitored and images were captured with the IncuCyte S3 every 2 hours over a period of 18 hours (microglia) or 24 hours (astrocytes). Two experiments with 6 replicates of each condition per experiment were quantified. For each well, 9 images were acquired, and fluorescence-positive cells were quantified using IncuCyte S3 software.
qPCR from in vitro cultures.
RNA was isolated from vehicle- or bile acid–treated microglia and astrocyte cultures using the RNeasy Plus Mini Kit (QIAGEN). First-strand cDNA was synthesized by reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad). Samples were quantified and amplified using SensiMix (Bioline), a SYBR-based reagent, and a CFX384 Touch Real-Time PCR Detection System (Bio-Rad). Primer sequences (Integrated DNA Technologies [IDT]) are listed in Supplemental Table 6. The Ct values for the target genes were normalized to the Actb reference gene (ΔCt) and experimental controls (ΔΔCt).
Assessment of oligodendrocyte killing by A1 astrocytes.
ACM were collected with complete Mini-EDTA–free Protease Inhibitor Cocktail (MilliporeSigma) and concentrated with Amicon Ultra-15 30-kDa filters (MilliporeSigma) by centrifugation at 4000 g for 20 minutes. Protein concentration was measured by Bradford assay.
Oligodendrocytes were isolated by sequential immunopanning. P6 to P8 mouse pup cerebral cortices were enzymatically and mechanically dissociated with papain (MACS, Miltenyi Biotec) to generate a single-cell suspension. The cell suspension was sequentially plated onto a series of negative-selection antibody-coated plates to remove endothelial cells (BSL1, catalog L-1100, Vector Laboratories), microglia (CD11b clone OX-42, Bio-Rad) and then onto a positive-selection plate for oligodendrocyte progenitors (PDGFR, BD Biosciences). Isolated oligodendrocyte progenitors were plated onto poly-d-lysine–coated coverslips in 24-well plates in oligodendrocyte base media (high-glucose DMEM, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, 4 mM l-glutamine, 5 μg/mL N-acetyl cysteine, 1× Sato, 1× B27, 5 μg/mL insulin, 10 ng/mL d-biotin, 1× trace elements B, 4.2 μg/mL forskolin) supplemented with 20 ng/mL PDGF-AA (Peprotech). Oligodendrocyte progenitors were allowed to proliferate and recover for several days, and media were replaced with differentiation media — oligodendrocyte base media supplemented with 40 ng/mL T3. After 24 hours of differentiation, 50 μg/mL concentrated ACM were added to differentiated oligodendrocytes with Cytotox Green (IncuCyte) and NucLight Rapid Red (IncuCyte). Plates were monitored in an IncuCyte S3 Live-Cell Analysis system incubator, and ×10 images were captured. The IncuCyte S3 program was used to set fixed threshold masks to quantify Cytotox and NucLight Rapid Red nuclear labeling. Images that met the defined density thresholds of 5% to 50% on phase contrast and 1% to 25% on Rapid Red nuclear labeling were used for quantification. Five experimental replicate sets of ACM (treatment with A0, A1, and A1 plus 70 μM TUDCA) with 3 to 4 wells per replicate were quantified. Data were analyzed using GraphPad Prism 8 (GraphPad Software). For each well, 3–14 images were quantified to calculate the mean and SEM of the ratio of cytotox confluence/rapid red confluence per well. The mean of the A0 cell death ratio was set to a baseline of 1, and A1 and A1 plus 70 μM TUDCA conditions were compared with the A0 baseline. A 2-way ANOVA was used to compare means between the well replicates and the experimental samples. An unpaired Welch’s t test was used to compare 2-sample group means.
EAE induction and treatment.
C57/BL6J mice were purchased from The Jackson Laboratory. GPBAR1-KO mice (52) were provided by Kristina Schoonjans (École Polytechnique Fédérale de Lausanne [EPFL], Lausanne, Switzerland) and bred in-house at Johns Hopkins University. Eight- to 11-week-old female mice were used in these experiments. In experiments comparing WT and GPBAR1-KO mice, both genotypes were mixed in shared cages before EAE induction.
