

Abstract
Purpose
To investigate characteristics of the foveal pit and the foveal avascular zone (FAZ) in patients with Alport syndrome (AS), a rare monogenetic disease due to mutations in genes encoding for collagen type IV.
Methods
Twenty-eight eyes of nine patients with AS, and five autosomal-recessive carriers and 15 eyes from 15 age-similar healthy control subjects were examined using optical coherence tomography (OCT) and OCT-angiography (OCT-A). Foveal configuration and FAZ measures including the FAZ area, circularity, and vessel density in the central 1° and 3° were correlated.
Results
Foveal hypoplasia was found in 10 eyes from seven patients with either genotype. In contrast, a staircase foveopathy was found in seven eyes of four X-linked AS patients. The average FAZ area did not differ significantly between AS patients and control subjects (mean ± SD 0.24 ± 0.24 mm2 vs. 0.21 ± 0.09 mm2; P = 0.64). Five eyes showed absence or severe anomalies of the FAZ with crossing macular capillaries that was linked to the degree of foveal hypoplasia on OCT images leading to a significant inverse correlation of FAZ area and foveal thickness (r = −0.88; P < 0.001). In contrary, female patients with X-linked mutations exhibited a significantly greater FAZ area (0.48 ± 0.30 mm2 vs. 0.21 ± 0.09 mm2; P = 0.007), in line with OCT findings of a staircase foveopathy.
Conclusions
The foveal phenotypic spectrum in AS ranges from foveal hypoplasia and absence of a FAZ to staircase foveopathy with an enlarged FAZ. Because the development of the FAZ and foveal pit are closely related, these findings suggest an important role for collagen type IV in foveal development and maturation.
Keywords: Alport syndrome, foveal hypoplasia, optical coherence tomography angio-graphy, foveal avascular zone
Alport syndrome (AS) was first described in 1927 by Arthur Cecil Alport who discovered the association between renal failure, hearing impairment, and later ophthalmologic symptoms in an affected family.1 AS is a rare inherited disease due to mutations in the genes coding for collagen IV affecting approximately 1–5:50,000.2,3 Biallelic mutations in the COL4A3 and COL4A4 genes located on chromosome 2 cause autosomal recessive AS and account for approximately 15% of all cases,4 whereas 85% of all patients with AS present an X-linked inheritance due to mutations in the COL4A5 gene on chromosome X.3
Autosomal-dominant inheritance has been reported but is subject of discussion.5,6 Female patients with a heterozygous X-linked mutation present with clinical symptoms delayed by about 30 years compared with X-linked affected men and are therefore termed patients.3
The common background of this multisystem disease is an alteration in collagen IV, which is a major component of basement membranes.7 In AS, the assembly of the collagen triple helix is impaired and more susceptible to proteolytic processes.8–10 The main site of pathology is the kidney because of the high amount of collagen IV in the glomerular basement membrane. As a result of the respective mutations in AS, the glomerular basement membrane—as a main contributor for the glomerular filtration barrier—is lamellated. This altered filtration barrier results in the early presentation of microhematuria or macrohematuria and proteinuria in AS patients, with further development of barrier disturbances and a reduced glomerular filtration rate eventually leading to end-stage renal failure.11,12
In the eye, the cornea, lens, and retina are typically affected because of the common abundancy of collagen IV in these tissues. The posterior polymorphous corneal dystrophy and anterior lenticonus due to affected basement membranes of the corneal endothelium and the lens epithelium are well-described ocular findings in AS patients.13–15
Previously published retinal findings include a dull macular reflex, a dot-and-fleck retinopathy, and a temporal thinning of the macular neuroretina. In some patients, a bull's-eye maculopathy has been reported.13,14,16,17
An involvement of Bruch's membrane (BrM) has been discussed because BrM comprises collagen type IV in its inner and outer layers.18 Moreover, collagen IV is also a component of the inner limiting membrane (ILM). Collagen type IV, especially the alpha 2 subdomain, has further been linked to the retinal vascular patterning during fetal development.19,20
In recent years, high-resolution spectral-domain optical coherence tomography (SD-OCT) and swept-source optical coherence tomography angiography (OCT-A) became widely available. In this context, numerous structural features related to the retinal vasculature such as the foveal avascular zone (FAZ) and parafoveal capillary density have been proposed as biomarkers for both ocular and systemic diseases including diabetes and neurodegenerative disease.21 Given that type IV collagen constitutes the main scaffold of the ILM, a refined investigation of the foveal pit morphology and FAZ seems warranted in diseases affecting collagen IV.
