

Abstract
Anaplastic thyroid cancer (ATC) is one of the most aggressive human malignancies, with an average life expectancy of ∼6 months from the time of diagnosis. The genetic and epigenetic changes that underlie this malignancy are incompletely understood. We found that ASH1-like histone lysine methyltransferase (ASH1L) is overexpressed in ATC relative to the much less aggressive and more common differentiated thyroid cancer. This increased expression was due at least in part to reduced levels of microRNA-200b-3p (miR-200b-3p), which represses ASH1L expression, in ATC. Genetic knockout of ASH1L protein expression in ATC cell lines decreased cell growth both in culture and in mouse xenografts. RNA-Seq analysis of ASH1L knockout versus WT ATC cell lines revealed that ASH1L is involved in the regulation of numerous cancer-related genes and gene sets. The pro-oncogenic long noncoding RNA colon cancer-associated transcript 1 (CCAT1) was one of the most highly (approximately 68-fold) down-regulated transcripts in ASH1L knockout cells. Therefore, we investigated CCAT1 as a potential mediator of the growth-inducing activity of ASH1L. Supporting this hypothesis, CCAT1 knockdown in ATC cells decreased their growth rate, and ChIP-Seq data indicated that CCAT1 is likely a direct target of ASH1L's histone methyltransferase activity. These results indicate that ASH1L contributes to the aggressiveness of ATC and suggest that ASH1L, along with its upstream regulator miR-200b-3p and its downstream mediator CCAT1, represents a potential therapeutic target in ATC.
Keywords: thyroid, cancer, gene expression, histone methylation, long noncoding RNA (lncRNA), microRNA, CCAT1, anaplastic thyroid cancer, ASH1-like histone lysine methyltransferase (ASH1L), epigenetics, miR-200b-3p, microRNA (miRNA)
Thyroid carcinoma, the most common endocrine malignancy (1), is divided into the major histological types papillary (∼90% of thyroid cancers), follicular (∼6%), anaplastic (∼1%), and medullary (∼2%). Papillary and follicular thyroid cancers (PTCs and FTCs) are generally well-differentiated tumors that can be treated successfully with surgery and radioiodine. Consequently, Surveillance, Epidemiology, and End Results data indicate that the overall 5-year survival of patients with thyroid cancer is very high, ∼98% (https://seer.cancer.gov/statfacts/html/thyro.html, Accessed December 20, 2019). In contrast, anaplastic thyroid cancer (ATC) is one of the most aggressive and least treatable human malignancies, with a median life expectancy of ∼6 months from the time of diagnosis (2). Thus, although ATC accounts for only ∼1% of thyroid cancer diagnoses, it accounts for as much as 39% of thyroid cancer deaths (2, 3). Although ATCs arise from thyroid follicular cells, they do not retain any of the biological characteristics of the normal thyroid, such as iodide uptake, thyroglobulin synthesis, or dependence on thyroid-stimulating hormone (TSH).
Multiple molecular and genetic factors have been implicated in the pathogenesis of ATC. Some ATCs are associated with differentiated thyroid cancer (DTC) components, and if the DTC contains a classic thyroid cancer driver mutation such as BRAF V600E, that mutation also is found in the ATC (4), consistent with dedifferentiation of DTC into ATC. However, some ATCs lack a DTC component, and in such cases the developmental pathway is obscure. Relative to DTCs, ATCs are more likely to harbor mutations in the TERT promoter (5–8), TP53 (9, 10), DNA damage response and cell cycle checkpoint genes such as ATM, and chromatin remodeling genes such as those in the SWI/SNF complex (7).
Several microRNAs (miRNAs) have been shown to have altered expression in ATC. For example, miR-146b, miR-221, miR-222, and miR-17-92 are up-regulated, and miR-200, miR-30, let-7d, and let-7g are repressed (reviewed in Ref. 11). Many of these changes may contribute to the aggressive biology of ATC. For example, evidence suggests that decreased miR-200 contributes to epithelial-to-mesenchymal transition, and decreased miR-30 contributes to invasion and migration.
In addition to genetic changes, epigenetic perturbations contribute to oncogenesis. Trithorax and Polycomb group proteins act antagonistically in the epigenetic regulation of gene expression. Trithorax group proteins usually activate gene expression and counteract Polycomb group protein-mediated gene silencing (12). ASH1-like histone lysine methyltransferase (ASH1L) is the mammalian homolog of Drosophila ash1, a member of the Trithorax group proteins (12, 13). ASH1L dimethylates histone H3 at lysine 36, forming H3K36me2 (14, 15), which tends to occur along gene bodies (16). Physiologically, ASH1L is thought to play important roles in nervous system development and function (17, 18) and maintenance of the hematopoietic stem cell population (19).
