

Abstract
The extracellular matrix (ECM) is the non-cellular component of tissues in the cardiovascular system and other organs throughout the body. It is formed of filamentous proteins, proteoglycans and glycosaminoglycans, which extensively interact and whose structure and dynamics are modified by cross-linking, bridging proteins and cleavage by matrix degrading enzymes. The ECM serves important structural and regulatory roles in establishing tissue architecture and cellular function. The ECM of the developing heart has unique properties created by its emerging contractile nature; similarly, ECM lining blood vessels is highly elastic in order to sustain the basal and pulsatile forces imposed on their walls throughout life. In this part 1 of a 4-part review series, we focus on the role, function and basic biology of ECM in both heart development and in the adult.
Keywords: Cardiovascular, matrix, collagen, development, homeostasis
Condensed abstract
The extracellular matrix (ECM) is the non-cellular component of tissues throughout the body and is formed of filamentous proteins, proteoglycans and glycosaminoglycans. The ECM serves important structural and regulatory roles in establishing tissue architecture and cellular function. The ECM of the developing heart has unique properties created by its emerging contractile nature; similarly, ECM lining blood vessels is highly elastic in order to sustain pulsatile forces throughout life. In this first part of a 4-part review series we focus on the role, function and basic biology of ECM in both heart development and in the adult.
As recently as the 1980’s, extracellular matrix (ECM) was considered an inert and static scaffold. Motivating this JACC review series, ECM is now understood to be an active and dynamic tissue component regulating multiple processes including cell migration, progenitor cell self-renewal and differentiation, tissue growth and morphogenesis, fibrosis and other processes (Central Illustration) (1–4). Moreover, ECM is a key element of the pathobiology of numerous cardiovascular disease processes.
Central Illustration. Organization of Cardiac and Vascular Extracellular Matrix (ECM).
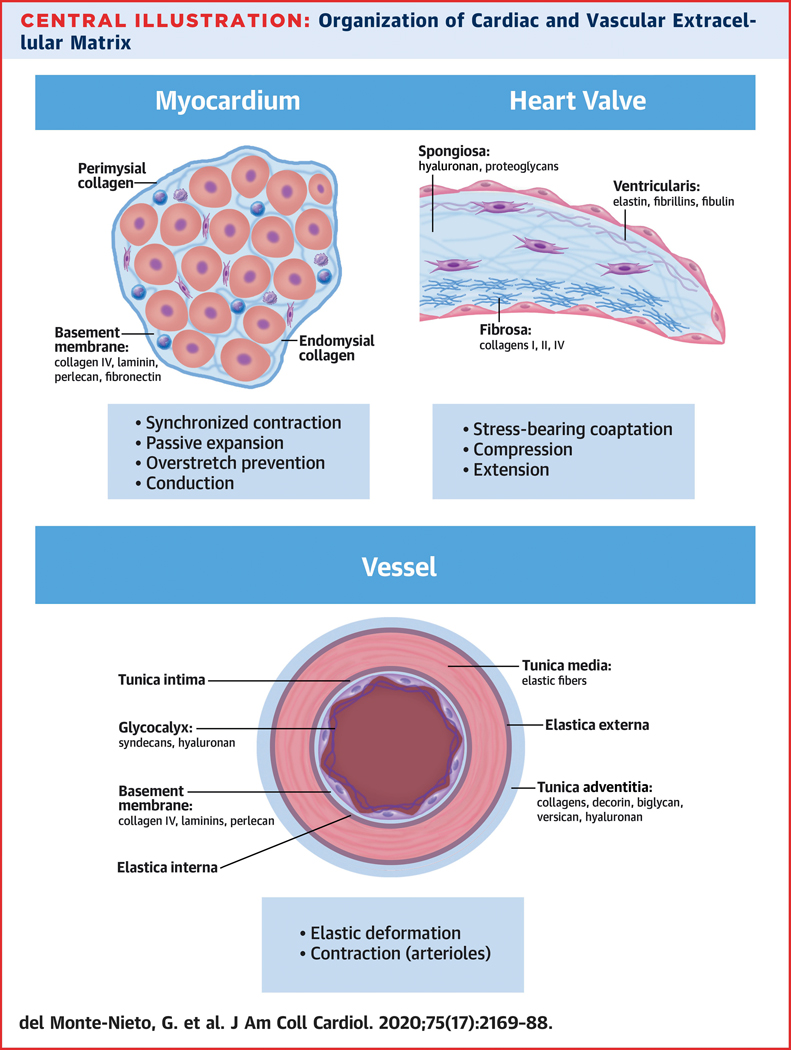
Cardiovascular tissues face different hemodynamic and mechanical requirements which have resulted in a unique building plan and highly specialized ECM and cellular compositions. Prominent sub-structures and ECM components are indicated.
In this 4-part review series titled “Extracellular Matrix in Cardiovascular Health and Disease,” we review the full breadth of ECM in the cardiovascular system (Table 1). This series should serve as a benchmark and resource for basic scientists and clinicians alike. Here, in part 1 we cover basic ECM biology in development and the adult, including its function, classification, normal biology, homeostatic turnover and role in aging.
Table 1.
Overview of 4-part review series: Extracellular Matrix in Cardiovascular Health and Disease.
| Part 1 | Basic Biology of Extracellular Matrix in the Cardiovascular System - Developmental Perspective. - Normal biology, cellular sources and homeostatic functioning of ECM in the adult |
| Part 2 | Extracellular Matrix in Vascular Disease - The composition of the vascular extracellular matrix - Extracellular matrix remodeling, arterial stiffness and atherosclerosis - Extracellular matrix and aortic aneurysm - Proteomics of the vascular extracellular matrix |
| Part 3 | Myocardial interstitial fibrosis in non-ischemic heart disease - Histological basis of myocardial interstitial fibrosis - Cellular and molecular mechanisms of myocardial interstitial fibrosis - Clinical consequences of myocardial interstitial fibrosis - Diagnosis of myocardial interstitial fibrosis - Treatment of myocardial interstitial fibrosis |
| Part 4 | Extracellular Matrix in Ischemic Heart Disease - ECM in the infarcted heart - ECM in chronic ischemic cardiomyopathy - Therapeutic opportunities: Targeting the ECM in ischemic heart disease |
ECM in the Cardiovascular System – A Developmental Perspective
Introduction
ECM composition is tissue-specific, and varies within a single tissue across development, homeostasis and disease. Passive and active biomechanical forces inevitably involve and are distributed through the ECM to directly influence cell signaling and function, referred to as mechanotransduction (5). The cardiovascular ECM is formed from filamentous and sheet-forming protein polymers such as collagens, elastins, fibullins and laminins. Glycosaminoglycan (GAG) polymers include hyaluronan (HA), chondroitin sulphate, heparan sulphate and keratan sulphate, with the sulphated forms becoming linked to proteoglycan core proteins before their secretion. Chondroitin sulphate proteoglycans, including versican and aggrecan, associate with HA and cartilage link protein (Crtl1), forming aggregates which have roles in ECM hydration (6). Heparan sulphate proteoglycans, including perlecan, bind to a range of growth factors and cytokines, modifying their functions (7,8). Bridging proteins such as fibronectin, proteoglycan link proteins (Haplns), periostin and fibulins facilitate and stabilize interactions between ECM molecules and ECM cellular receptors including integrins (5). A variety of collagen cross-linking proteins modify filamentous ECM, and members of the matrix metalloprotease (MMP) and a disintegrin and metalloprotease with thrombospondin motifs (ADAMTS) families are responsible for ECM cleavage, facilitating ECM dynamics and turnover (9).
Using model organisms, many mutations in cardiovascular ECM genes have been characterized and shown to have no phenotype during development, highlighting a high degree of functional redundancy and ECM network compensation (9). However, a subset of ECM genes is essential for cardiovascular morphogenesis and integrity; below we illustrate several specific examples.
ECM in vascular development
The developing vascular system forms by vasculogenesis, the de novo generation of cord-like vascular structures from endothelial progenitors (angioblasts). This primitive vessel network expands by angiogenesis, whereby new vessels form by sprouting of preexisting vascular beds. Within established vessels, quiescent endothelial cells are associated with a basement membrane (BM) formed predominantly from laminins, collagens IV and XVIII, perlecan, and other ECM components (Figure 1D). During angiogenic remodeling, endothelial cells break the BM and migrate into surrounding tissues interacting with interstitial ECM components including fibronectin, vitronectin, thrombospondins, tenascins and different collagens (10). Depending on the vessel state, a spectrum of different ECM receptors (mainly integrins) are expressed on endothelial cell membranes.
Figure 1. Overview of the main cardiac developmental processes regulated by Extracellular Matrix (ECM) components or ECM degradation.
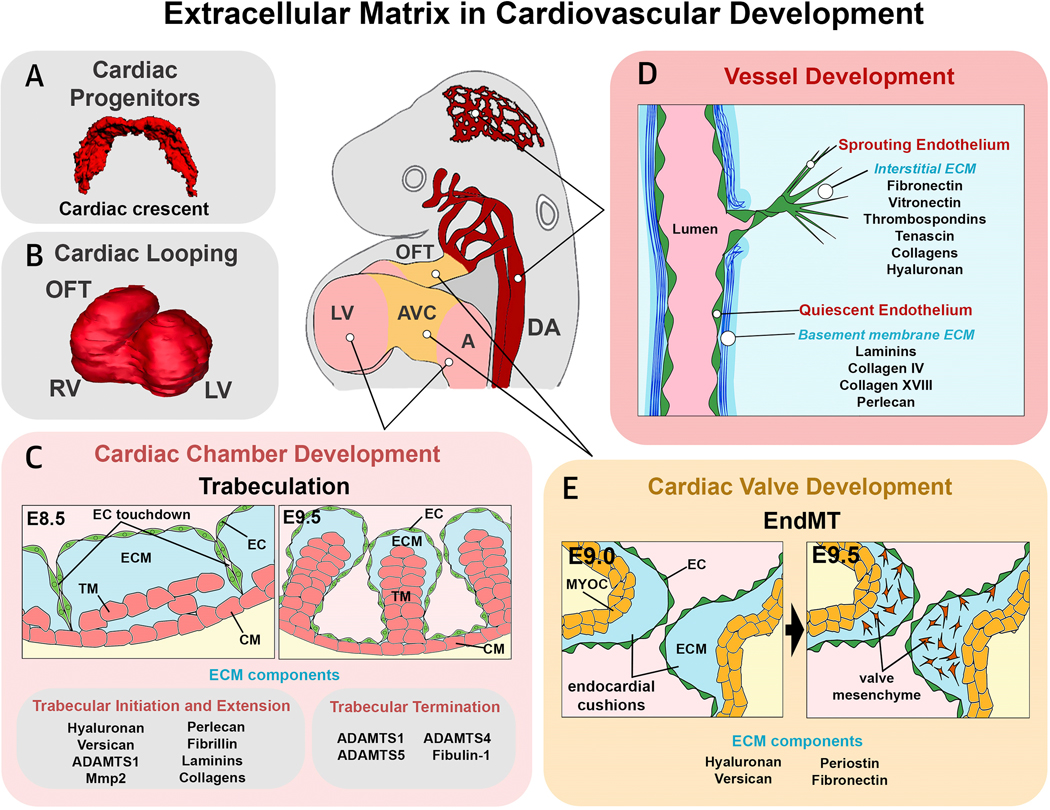
(A) Early cardiac progenitor formation; (B) cardiac looping; (C) cardiac chamber development and trabeculation; (D) vessel development; (E) cardiac valve development. OFT, outflow tract; RV, right ventricle; LV, left ventricle; AVC, atrioventricular canal; A, atrium; DA, dorsal aorta; TM, trabecular myocardium; ECM, extracellular matrix; CM, compact myocardium; EC, endocardium; MYO, myocardium.