Methods
Cohort Characteristics
The cross-sectional prospective case-control study was performed between March 2018 to March 2019. Individuals were recruited in the clinic for rare retinal diseases of the Department of Ophthalmology, University Hospital Bonn, Germany. The study was performed in adherence to the declaration of Helsinki. Institutional review board approval (Approval 133-15, Ethikkommission, Medizinische Fakultät, Rheinische Friedrich-Wilhelms Universität Bonn) and patients’ written consent were obtained.
All patients with AS were genetically examined prior to before our study and had a plausible genotype. Five female patients with an X-linked mutation were included. In accordance with previous publications, we defined these subjects as patients.3
Exclusion criteria included severe ocular comorbidities, previous intraocular surgery other than cataract surgery, a history of preterm birth, and high refractive errors (>4 dpt spherical equivalent). Two eyes were excluded because of previous vitreoretinal surgery.
All patients underwent a complete ophthalmologic examination including refraction, Best Corrected Visual Acuity (BCVA) testing, intraocular pressure measurement, anterior segment and dilated fundus examination by three ophthalmologists (K.H., P.H., K.L.) for anterior and posterior abnormalities. As control subjects, we included 15 eyes of 15 subjects without any eye disease or known systemic condition.
All patients underwent an extensive imaging protocol including color fundus photography (Visucam; Zeiss, Oberkochen, Germany), SD-OCT (Spectralis OCT 2; Heidelberg Engineering, Heidelberg, Germany), and fundus autofluorescence imaging (HRA2, Heidelberg Engineering, Heidelberg, Germany). OCT-A was performed in all patients using the PlexElite 9000 (Zeiss) including a 3 × 3 mm, 6 × 6 mm, and 9 × 9 mm macular scan. SD-OCT images were acquired using the high-resolution mode (5.7 µm/pixel) in an area of 30° × 25° with 121 B-Scans.
Image Processing and Data Analysis
The preset instrument parameters were used to define the superficial and deep capillary plexus in the OCT-A images. Correct segmentation was verified by an experienced examiner and - when necessary - manually corrected. For further processing (i.e., binarizing of the OCT-A images for quantification of vessel densities), the OCT-A data were uploaded to the Advanced Retina Imaging (ARI) network hub (Zeiss, Oberkochen, Germany). In this platform, the Zeiss proprietary Macular density algorithm (version 07.1) was applied, which provides a binarized perfusion density image as previously described.22 Vessel density was then quantified on the resulting binarized OCT-A images using ImageJ (Bethesda, MD, USA).
The FAZ, the circularity of the FAZ, and the vessel density of the central 1° and 3° were manually measured using ImageJ. The FAZ was defined to be abnormal, if there was no detectable FAZ or if vessels crossed the FAZ. Small FAZ areas were defined to be all those with an area of <0.18 mm2, as defined in previous studies.23
The quantification of foveal thickness, temporal thinning, and foveal pit width in the structural OCT images were analyzed using the built-in segmentation and analysis software of the Heidelberg Eye Explorer (Version 2.3.3; Heidelberg Engineering). Correct segmentation was verified by an experienced examiner and—when necessary—manually corrected.
OCT images were quantified regarding foveal thickness by means of OCT-volume scans (121 B-scans, 30° horizontal × 25° vertical; Spectralis; Heidelberg Engineering) using the Heidelberg Eye Explorer. To measure the temporal and nasal retinal thickness, the foveal horizontal scan was analyzed for the highest elevation of the retina in the parafoveal regions. B-scan segmentation was manually corrected if needed. Nasal-to-temporal difference was calculated by subtraction for each individual.
A grading regarding the foveal pit was realized according to the classification of Thomas et al.24 In this grading, the normal foveal pit is characterized by the extrusion of the plexiform layers, a discernable foveal pit, the lengthening of the photoreceptor outer segments and a widening of the outer nuclear layer. Every grade displays the absence of these characteristics in that order from 1 to 4. OCT-Images shown in Figures 1 and 2 were rectified along the BrM for better comparison between patients and control.
Figure 1.