ASH1L has been identified as a critical component of an oncogenic complex that drives mixed-lineage leukemia. In this malignancy, ASH1L “writes” H3K36me2 marks, which are “read” by reader proteins such as lens epithelium-derived growth factor, leading to activation of critical leukemia drivers such as HOX genes (20). ASH1L also has been implicated in the pathogenesis of a subset of acute myeloid leukemias (21). Although less well studied in other malignancies, ASH1L frequently shows high-level amplification in breast cancer, and high mRNA levels are associated with shortened survival (22). ASH1L also is overexpressed in hepatocellular carcinoma (23, 24). A single publication has tied ASH1L to thyroid cancer (25). This study provided evidence that FTCs have increased expression of ASH1L protein (but not RNA) relative to PTCs and that increased ASH1L protein is a consequence of reduced expression of miR-142-3p, which decreases the translation of ASH1L mRNA.
In the present study, we demonstrate that ASH1L protein abundance is much greater in ATCs than PTCs. Genetic knockout (KO) of ASH1L protein expression in ATC cell lines inhibits cell proliferation in vitro and xenograft tumor formation in vivo. We identify ASH1L-responsive genes, including Colon cancer associated transcript 1 (CCAT1), that are potential mediators of ASH1L's oncogenic actions in ATC. Furthermore, we demonstrate that miR-200b-3p, which is expressed at very low levels in ATC relative to PTC and FTC (26, 27), inhibits ASH1L expression. Therefore, decreased levels of miR-200b-3p likely underlie increased ASH1L protein abundance in ATC. The data together suggest that ASH1L, miR-200-3p, and CCAT1 are potential therapeutic targets in ATC.
Results
ASH1L is highly expressed in ATC
Published data support the hypothesis that ASH1L protein is overexpressed in FTC compared with PTC (25). Given that ATC presents a much more intractable clinical problem, we tested whether ASH1L might play a role in this disease. We performed Western blotting of nuclear extracts from six fresh frozen ATCs and five PTCs (Fig. 1A). Quantification of the ASH1L bands normalized to lamin B1 shows that the median ASH1L protein expression is at least 5-fold greater in ATCs than PTCs (p = 0.0022) (Fig. 1B). In addition, we found that ASH1L is expressed at similar or greater levels in the ATC cell lines BHT-101, SW1736, and JEM493, compared with the FTC cell lines FTC-133, FTC-236, FTC-238, TT2609-C02, and 8505C (Fig. 1, C and D). These data suggest that ASH1L may play a role in the aggressive biology of ATC. In the published study on ASH1L in FTC (25), Western blots showed that the ASH1L immunoreactive bands were smaller than expected and were cytoplasmic. However, in our studies, ASH1L is nuclear, and we have found no evidence of smaller ASH1L species, defined as immunoreactive bands that disappear upon genetic ASH1L KO.
Figure 1.
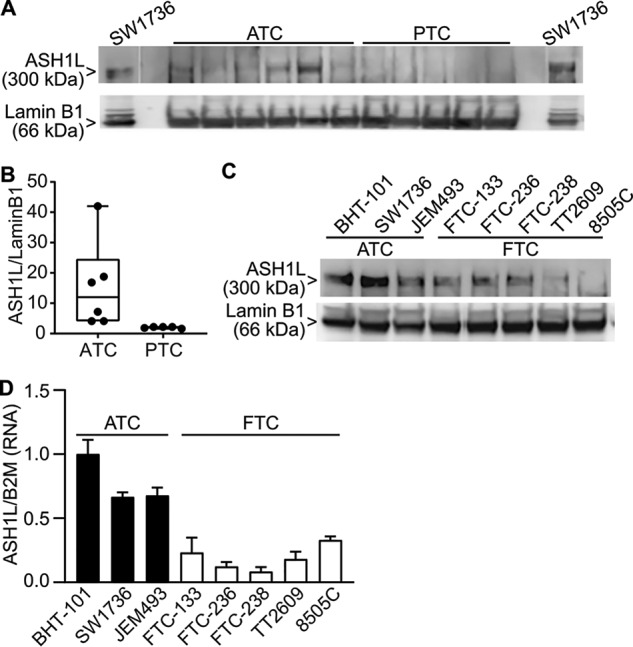
ASH1L is more highly expressed in ATC than in well-differentiated thyroid cancer. A, Western blotting of ASH1L protein expression in nuclear extracts from 6 primary ATCs and 5 primary PTCs. The first and last lanes contain nuclear extracts from the ATC cell line SW1736, used as a positive control to identify ASH1L, based on size of the band (300 kDa) and disappearance with CRISPR-Cas9 KO (see Figs. S2 and 2). Blank lanes were run between the SW1736 samples and the primary tumor samples. Lamin B1 (66 kDa) was used as a control protein. B, box-and-whiskers plot quantification of ASH1L abundance normalized to lamin B1 from panel A. ASH1L protein is more highly expressed in ATC than PTC (p = 0.0022; Mann-Whitney test, 2-tailed). C, Western blotting of ASH1L protein expression in nuclear extracts from 3 ATC cell lines and 5 FTC cell lines. Lamin B1 was used as a control protein. D, RT-qPCR of ASH1L mRNA expression from 3 ATC cell lines and 5 FTC cell lines. B2M was used as a control mRNA. Results are means ± S.D. (n = 3) normalized to the BHT-101 mean, which was set to 1. ASH1L is more highly expressed in the ATC cell lines than the FTC cell lines (p = 0.001, 2-tailed t test).