Most mutant models for ECM components and receptors do not show developmental angiogenic defects (11,12). However, germline deletion of the fibronectin gene, and deletion of alternatively spliced EIIIA and EIIIB exons show defects and hemorrhage in multiple vascular beds (13). Mutation of α4/α5 and β1 integrin chains, constituting fibronectin receptors, also show early embryonic lethality associated with vascular and heart defects (14,15). Laminin α4 mutants show disrupted capillary BMs and hemorrhage at fetal stages (16), whereas fibulin-4 mutants show aortic narrowing and tortuosity due to the presence of abnormal rod-like elastin filaments (17).
As vessels remodel and enter new territories, the stiffness of the ECM around vascular projections changes, and the associated changes to mechanotransduction can alter the activity of transcription factors such as GATA2 that regulate vessel integrity, growth, metabolism and morphogenesis (18). ECM-integrin interactions and heparan sulphate have been linked to activation of angiokines such as vascular endothelial cell growth factor (VEGF), as well as the localization of vascular adhesion receptors such as VE-cadherin (19,20). In zebrafish, mutation of the transmembrane protein Tmem2, which degrades HA, led to excessive accumulation of HA in the heart and around vessels, blocking VEGF signaling and angiogenic development (21). This phenotype was rescued by enforced degradation of HA, or delivery of oligomeric HA or VEGF-C to embryos, suggesting a model in which dynamic degradation of HA augments VEGF signaling, perhaps by liberating VEGF molecules from the ECM or facilitating ligand-receptor interactions.
Cardiac progenitor cell migration and heart tube development
During formation of the primitive vertebrate heart tube, myocardial and endocardial progenitors condense anteriorly. In amniotes, this region is called the “cardiac crescent” ( Figure 1A). Cardiac progenitors lie adjacent to the anterior endoderm separated by a highly-hydrated embryonic ECM. Bilaterally-organized cardiac progenitor cells then migrate towards the embryonic midline and fuse to give rise to the primary heart tube composed of myocardial and endocardial layers separated by a thick ECM called cardiac jelly.
Studies in chick, mouse and zebrafish embryos show that progenitor migration requires fibronectin (22–24), a major modular component of cardiac ECM that binds to multiple integrins, heparan sulphate proteoglycans, collagens and fibrins, and is implicated in cell-substratum adhesion influencing cell shape, migration and division. Fibronectin may bind platelet-derived growth factor (PDGF) which is secreted from endoderm and acts as a guidance molecule for the medial migration of cardiac progenitors (25). In zebrafish, fibronectin secreted from endocardium is deposited at the midline, and around cardiomyocyte progenitors as they migrate medially between endoderm and an extra-embryonic tissue layer called the yolk syncytial layer (26). Fibronectin does not have an instructive role in migration; rather, it is necessary for the epithelialization and maturation of cardiomyocyte precursors (24).
Soon after formation of the primitive heart tube, the heart undergoes cardiac looping, which transforms the approximately straight primary heart tube into an elongated looped tube (Figure 1B). During cardiac looping, different regions acquire more definitive character as chamber versus non-chamber myocardium, and chamber-associated versus valve-forming endocardium (27). The pathways that guide heart looping have their origins at earlier developmental stages when bilateral embryonic symmetry is broken (28). Although differential ECM distribution has no instructive role during heart looping (29), the early inhibition of MMP2 activity leads to defects in heart migration or fusion (leading to cardia bifida). However, later MMP2 inhibition blocks heart looping by interrupting regional ECM degradation necessary for breakdown of the dorsal mesocardium, a transient myocardial ligament which tethers the early heart tube to more dorsal embryonic tissues (30) and which is critical for development of torsional strain during cardiac looping (31).
Cardiac valve development
In newly-formed hearts, the thickness of the embryonic cardiac jelly ECM is homogeneous, but regional differences subsequently arise. In valvulogenic regions (Figure 1E), active ECM synthesis by myocardium and endocardium, and lack of ECM degradation, lead to formation of prominent acellular ECM swellings between the endocardial and myocardial layers called endocardial cushions (32). Endocardial cushions act initially as primitive valves and are the primordia of the definitive heart valve leaflets as they become cellularized by endothelial to mesenchymal transition (EndMT), producing a pro-fibrotic valve mesenchyme (32,33). Cushions also play roles in chamber septation, knitting together the different tissue elements contributing to the AV septal complex.
The proper synthesis and structural organization of ECM within endocardial cushions is critical for valve development and maturation (Figure 1E). HA is the most abundant GAG in the developing heart and is involved in valvular endothelial cell proliferation and EndMT (6). HA deficiency caused by excessive degradation (34) or loss-of-function (LOF) mutation in Has2 and Ugdh genes (involved in synthesis of HA and other GAGs (35,36)) lead to cushion and valve defects, whereas excess HA leads to excessive EndMT (37). HA interacts with ErbB2/ErbB3, the endocardial signaling receptor for neuregulin1 involved in regulation of endocardial EndMT and cushion ECM synthesis (38), and likely regulates its function because neuregulin1 and ERBB2 mouse mutants die around E10.5 showing severe heart defects associated with reduced cardiac cushion and ventricular chamber ECM, a phenotype similar to that of Has2 and versican mutants (35,39–41). Versican is also involved in valve maturation (42). Interestingly, periostin, a matricellular bridging protein that promotes fibrogenesis in cardiac valves and other fibrous cardiac structures also protects AV valve mesenchyme against inappropriate differentiation to a myocardial fate (43). Periostin also protects outflow tract valve mesenchyme against calcification by suppressing the expression of osteogenic transcription factor Runx2 (44). In a further advance, phagocytic macrophages derived from hemogenic endocardium were shown to be important for the remodeling of endocardial cushion ECM (45).
Valve development is a striking example of how biomechanical forces guide tissue morphogenesis. Valve endothelial cells respond to complex flow parameters by expressing the flow-dependent transcription factor Klf2, regulating downstream Notch and canonical Wnt signaling pathways in spatially-distinct patterns (46,47). These govern mesenchymal cell proliferation and cushion remodeling. As the zebrafish atrio-ventricular valve matures, Has2 expression (governing HA synthesis) is reduced, while fibronectin expression is increased, corresponding to the transition from a hydrostatic to fibrotic environment in forming valve leaflets (48). Fibronectin deposition is thus flow-dependent and essential for valve formation.
Human pre-valvular endothelial cells were recently generated from induced pluripotent stem cells by directed differentiation (49). Upon stimulation with BMP2, they underwent EndMT and expressed ECM and other markers typical of the different stratification layers (fibrosa, spongiosa, ventricularis) seen during valve maturation. This system was used to study pathways contributing to mitral valve prolapse and myxomatous degeneration (49), demonstrating the promise of these cells for valve disease modeling.
Cardiac chamber formation and trabeculation
During heart looping, the future cardiac ventricles undergo differentiation and proliferation, promoting chamber expansion (ballooning) (Figure 1B) (50). Soon after, the process of ventricular trabeculation gives rise to a sponge-like specialized myocardium on the luminal side of the chamber wall that is critical for increasing pumping efficiency of the developing heart and surface area for nutrient and oxygen exchange (Figure 1C) (39). The trabecular network expands in size and complexity until E14.5 in mice (39,51). Thereafter, the process of myocardial compaction is initiated, leading to simplification of the trabecular layer through its integration into the chamber wall (52).
Soon after the formation of the primitive heart tube, the regions destined to become the cardiac chambers undergo a progressive reduction of cardiac jelly (Figure 1C). In most reptiles, birds, and mammals, reduction of ECM in the forming atria is virtually complete; however atrial ECM is maintained in zebrafish where it plays a role in inhibiting trabeculation (53). Historically, the main genetic studies implicating cardiac jelly in regulation of chamber development came from analysis of LOF mutants in Has2 (35) and versican (40). Homozygotes of these mutant strains show a total reduction of cardiac jelly in forming chambers, and severely defective trabeculation; however, these studies focused largely on the roles of Has2 and versican in valve development (35,40). LOF mutation of perlecan, a heparan sulphate proteoglycan involved in formation of the cardiomyocyte BM, leads to cardiac chamber defects involving fenestration of the ventricular myocardial wall (54). Many other ECM components are expressed in the developing ventricular chambers including laminins, fibrillin and collagens. However, no cardiac chamber phenotypes have been seen in mutant models.
Several studies have highlighted the importance of ECM degradation by metalloproteases (ADAMTS1, ADAMTS4 and ADAMTS5) (51,55) and the metalloprotease co-factor fibulin-1 (56) in the process of trabecular termination (Figure 1C). LOF mutants of Adamts1 and fibulin-1 lead to a hyper-trabeculation phenotype, associated with persistence of trabecular growth beyond the normal termination stage (51,56), whereas ectopic expression of Adamts1, Adamts4 or Adamts5 promotes premature termination of trabecular growth associated with total degradation of chamber ECM (51,55). These results highlight the importance of ECM degradation at the terminal stages of trabecular development, and link chamber ECM to regulation of trabecular growth.
The importance of ECM dynamics during the early phases of chamber development were only recently described (39). del Monte-Nieto et al. determined that trabecular localization, architecture and growth are regulated by a fine balance between ECM degradation and synthesis, directed from different regions of the chamber endocardium (Figure 1C). The restricted activity of the Notch pathway in chamber endocardium controls ECM degradation, leading to formation of numerous endocardial sprouts that tunnel through the cardiac jelly ECM to contact the myocardial layer. The distribution of endocardial sprouts creates a series of endocardial domes delimiting cardiac jelly-rich areas called ECM bubbles, within which the trabecular myocardium expands. Formation of trabeculae likely also involves the extrusion of cardiomyocytes from the outer compact layer, as seen in zebrafish (57); however, the specific architecture of the trabecular projections in mammals is determined by endocardial cell sprouting behaviors leading to formation of endocardial domes and ECM bubbles. ECM degradation regulated by the Notch pathway occurs through transcriptional control of metalloprotease genes including Adamts1, and is counterbalanced by ECM synthesis regulated by neuregulin1, also expressed in endocardium but acting through its Erbb2/Erbb4 receptor complex on trabecular cardiomyocytes to promote expression of Has2 and versican at the trabecular tips. This finely regulated molecular control of ECM dynamics during trabeculation is governed by the opposing roles of the Notch and neuregulin1 pathways. Disruption of ECM degradation by mutation of Notch1 leads to a lack of endocardial sprouting, excess ECM and disorganized trabecular myocardial growth, whereas disruption of ECM synthesis by mutation of neuregulin1 leads to reduction of ECM and arrest of trabecular growth (39).
As trabeculation proceeds and the endocardium establishes progressively closer contact with myocardium, there is ongoing ECM degradation at the base of forming trabeculae, whereas ECM synthesis is maintained at the tips of trabeculae (Figure 1C). ECM degradation is dominant and progressive, eventually eliminating most of the chamber ECM at the termination phase as endocardium and myocardium become closely associated across the whole trabecular network (39,51) (Figure 1C). Therefore, in summary there is a critical role for cardiac ECM as an integral component of trabecular architectural patterning and growth during chamber development (39). A summary of differing ECM molecules and components expressed during cardiovascular development, and the associated defects that arise with their perturbation (i.e. LOF), is presented in Supplemental Table 1.