EAE was induced by subcutaneous injection into 2 sites on the abdomen of 150 μg myelin oligodendrocyte glycoprotein (MOG35–55) peptide (Johns Hopkins Peptide Synthesis Core Facility) emulsified in complete Freund’s adjuvant (Difco). A total of 250 ng pertussis toxin (List Biologicals) was administered by intraperitoneal injection at the time of immunization and 48 hours later. Mice were monitored for signs of EAE and scored on the basis of the following criteria: 0, no disease; 1, limp tail; 2, hind limb weakness; 3, hind limb paralysis; 4, forelimb weakness in addition to hind limb paralysis; 5, death due to EAE.
At the onset of clinical disease, mice were randomized to daily treatment with either 500 mg/kg TUDCA (dissolved in PBS) or vehicle (PBS) administered by oral gavage. Mice were treated until day 28 after immunization or in certain experiments for a duration of 1 week. Scoring of disease severity was performed by raters blinded to the treatment allocation and genotype.
Analysis of immune infiltrate in EAE.
Mice were euthanized with isoflurane and then cardiac perfused with cold HBSS without cations. Spinal cords were flushed from the column with hydrostatic pressure. Tissue was mechanically dissociated by passing through a 16-g needle and then enzymatically digested with collagenase IV (0.5 mg/mL, Worthington) and DNAse I (100 U/mL, Worthington) for 30 minutes at 37°C with mixing after 15 minutes. After the enzymes were washed out, cells were separated from myelin debris by resuspending the tissue in a 30% Percoll (GE)/HBSS solution followed by centrifugation for 10 minutes at 750 g with no brake. Debris was aspirated off, and the cellular pellet was collected and then stimulated with cell stimulation cocktail plus protein transport inhibitors (Thermo Fisher Scientific) for 5 hours at 37°C in complete IMDM-based media (Gibco, Thermo Fisher Scientific).
Following stimulation, cells were washed and then stained with Viobility 405/520 (Miltenyi Biotec) and Fc blocked with TruStain Fc Block (BioLegend). Cells were then washed and stained with surface antibodies for 30 minutes at room temperature in the dark in FACS buffer (2% FBS, 2 mM EDTA in PBS). The antibodies used are listed in Supplemental Table 10. Cells were then fixed and permeabilized with FoxP3 Fix/Perm Buffer (eBioscience) for 20 minutes and then washed in FoxP3 permeabilization buffer (eBioscience) before intracellular staining in FoxP3 permeabilization buffer for 1 hour at room temperature in the dark. Cells were then washed again with permeabilization buffer, washed in FACS buffer, and then resuspended in FACS buffer and run on a MACSQuant 10 flow cytometer (Miltenyi Biotec). Data analysis was performed with FlowJo 10 (BD Biosciences). Fluorescence minus one (FMO) controls were used to guide analysis.
T cell proliferation assay.
Ten-week-old male and female C57/BL6 naive mice were euthanized with isoflurane followed by cervical dislocation, and spleens were collected. Spleens were then mechanically dissociated by mashing over a 100-μM cell strainer. RBCs were lysed with RBC Lysis Buffer (BioLegend). From the remaining leukocytes, naive CD4+ T cells were enriched by negative selection with a CD4– selection kit (BioLegend), and, after purity was confirmed by flow cytometry, cells were labeled with Cell Proliferation Dye eFluor 450 (Thermo Fisher Scientific) and cultured for 3 days in complete RPMI (cRPMI) media in a 96-well dish coated with 2 μg/mL anti-CD3 (BD Biosciences) and 2 μg/mL soluble anti-CD28 (BD Biosciences) in the presence or absence of TUDCA. After 3 days in culture, cells were collected and stimulated with cell stimulation cocktail plus protein transport inhibitors (Thermo Fisher Scientific) for 5 hours at 37°C in cRPMI media. Following stimulation, cells were washed and stained with Viobility 405/520 (Miltenyi Biotec) and then washed and stained with surface markers in FACS buffer for 30 minutes at room temperature. Cells were washed again and fixed and permeabilized with FoxP3 fixation/permeabilization buffer (Thermo Fisher Scientific), and then washed in FoxP3 permeabilization buffer (Thermo Fisher Scientific) and stained with intracellular antibodies for 1 hour in permeabilization buffer in the dark at room temperature. Next, cells were washed in permeabilization buffer followed by FACS buffer and then resuspended in FACS buffer and run on a MACSQuant 10 flow cytometer (Miltenyi Biotec). FMO controls were used for guiding analysis as needed, and data were analyzed with FlowJo 10 software (BD Biosciences).