ASH1L is required for ATC cell growth
We used two shRNAs to knock down ASH1L mRNA and protein in BHT-101 cells by ∼50% (Fig. S1A), which resulted in decreased growth rates assessed by MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay (Fig. S1B). We injected 5 × 106 ASH1L shRNA (referred to here as shASH1L) or control shRNA (referred to here as shControl) BHT-101 cells subcutaneously into each of 6 NOD-SCID mice. Tumors formed in all mice, but the tumor growth rate was ∼50% lower with the shASH1L cells, and those tumors were ∼50% smaller by weight at sacrifice than those of the shControl cells (Fig. S1, C to E). Given these encouraging results, we used CRISPR-Cas9 to knock out ASH1L expression in BHT-101 cells.
CRISPR-Cas9 and four guide RNAs (gRNAs) that target different regions of exon 3 or 11 were used to create four BHT-101 cell lines with premature stop codons in both alleles of ASH1L. The positions of the stop codons are shown schematically in Fig. 2A, along with several functional domains of the protein (14). The minimum catalytic domain is the SET domain, and all stop codons terminate translation before this domain. Sanger sequencing confirmed the following. In clone 1, one allele encodes ASH1L amino acids 1–413 followed by the novel sequence QV*, and the other encodes amino acids 1–413 followed by the novel sequence GRSDQ* (the asterisk signifies a stop codon). In clone 2, both alleles encode ASH1L amino acids 1–610 followed immediately by a stop codon. In clone 3, one allele encodes ASH1L amino acids 1–2129 followed by the novel sequence PEDTEA*, and the other encodes amino acids 1–2129 followed by the novel sequence TEDTEA*. In clone 4, one allele encodes ASH1L amino acids 1–2174 followed by the novel sequence RGGRQ*, and the other encodes amino acids 1–2169 followed by a stop codon. Since these proteins terminate before translation of the catalytic domain, they would not be enzymatically active even if expressed. However, the only antibody that works well in Western blots (A301-749A; Bethyl) targets the very C terminus of the protein, so we cannot assess the expression of putative prematurely truncated species. As expected, all 4 clones show complete loss of ASH1L protein by Western blotting (Fig. 2B). All 4 clones have ∼50% decreased ASH1L mRNA levels, likely reflecting nonsense-mediated decay (assessed by real-time quantitative PCR (RT-qPCR); data not shown). Importantly, all four ASH1L KO clones grow slowly in culture, assessed by MTT assay (Fig. 2C, left) and validated by direct cell counting (Fig. 2C, right). These data confirm that growth of the BHT-101 ATC cell line in culture depends on ASH1L.
Figure 2.