Excitation/contraction coupling of cardiomyocytes
In mammalian heart development, cardiac progenitor cells within the cardiac crescent initiate asynchronous Ca2+ oscillations prior to their migration to the midline. Synchronized contraction wavefronts are initiated as the heart tube is formed (58). As discussed above, progenitor migration is accompanied by cardiomyocyte maturation involving increased cell polarization, sarcomere assembly and cell volume, as well as maturation of the cardiac electrical system, whose activity is essential for the maturation process (58). As the embryonic heart matures, its stiffness increases 10-fold, aligned with increases in synthesis of HA, collagens and other ECM components, and mechanosensitive adhesion complexes that include integrins, talin and vinculin (3,59,60). The stiffness of the ECM at progressive developmental stages appears to be optimized for sarcomeric gene expression and protein assembly, as well as sarcomere spacing, connectivity and contraction, and tissue conduction (3,58). Collagen synthesis and organization is likely to be further enhanced by biomechanical strain generated within the ECM as a result of contraction. Controlled softening of the ECM by collagenase treatment, or stiffening by cross-linking, suppresses cardiomyocyte beating (3). Recently, Liu and colleagues proposed that in early heart development, contraction wavefronts are propagated mechanically not electrically (61). In their model, embryonic cardiomyocytes represent mechanically excitable inclusions in the ECM, and depolarization occurs when local biomechanical strain reaches a threshold. As such, strain and contraction “diffuse” through the system to generate the contraction wavefront, before maturation of cardiomyocyte electrical coupling.
Control of fetal cardiomyocyte proliferation by fibroblast ECM
Cardiomyocyte proliferation occurs during trabeculation and later thickening of the compact layer, and is controlled by mitogenic factors secreted from both epicardium and endocardium (62). As described above, this process requires correct ECM dynamics (39). During later fetal development, the cardiac chamber walls become infiltrated by fibroblasts, which have their origins in the epicardium (63). Through this process, cardiomyocytes become surrounded by a connective tissue that is responsible for the synthesis and modulation of the fetal and adult cardiac ECM scaffold. Cardiac fibroblasts are recognized as having many roles as sentinels, tissue architects, paracrine signaling hubs and lineage precursors. Fetal cardiac fibroblasts stimulated proliferation of embryonic cardiomyocytes in in vitro co-cultures, whereas adult fibroblasts stimulated hypertrophy (60), highlighting the importance of fibroblast-CM paracrine signaling throughout heart development. Genes encoding fibronectin, collagen, tenascin C and periostin were among those more highly expressed in fetal compared with adult fibroblasts, and synthetic matrices composed of integrin ligands such as collagen and fibronectin also stimulated cardiomyocyte proliferation, with knockdown of the genes for fibronectin and collagen3a1 reducing cardiomyocyte proliferation (60). These ECM components are in fact necessary for the activity of mitogens secreted from fetal fibroblasts, including heparin-binding epidermal growth factor-like growth factor (HB-EGF) and fibroblast growth factor (FGF) (60). Downstream, fibroblast ECM components act through the cardiomyocyte-specific fetal integrin β1A isoform connecting to MAP kinase and PI3 kinase pathways. Thus, in the fetal heart, integrin ligands and other ECM components interact to coordinate the activity of cardiomyocyte mitogens secreted by fibroblasts.
Role of ECM in fetal development and congenital heart disease
Human ECM gene variants have been causatively linked to congenital heart disease, generally in association with complex syndromes (9). For example, patients with mutations in the fibrillin 1 gene (FBN1) associated with the connective tissue disorder Marfan’s syndrome, show thickening of the AV valves and mitral and/or tricuspid valve prolapse, and aortic aneurysm. Patients carrying variants in the CHST3 and CHST14 genes (encoding enzymes involved in sulphation of GAGs) have syndromes including ventricular and atrial septal defects, respectively. Numerous mouse ECM mutants and mutant combinations also develop congenital heart disease-like phenotypes during fetal stages, highlighting the important but sometime subtle roles played by ECM components during development (Supplemental Table 1).
ECM, postnatal heart growth and regeneration
Immediately after birth, mouse cardiomyocytes retain some proliferative ability associated with a robust regenerative response to apical surgical resection or induced myocardial infarction (64). The main mechanism of cardiomyocyte renewal involves de-differentiation and proliferation of surviving cardiomyocytes, as also seen in zebrafish hearts, which retain regenerative ability into adulthood (65). However, regenerative ability in mice wanes over the first postnatal week (64) and this is believed to be due to a decline in cardiomyocyte proliferation in favor of bi-nucleation, reflecting the transition to hypertrophic growth (66,67). However, loss of regenerative ability occurs as early as postnatal day (P)2 in mice or P3 in pigs, in advance of changes in cardiomyocyte cell cycle activity (66,68). ECM is known to undergo major changes during this period and is being increasingly investigated for its roles in regeneration. There is a rapid increase in the elastic modulus of the mouse ventricle between P1 and P2 from 12kPa to 39kPa (66,69), which coincides with an increase in expression of ECM and cell-ECM interaction genes and proteins (66). Reduction of ECM stiffness led to a prolongation of the neonatal regenerative window until at least P3 (66), suggesting that the sharp increase in cardiac ECM stiffness postnatally is inhibitory for heart regeneration. However, periostin, a matricellular protein that contributes to the viscoelastic properties of tissues through its role in promoting ECM synthesis and fibrillogenesis (43,70), is also essential for neonatal cardiac regeneration by constraining GSK3β signaling, impacting cardiomyocyte proliferation, angiogenesis and monocyte recruitment (71).
Agrin is a large ECM heparan sulphate proteoglycan that is implicated in self-renewal, proliferation and differentiation of a variety of cell types (72). Agrin is expressed in mammalian fetal hearts but is down-regulated during the first postnatal week (64), and fetal hearts lacking agrin show higher cardiomyocyte contraction frequencies, a phenotype that is reversed by recombinant agrin (73). In this setting, agrin binds to the α3 subunit of the membrane Na, K-ATPase, inhibiting its function (73). In a screen for pro-regenerative ECM components, agrin peptides were found to be enriched in cell-free ECM preparations of P1 hearts that were capable of stimulating proliferation of normally quiescent P7 cardiomyocytes (72). Genetic studies showed the importance of agrin for cardiomyocyte cell cycle activity and heart regeneration in the immediate neonatal period (72,73). Furthermore, direct injection of a recombinant agrin peptide into the neonatal heart extended the window of postnatal regeneration until at least P7 (72). The receptor for agrin in this context is dystroglycan-1 (Dag1), a component of the membrane-localized dystroglycan complex, which links ECM to the actin cytoskeleton. Binding of agrin to Dag1 stimulates ERK signaling and partial disassembly of the dystroglycan complex, leading to release from the membrane of the transcription factor Yap, a central transcriptional mediator of cell growth and organ size (72). In non-dividing cardiomyocytes, Yap is normally tethered to the membrane dystroglycan complex via Dag1, sequestering it away from the nucleus (74). Recombinant agrin can also promote regeneration of adult murine hearts though stimulating cardiomyocyte proliferation (72), which may have significance for cardiac regenerative medicine.
Summary of the role of ECM in cardiovascular development
These vignettes are by no means comprehensive, however, they serve to highlight the indispensable role of ECM during heart and vessel development. Indeed, as already discussed, the importance of ECM in early cardiovascular development has only been fully appreciated in the last 2 decades, when we have moved from the perspective of a static role as an inert scaffold that maintains tissue turgor, to a dynamic role that includes regulating critical cell and tissue functions. This is a theme that will be revisited throughout this review series.
Normal biology, cellular sources and homeostatic functioning of ECM in the adult
In the adult, ECM is a complex, dynamic and multi-component network that is critical for the proper functioning of differing cardiovascular tissues and organs, and which confers specific mechanical functions in response to different physiologic and pathophysiologic stimuli (Central Illustration). Mechanical stress, wall tension, shear stress and pressure gradients in different vascular beds are important regulators of ECM composition and supramolecular structure. In addition, ECM signals to cardiovascular cells through specific receptors such as integrins or CD44 and provides binding sites for growth factors and cytokines. This signaling is thought to critically determine the function of cardiovascular cells such as fibroblasts and even cardiomyocytes. Many pathologies such as atherosclerosis, diabetes, pressure overload and ischemic heart disease, as well as aspects of aging, lead to and are partly driven by pathophysiologic ECM remodeling.
Cardiac and vascular ECM networks in the adult contain collagens, various proteoglycans, matricellular proteins, and GAGs such as hyaluronan (HA). For simplicity, ECM is often divided into structural ECM (e.g. collagens and elastin) and non-structural ECM. However, different aspects of collagen fibril formation such as fibrillogenesis, alignment of collagen fibrils, lateral fusion and collagen density are fine-tuned by non-fibrillar collagens and collagen binding proteoglycans. Similarly, the HA matrix network depends on the integration of HA binding proteins (hyaladherins) such as versican. Therefore, many of the so-called non-structural ECM components critically affect ECM structure.
Another level of structural and likely functional complexity arises from glycosylation patterns of the cardiac and vascular glycoproteins and proteoglycans. The best-known ECM receptors are the integrin heterodimers that are activated upon engagement of ECM ligands and which signal through focal adhesions into cells (outside-in signaling). Other ECM receptors include discoidin domain receptors which recognize collagen, CD44 which recognizes osteopontin, HA and versican as ligands, and toll like receptors which recognize ECM fragments after damage and ECM degradation. Furthermore, ECM molecules can be linked covalently or non-covalently. Covalent cross-linking is initiated by transglutaminases, lysyl oxidases, lysyl oxidase-like enzymes and lysyl hydroxylases (75). HA can be cross-linked by tumor necrosis factor (TNF)-stimulated gene 6 (TSG6), and pentraxin 3 (76) (Figure 2). Enhanced collagen cross-linking is associated with increased ventricular and vascular stiffness (77). These modifications change both mechanical and signaling properties.
Figure 2. Hyaluronan and Hyaluronan Binding Proteins.