Phenotypic spectrum of persisting inner retinal layers in Alport syndrome. A normal foveal configuration as shown in the upper panel, including extrusion of the plexiform layer, widening of the outer nuclear layer (ONL) and lengthening of the photoreceptor outer segments (OS). The chosen examples of Alport patients show different manifestations of persisting inner retinal layers. While continuation of the plexiform layers is the most frequent characteristic (A–H), more extreme examples exhibit additionally inner nuclear layers leading to a hypoplastic foveal pit (B–F). The different forms represent the specific layer not yielding the foveal pit in the respective OCT image: Circle = nerve fiber layer, rectangle = inner plexiform layer, triangle = inner nuclear layer, star = outer plexiform layer.
Figure 2.

Phenotypic spectrum of “staircase-foveopathy” in Alport syndrome. A normal foveal configuration as shown in the upper panel (A), whereas the exemplary patients show irregular depletion of the inner retinal layers, beginning in parafoveal areas nasally and temporally. The thinning is not homogenous but in a “staircase”-like manner (white arrows). Despite this broadly allocated thinning, the outer retina is preserved in all cases. The (apparently) higher reflectivity of the choroid in patients compared to controls is related to the enhanced depth imaging mode used in patients, which visualizes the thinner choroids in patients more clearly than the regular OCT mode.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) and Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). After a normal distribution for each group was confirmed, two-tailed t-test was applied for further analysis. The Mann-Whitney U test was used to compare data with non-normal distributions. For comparison of variability of cohorts, Levene’s test was used. P values <0.05 were considered statistically significant.
Results
Cohort
A total of 28 eyes of 14 AS patients (aged 36.4 ± 21.7 years [mean ± SD]; range 5–69 years) and five autosomal-recessive carriers (39.7 ± 15.3 years) were investigated and compared to 15 eyes of 15 age-similar control subjects (aged 38.1 ± 15.1 [mean ± SD]; range 21–58 years). We further included two male genetically confirmed X-linked AS patients who have been in our clinic before our study. The same imaging protocol (without OCT-A) has been performed in these patients.
Mean patient visual acuity was 0.3 ± 0.2 (logMAR) among the patients, whereas it was 0.00 ± 0.09 in control subjects (P < 0.001). Two eyes of two female X-linked AS patients were excluded due to previous vitreoretinal surgery of a macular hole and retinal detachments, respectively. Thus 26 eyes were included in the study.
Pathogenic mutations in the COL4A5 gene on chromosome X were found in 7 patients (54%), of which three male patients were diagnosed with X-linked AS, one female was diagnosed with homozygous X-linked AS, and three females (12%) were heterozygous X-linked patients. Pathogenic autosomal-recessive mutations (COL4A3 or COL4A4) were found in seven subjects and patients (46%). Two male patients were diagnosed with autosomal-recessive AS and in one of them a consanguineous background was confirmed. Five subjects were confirmed as autosomal-recessive carriers of one pathogenic mutation after testing due to AS in a family member.
Foveal Pit and Foveal Thickness
In the Alport group, a total of nine eyes revealed persisting inner retinal layers on OCT images (Fig. 1). Using the classification by Thomas et al.,24 two eyes revealed a grade 2 hypoplasia with absent extrusion of the plexiform layers and absent foveal pit whereas seven eyes showed a grade 1 hypoplasia with only missing extrusion of the plexiform layers, which was termed “persisting inner retinal layers” in previous studies.
In extreme cases (Fig. 1, B and C) the persisting inner retinal layers comprise the nerve fiber layer (white circle), inner plexiform layer (white rectangle), inner nuclear layer (white triangle) and the outer plexiform layer (white star). This is associated with a marked foveal hypoplasia. Also note the temporal retinal thinning compared with the nasal retina. However, in moderate cases (Fig. 1, D–F) a continuous inner nuclear layer is still observable between the two plexiform layers, but the foveal pit is steeper and the nerve fiber layer cannot be detected with certainty in the very center.
Mild cases (Fig. 1, G, H) show a single layer of hyperreflective tissue comparable to the plexiform layers. The inner nuclear layer is discontinued in the area of the foveal pit; however, the layer shows an irregular and asymmetric pattern in the perifoveal area.
Mean foveal thickness did not differ significantly between AS patients or autosomal-recessive carriers (229.00 ± 47.88 µm for AS patients; P = 0.69; 234.50 ± 24.46 µm for autosomal-recessive carriers, P = 0.21) compared with control subjects (223.73 ± 17.67 µm). However, the interindividual variation was significantly higher in AS patients and autosomal-recessive carriers as compared with control subjects (f-ratio = 4.45, P = 0.043).