ATC cell growth depends on ASH1L expression. A, Schematic of the ASH1L protein, with asterisks marking the positions of frameshift-induced stop codons in BHT-101 cell clones 1, 2, 3, and 4, created using CRISPR-Cas9. The schematic also shows ASH1L functional domains. The SET domain is the minimal catalytic domain. AWS, associated with SET; BAH, bromo-adjacent homology. B, Western blotting of nuclear extracts showing loss of ASH1L protein expression in the 4 BHT-101 ASH1L KO cell lines. The ASH1L band in the WT cells is 300 kDa. A nonspecific band (NS) migrates slightly faster. Lamin B1 (66 kDa) was used as a control protein. C, ASH1L KO cell lines grow more slowly in culture than the BHT-101 WT cells. Left, MTT assay showing the means ± S.D. for sextuplicate wells at each time point. All KO lines differ from the WT at days 4 and 6 (p < 0.001, Dunnett's test). Right, direct cell counts of a parallel experiment in which the cells in sextuplicate wells were trypsinized, pooled and counted in a Luna-FL automated fluorescence cell counter with acridine orange and propidium iodide. D, six NOD-SCID mice received flank injections of 5 × 106 BHT-101 WT cells or KO clone 4 or 1. The mice harboring the WT cells were sacrificed at day 29 postinjection due to the formation of large tumors. The mice harboring KO clone 4 were harvested at day 48, at which point they had small tumors. The mice harboring KO clone 1 were harvested at day 42, at which point they had no visible tumors. Each symbol in the graph represents the tumor from an individual mouse, and the bars indicate the means ± S.D. The KO clone 4 tumors were smaller than the WT tumors despite being harvested 19 days later (p < 0.0001, 2-tailed t test). Bottom, WT and KO clone 4 tumors; PHD, plant homeodomain.
To assess whether ASH1L dependence is a property of ATC cell lines in addition to BHT-101, we also studied the ATC cell lines SW1736 and JEM493. Both BHT-101 and SW1736 contain a BRAF V600E mutation, and JEM493 contains HRAS Q61R. We used CRISPR-Cas9 and the same gRNAs that were used for BHT-101 cells to create frameshift mutations in both ASH1L alleles of SW1736 and JEM493 cells. Western blots confirmed loss of ASH1L protein (Fig. S2A). ASH1L KO decreased the growth rates of SW1736 and JEM493 cells (Fig. S2B), indicating that ASH1L dependence is not unique to BHT-101 cells and does not require BRAF V600E.
We focused on BHT-101 cells to investigate the role of ASH1L in ATC cell growth in vivo. We injected 5 × 106 WT BHT-101 cells and KO clones 1 and 4 into the flanks of NOD-SCID mice (6 animals each). As shown in Fig. 2D, all 6 mice receiving WT cells were sacrificed at day 29 postinjection due to the formation of large tumors, 0.91 ± 0.17 g (mean ± S.D.). In contrast, the clone 4 mice were sacrificed at day 48, at which time they had small tumors, 0.15 ± 0.11 g (p < 0.0001 versus WT, 2-tailed t test). The clone 1 mice were sacrificed at day 42 and had no detectable tumors at that time. These results indicate that BHT-101 cell growth in vivo is highly ASH1L dependent.
ASH1L regulates a diverse set of genes in ATC
To identify ASH1L-regulated genes, an RNA-Seq analysis was performed on mRNA from the 4 BHT-101 ASH1L KO clones, compared with 3 separate passages of the WT BHT-101 cells. We used a stringent definition of differentially expressed genes as those with a false discovery rate (FDR) of <0.05 and an absolute fold change (FC) of >2 in the same direction in all 4 KO lines versus the WT cells. This resulted in 53 down-regulated genes and 103 induced genes in the KO cells, implying that ASH1L induces 53 genes and represses 103. A heat map of these differentially expressed genes is shown in Fig. 3. The 15 genes most significantly down-regulated by ASH1L KO, and the 15 genes most significantly induced, are shown in Table 1, and the complete list is provided in Table S1. We used RT-qPCR to confirm the RNA-Seq data for 10 of the differentially expressed genes, and the results were uniformly consistent (Table S1, last column).
Figure 3.

Heat map of differentially expressed genes in BHT-101 ASH1L KO cell lines versus WT cells. CRISPR-Cas9 was used to create 4 cell lines with premature stop codons in different positions within ASH1L prior to the catalytic domain. RNA expression of these 4 KO cell lines was compared with that of 3 separate passages of BHT-101 WT cells. Differentially expressed genes are defined as those with FDR of <0.05 and absolute FC of >2 in all 4 KO cell lines compared with WT cells, resulting in 51 down-regulated genes and 103 induced genes in the KO cells.
Table 1.