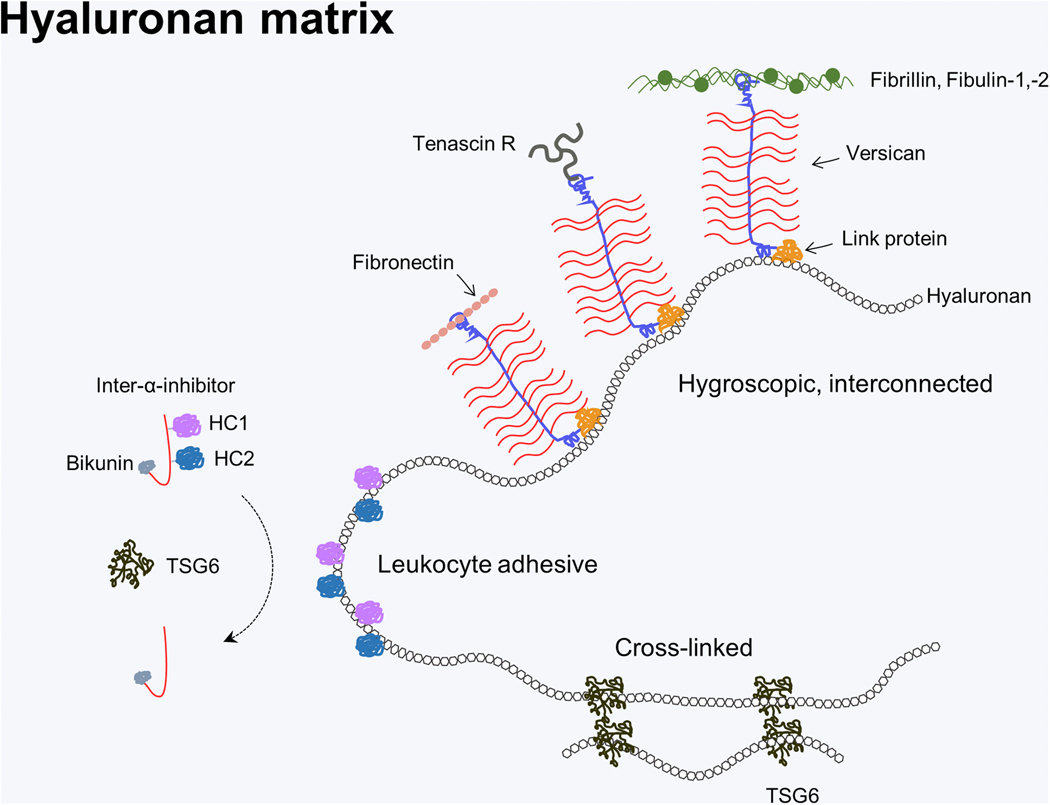
Hyaluronan (HA) matrix diversification via HA binding proteins highlights its multifunctionality. HA’s function is diversified by HA binding proteins that specifically bind via a link module domain. For example, interaction with versican, a large chondroitin sulfate proteoglycan can lead to large hydrated aggregates that allow cell migration and proliferation. Versican can also bring together other ECM components (e.g. fibronectin, tenascin-R, and microfibrils) via its C terminal domain (106). During inflammation HA can be covalently modified by the addition of heavy chains 1, −2 (HC1, −2) from inter-α-inhibitor, a process mediated by TSG6, resulting in an ECM that is highly adhesive for leukocytes. HA can also be cross-linked by TSG6 resulting in a more condensed ECM, of which the full implications are unknown (76).
Building on these themes, in this section of this review, important functions of adult cardiovascular ECM in physiological conditions are addressed. Furthermore, we point out new opportunities to explore ECM function using single-cell RNA sequencing (scRNAseq) and other approaches.
Normal biology and classification of ECM in the cardiovascular system
Heart
In the heart, cardiomyocytes are inter-connected by intercalated disks to form a multicellular syncytium allowing coordinated myocardial excitation and contraction. Cardiomyocytes are enclosed by a network of BM proteins including networks of collagen IV, laminin, perlecan and fibronectin (Central Illustration). Cardiomyocytes and endomysial fibroblasts sense the ECM network through integrin ECM receptors, thereby receiving signaling cues regulating differentiation, proliferation, migration and excitation. Endomysial and perimysial collagens provide a structured microenvironment to cardiomyocytes, imparting stiffness to the left ventricular wall and supporting force transmission (78). This is also reflected by the fact that collagen is the most abundant ECM component in the heart. Changes in cardiac collagen networks have profound effects on myocardial contraction, relaxation and diastolic stiffness (79) as well as electrical conduction (80). The collagenous ECM of the endomysium and perimysium also serves as a scaffold for non-cardiomyocytes such as microvascular endothelial cells and fibroblasts. Collagen type I is the main collagen and confers tensile strength. Thin fibers of collagen type III contribute more to elasticity of the cardiac ECM (81). Collagen is secreted by fibroblasts as a soluble precursor molecule that is cleaved at both the N-and C-terminus by specific proteinases as a prerequisite for fibrillogenesis. Collagen type I is a triple helical protein that self assembles into fibrils in the presence of fibronectin, integrins and collagen V. The fibril assembly occurs extracellularly at the plasma membrane which enables cells (i.e. cardiac fibroblasts) to tightly control fibrillogenesis. Importantly, ~50 collagen binding proteins are known, which are presumably used to form different fibril patterns (82).
Cardiac valves
Cardiac valve leaflets consist of three layers; the ventricularis, spongiosa and fibrosa, which are surrounded by a layer of valvular endothelial cells and populated with valvular interstitial cells (Central Illustration). Each layer has a unique ECM composition to facilitate its specific function. The ventricularis layer, nearest the inflow, is predominantly comprised of elastic fibers, consisting of an elastin core surrounded by a microfibril sheath containing fibrillin-1, fibrillin-2 and fibulins. This imparts flexibility to stretch and recoil during the cardiac cycle (83). The middle spongiosa layer is mainly comprised of HA (84) and proteoglycans; versican, decorin and biglycan (85). HA and proteoglycan matrices cushion blood pressure forces, assist in re-alignment of collagen and elastin fibers, and resist delamination (86). The fibrosa layer, nearest the outflow, is rich in fibrillar collagens I, III and V, which impart stiffness and ensure leaflet integrity (83). Collagen type I is the predominant collagen and is mostly restricted to the fibrosa, while collagen III is more widely expressed. Additionally, a sub-endothelial BM layer is present, which consists of collagen types I and IV, as well as laminin. There are also minor ECM components expressed throughout the leaflet including vitronectin and fibronectin (87) as well as osteonectin and periostin (83,88). Recently, proteomic mapping of 16 different heart regions indicated that valves have the highest percentage of protein expression dedicated to ECM as compared to other heart regions (89).
Blood vessels
In blood vessels, tissue morphology and ECM composition are adapted to fulfill specific functions in different vascular beds: large elastic arteries (aorta and great vessels), muscular arteries and arterioles, capillaries, venules and veins (90). The generic building plan of blood vessels consists of the tunica intima, tunica media and tunica adventitia (Central Illustration). The layers are connected by the membrana elastica interna and membrana elastica externa. The tunica intima represents the luminal lining of endothelial cells that are attached to the BM. The tunica media contains contractile smooth muscle cells (SMC) and allows active vasoconstriction and relaxation, while the tunica adventitia attaches the vessel with the surrounding connective tissue. Whereas in contractile arteries the tunica media is thickest, in veins the tunica adventitia is most pronounced.
Endothelial glycocalyx extends from the endothelial surface into the lumen (Central Illustration). It plays a protective role by supporting endothelial barrier function, preventing platelet adhesion, and facilitating the rolling of immune cells on the endothelial surface (91). A variety of pathophysiologic stimuli cause glycocalyx shedding, which is likely one of the first steps in immune cell extravasation and thrombotic complications (92). Endothelial glycocalyx is composed of heparan sulfate GAGs (syndecans) and HA (93). In addition, the polyanionic glycocalyx traps a variety of heparin binding proteins and hyaladherins, as well as cationic proteins.
The BM of blood vessels is composed of collagens type IV and –XVIII, laminins, nidogens/entactins and perlecan. Characteristically, von Willebrand-factor is present in the endothelial BM which initiates platelet adhesions and blood coagulation via factor VIII when the endothelial layer becomes disrupted. Collagen IV plays a key role in assuring BM stability. Laminins, in particular Laminin 411, are also critical constituents of the BM in forming networks. The two networks (laminin and collagen IV) are connected by nidogens. Perlecan stabilizes BMs by interactions with the above-mentioned ECM constituents. In addition, perlecan interacts with heparin binding growth factors and cytokines, and protects the BM from proteolytic degradation. The ECM of BM signals through integrins to endothelial cells and regulates pivotal endothelial functions such as adhesion molecule expression, tight junction formation, endothelial metabolism and prostacyclin synthesis. The BM is impermeable to cells, with the exception of leukocytes.
The tunica media provides elasticity to arteries, facilitating their pulsatile stretch. Therefore, the aorta and great vessels contain large amounts of elastic fibers, while more distal arteries have pronounced SMC layers. Elastin networks with intervening SMCs are assembled concentrically, which allows an active regulation of vascular tone and luminal diameter. In muscular arteries, elastin is not deposited as sheets but more in the form of fibers. In contrast, capillaries have no regular tunica media but instead a sheet of scattered pericytes that are encapsulated in BM.
In the tunica adventitia, a loose ECM is formed containing collagens, collagen binding proteoglycans (e.g. biglycan and decorin) and HA. In addition, large amounts of versican, a HA-binding proteoglycan, are present. The adventitia also harbors vasa vasorum, fibroblasts, stem cells and immune cells.
Below, typical ECM components of healthy myocardium, heart valves and blood vessels are introduced.
Collagens
Collagens of the heart can be subdivided into fibrillar and non-fibrillar collagens. Major fibrillar forms are collagen type I, -III, -V and -XI (94). Collagen type I and –III are the main collagens in the heart and blood vessels (81). Collagen type I forms thick rod-like fibers (50–150nm diameter) conferring high tensile strength. Certain mutations of collagen type I alter diastolic compliance, such as in osteogenesis imperfecta mice. On the contrary, fine fibrils used for highly flexible reticular networks are built from collagen type III, which is typical also for skin, fetal tissue and blood vessels. Collagen type III is mixed with type I in the heart and the type I/type III ratio influences mechanical properties. Accordingly, the proportion of type III is highest in the epimysium and decreases in perimysium. In addition, the proportion of type I collagen increases with age, which contributes to ECM stiffening (81). Collagen type V is found in the interstitium and regulates collagen fibrillogenesis, whereas collagen type XI has the same function in heart valves. The non-fibrillar collagen type IV forms an open network with other ECM molecules of the BM, and is important for BM cell adhesion and molecular transport. Collagen type VIII is expressed at low levels and might contribute to connecting the BM with elastic laminae and microfibrils, and the elastic laminae with collagen fibrils. Collagen type VI is expressed in the adult heart, and in the media and adventitia of blood vessels, although its functions are not well understood. Collagen type XV is indispensable for normal cardiac function as shown by knockout mice that develop vascular permeability, perturbed cardiomyocyte alignment and abnormal interstitial ECM formation resulting in cardiomyopathy. The role of collagen type XVIII in the cardiovascular system is not well understood (81,95).
Laminins
Laminins are glycoproteins that are required for BM assembly and function both in the heart and vasculature. Laminin is assembled from various alpha-, beta- and gamma chains that form 15 of the 60 possible heterotrimers. The chains are covalently linked to stabilize the heterotrimer. The three short arms and a long coiled-coil structure formed from all three chains to create a cross-like molecule of 700–900 kDa (96). Laminin isoforms form networks and through their integrin binding domain mediate cell adhesion (i.e. ligand for α3β1 α6β1 α6β4 α7β1 integrins) (97).
Elastin
Elastin is synthesized as tropoelastin, which is processed extracellularly to elastin. Supramolecular elastin aggregates are then cross-linked by lysyl oxidases at lysine-rich regions. Elastic fibers consist of an elastin rich core surrounded by a sheath of microfibrils composed of several components including fibrillin-1. This elastin can be extended more than two-fold, which is important to enable arteries to extend and retract in response to arterial pulse waves, and thereby pushing blood forward in diastole. In addition, elastin and polymeric collagen inhibit SMC proliferation in the vessel wall. Interestingly, fibrillin-1 mutations cause Marfan syndrome that manifests by dilation and dissection of large arteries. Similarly, the fibulins and EMILINs are critical for proper formation of the elastin network in the vessel wall, e.g. fibulin-5 null mice have tortuous and elongated aortas (98).