Immunohistochemistry of EAE tissue.
EAE mice were deeply anesthetized and then perfused transcardially with chilled PBS followed by 4% PFA. Spinal columns were dissected and postfixed in 4% PFA overnight followed by cryoprotection with 30% sucrose for 48 hours. The spinal cords were then dissected and embedded in OCT before freezing. Tissues were sectioned (10-μm sections) on a cryostat and mounted on glass slides (Super-frost Plus; Thermo Fisher Scientific). Sections were blocked and permeabilized in PBS containing 5% normal goat serum (NGS) and 0.4% Triton X-100 for 1 hour at room temperature followed by incubation overnight at 4°C in PBS containing 3% NGS, 0.1% Triton X-100, and primary antibody (Supplemental Table 7). Sections were incubated in secondary antibodies conjugated to Alexa fluorophores (1:1000, Invitrogen, Thermo Fisher Scientific) for 1 hour at room temperature before mounting in anti-fade reagent with DAPI (Prolong Gold Anti-fade, Gibco, Thermo Fisher Scientific). Antibody validation is shown in Supplemental Figure 7.
Black Gold staining (Black Gold II Myelin Staining Kit, MilliporeSigma) was performed according to the manufacturer’s instructions. In short, slides were incubated in 0.3% Black Gold at 60°C for 12 to 20 minutes until the thinnest fibers were stained. The slides were then fixed in 1% sodium thiosulfate for 3 minutes, counterstained with 0.1% Cresyl Violet (MilliporeSigma) for 3 minutes, dehydrated using a series of gradated alcohols, cleared in xylene, and coverslipped with VectaMount Permanent Mounting Media (Vector Laboratories).
Slides were imaged alternatively on a Keyence BZ-X710 All-in-One Fluorescence Microscope (bright-field, ×20), a Zeiss Axio Observer Z1 epifluorescence microscope (×20), and a Zeiss LSM 800 confocal microscope (Apochromat ×20/0.8; 516 × 516 pixels). Imaging was performed on 5–6 sections per mouse in 5–8 mice in each group. ImageJ software (NIH) was used to create a binary image of the staining and to subsequently quantify the staining intensity and area.
Statistics.
For untargeted and targeted metabolomics data, we excluded any metabolites that had greater than 30% missing values and then normalized and scaled the metabolite’s relative abundance/absolute concentration. The preprocessed data were then used to generate pathway deregulation scores for primary analyses using the Pathifier package in R (53). The pathway deregulation scores indicate the extent to which the pathway in an individual deviates from the normal “healthy” state. To do this, each sample was projected in multidimensional space (the number of dimensions is equal to the number of metabolites in the pathway), and a principal components curve was constructed to capture the variation in this set of points. Each individual was projected onto this curve, and the pathway deregulation score was then derived from the distance along this curve between an individual’s projection and the projections for the control subjects. The scores ranged from 0 to 1, with higher scores indicating greater abnormality. The individual metabolite abundances and the pathway deregulation scores were then compared between groups using multivariate linear regression models adjusted for age, sex, and race. The P values were not adjusted for multiple comparisons, since multiple cohorts were used to validate our findings. We also generated heatmaps of Z scores of the relative abundances of each of the bile acid metabolites using Morpheus (http://software.broadinstitute.org/morpheus). We used Stata version 15 for regression analyses and GraphPad Prism 8.0 to generate all graphs. The –ΔΔCt values for astrocyte gene expression were compared between culture conditions using a Kruskal-Wallis test with Dunn’s multiple comparisons test. Heatmaps for gene expression profiles of astrocyte cultures were also created using Morpheus. Gene expression values from microglial PCR data were compared using 1-way ANOVA between groups with Dunnett’s correction for multiple comparisons. We compared EAE scores between TUDCA and vehicle groups in WT and GPBAR1-KO mice using a 2-tailed Mann-Whitney U test. We compared quantification of staining for various markers between the groups using an unpaired, 2-tailed Student’s t test. A P value of less than 0.05 was considered significant.