Most significantly down-regulated and up-regulated genes in ASH1L KO BHT-101 cells compared to WT BHT-101 cellsa
| Gene symbol | Log2 FC | FDR |
|---|---|---|
| SLC6A15 | −8.01 | 2.04E−39 |
| TSSC2 | −5.93 | 4.12E−22 |
| ONECUT3 | −5.27 | 2.21E−18 |
| GLB1L3 | −4.87 | 6.19E−14 |
| HAPLN1 | −5.08 | 1.76E−10 |
| VSTM2L | −2.45 | 3.11E−09 |
| MORC4 | −2.14 | 7.18E−08 |
| FADS2 | −1.62 | 2.31E−06 |
| ANKRD1 | −2.10 | 2.94E−06 |
| SCARA3 | −1.60 | 4.30E−05 |
| CCAT1 | −6.10 | 7.52E−05 |
| NNMT | −2.88 | 8.76E−05 |
| PEAR1 | −2.06 | 1.00E−04 |
| TWSG1 | −1.60 | 2.48E−04 |
| TRIB3 | −1.66 | 3.60E−04 |
| RNF212 | 3.31 | 2.81E−17 |
| AC091801.1 | 8.04 | 4.76E−16 |
| SLC1A1 | 3.12 | 8.13E−15 |
| INPP4B | 4.14 | 6.90E−11 |
| PTCHD4 | 5.81 | 1.60E−10 |
| NUP210 | 1.58 | 3.24E−10 |
| IGFBP7 | 4.53 | 7.18E−08 |
| LRRC6 | 2.84 | 1.06E−07 |
| MMP16 | 5.58 | 1.30E−07 |
| GLI3 | 2.58 | 2.23E−07 |
| FBN1 | 1.80 | 2.48E−07 |
| PRICKLE1 | 5.76 | 1.01E−06 |
| ZNF827 | 2.01 | 1.01E−06 |
| LDOC1 | 3.14 | 2.31E−06 |
| SPOCK1 | 6.36 | 2.62E−06 |
a The 15 most significantly up-regulated and 15 most significantly down-regulated genes are shown. Genes are sorted by FDR (repressed genes followed by induced genes).
We focused on genes down-regulated in ASH1L KO cells, since genes directly regulated (induced) by the high levels of ASH1L in ATC should be in this group. A subset of these genes with known pro-oncogenic associations is listed in Table 2 and is briefly described. CCAT1, TWSG1, HAPL1, and NNMT are overexpressed in various malignancies in which they promote cancer cell proliferation, migration, and invasion (28–32). CCAT1 encodes a long noncoding RNA (lncRNA). Due to its magnitude of change with ASH1L KO (68-fold down-regulated) and the strength of its association with cancer, the role of CCAT1 in ATC was explored further (see below). TWSG1 encodes a protein that is involved in bone morphogenetic protein signaling and is overexpressed in PTC. HAPL1 encodes an extracellular matrix protein that exerts protumorigenic effects in mesothelioma (30) and hepatocellular carcinoma (33). NNMT encodes nicotinamide N-methyltransferase, which, when overexpressed, results in hypomethylation of several activating and repressive histone marks (34) and therefore can mediate indirect effects of ASH1L on gene expression. PRDM9 encodes a protein that catalyzes histone H3K4 trimethylation, which is associated with gene activation and is induced in multiple cancers (35). Therefore, like NNMT, increased PRDM9 can mediate indirect effects of ASH1L on gene expression. MORC4 encodes a protein that is pro-oncogenic and antiapoptotic in breast cancer (36). TRIB3 encodes a protein that is overexpressed in several cancers, including colon cancer, where it promotes disease progression through interaction with beta-catenin and TCF4 (37). IL6 is overexpressed in a subset of ATCs (38) and encodes an important protumorigenic cytokine in the tumor microenvironment (39).
Table 2.
Selected genes with pro-oncogenic associations that are down-regulated in BHT-101 ASH1L KO cellsa
| Gene symbol | Gene name | FDR | Rank by FDR | Log2 FC | Rank by fold repression |
|---|---|---|---|---|---|
| HAPL1 | Hyaluronan and proteoglycan link protein 1 | 1.76 × 10−10 | 5 | −5.08 | 5 |
| MORC4 | MORC family CW-type zinc finger 4 | 7.18 × 10−8 | 7 | −2.14 | 23 |
| CCAT1 | Colon cancer associated transcript 1 | 7.52 × 10−5 | 11 | −6.10 | 2 |
| NNMT | Nicotinamide N-methyltransferase | 8.76 × 10−5 | 12 | −2.88 | 14 |
| TWSG1 | Twisted gastrulation BMP signaling modulator 1 | 2.48 × 10−4 | 14 | −1.60 | 42 |
| TRIB3 | Tribbles pseudo-kinase 3 | 3.60 × 10−4 | 15 | −1.66 | 39 |
| IL6 | Interleukin 6 | 0.0013 | 22 | −1.87 | 31 |
| PRDM9 | PR/SET domain 9 | 0.0069 | 35 | −4.37 | 8 |
a Genes are sorted by FDR.