Nonetheless, the nasal-to-temporal retinal thickness difference was significantly higher in AS patients, with a mean difference of 58.67 ± 29.69 µm compared with control subjects (18.67 µm, P < 0.0001). Autosomal-recessive carriers showed very similar results as compared with control subjects (18.20 ± 10.22 µm).
A “staircase-foveopathy” and mild phenotypes with beginning inner retinal dimples were found in seven eyes of four patients, three male and one female, solely affected by the X-linked subform of AS.
Staircase Foveopathy and Enlarged FAZ
Staircase foveopathy, defined as irregular loss of inner retinal layers in the central macular region, leading to a broadly thinned retina with a stair-like appearance, can be visualized using OCT imaging. Because of the thinning, the center of the foveal pit is not clearly discernable.17
In our cohort, seven eyes revealed a staircase configuration of different extent (Fig. 2). OCT-A was assessed in only three of these eyes. The mean FAZ area in this group was 0.57 ± 0.22 mm2 and thus significantly larger than in control subjects (0.2120 ± 0.09 mm2; P < 0.001). Descriptively, all eyes revealed a scattered vasculature around the enlarged FAZ.
Although the temporal retina is in general thinner in AS patients, all horizontal retinal sectors of the ETDRS grid are significantly thinner in patients exhibiting staircase foveopathy (see Fig. 3).
Figure 3.

Comparison of retinal thickness using the ETDRS-grid. (A) ETDRS-grid sectors for a right eye applied in B and C. For left eyes, the vertically mirrored version was applied. (B) Retinal thickness in the respective ETDRS-grid sector for all AS patients compared with control subjects. Although AS patients show greater interindividual variability in all sectors, the retina in AS patients is significantly thinner in the temporal area. (C) Retinal thickness in the respective ETDRS-grid sector for patients exhibiting staircase foveopathy. The retina is significantly thinner in all horizontal sectors.
FAZ-Alterations in Alport Syndrome Patients
FAZ area in the superior plexus was 0.24 ± 0.24 mm2 vs. 0.21 ± 0.09 mm2 for control subjects (P = 0.63) with high interindividual variability in the AS group (Figure 7). Autosomal-recessive carriers had very similar FAZ-areas and standard deviation compared with control subjects (0.21 ± 0.10 mm2 vs. 0.21 ± 0.09 mm2; P = 0.58). However, six eyes of AS patients (24%) revealed abnormalities of the FAZ and the central foveal capillary network with variable extent and different from any control FAZ. 12 eyes (46%) revealed small FAZs (<0.18 mm2), whereas only two subjects in the control group showed a FAZ smaller than <0.18 mm2 (P < 0.001).
Figure 7.
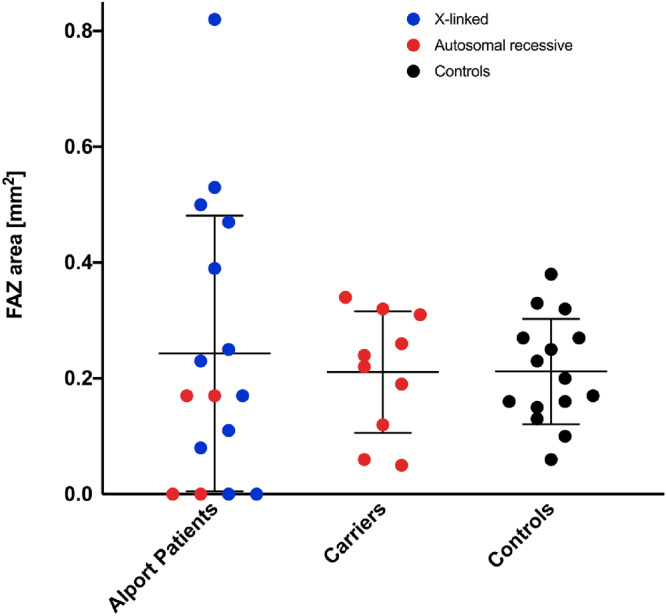
FAZ area in patients, controls and autosomal-recessive carriers. Plot showing the FAZ area size in Alport patients, autosomal-recessive carriers and controls. Blue dots show phenotypes due to an X-linked mutation, and red dots represent autosomal-recessive inheritance. Control subjects are shown as black dots. While absent FAZ is present for X-linked and autosomal recessive phenotypes, enlarged FAZ sizes are only exhibited due to of X-linked mutations. When analyzing the OCTs of the largest FAZ areas in this graph all presented with a staircase foveopathy. Autosomal-recessive carriers show overall similar FAZ areas compared with control subjects.