Fibronectin
Fibronectin is a glycoprotein secreted by many cell types including cardiac fibroblasts and endothelial cells, and can act as a template for assembly of fibrillin-1 (99) and latent transforming growth factor-beta-binding proteins (LTBPs) (100). After integrin-dependent polymerization, fibronectin is required for collagen fibril assembly (82,101). Furthermore, fibronectin activates important cellular responses through integrin signaling (α5β1 and αv-class heterodimers) such as adhesion, proliferation, migration and differentiation.
Hyaluronan (HA)
HA is linear glycosaminoglycan composed of alternating β-(1,4)-N-acetyl-D-glucosamine and β-(1,3)-D-glucuronic acid containing disaccharides. It is synthesized at the plasma membrane by hyaluronan synthases and is not modified by sulfation or acetylation. Therefore, HA is identical in all mammals and evolutionarily lower animals. It is an important component of the microenvironment and is of extremely high molecular mass (up to 7 MDa) and large hydrodynamic volume. HA is a main component of adventitial and valve ECM. HA binds to several receptors at the surface of mesenchymal and immune cells. The best-known HA receptors are CD44, receptor of HA mediated motility (RHAMM) and LYVE1. These receptors regulate the phenotypes of fibroblasts, immune cells, endothelial cells and SMCs (102–104). Furthermore, the binding of different proteins and ECM constituents to HA, such as HA binding proteoglycans versican or TSG6, modify the HA matrix and skew it into different functions (Figure 2).
Proteoglycans
Cell surface proteoglycans:
Syndecans 1–4 are transmembrane heparan-/chondroitin sulfate proteoglycans with unique extracellular domains that signal through a short intracellular component in concert with growth factors and other ECM molecules (105). Functionally, syndecan is thought to modulate heparin binding growth factor activity and has an anti-inflammatory function. Additionally, glypicans 1–6, are glycosyl-phosphatidylinositol (GPI) anchored heparan sulfate proteoglycans, involved in Wnt, Hedgehog, FGF and BMP signaling.
HA-binding proteoglycans:
Versican binds through its G1 domain specifically to HA and via its G3 domain to other ECM molecules. Thereby, versican/HA complexes can form large multi-molecular aggregates, which are detected mainly in the pericardium and also the adventitia of blood vessels including the coronary arteries. Versican is present as various splice variants (V0, V1, V2, V3) that vary in the number of chondroitin sulfate side chains attached to the core protein. The physiological role in the healthy adult heart and vasculature has not been defined, however, it is critical for embryonic development and response to injury in the cardiovascular system (106).
BM proteoglycans:
Perlecan is the largest heparan sulfate proteoglycan and has important functions in healthy BM, being critical for BM assembly and during cardiac development. Furthermore, the heparan sulfate side chains enable multiple interactions with heparin binding proteins such as growth factors and chemokines (107).
Small leucine rich proteoglycans (SLRPs):
SLRPs are divided into five different classes based on their structure. SLRPs are involved both in cardiac and vascular homeostasis and remodeling. Knowledge about their role in physiological conditions is limited and best understood for class I and class II SLRPs. The class I SLRPs decorin and biglycan carry one or two chondroitin sulfate and dermatan sulfate side chains, respectively. Important functions include regulation of growth factor availability and activity (i.e. transforming growth factor-β1 (TGF-β1)), as well as modifying the assembly of collagen fibrils and fibronectin. The loss of decorin and biglycan disturbs collagen fibrillogenesis, leading to skin fragility and bone ECM disturbance. Furthermore, the dermatan sulfate side chains of biglycan balance thrombin activity by activation of heparin cofactor 2 (108).
Matricellular proteins:
Thrombospondins 1–5, Secreted Protein Acidic and Rich in Cysteine (SPARC), tenascins, osteopontin, periostin and the Cyr61/CTGF/NOV (CCN) protein family are important positive and negative regulators in vascular and cardiac physiology. For example, thrombospondins 1, 2 and 5 are known to regulate collagen fibril assembly, while thrombospondins 1 and 2 are negative regulators of physiological angiogenesis and cardiac remodeling (reviewed in (109)).
Cellular sources of ECM in the adult cardiovascular system
Cells within the heart
Most of the cardiac mass is attributable to cardiomyocytes. However non-myocyte cells outnumber cardiomyocytes, and recent data indicate that both fibroblasts and endothelial cells together are the predominant cells in the heart (110). Resident cardiac fibroblasts derive during development from the epicardium and endocardium, with a minor proportion possibly from neural crest cells. Cardiac fibroblasts reside in the endomysium and fulfill a key role in maintaining the physiologic ECM environment around cardiomyocytes (111). Elaboration of ECM by cardiac fibroblasts is regulated by mechanical, electrical and neurohormonal stimulation (112,113). Other cell types within the heart include mast cells and resident immune cells like tissue resident macrophages (114) that contribute to tissue and likely also ECM homeostasis by interactions with fibroblasts and ECM degradation (115). This homeostatic function of resident macrophages is in contrast to that of monocyte-derived macrophages that are typically involved in the response to pathophysiologic stimuli. Furthermore, prototypical vascular cells as detailed below contribute to the cardiac ECM microenvironment in health and disease.
Vascular cells
Endothelial cells form the luminal lining in blood vessels and maintain a glycocalyx extending into the lumen and a BM at the basolateral side. In the tunica media of blood vessels, SMCs are primarily responsible for production of ECM. SMC of the tunica media are surrounded by a BM and have a differentiated contractile and non-proliferative phenotype. The contractile function depends on alpha-smooth muscle actin expression and interaction with collagen and laminin binding integrins. Their phenotype switches to a secretory and proliferative phenotype in response to growth factors, inflammatory mediators and remodeling of the mature healthy ECM environment (elastin and collagen degradation) (116). Pericytes are found in the circumference of capillaries. Furthermore, immune cells enter and leave the vascular walls. In principle many immune cells such as macrophages are able to synthesize and degrade vascular ECM, but to what extend this occurs in healthy conditions is not clear yet (104,117).
Single-cell RNA sequencing of cardiac cells
The physiologic functions of many ECM molecules in healthy cardiovascular organs, particularly those that are less abundant, remain to be discovered. Furthermore, most ECM-related studies focus on phenotypes in disease models, and not homeostasis. In addition, ECM proteomics has only recently become possible, and has not been widely used to describe the ECM of healthy tissues (118). As a solution, single-cell RNA sequencing (scRNAseq) appears poised to address this issue and recently, two groups provided scRNAseq data from the cardiovascular systems of adult mice (119,120). Below we have used data from one of these (119) to explore the cardiac ECM and ECM-associated transcriptome. Importantly, this study revealed 5 different sub-populations of cardiac fibroblasts, including a population devoted to antagonizing WNT signaling (F-WntX). Figure 3 gives an overview of the main cell populations, including subtypes of fibroblasts, endothelial cells, resident macrophages, T-cells, B-cells and vascular cells, as well as proliferating cells in adult murine hearts under normal homeostasis. The heat map of ECM GO terms not only shows that ECM genes are most strongly expressed in fibroblasts, but also allows us to appreciate the vast number of genes involved in cardiac ECM. Furthermore, it demonstrates that, to some extent, almost all cardiac non-myocyte populations contribute to cardiac ECM gene expression (Figure 3A, B). As expected, collagen type I expression is highest in cardiac fibroblast clusters. Collagen type IV as part of the BM is expressed by both endothelial cells and fibroblasts. TGF-β1 as an example of an ECM promoting growth factor is widely expressed by endothelial cells, B-cells and resident macrophages, highlighting inter-cell communication. This analysis can also be used to compare the expression of single ECM genes in different cell populations to stimulate new hypotheses about ECM regulation and function. For example, Lamb3 (laminin subunit beta-3), appears to be expressed primarily by B-cells (Figure 3C). A closer view on the expression patterns of the collagens and “glyco-matrix” is given in Figure 4. The expression pattern of collagens fits with the current paradigm that collagen types I, III, IV, V and VI are the predominant collagens of healthy myocardium. Interestingly, fibroblast sup-populations show heterogenous expression levels of collagens, e.g. Col6a1, Col6a2 and Col6a3, which might relate to their specific phenotypes (Figure 4B). Similarly, glycoproteins and proteoglycans are expressed mainly in fibroblasts, however, there is differential expression in other cell types. For example, the expression of Lyve-1, an HA receptor, is strongly associated with tissue resident macrophages (Figure 4B).
Figure 3. Cellular sources of ECM production in cardiac tissue revealed by single-cell RNA sequencing.
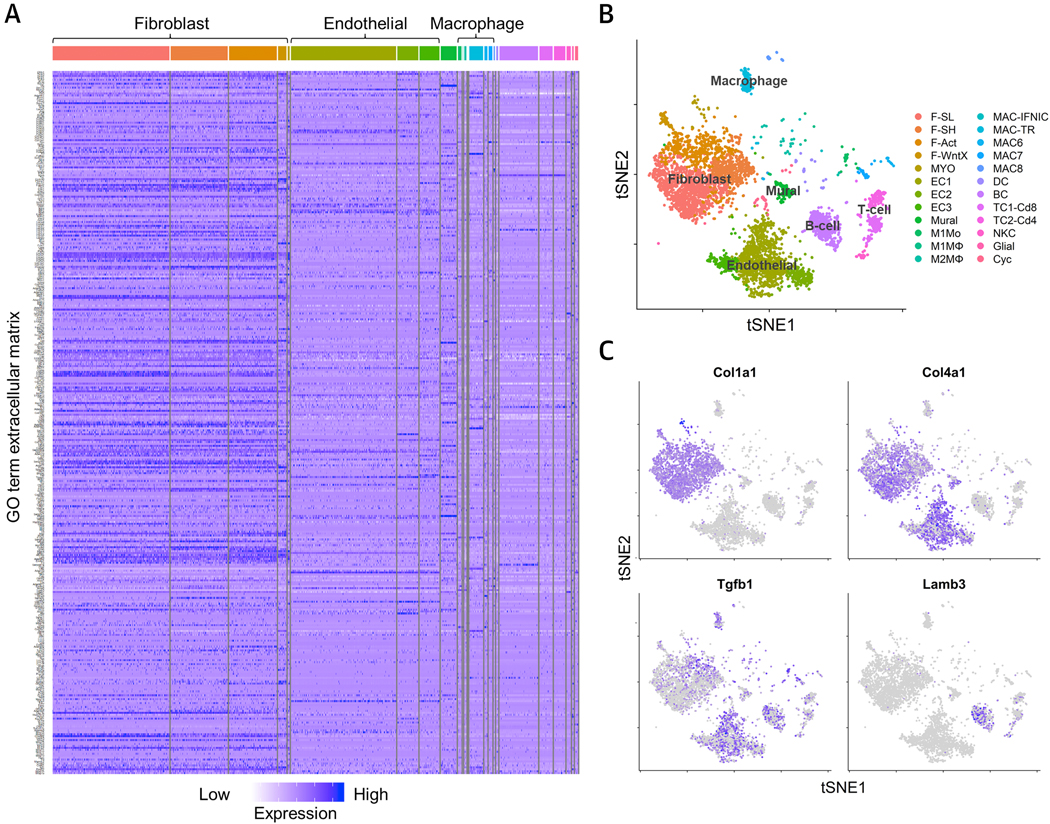
ScRNAseq of the total interstitial, non-cardiomyocyte, cell population of control hearts. (A) Heatmap of ECM gene ontology (GO) term expression across all populations, confirming the highest expression of ECM is generated by fibroblasts. (B) t-distributed stochastic neighbor embedding (tSNE) plot of cell population clusters showing that the major cell types, i.e. fibroblasts and endothelial cells, can be subdivided into 5 distinct fibroblast sub-populations (F-SL, F-SH, F-Act, F-WntX and MYO) and 3 distinct endothelial cell populations (EC1, −2, −3). (C) tSNE plots showing the expression patterns of collagen type I, collagen type IV, TGF-β1 and laminin subunit beta-3. Figure generated using data made available by Farbehi et al. (119). F-SL, fibroblast-Sca1-low; F-SH, fibroblast-Sca1-high; F-Act, fibroblast-activated; F-WntX, fibroblast-Wnt expressing; MYO, myofibroblast; EC1–3, endothelial cell 1–3; M1Mo, M1 monocyte; M1MФ, M1 macrophage; M2MФ, M2 macrophage; MAC-IFNIC, macrophage-IFN inducible cell; MAC-TR, macrophage-tissue resident; MAC6–8, macrophage 6–8; DC, dendritic cell; BC, B-cell; TC1-Cd8, T-Cell 1-Cd8+; TC2-Cd4, T-cell 2-Cd4+; NKC, natural killer cell; Cyc, cycling cell.