Study approval.
All participants at both centers provided informed consent before their participation in the studies, which were approved by the IRBs at each site (Johns Hopkins University and UCSF). For all animal experiments, we adhered to the guidelines in the NIH’s Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). All mice were maintained in a federally approved animal facility at Johns Hopkins University, and all animal experimental protocols were approved by the IACUC of Johns Hopkins University.
Author contributions
PB was involved in conceptualization, data curation, formal analysis, funding acquisition, investigation, supervision, visualization, and writing of the original draft. LM was involved in data curation, investigation, validation, and review and editing of the manuscript. EH was involved in investigation, visualization, and review and editing. KCF was involved in investigation, formal analysis, and review and editing. MDS, KM, SK, AAR, JGC, AT, SS, KV, CV, MG, HNL, KM, MT, ESS, and RD were involved in investigation, visualization, and review and editing. BN was involved in investigation, validation, and review and editing. EMM and EW were involved in investigation, supervision, and review and editing. PAC was involved in conceptualization, methodology, funding acquisition, supervision, and review and editing of the manuscript.
Supplementary Material
Acknowledgments
PB was supported by a John F. Kurtzke Clinician Scientist Development Award from the American Academy of Neurology and a Career Transition Award (TA-1503-03465) from the National Multiple Sclerosis Society. KF was supported by 1K01MH121582-01 from the National Institute of Mental Health (NIMH), NIH, and TA-1805-31136 from the National Multiple Sclerosis Society (NMSS). RD, CV, and AT are supported by the National Institute of Neurological Disorders and Stroke (NINDS), NIH (NS096148 and NS097303). This study was partly supported by a Marilyn Hilton Award for Innovation in MS research (to PAC) and a research grant from the NMSS (RG-1707-28601). We would also like to thank Krisitina Schoonjans for providing the GPBAR1-KO mice.
Version 1. 03/17/2020
In-Press Preview
Version 2. 05/26/2020
Electronic publication
Version 3. 07/01/2020
Print issue publication
Footnotes
Conflict of interest: PB has received honoraria from Sanofi Genzyme, and GSK and has received research funding from EMD Serono and Amylyx Pharmaceuticals. EMM has received research support from Biogen, Genzyme, Teva Pharmaceutical Industries, and Sun Pharma. EW has received personal compensation from the American Academy of Neurology (AAN), the Corpus, and research support from Roche, Biogen Idec, and Novartis. PAC has received personal compensation from Biogen and Disarm Therapeutics, as well as research support from Annexon Biosciences, Biogen, MedImmune, and Novartis.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(7):3467–3482.https://doi.org/10.1172/JCI129401.
Contributor Information
Pavan Bhargava, Email: pbharga2@jhmi.edu.
Matthew D. Smith, Email: matthewsmith@jhu.edu.
Leah Mische, Email: leah.mische@gmail.com.
Kathryn C. Fitzgerald, Email: kfitzg13@jhmi.edu.
Sol Kim, Email: skim440@jhmi.edu.
Jaime Gonzalez-Cardona, Email: jgonza54@jhmi.edu.
Christina Volsko, Email: cmvolsko@gmail.com.
Ajai Tripathi, Email: tripata@ccf.org.
Sonal Singh, Email: ssingh79@jhu.edu.
Kesava Varanasi, Email: kvarana1@jhu.edu.
Hannah-Noelle Lord, Email: hlord3@jhmi.edu.
Michelle Taylor, Email: mtayl125@jhmi.edu.