Patients with persisting inner retinal layers consequently revealed smaller or absent FAZ and differed as a subgroup significantly from control subjects (0.04 ± 0.06 mm2, P < 0.001). Patients exhibiting a staircase foveopathy had significantly larger FAZ areas (0.47 ± 0.31 mm2) compared to control subjects (P = 0.0069), consistent with missing inner retinal layers in OCT imaging. These findings are in coherence with the negative correlation of central retinal thickness and FAZ area, shown in Figure 4C. FAZ-circularity was significantly lower not only in patients (0.512 ± 0.26; P = 0.0083) but interestingly also in AR carriers (0.04 ± 0.11; P = 0.0045) compared with control subjects (0.73 ± 0.08).
Figure 4.

( A – D ) Foveal thickness, nasal-to-temporal thickness and relation to FAZ area. (A) Average foveal thickness is similar in Alport patients, autosomal-recessive carriers and controls, whereas Alport patients exhibit a higher interindividual variability. (B) Nasal-to-temporal retinal thickness difference is significantly elevated in Alport Patients. Again, autosomal-recessive carriers and controls do not differ significantly. (C) FAZ area is highly correlated with the foveal thickness in AS patients. (D) shows that a more Alport-specific characteristic, the nasal-to-temporal thickness difference, does not exhibit a clear correlation to the FAZ area.
Vessel density in the central 1° was significantly higher in AS patients (31.5% ± 5.4%) compared with in control subjects (22.4% ± 9.9%; P = 0.0149). However, vessel density in the central 3° was only slightly higher for AS patients (37.7% ± 4.2%) than in control subjects (39.4% ± 2.8%; P = 0.0903).
Intriguingly, when correlating the FAZ size with OCT parameters, the foveal thickness is a better predictor for the FAZ area in the superior capillary plexus for AS patients (r = –0.88; P = 0.00078) than the AS-characteristic nasal-to-temporal thickness difference (r = –0.279, P = 0.38, Fig. 4). FAZ size in AS patients showed a mild correlation with visual acuity (r = 0.55, P = 0.025) and a weak positive relation to age (r = 0.44, P = 0.02).
Characteristics of Patients With Absent or Partially Vascularized FAZ
The interaction of foveal pit formation, FAZ vascularization and genetic and environmental factors is complex, making a correlation in a small cohort of a rare disease difficult. For reasons of comprehensibility of this complex phenotype, we decided to describe four patients with the most striking abnormalities in FAZ and foveal pit formation to show the variety of altered characteristics in Alport syndrome (Fig. 567). A comparison of superficial and deep plexus in these patients is provided in Figure 6.
Figure 5.

Multimodal imaging of foveal vasculature and abnormalities of the foveal avascular zone by optical coherence tomography angiography (OCT-A), OCT and color fundus photography. Patient 1. Young patient with bi-allelic AR-mutations in COL4A4. OCT reveals foveal hypoplasia grade 1 in the right and grade 2 in the left eye. OCTA reveals an absent FAZ in both eyes with unusually thick vessels extending toward the foveal center. Auxiliary findings were a central dot-and-fleck retinopathy and a dull macular reflex. Patient 2. Female patient with a bi-allelic X-linked mutation. OCT reveals persisting inner retinal layers. Coherently OCTA images reveal crossing capillaries in the FAZ although less dense than in A. Patient 3. 7-year-old boy with temporally FAZ-crossing vessels and a perifoveal scattered vascular complex. IR-Images show a macular dot-and-fleck-retinopathy. Patient 4. Staircase foveopathy on OCT linked to a decreased circularity and loose perifoveal vessels on OCTA. Distinct dot-and-fleck retinopathy are visible on IR Images and fundus photography reveals discolored areas around the lower temporal vessel arch.
Figure 6.

Superior and deep capillary plexus as perfusion trace images. Although the FAZ is absent or diminished in the superior plexus, a clear FAZ in the deep plexus was present in all cases. However, the deep capillary plexus seems to be unusually elongated in the horizontal axis.