ASH1L regulates gene sets associated with oncogenesis
Functional enrichment testing of the RNA-Seq data comparing the 4 ASH1L KO cells lines to the WT cells identified 85 Gene Ontology (GO) biological processes and KEGG pathways, as well as 47 previously described custom cancer-related gene sets from MSigDB (40), with FDR of <0.05 (Table S2). Many of the GO terms and KEGG pathways are involved in cancer-related processes such as cell death, extracellular-matrix organization, cell-cell adhesion, glycolysis, fatty acid metabolism, and nucleic acid metabolism; a subset of these is shown in Fig. 4. In addition, the custom cancer-related gene sets link ASH1L to TP53 function and RAF signaling (Fig. 4), both of which are potentially important, since ATCs commonly have TP53 mutations and increased mitogen-activated protein kinase (MAPK) activity, even if BRAF is WT (7) (BHT-101 cells harbor TP53 I251T and BRAF V600E mutations).
Figure 4.

Examples of gene sets enriched in a comparison of gene expression in BHT-101 ASH1L KO cells versus WT cells.
CCAT1 is a downstream mediator of ASH1L function in ATC
As noted previously (Table 1), the lncRNA CCAT1 was the second most highly down-regulated transcript in the BHT-101 ASH1L KO cells, 68-fold repressed (FDR = 7.52 × 10−5), indicating that it is strongly induced (directly or indirectly) by ASH1L. CCAT1 is overexpressed in numerous cancers, in which it induces cell proliferation, migration, and invasion (29). For these reasons, we investigated CCAT1 as a potential mediator of ASH1L function in ATC. In primary ATC tumors, the expression level of CCAT1 correlates with ASH1L protein abundance (Fig. 5A) (R2 = 0.75; p = 0.026), as would be expected if ASH1L induces CCAT1 expression in this disease.
Figure 5.

CCAT1 is an ASH1L target gene and a mediator of ASH1L function. A, ASH1L protein expression normalized to lamin B1 (data from Fig. 1) was compared with CCAT1 lncRNA expression in the same tumors, measured by RT-qPCR and normalized to Pgk1. The expression of CCAT1 correlates with that of ASH1L protein (R2 = 0.75; p = 0.026). B, two independent shRNAs were used to knock down CCAT1 expression in BHT-101 cells, compared with shControl cells. CCAT1 lncRNA levels were determined by RT-qPCR, normalized to B2M, and expressed relative to the shControl value, which was set to 1. The bars indicate means ± S.D. (n = 3). Both shCCAT1-1 and -2 differ significantly from shControl (p < 0.0001; Dunnett's test). C, BHT-101 cells with CCAT1 knockdown grow more slowly than shControl cells, assessed by MTT assay. Each data point represents the means ± S.D. for sextuplicate wells. Both shCCAT1 cell lines differ significantly from shControl at days 3 and 4 (p < 0.0001, Dunnett's test). D, CCAT1 is a direct target gene of ASH1L. ChIP-Seq was performed on BHT-101 WT cells and the ASH1L KO clones 1 and 4 using an antibody for H3K36me2, the histone mark catalyzed by ASH1L. In 3 separate experiments, a portion of the CCAT1 gene (marked by blue bars) is dimethylated at H3K36 in the WT cells (WT), but the H3K36me2 peaks are lost in all 3 experiments in the KO#1 cell line and in 2 of 3 experiments in the KO#4 cell line (KO#4 experiment 3 peaks are indicated by pink bars). Peaks were called using MACS2 (FDR < 0.05).
We used two independent shRNAs to knock down CCAT1 in BHT-101 cells. As determined by RT-qPCR, shCCAT1-1 and shCCAT1-2 decreased CCAT1 lncRNA to 11 and 23% of its level in shControl cells, respectively (Fig. 5B). Notably, shCCAT1-1 markedly decreased cell growth and shCCAT1-2 had a modest effect (Fig. 5C), consistent with the relative levels of knockdown (both shCCAT1 cell lines differ significantly from shControl at days 3 and 4; p < 0.0001, Dunnett's test). These data suggest that CCAT1 mediates at least part of the cell growth-promoting effect of ASH1L.
We used ChIP-Seq to test the hypothesis that CCAT1 is a direct ASH1L target gene. Since the ASH1L antibody is not of ChIP quality, we performed ChIP with an antibody to H3K36me2, the histone mark catalyzed by ASH1L. As expected, in three separate experiments, the CCAT1 gene in WT BHT-101 cells showed high H3K36 dimethylation, especially in the intron (Fig. 5D). In contrast, CCAT1 was devoid of H3K36me2 peaks in all three experiments using ASH1L KO clone 1 cells, and in two of the three experiments using KO clone 4 cells (Fig. 5D). It is not known why H3K36 dimethylation persisted in one of the three KO clone 4 experiments; however, this KO clone displayed the least-reduced growth rate (Fig. 2) and the ChIP-Seq result could reflect compensation by unknown mechanisms.