Marjan Gharagozloo, Email: mgharag1@jhmi.edu.
Elias S. Sotirchos, Email: ess@jhmi.edu.
Bardia Nourbakhsh, Email: bnourba1@jhmi.edu.
Ranjan Dutta, Email: duttar@ccf.org.
Emmanuelle Waubant, Email: emmanuelle.waubant@ucsf.edu.
Peter A. Calabresi, Email: pcalabr1@jhmi.edu.
References
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378(2):169–180. doi: 10.1056/NEJMra1401483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isobe N, et al. An ImmunoChip study of multiple sclerosis risk in African Americans. Brain. 2015;138(Pt 6):1518–1530. doi: 10.1093/brain/awv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Multiple Sclerosis Genetics Consortium (IMSGC) et al. Evidence for polygenic susceptibility to multiple sclerosis--the shape of things to come. Am J Hum Genet. 2010;86(4):621–625. doi: 10.1016/j.ajhg.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascherio A, et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014;71(3):306–314. doi: 10.1001/jamaneurol.2013.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhargava P, Calabresi PA. Metabolomics in multiple sclerosis. Mult Scler. 2016;22(4):451–460. doi: 10.1177/1352458515622827. [DOI] [PubMed] [Google Scholar]
- 6.Wikoff WR, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA. 2009;106(10):3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhargava P, Fitzgerald KC, Calabresi PA, Mowry EM. Metabolic alterations in multiple sclerosis and the impact of vitamin D supplementation. JCI Insight. 2017;2(19):95302. doi: 10.1172/jci.insight.95302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villoslada P, et al. Metabolomic signatures associated with disease severity in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2017;4(2):e321. doi: 10.1212/NXI.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickens AM, et al. A type 2 biomarker separates relapsing-remitting from secondary progressive multiple sclerosis. Neurology. 2014;83(17):1492–1499. doi: 10.1212/WNL.0000000000000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30(3):332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMillin M, DeMorrow S. Effects of bile acids on neurological function and disease. FASEB J. 2016;30(11):3658–3668. doi: 10.1096/fj.201600275R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo C, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45(4):802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Hucke S, et al. The farnesoid-X-receptor in myeloid cells controls CNS autoimmunity in an IL-10-dependent fashion. Acta Neuropathol. 2016;132(3):413–431. doi: 10.1007/s00401-016-1593-6. [DOI] [PubMed] [Google Scholar]
- 14.Ho PP, Steinman L. Obeticholic acid, a synthetic bile acid agonist of the farnesoid X receptor, attenuates experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2016;113(6):1600–1605. doi: 10.1073/pnas.1524890113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis ND, et al. A GPBAR1 (TGR5) small molecule agonist shows specific inhibitory effects on myeloid cell activation in vitro and reduces experimental autoimmune encephalitis (EAE) in vivo. PLoS ONE. 2014;9(6):e100883. doi: 10.1371/journal.pone.0100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanguas-Casás N, Barreda-Manso MA, Nieto-Sampedro M, Romero-Ramírez L. TUDCA: an agonist of the bile acid receptor GPBAR1/TGR5 with anti-inflammatory effects in microglial cells. J Cell Physiol. 2017;232(8):2231–2245. doi: 10.1002/jcp.25742. [DOI] [PubMed] [Google Scholar]
- 17.Gaspar JM, Martins A, Cruz R, Rodrigues CM, Ambrósio AF, Santiago AR. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience. 2013;253:380–388. doi: 10.1016/j.neuroscience.2013.08.053. [DOI] [PubMed] [Google Scholar]
- 18.Mortiboys H, Furmston R, Bronstad G, Aasly J, Elliott C, Bandmann O. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology. 2015;85(10):846–852. doi: 10.1212/WNL.0000000000001905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elia AE, et al. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol. 2016;23(1):45–52. doi: 10.1111/ene.12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MahmoudianDehkordi S, et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease — an emerging role for gut microbiome. Alzheimers Dement. 2019;15(1):76–92. doi: 10.1016/j.jalz.2018.07.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhargava P, Mowry EM. Gut microbiome and multiple sclerosis. Curr Neurol Neurosci Rep. 2014;14(10):492. doi: 10.1007/s11910-014-0492-2. [DOI] [PubMed] [Google Scholar]