In patient 1 (Fig. 5, first and second row), no FAZ at all was detectable in both eyes. OCT-A revealed dense vascularization with extension of thicker vessels deriving directly from the great arch vessels reaching the very central foveal area. Analyzing the OCT-scan, these findings were associated with a misshaped and hypoplastic foveal pit grade 2 in the right eye and grade 4 in the left eye (first row, second column). Persisting inner retinal layers and pronounced temporal retinal thinning were further findings. Visual acuity was reduced to 0.2 logMAR in the right eye and 0.1 logMAR in the left eye for which no other morphologic explanation (clear media and a normal shaped lens) was found. This patient was genetically found to have a homozygous mutation in the COL4A4 gene on chromosome 2 due to consanguinity of the parents. His brother and mother were heterozygous carriers of one mutation and showed a normal FAZ and regular foveal pit configuration.
Patient 2 showed a dense vascularization in both foveae with only a minimal FAZ in the left eye (Fig. 5, third and fourth row). In contrast to patient 1, this patient did not show any structural foveal pit anomalies in OCT scans. Fundus examination and infrared imaging as well as fundus autofluorescence (not shown) were without pathologic findings. Due to a point mutation on chromosome 2 (c.1871 G>A), which led to end-stage renal failure in male relatives of this patient, drastic renal involvement was predicted but so far, the 52-year-old patient only suffered from mild microhematuria. Visual acuity was 0.00 logMAR (20/20) in both eyes, and ophthalmologic examination was without further pathologic findings apart from the densely vascularized fovea in OCT-A.
Patient 3 (Fig. 5, fifth row) revealed foveal macular capillaries in the right eye, whereas the left eye had a normal FAZ. This 7-year-old boy with X-linked AS showed a significant temporal retinal thinning in OCT and alterations of the foveal pit, especially a disorganization of the inner retinal layers passing into the foveal pit. Visual acuity was good (0.00 logMAR, 20/20 in both eyes) in both eyes.
Patient 4 (Fig. 5, sixth row) showed a discernable FAZ but presented with capillaries crossing its center. OCT revealed a staircase foveopathy as described in one patient of AS before.17 Infrared reflectance image showed a ring-shaped dot-and-fleck retinopathy, localized at the transition zone of normal retinal layers to the staircase formation. Fundus autofluorescence showed hyperreflective areas centrally and close to the superior vessel arch that were not observed in any other AS patient and have as yet not been reported in the literature.
Discussion
Our study revealed a variety of abnormalities of the foveal configuration, as well as the shape and circularity of the FAZ in patients with AS. Specifically, two opposing phenotypic variants were observed. Patients with AS commonly exhibited foveal hypoplasia as characterized by a small or absent FAZ, misshaped foveal pit, increased foveal thickness and persisting inner retinal layers. On the other hand, a subset of patients exhibited staircase-foveopathy associated with an ill-defined enlarged FAZ. Thus, the spectrum of structural findings in AS and its relationship to the genotype appears to be more complex than previously assumed.
Foveal hypoplasia and absence of the FAZ has been observed in multiple ocular diseases including albinism, aniridia, retinopathy of prematurity, incontinentia pigmenti, achromatopsia, familial exudative vitreoretinopathy and Stickler syndrome.24–27 However, this study represents to the best of our knowledge the first description of foveal hypoplasia and absence of the FAZ in AS. Interestingly, similar findings including foveal hypoplasia, persisting inner retinal layers and smaller or absent FAZ were reported for patients with Stickler syndrome.28 Stickler syndrome is an inherited disease due to mutations mostly in the COL2A1 and COL11A1 genes, encoding for collagen II and XI, respectively. These similar findings in two monogenetic diseases with mutations in genes coding for collagen formation and assembly highlight the importance of collagen as an extracellular component in correct foveal development.