ASH1L KO causes extensive loss of H3K36 dimethylation
Given that multiple enzymes are capable of catalyzing H3K36 dimethylation, we hypothesized that ASH1L KO would result in less than global loss of H3K36me2 across the BHT-101 cell epigenome. This was confirmed by analysis of the H3K36me2 ChIP-Seq data, which showed that the WT cells contain 80,542 peaks, 48,417 (60%) of which are retained in the ASH1L KO cells and 32,125 (40%) of which are lost. Interestingly, 8942 peaks were present only in the KO cells, likely reflecting secondary changes in the activities of other histone methyltransferases. Thus, there is a net loss of ∼23,000 H3K36me2 peaks in the ASH1L KO cells.
To investigate H3K36me2 peak changes in the full set of differentially expressed genes shown in Fig. 3 and Table S1, we identified peaks present only in the WT group, not in the KO clone groups, and annotated those peaks to known genes. Of 32,125 differential H3K36me2 peaks which were present only in the WT group, 25,005 (∼78%) were annotated to 6,831 genes, and 129 of them were annotated to 39 differentially expressed genes (Table S3). More specifically, 24 of 53 (45%) genes down-regulated by ASH1L KO contained peaks unique to the WT cells, compared with only 15 of 103 (15%) up-regulated genes (p < 0.0001, 2-tailed Fisher's exact test). This difference is consistent with the fact that ASH1L is generally associated with the induction of gene expression.
miR-200b regulates ASH1L protein expression in ATC
Published data provide evidence that ASH1L is highly expressed at the protein level, but not at the mRNA level, in FTC, implying that ASH1L protein abundance is regulated posttranslationally (25). We found similar results in primary ATC tumors, in which there is no significant correlation between ASH1L protein abundance and mRNA expression (Fig. 6A). Since miRNAs can inhibit the translation of mRNAs, we examined whether the high level of expression of ASH1L protein in ATC might be explained by reduced expression of a miRNA. Several miRNAs have reduced expression in ATC compared with PTC and FTC (reviewed in Ref. 11). One of these, miR-200b-3p, has putative binding sites in the 3′ UTR of ASH1L mRNA at nucleotides (nt) 10549–10555 and 12010–12015 (NM_018489.3) as predicted by the TargetScan (RRID:SCR_010845) (41) and miRDB (RRID:SCR_010848) (42) web tools. Consistent with published data (26, 27), we confirmed that miR-200b-3p expression is low in primary ATCs compared with that in PTCs (Fig. 6B). We then used lentivirus to overexpress miR-200b in BHT-101 cells and JEM493 cells (Fig. 6C) and found that this decreased ASH1L protein abundance by ∼50% (Fig. 6D) but had no effect on ASH1L mRNA expression (Fig. 6E). These results support the hypothesis that the low level of miR-200b-3p in ATC compared with PTC and FTC likely underlies the greater abundance of ASH1L protein in ATC.
Figure 6.

ASH1L protein abundance is regulated by miR-200b-3p. A, ASH1L protein abundance in primary ATC tumors (data from Fig. 1) was compared with ASH1L mRNA levels in the same tumors, assessed by RT-qPCR, and normalized to Pgk1. ASH1L protein and mRNA expression levels do not correlate significantly in these tumors (p = 0.56). B, expression of miR-200b-3p is reduced in primary ATCs compared with PTCs (p = 0.0087, Mann Whitney test). Expression levels in the ATCs and PTCs from Fig. 1 were measured by RT-qPCR and normalized to RNU38B. C, lentivirus was used to overexpress miR-200b in BHT-101 cells and JEM493 cells. miRNA levels were measured by RT-qPCR, normalized to RNU38B and relative to control cells set to 1. The bars represent means ± S.D. (n = 3). D, overexpression of miR-200b results in decreased ASH1L protein abundance in BHT-101 and JEM493 cells, assessed by Western blotting. Lamin B1 was a loading control. The ASH1L band is 300 kDa, and lamin B1 is 66 kDa. Quantitation of the ASH1L/lamin B1 band ratios is shown below the Western blots, relative to the control cells set to 1. E, overexpression of miR-200b does not decrease ASH1L mRNA expression in BHT-101 or JEM493 cells. ASH1L mRNA expression was measured by RT-qPCR, normalized to B2M, and expressed relative to the value for control cells, which was set to 1. The bars represent means ± S.D. (n = 3).