Figure 4. Gene expression profile of major ECM- and ECM-associated components in the non-myocytic cell populations of healthy adult myocardium.
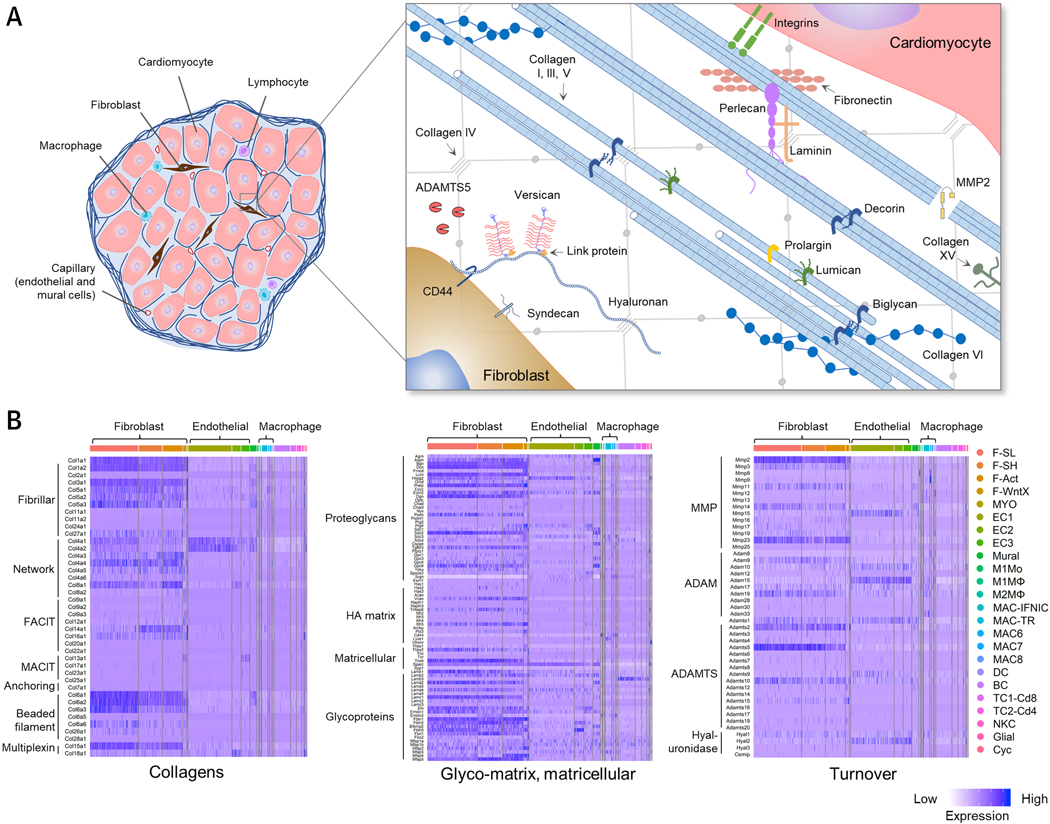
(A) Graphical representation of the cellular composition and ECM organization in the myocardium. (B) Heatmaps of ECM gene expression from scRNAseq analysis of control hearts. Figure generated using data made available by Farbehi et al. (119). Abbreviations as per Figure 3.
Homeostatic turnover and role in aging
The turnover of collagenous and non-collagenous ECM is mediated by the endopeptidase MMPs, and is balanced by tissue inhibitors of MMPs (TIMPs). In addition, other proteases contribute to ECM turnover such those related to the ADAMTS family, and glycolytic enzymes like glycosaminoglycan degrading enzymes (e.g. hyaluronidases). In the adult, MMPs are thought to be responsible for the majority of cardiovascular ECM turnover (primarily collagens), whereas the ADAMTS family plays a larger role in the embryo. MMPs are expressed by cardiomyocytes, fibroblasts, SMCs, and endothelial cells (121). MMP activation is achieved by removal of their N-terminal propeptide, a tightly regulated process typically catalyzed by autoproteolysis, serine proteases (e.g. plasmin) or by other MMPs (e.g. MMP3 activation of pro-MMP1) (122). A list of major cardiovascular ECM-degrading enzymes and their substrates can be found in Table 2. Looking back to the scRNAseq data, a unique expression pattern of ECM turnover genes among different cell types can be appreciated (Figure 4B). For example, fibroblast populations are characterized by high expression of Mmp2, Mmp23, Adamts2 and Adamts5.
Table 2.
Overview of enzymes and substrates involved in cardiovascular ECM turnover. References for ECM substrates can be found in the following reviews (122,128–132) unless noted in the table.
| Class | Type | Name | ECM substrates |
|---|---|---|---|
| MMP | Collagenases | MMP1 | Collagens (I, II, III, VII, X), gelatin, tenascin, perlecan, entactin, aggrecan, link protein |
| MMP8 | Collagens (I, II, III,); gelatin, aggrecan, link protein | ||
| MMP13 | Collagens (I, II, III, IV, IX, X, XIV), aggrecan, perlecan, tenascin, fibronectin, osteonectin, laminin | ||
| MMP18 | Collagen I, gelatin | ||
| Gelatinases | MMP2 | Gelatin, collagens (I, II, III, IV, V, XI), vitronectin, fibronectin, laminin | |
| MMP9 | Gelatin (III, IV, V), entactin, aggrecan, elastin, link protein, vitronectin | ||
| Stromelysins | MMP3 | Collagens (III, IV, X), aggrecan, decorin, gelatins, tenascin, link protein, perlecan, fibronectin, laminin | |
| MMP10 | Collagens (III, IV, V), aggrecan, fibronectin, laminin, link protein | ||
| MMP11 | Collagen IV, gelatin, fibronectin, laminin | ||
| Matrilysins | MMP7 | Collagens (I, IV), gelatin, decorin, elastin, fibronectin, vitronectin, laminin, tenascin | |
| MMP26 | Collagen IV, fibronectin, gelatin | ||
| Membrane Type | MMP14 (MT1-MMP) | Collagens (I, II, III), gelatin, aggrecan, fibronectin, fibrin, laminin | |
| MMP15 (MT2-MMP) | Fibrin, fibronectin, tenascin, nidogen, aggrecan, perlecan, laminin | ||
| MMP16 (MT3-MMP) | Collagen III, gelatin, fibronectin, fibrin | ||
| MMP24 (MT5-MMP) | Gelatin, fibronectin, proteoglycans | ||
| GPI Anchored | MMP17 (MT4-MMP) | Gelatin, fibrinogen | |
| MMP25 (MT6-MMP) | Collagen IV, gelatin, fibrin, SPARC | ||
| Metalloelastase | MMP12 | Collagen IV, fibronectin, laminins, vitronectin, proteoglycans, chondroitin sulfate, elastin | |
| Enamelysin | MMP20 | Collagen V | |
| Other | MMP19 | Collagen IV, nidogen, laminins, fibronectin | |
| MMP23 (CA-MMP) | Auto-proteolysis, unknown | ||
| MMP27 | Unknown | ||
| MMP28 | Unknown | ||
| ADAM | ADAM8 | Fibronectin (133) | |
| ADAM9 | Laminin | ||
| ADAM12 | Gelatin (134) | ||
| ADAMTS | Aggrecanase | ADAMTS1 | Aggrecan, versican, syndecan-4, nidogen-1, −2, gelatin |
| ADAMTS4 | Aggrecan, versican, biglycan, reelin, brevican, matrilin-3, oligomeric matrix protein | ||
| ADAMTS5 | Aggrecan, versican, reelin, biglycan, matrilin-4, brevican | ||
| ADAMTS8 | Aggrecan | ||
| ADAMTS9 | Aggrecan, versican, fibronectin | ||
| ADAMTS15 | Aggrecan, versican | ||
| ADAMTS20 | Versican | ||
| Procollagen | ADAMTS2 | Procollagens (I, II, III, V), fibronectin | |
| N-propeptidases | ADAMTS3 | Procollagen II, biglycan, fibronectin, latent transforming growth factor-beta-binding protein (LTBP1) | |
| ADAMTS14 | Procollagen I and III, fibronectin, LTBP1 | ||
| cartilage oligomeric matrix protein (COMP) -cleaving | ADAMTS7 | COMP | |
| ADAMTS12 | COMP | ||
| vWF proteinase | ADAMTS13 | von-Willebrand (vWF) factor | |
| Other | ADAMTS6 | Unknown | |
| ADAMTS10 | Fibrillin-1, −2 | ||
| ADAMTS16 | Aggrecan, fibronectin | ||
| ADAMTS17 | Unknown | ||
| ADAMTS18 | Unknown | ||
| ADAMTS19 | Unknown | ||
| Hyaluronidases | Hyal-1 | Hyaluronan, chondroitin, chondroitin sulfate | |
| Hyal-2 | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Hyal-3 | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Hyal-4 | Chondroitin, chondroitin sulfate, hyaluronan | ||
| Hyal-5 | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Hyal-6 | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Cemip | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Cemip2 | Hyaluronan, chondroitin, chondroitin sulfate | ||
| Spam1 | Hyaluronan, chondroitin, chondroitin sulfate | ||
As a paradigm covered in depth in part 3 of this review series, the aged ‘healthy’ heart shows progressive interstitial fibrosis and ECM stiffening, which is likely driven by age dependent local changes such as myocyte loss and subsequent hypertrophy of remaining myocytes as well as increased peripheral vascular stiffness and increased afterload. As a consequence, left ventricular hypertrophy, impaired ventricular relaxation and diastolic dysfunction develop which ultimately lead to heart failure with preserved ejection fraction commonly diagnosed in the ‘healthy’ elderly patient.