The foveal development begins during the twenty-fifth fetal week and is fully completed about 45 weeks after birth.29 Pit formation occurs by centrifugal displacement of inner retinal structures, with cone photoreceptors moving toward the center.30
Because the genetic background of AS concerns collagen type IV, a major component in basement membranes, an important role of this collagen for the macular development appears likely. A close relationship is also present between collagen IV and angiogenesis because endothelial cells develop podosomes, leading to collagen IV degradation within the vascular basal laminar during the sprouting process in retinal angiogenesis.31
On the other hand, a subset of AS patients revealed an enlarged FAZ in the presence of a staircase-foveopathy. This very unusual characteristic of the inner layers in the central retina with preserved outer retinal layers has been described in only one previous case report without genotyping which also proposed the descriptive term.17 In our cohort, seven eyes revealed this peculiar finding, and all affected patients had an X-linked genetic background. This alteration was limited to adult patients, whereas younger patients of any genotype did not reveal this phenotypic characteristic. Although we did not assess longitudinal variations, it may be speculated that this phenotype constitutes a progressive degenerative process rather than a stationary alteration. This hypothesis would be supported by a study of Pöschl et al.,32 who in a mouse model showed that the absence of collagen IV has only limited impact on basement membrane development, since laminin is sufficient to form the basic matrix but cannot act as an equivalent long-term scaffold. However, the investigations showed that in the long run, the stability and functionality is not maintained and might lead to damage of the adjacent tissues.32
Further clinical support is given by a study of foveal changes after ILM peeling.33 Here, so-called iatrogenic “inner retinal dimples” potentially parallel dot-and-fleck retinopathy and precede staircase foveopathy in AS. These inner retinal dimples typically manifest within 12 months after surgical ILM peeling and are characterized by notches in the inner retinal layers.33 A putative pathologic association is the ILM as a main component for preservation of inner retinal layers, perpetuating their stability and integrity, which leads to disturbances after ILM-surgery in vulnerable subjects.
In the future, both extended cross-sectional and longitudinal natural-history studies are needed. For a distinct genotype-phenotype correlation, higher numbers of patients in each cohort are necessary. Longitudinal observations have to untangle the role of the staircase foveopathy and whether this condition is a stable characteristic or a progressive degenerative process. Furthermore, interdisciplinary studies can help to enlighten the ocular pathologic findings in the context of nephrologic findings and renal failure.
In summary, multifaceted foveal pit abnormalities represent a common finding in AS patients. The abnormalities include foveal hypoplasia to variable degrees with absence of a FAZ (all genetic subgroups), as well as staircase foveopathy with an enlarged FAZ (X-linked genetic disease background). The temporal retinal thinning can be correlated to a misshaped foveal pit, which in turn is in its development closely linked to the FAZ formation. These new findings give further insight not only into the AS phenotype but also into the importance of collagen IV on macular development, maturation and maintenance.
Acknowledgments
Supported by BONFOR GEROK Program, Faculty of Medicine, University of Bonn, Grant No O-137.0022 and O-137.0025 (MP), and BONFOR GEROK Program, Faculty of Medicine, University of Bonn, Grant No O-137.0028 (MWMW). The authors alone are responsible for the content and writing of the paper.
Disclosure: K. Hess, Carl Zeiss Meditec (F), Heidelberg Engineering (F); M. Pfau, Carl Zeiss Meditec (F), Heidelberg Engineering (F); M.W.M. Wintergerst, Carl Zeiss Meditec (F), Heidelberg Engineering (F); K.U. Loeffler, None; F.G. Holz, Carl Zeiss Meditec (F, R), Heidelberg Engineering (C, F, R); P. Herrmann, Carl Zeiss Meditec (F), Heidelberg Engineering (F)
References
- 1. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. 1927; 1: 504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000; 58: 925–943. [DOI] [PubMed] [Google Scholar]
- 3. Savige J, Colville D, Rheault M et al.. Alport syndrome in women and girls. Clin J Am Soc Nephrol. 2016; 11: 1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol. 2013; 24: 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018; 3: 1239–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013; 24: 364–375. [DOI] [PubMed] [Google Scholar]
- 7. Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003; 348: 2543–2556. [DOI] [PubMed] [Google Scholar]