- 22.Jangi S, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015. doi: 10.1038/ncomms12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cekanaviciute E, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. 2017;114(40):10713–10718. doi: 10.1073/pnas.1711235114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh N, et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc Natl Acad Sci USA. 2018;115(2):E302–E309. doi: 10.1073/pnas.1716032115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangalam A, et al. Profile of circulatory metabolites in a relapsing-remitting animal model of multiple sclerosis using global metabolomics. J Clin Cell Immunol. 2013;4(3) doi: 10.4172/2155-9899.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol. 2017;101(1):47–64. doi: 10.1007/s00253-016-8006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Launay N, et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017;133(2):283–301. doi: 10.1007/s00401-016-1655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosa AI, et al. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(9):2171–2181. doi: 10.1016/j.bbadis.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Miron VE, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giles DA, et al. Myeloid cell plasticity in the evolution of central nervous system autoimmunity. Ann Neurol. 2018;83(1):131–141. doi: 10.1002/ana.25128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothhammer V, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724–728. doi: 10.1038/s41586-018-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cantarel BL, et al. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J Investig Med. 2015;63(5):729–734. doi: 10.1097/JIM.0000000000000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tremlett H, et al. Gut microbiota in early pediatric multiple sclerosis: a case-control study. Eur J Neurol. 2016;23(8):1308–1321. doi: 10.1111/ene.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. 2016;6:28484. doi: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48(11):1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarling EJ, et al. RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J Clin Invest. 2017;127(10):3741–3754. doi: 10.1172/JCI94029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puleston DJ, Villa M, Pearce EL. Ancillary activity: beyond core metabolism in immune cells. Cell Metab. 2017;26(1):131–141. doi: 10.1016/j.cmet.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGillicuddy FC, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119(8):1135–1145. doi: 10.1161/CIRCULATIONAHA.108.810721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jena PK, Sheng L, Di Lucente J, Jin LW, Maezawa I, Wan YY. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018;32(5):2866–2877. doi: 10.1096/fj.201700984RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hucke S, et al. The farnesoid-X-receptor in myeloid cells controls CNS autoimmunity in an IL-10-dependent fashion. Acta Neuropathol. 2016;132(3):413–431. doi: 10.1007/s00401-016-1593-6. [DOI] [PubMed] [Google Scholar]
- 42.Kida T, Tsubosaka Y, Hori M, Ozaki H, Murata T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(7):1663–1669. doi: 10.1161/ATVBAHA.113.301565. [DOI] [PubMed] [Google Scholar]
- 43.Albrecht S, et al. Activation of FXR pathway does not alter glial cell function. J Neuroinflammation. 2017;14(1):66. doi: 10.1186/s12974-017-0833-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menni C, et al. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol. 2013;42(4):1111–1119. doi: 10.1093/ije/dyt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Angelin B, Björkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest. 1982;70(4):724–731. doi: 10.1172/JCI110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angelin B, Björkhem I. Postprandial serum bile acids in healthy man. Evidence for differences in absorptive pattern between individual bile acids. Gut. 1977;18(8):606–609. doi: 10.1136/gut.18.8.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nourbakhsh B, Bhargava P, Tremlett H, Hart J, Graves J, Waubant E. Altered tryptophan metabolism is associated with pediatric multiple sclerosis risk and course. Ann Clin Transl Neurol. 2018;5(10):1211–1221. doi: 10.1002/acn3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 49.Dutta R, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59(3):478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 50.Dutta R, et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol. 2011;69(3):445–454. doi: 10.1002/ana.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017;133(1):13–24. doi: 10.1007/s00401-016-1653-y. [DOI] [PubMed] [Google Scholar]
- 52.Thomas C, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proc Natl Acad Sci USA. 2013;110(16):6388–6393. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.