ECM-immune interactions
Evidence is growing that the ECM modulates immune reactions and that it may be an important modulator of immunological surveillance. During homeostasis, intact ECM does not activate immune cells or immune responses and thereby contributes to an anti-inflammatory environment. However, in response to pathophysiologic stimuli, ECM degradation and fragmentation lead to the release of ECM fragments that activate inflammatory responses either through toll like receptors or altered signaling properties. Furthermore, recruitment of immune cells is stimulated by matrix fragments (matrikines) (123).
The interactive networks of ECM molecules can also be remodeled to acquire different functions in health and disease. For example, HA binding proteins can be recruited into the HA matrix which affect migratory and proliferative phenotypes, e.g. of fibroblasts, or even transform it into a pro-inflammatory ECM that retains monocytes and macrophages (Figure 2) (124). Of this family of HA binding and modifying proteins, many are expressed by cardiac fibroblasts as suggested by the scRNAseq data, including aggrecan (Acan), versican (Vcan), link protein 1, −3 (Hapln1, −3), TSG-6 (Tnfaip6), inter-α-inhibitor heavy chains 2,3,4,5 (Itih2, −3, −4, −5), bikunin (Ambp), and pentraxin 3 (Ptx3) (Figure 4B). HA is also an important part of the immune synapse thereby stimulating T-cell responses (125). Furthermore, GAG side chains of perlecan, syndecans and class I and II SLRPs (decorin, biglycan) activate anti-thrombin III and heparin cofactor 2, respectively (108,126) and inhibit thrombin activity which plays an important pro-inflammatory role next to its role in hemostasis (127). Finally, through heparan sulfate and dermatan sulfate GAGs, intact ECM is able to store growth factors and cytokines that can further modulate immune-related and other processes.
Conclusions
The last two decades have seen enormous advances in our understanding of ECM physiology and pathobiology. Historically, ECM was thought to be static thereby providing the “mortar” for cells, but it is now recognized that the ECM is highly dynamic in response to physiologic and pathophysiologic stimuli and confers very specific signaling and mechanical functions. These important paradigm shifts have seen many new insights arise in both development and in the adult, however, by leveraging novel tools like scRNAseq and high-throughput proteomics, it is anticipated that a great deal more remains to be discovered.
Supplementary Material
Highlights.
-
-
The extracellular matrix (ECM) is the non-cellular component of tissues throughout the body, and is formed of filamentous proteins, proteoglycans and glycosaminoglycans, which extensively interact and whose structure and dynamics are modified by cross-linking.
-
-
While historically ECM was thought to be static thereby providing the “mortar” for cells, on the contrary, it is now recognized that the ECM is highly dynamic during development and in response to physiologic and pathophysiologic stimuli, conferring very specific signaling and mechanical functions.
-
-
By leveraging novel tools like single cell RNA sequencing and high-throughput proteomics, it is anticipated that a great deal more remains to be discovered, which may lead to future novel therapeutic opportunities.
Acknowledgements:
del Monte-Nieto acknowledges research support from the Heart Foundation of Australia (FLF1–102036), the Australian Research Council (ARC DP190101475) and the Australian Regenerative Medicine Institute, which is supported by grants from the State Government of Victoria and the Australian Government. Fischer acknowledges research support from the German Research Foundation (DFG) (SFB1116, IRTG1902, TRR259). Harvey acknowledges support from the National Health and Medical Research Council (NHMRC) of Australia (1074386; 573732, 573705, 1118576), the Australian Research Council (ARC DP190101475, DP160104858), the New South Wales (NSW) Government Ministry of Health (20:20 campaign; Cardiovascular Disease Senior Scientist Grant). Kovacic acknowledges research support from the National Institutes of Health (R01HL130423, R01HL135093).
Abbreviations
- ADAMTS
a disintegrin and metalloprotease with thrombospondin motifs
- BM
basement membrane
- ECM
extracellular matrix
- EndMT
endothelial to mesenchymal transition
- GAG
glycosaminoglycans
- HA
hyaluronan
- LOF
loss-of-function
- MMP
matrix metalloprotease
- PDGF
platelet-derived growth factor
- scRNAseq
single-cell RNA sequencing
- SLRP
small leucine rich proteoglycans
- SMC
smooth muscle cell
- TGF-β
transforming growth factor-β
- TNF
tumor necrosis factor
- TSG6
(TNF)-stimulated gene 6
- VEGF
vascular endothelial cell growth factor
Footnotes
Disclosures: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 2006;126:677–89. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert PM, Havenstrite KL, Magnusson KE et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 2010;329:1078–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majkut S, Idema T, Swift J, Krieger C, Liu A, Discher DE. Heart-specific stiffening in early embryos parallels matrix and myosin expression to optimize beating. Curr Biol 2013;23:2434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poh YC, Chen J, Hong Y et al. Generation of organized germ layers from a single mouse embryonic stem cell. Nat Commun 2014;5:4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2010;2:a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toole BP, Yu Q, Underhill CB. Hyaluronan and hyaluronan-binding proteins. Probes for specific detection. Methods Mol Biol 2001;171:479–85. [DOI] [PubMed] [Google Scholar]
- 7.Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn 1997;210:130–45. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Loeb JA. Neuregulin-heparan-sulfate proteoglycan interactions produce sustained erbB receptor activation required for the induction of acetylcholine receptors in muscle. J Biol Chem 2001;276:38068–75. [DOI] [PubMed] [Google Scholar]
- 9.Lockhart M, Wirrig E, Phelps A, Wessels A. Extracellular matrix and heart development. Birth Defects Res A Clin Mol Teratol 2011;91:535–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res 2005;97:1093–107. [DOI] [PubMed] [Google Scholar]
- 11.Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost 2007;5 Suppl 1:32–40. [DOI] [PubMed] [Google Scholar]
- 12.Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol 2010;341:126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol 2007;311:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis SE, Goh KL, Hodivala-Dilke K et al. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol 2002;22:927–33. [DOI] [PubMed] [Google Scholar]
- 15.Yang JT, Rayburn H, Hynes RO. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 1995;121:549–60. [DOI] [PubMed] [Google Scholar]
- 16.Thyboll J, Kortesmaa J, Cao R et al. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol 2002;22:1194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaughlin PJ, Chen Q, Horiguchi M et al. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol 2006;26:1700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frye M, Taddei A, Dierkes C et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat Commun 2018;9:1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 2011;437:169–83. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto H, Ehling M, Kato K et al. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun 2015;6:6429. [DOI] [PubMed] [Google Scholar]
- 21.De Angelis JE, Lagendijk AK, Chen H et al. Tmem2 Regulates Embryonic Vegf Signaling by Controlling Hyaluronic Acid Turnover. Dev Cell 2017;40:421. [DOI] [PubMed] [Google Scholar]
- 22.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993;119:1079–91. [DOI] [PubMed] [Google Scholar]
- 23.Linask KK, Lash JW. A role for fibronectin in the migration of avian precardiac cells. I. Dose-dependent effects of fibronectin antibody. Dev Biol 1988;129:315–23. [DOI] [PubMed] [Google Scholar]
- 24.Trinh LA, Stainier DY. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell 2004;6:371–82. [DOI] [PubMed] [Google Scholar]
- 25.Bloomekatz J, Singh R, Prall OW et al. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakaguchi T, Kikuchi Y, Kuroiwa A, Takeda H, Stainier DY. The yolk syncytial layer regulates myocardial migration by influencing extracellular matrix assembly in zebrafish. Development 2006;133:4063–72. [DOI] [PubMed] [Google Scholar]
- 27.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev 2003;83:1223–67. [DOI] [PubMed] [Google Scholar]
- 28.Hamada H, Tam PP. Mechanisms of left-right asymmetry and patterning: driver, mediator and responder. F1000Prime Rep 2014;6:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldwin HS, Solursh M. Degradation of hyaluronic acid does not prevent looping of the mammalian heart in situ. Dev Biol 1989;136:555–9. [DOI] [PubMed] [Google Scholar]
- 30.Linask KK, Han M, Cai DH, Brauer PR, Maisastry SM. Cardiac morphogenesis: matrix metalloproteinase coordination of cellular mechanisms underlying heart tube formation and directionality of looping. Dev Dyn 2005;233:739–53. [DOI] [PubMed] [Google Scholar]
- 31.Le Garrec JF, Dominguez JN, Desgrange A et al. A predictive model of asymmetric morphogenesis from 3D reconstructions of mouse heart looping dynamics. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markwald RR, Fitzharris TP, Smith WN. Sturctural analysis of endocardial cytodifferentiation. Dev Biol 1975;42:160–80. [DOI] [PubMed] [Google Scholar]
- 33.Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat 1977;148:85–119. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura A, Manasek FJ. An experimental study of the relation of cardiac jelly to the shape of the early chick embryonic heart. J Embryol Exp Morphol 1981;65:235–56. [PubMed] [Google Scholar]
- 35.Camenisch TD, Spicer AP, Brehm-Gibson T et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 2000;106:349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh EC, Stainier DY. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 2001;293:1670–3. [DOI] [PubMed] [Google Scholar]
- 37.Bernanke DH, Markwald RR. Effects of hyaluronic acid on cardiac cushion tissue cells in collagen matrix cultures. Tex Rep Biol Med 1979;39:271–85. [PubMed] [Google Scholar]
- 38.Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med 2002;8:850–5. [DOI] [PubMed] [Google Scholar]
- 39.Del Monte-Nieto G, Ramialison M, Adam AAS et al. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 2018;557:439–445. [DOI] [PubMed] [Google Scholar]
- 40.Hatano S, Kimata K, Hiraiwa N et al. Versican/PG-M is essential for ventricular septal formation subsequent to cardiac atrioventricular cushion development. Glycobiology 2012;22:1268–77. [DOI] [PubMed] [Google Scholar]
- 41.Negro A, Brar BK, Lee KF. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog Horm Res 2004;59:1–12. [DOI] [PubMed] [Google Scholar]
- 42.Kern CB, Twal WO, Mjaatvedt CH et al. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn 2006;235:2238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR. The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology. J Cell Commun Signal 2009;3:275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol Genomics 2009;39:160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shigeta A, Huang V, Zuo J et al. Endocardially Derived Macrophages Are Essential for Valvular Remodeling. Dev Cell 2019;48:617–630 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goddard LM, Duchemin AL, Ramalingan H et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev Cell 2017;43:274–289 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paolini A, Abdelilah-Seyfried S. The mechanobiology of zebrafish cardiac valve leaflet formation. Curr Opin Cell Biol 2018;55:52–58. [DOI] [PubMed] [Google Scholar]
- 48.Steed E, Faggianelli N, Roth S, Ramspacher C, Concordet JP, Vermot J. klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nat Commun 2016;7:11646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neri T, Hiriart E, van Vliet PP et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat Commun 2019;10:1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christoffels VM, Habets PE, Franco D et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev Biol 2000;223:266–78. [DOI] [PubMed] [Google Scholar]