- 8. Lee JM, Nozu K, Choi DE, Kang HG, Ha IS, Cheong HI. Features of autosomal recessive Alport syndrome: a systematic review. J Clin Med. 2019; 8: 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nozu K, Nakanishi K, Abe Y et al.. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. 2019; 23: 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hertz JM, Thomassen M, Storey H, Flinter F. Clinical utility gene card for: Alport syndrome—update 2014. Eur J Hum Genet. 2015; 23: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kruegel J, Rubel D, Gross O. Alport syndrome—insights from basic and clinical research. Nat Rev Nephrol. 2013; 9: 170–178. [DOI] [PubMed] [Google Scholar]
- 12. Hamano Y, Grunkemeyer JA, Sudhakar A et al.. Determinants of vascular permeability in the kidney glomerulus. J Biol Chem. 2002; 277: 31154–31162. [DOI] [PubMed] [Google Scholar]
- 13. Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015; 10: 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Savige J, Wang Y, Crawford A et al.. Bull's eye and pigment maculopathy are further retinal manifestations of an abnormal Bruch's membrane in Alport syndrome. Ophthalmic Genet. 2017; 38: 238–244. [DOI] [PubMed] [Google Scholar]
- 15. Savige J, Liu J, DeBuc DC et al.. Retinal basement membrane abnormalities and the retinopathy of Alport syndrome. Invest Ophthalmol Vis Sci. 2010; 51: 1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ahmed F, Kamae KK, Jones DJ et al.. Temporal macular thinning associated with X-linked Alport syndrome. JAMA Ophthalmol. 2013; 131: 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanojcic N, Raja MS, Burton BJ. Choroidal thinning and “stair-case” foveal sign in a patient with Alport syndrome. Retin Cases Brief Rep. 2014; 8: 52–55. [DOI] [PubMed] [Google Scholar]
- 18. Swaminathan SS, Shah P, Zheng F, Gregori G, Rosenfeld PJ. Detection of choriocapillaris loss in Alport syndrome with swept-source OCT angiography. Ophthalmic Surg Lasers Imaging Retina. 2018; 49: 138–141. [DOI] [PubMed] [Google Scholar]
- 19. Hendrickson A, Possin D, Vajzovic L, Toth CA. Histologic development of the human fovea from midgestation to maturity. Am J Ophthalmol. 2012; 154: 767–778.e762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kozulin P, Natoli R, O'Brien KM, Madigan MC, Provis JM. Differential expression of anti-angiogenic factors and guidance genes in the developing macula. Mol Vis. 2009; 15: 45–59. [PMC free article] [PubMed] [Google Scholar]
- 21. Al-Sheikh M, Akil H, Pfau M, Sadda SR. Swept-source OCT angiography imaging of the foveal avascular zone and macular capillary network density in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2016; 57: 3907–3913. [DOI] [PubMed] [Google Scholar]
- 22. Rabiolo A, Gelormini F, Sacconi R et al.. Comparison of methods to quantify macular and peripapillary vessel density in optical coherence tomography angiography. PLoS One. 2018; 13: e0205773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen C, Liu C, Wang Z et al.. Optical coherence tomography angiography in familial exudative vitreoretinopathy: clinical features and phenotype-genotype correlation. Invest Ophthalmol Vis Sci. 2018; 59: 5726–5734. [DOI] [PubMed] [Google Scholar]
- 24. Thomas MG, Kumar A, Mohammad S et al.. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011; 118: 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pakzad-Vaezi K, Keane PA, Cardoso JN, Egan C, Tufail A. Optical coherence tomography angiography of foveal hypoplasia. Br J Ophthalmol. 2017; 101: 985–988. [DOI] [PubMed] [Google Scholar]
- 26. Mintz-Hittner HA, Knight-Nanan DM, Satriano DR, Kretzer FL. A small foveal avascular zone may be an historic mark of prematurity. Ophthalmology. 1999; 106: 1409–1413. [DOI] [PubMed] [Google Scholar]
- 27. Yanni SE, Wang J, Chan M et al.. Foveal avascular zone and foveal pit formation after preterm birth. Br J Ophthalmol. 2012; 96: 961–966. [DOI] [PubMed] [Google Scholar]
- 28. Matsushita I, Nagata T, Hayashi T et al.. Foveal hypoplasia in patients with Stickler syndrome. Ophthalmology. 2017; 124: 896–902. [DOI] [PubMed] [Google Scholar]
- 29. Hendrickson AE, Yuodelis C.. The morphological development of the human fovea. Ophthalmology. 1984; 91: 603–612. [DOI] [PubMed] [Google Scholar]
- 30. Yuodelis C, Hendrickson A.. A qualitative and quantitative analysis of the human fovea during development. Vision Res. 1986; 26: 847–855. [DOI] [PubMed] [Google Scholar]
- 31. Spuul P, Daubon T, Pitter B et al.. VEGF-A/notch-induced podosomes proteolyse basement membrane collagen-IV during retinal sprouting angiogenesis. Cell Rep. 2016; 17: 484–500. [DOI] [PubMed] [Google Scholar]
- 32. Pöschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004; 131: 1619–1628. [DOI] [PubMed] [Google Scholar]
- 33. Fukukita H, Ito Y, Iwase T et al.. Inner macular changes after vitrectomy with internal limiting membrane peeling for rhegmatogenous retinal detachment: similarity with Alport syndrome. Retina. 2018; 39: 2332–2340. [DOI] [PubMed] [Google Scholar]