- 51.Stankunas K, Hang CT, Tsun ZY et al. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell 2008;14:298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tian X, Li Y, He L et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat Commun 2017;8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rasouli SJ, Stainier DYR. Regulation of cardiomyocyte behavior in zebrafish trabeculation by Neuregulin 2a signaling. Nat Commun 2017;8:15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res 2002;91:158–64. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Z, Rawnsley DR, Goddard LM et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev Cell 2015;32:168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooley MA, Fresco VM, Dorlon ME et al. Fibulin-1 is required during cardiac ventricular morphogenesis for versican cleavage, suppression of ErbB2 and Erk1/2 activation, and to attenuate trabecular cardiomyocyte proliferation. Dev Dyn 2012;241:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cherian AV, Fukuda R, Augustine SM, Maischein HM, Stainier DY. N-cadherin relocalization during cardiac trabeculation. Proc Natl Acad Sci U S A 2016;113:7569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyser RC, Miranda AM, Chen CM, Davidson SM, Srinivas S, Riley PR. Calcium handling precedes cardiac differentiation to initiate the first heartbeat. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science 2009;323:638–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ieda M, Tsuchihashi T, Ivey KN et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell 2009;16:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiou KK, Rocks JW, Chen CY et al. Mechanical signaling coordinates the embryonic heartbeat. Proc Natl Acad Sci U S A 2016;113:8939–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith TK, Bader DM. Signals from both sides: Control of cardiac development by the endocardium and epicardium. Semin Cell Dev Biol 2007;18:84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore-Morris T, Cattaneo P, Puceat M, Evans SM. Origins of cardiac fibroblasts. J Mol Cell Cardiol 2016;91:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porrello ER, Mahmoud AI, Simpson E et al. Transient regenerative potential of the neonatal mouse heart. Science 2011;331:1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kikuchi K, Holdway JE, Werdich AA et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010;464:601–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Notari M, Ventura-Rubio A, Bedford-Guaus SJ et al. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci Adv 2018;4:eaao5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen Med 2016;1:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mahmoud AI, Porrello ER. Upsizing Neonatal Heart Regeneration. Circulation 2018;138:2817–2819. [DOI] [PubMed] [Google Scholar]
- 69.Jacot JG, Martin JC, Hunt DL. Mechanobiology of cardiomyocyte development. J Biomech 2010;43:93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norris RA, Damon B, Mironov V et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 2007;101:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Z, Xie J, Hao H et al. Ablation of periostin inhibits post-infarction myocardial regeneration in neonatal mice mediated by the phosphatidylinositol 3 kinase/glycogen synthase kinase 3beta/cyclin D1 signalling pathway. Cardiovasc Res 2017;113:620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bassat E, Mutlak YE, Genzelinakh A et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017;547:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hilgenberg LG, Pham B, Ortega M et al. Agrin regulation of alpha3 sodium-potassium ATPase activity modulates cardiac myocyte contraction. J Biol Chem 2009;284:16956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morikawa Y, Heallen T, Leach J, Xiao Y, Martin JF. Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 2017;547:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyre DR, Weis MA, Wu JJ. Advances in collagen cross-link analysis. Methods 2008;45:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baranova NS, Nileback E, Haller FM et al. The inflammation-associated protein TSG-6 cross-links hyaluronan via hyaluronan-induced TSG-6 oligomers. J Biol Chem 2011;286:25675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.López B, Querejeta R, González A, Beaumont J, Larman M, Díez J. Impact of treatment on myocardial lysyl oxidase expression and collagen cross-linking in patients with heart failure. Hypertension 2009;53:236–42. [DOI] [PubMed] [Google Scholar]
- 78.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989;13:1637–52. [DOI] [PubMed] [Google Scholar]
- 79.Baicu CF, Stroud JD, Livesay VA et al. Changes in extracellular collagen matrix alter myocardial systolic performance. Am J Physiol Heart Circ Physiol 2003;284:H122–32. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 2010;7:1438–45. [DOI] [PubMed] [Google Scholar]
- 81.Medugorac I, Jacob R. Characterisation of left ventricular collagen in the rat. Cardiovasc Res 1983;17:15–21. [DOI] [PubMed] [Google Scholar]
- 82.Engvall E, Ruoslahti E, Miller EJ. Affinity of fibronectin to collagens of different genetic types and to fibrinogen. J Exp Med 1978;147:1584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wiltz D, Arevalos A, Balaoing LR et al. Extracellular Matrix Organization, Structure, and Function In: Aikawa E, editor Calcific Aortic Valve Disease: IntechOpen, 2013. [Google Scholar]
- 84.Murata K Acidic glycosaminoglycans in human heart valves. J Mol Cell Cardiol 1981;13:281–92. [DOI] [PubMed] [Google Scholar]
- 85.Grande-Allen KJ, Osman N, Ballinger ML, Dadlani H, Marasco S, Little PJ. Glycosaminoglycan synthesis and structure as targets for the prevention of calcific aortic valve disease. Cardiovasc Res 2007;76:19–28. [DOI] [PubMed] [Google Scholar]
- 86.Stella JA, Sacks MS. On the biaxial mechanical properties of the layers of the aortic valve leaflet. J Biomech Eng 2007;129:757–66. [DOI] [PubMed] [Google Scholar]
- 87.Latif N, Sarathchandra P, Taylor PM, Antoniw J, Yacoub MH. Localization and pattern of expression of extracellular matrix components in human heart valves. J Heart Valve Dis 2005;14:218–27. [PubMed] [Google Scholar]
- 88.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res 2009;105:408–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doll S, Dressen M, Geyer PE et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat Commun 2017;8:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eble JA, Niland S. The extracellular matrix of blood vessels. Curr Pharm Des 2009;15:1385–400. [DOI] [PubMed] [Google Scholar]
- 91.Dogne S, Flamion B, Caron N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler Thromb Vasc Biol 2018;38:1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang X, Sun D, Song JW et al. Endothelial cell dysfunction and glycocalyx - A vicious circle. Matrix Biol 2018;71–72:421–431. [DOI] [PubMed] [Google Scholar]
- 93.Nagy N, Freudenberger T, Melchior-Becker A et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: novel insights into the role of hyaluronan synthesis. Circulation 2010;122:2313–22. [DOI] [PubMed] [Google Scholar]
- 94.Bishop JE, Laurent GJ. Collagen turnover and its regulation in the normal and hypertrophying heart. Eur Heart J 1995;16 Suppl C:38–44. [DOI] [PubMed] [Google Scholar]
- 95.Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res 2019;125:117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hohenester E Structural biology of laminins. Essays Biochem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nishiuchi R, Takagi J, Hayashi M et al. Ligand-binding specificities of laminin-binding integrins: a comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol 2006;25:189–97. [DOI] [PubMed] [Google Scholar]
- 98.Yanagisawa H, Davis EC, Starcher BC et al. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 2002;415:168–71. [DOI] [PubMed] [Google Scholar]
- 99.Sabatier L, Chen D, Fagotto-Kaufmann C et al. Fibrillin assembly requires fibronectin. Mol Biol Cell 2009;20:846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dallas SL, Sivakumar P, Jones CJ et al. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem 2005;280:18871–80. [DOI] [PubMed] [Google Scholar]
- 101.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol 2008;20:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Slevin M, Krupinski J, Gaffney J et al. Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol 2007;26:58–68. [DOI] [PubMed] [Google Scholar]
- 103.Huebener P, Abou-Khamis T, Zymek P et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 2008;180:2625–33. [DOI] [PubMed] [Google Scholar]
- 104.Petz A, Grandoch M, Gorski DJ et al. Cardiac Hyaluronan Synthesis Is Critically Involved in the Cardiac Macrophage Response and Promotes Healing After Ischemia Reperfusion Injury. Circ Res 2019;124:1433–1447. [DOI] [PubMed] [Google Scholar]
- 105.Fears CY, Woods A. The role of syndecans in disease and wound healing. Matrix Biol 2006;25:443–56. [DOI] [PubMed] [Google Scholar]
- 106.Wight TN. Provisional matrix: A role for versican and hyaluronan. Matrix Biol 2017;60–61:38–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iozzo RV, Zoeller JJ, Nystrom A. Basement membrane proteoglycans: modulators Par Excellence of cancer growth and angiogenesis. Mol Cells 2009;27:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Grandoch M, Kohlmorgen C, Melchior-Becker A et al. Loss of Biglycan Enhances Thrombin Generation in Apolipoprotein E-Deficient Mice: Implications for Inflammation and Atherosclerosis. Arterioscler Thromb Vasc Biol 2016;36:e41–50. [DOI] [PubMed] [Google Scholar]
- 109.Schellings MW, van Almen GC, Sage EH, Heymans S. Thrombospondins in the heart: potential functions in cardiac remodeling. J Cell Commun Signal 2009;3:201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pinto AR, Ilinykh A, Ivey MJ et al. Revisiting Cardiac Cellular Composition. Circ Res 2016;118:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tallquist MD. Cardiac fibroblasts: from origin to injury. Curr Opin Physiol 2018;1:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eghbali M, Blumenfeld OO, Seifter S et al. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization. J Mol Cell Cardiol 1989;21:103–13. [DOI] [PubMed] [Google Scholar]
- 113.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther 2009;123:255–78. [DOI] [PubMed] [Google Scholar]
- 114.Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol 2002;118:41–9. [DOI] [PubMed] [Google Scholar]
- 115.Epelman S, Lavine KJ, Beaudin AE et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shankman LS, Gomez D, Cherepanova OA et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chang MY, Chan CK, Braun KR et al. Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J Biol Chem 2012;287:14122–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barallobre-Barreiro J, Didangelos A, Schoendube FA et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 2012;125:789–802. [DOI] [PubMed] [Google Scholar]
- 119.Farbehi N, Patrick R, Dorison A et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Skelly DA, Squiers GT, McLellan MA et al. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep 2018;22:600–610. [DOI] [PubMed] [Google Scholar]
- 121.Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci 2001;68:799–814. [DOI] [PubMed] [Google Scholar]
- 122.Takawale A, Sakamuri SS, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr Physiol 2015;5:687–719. [DOI] [PubMed] [Google Scholar]
- 123.Grandoch M, Bollyky PL, Fischer JW. Hyaluronan: A Master Switch Between Vascular Homeostasis and Inflammation. Circ Res 2018;122:1341–1343. [DOI] [PubMed] [Google Scholar]
- 124.de la Motte CA, Hascall VC, Drazba J, Bandyopadhyay SK, Strong SA. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-alpha-trypsin inhibitor is crucial to structure and function. Am J Pathol 2003;163:121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bollyky PL, Evanko SP, Wu RP et al. Th1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell Mol Immunol 2010;7:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang L, Beeler DL, Lawrence R et al. 6-O-sulfotransferase-1 represents a critical enzyme in the anticoagulant heparan sulfate biosynthetic pathway. J Biol Chem 2001;276:42311–21. [DOI] [PubMed] [Google Scholar]
- 127.Posma JJ, Posthuma JJ, Spronk HM. Coagulation and non-coagulation effects of thrombin. J Thromb Haemost 2016;14:1908–1916. [DOI] [PubMed] [Google Scholar]
- 128.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 2003;92:827–39. [DOI] [PubMed] [Google Scholar]
- 129.Yang CY, Chanalaris A, Troeberg L. ADAMTS and ADAM metalloproteinases in osteoarthritis - looking beyond the ‘usual suspects’. Osteoarthritis Cartilage 2017;25:1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008;29:258–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol 2015;16:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stern R, Jedrzejas MJ. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev 2006;106:818–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zack MD, Malfait AM, Skepner AP et al. ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala(271). Arthritis Rheum 2009;60:2704–13. [DOI] [PubMed] [Google Scholar]
- 134.Roy R, Wewer UM, Zurakowski D, Pories SE, Moses MA. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem 2004;279:51323–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.