Discussion
ATC is one of the most aggressive and least treatable human malignancies, with an average life expectancy of only ∼6 months from the time of diagnosis (2). This contrasts with the much more common differentiated thyroid cancers (PTC and FTC), which are usually curable with surgery and radioiodine. ATCs are undifferentiated tumors that do not express classical thyroid markers such as the TSH receptor, sodium iodide symporter, and thyroglobulin. Oncogenic mutations that underlie differentiated thyroid cancers such as BRAF V600E also often occur in ATCs, but the accumulation of additional genetic and epigenetic changes leads to much more aggressive behavior. A more complete understanding of these changes is necessary to identify novel approaches to therapy.
ASH1L is a histone methyltransferase that dimethylates H3K36, an epigenetic mark that tends to occur along gene bodies and is associated with gene activation (14, 15, 43, 44). Recent evidence has implicated ASH1L in the pathogenesis of a subset of leukemias (20, 21). ASH1L also has been shown to be overexpressed in breast and hepatocellular carcinomas, although whether it plays a role in the biology of these tumors is uncertain (22, 24). Of particular interest, ASH1L has been reported to be overexpressed at the protein level, but not the mRNA level, in FTC compared with PTC (25), suggesting a potential link between this histone methyltransferase and a subset of thyroid cancers. An unexpected aspect of the findings in FTC is that the ASH1L protein was detected by Western blotting as smaller-than-expected bands that were cytoplasmic.
Here, we show that ASH1L protein is overexpressed in the nuclei of primary ATCs compared with PTCs. The genetic loss of ASH1L in ATC cell lines results in decreased growth rates in culture. For these experiments, we used CRISPR-Cas9 to create premature stop codons at 4 different locations in the ASH1L sequence, all of which precede the catalytic SET domain. Since the ASH1L antibody recognizes an epitope at the carboxyl terminus of the protein, we are unable to determine if the truncated proteins are expressed to any significant degree. Even though all KO clones decrease cell growth, the magnitude of the effect varies. This may suggest that the truncated proteins are expressed and that they have distinct biological activities, a point that is worthy of further investigation to better understand ASH1L function. As shown in Fig. 2A, ASH1L is a large protein, and multiple regions may have noncatalytic roles in protein-protein interactions or other functions. In fact, recent evidence suggests that ASH1L may play an important noncatalytic role in hematopoiesis (45).
We found that ASH1L KO impairs the growth of mouse xenograft tumors much more profoundly than it impairs the growth of the same cells in culture. This might suggest that ASH1L has a proangiogenic effect, which would be relevant only in vivo. Alternatively, ASH1L might interact with the tumor microenvironment to enhance in vivo growth. For example, tumor-associated macrophages are felt to play an important role in supporting the aggressive behavior of ATCs (46, 47), and it is possible that ASH1L is involved in the recruitment of these cells.
When a very stringent definition of differentially expressed genes was used, analysis of RNA-Seq data identified 53 genes with decreased expression in ASH1L KO cells and 103 genes with increased expression, implying that ASH1L induces the expression of 53 genes and represses 103 in ATC. Since H3K36me2 activates gene expression, direct target genes of ASH1L will be in the set of 53 that are down-regulated in the ASH1L KO cells. This set includes numerous genes with known pro-oncogenic actions. Of particular note are PRDM9 and NNMT, both of which lead to epigenetic changes and therefore can mediate indirect effects of ASH1L on gene expression. In addition, the induction of IL6 could play a role in the recruitment of tumor-associated macrophages. Thus, because ASH1L is an epigenetic modifier, increased activity of this single enzyme has the potential to active numerous pathways that contribute to oncogenesis in diverse ways.
However, we focused on CCAT1 as a potentially important ASH1L target gene due to the magnitude of the decrease in CCAT1 lncRNA expression in ASH1L KO cells (68-fold repressed) and the numerous pro-oncogenic associations of CCAT1 (reviewed in Ref. 29). Supporting this potential role, we found that shRNA knockdown of CCAT1 decreases the growth rate of ATC cells. We also found evidence that CCAT1 is a direct ASH1L target gene, since CCAT1 is broadly dimethylated at H3K36 in BHT-101 WT cells, but this dimethylation is lost following ASH1L KO. This broad pattern of H3K36 dimethylation is similar to the ASH1L effect in leukemic cells (20).
In nonthyroid cancers, CCAT1 has been shown to act as a scaffolding molecule and/or to bind to and inhibit the activity of various miRNAs, leading to pro-oncogenic effects, including increased MAPK activity, MYC expression, and beta-catenin signaling (29, 48–50). These particular effects may be relevant to ATCs, which commonly have increased MAPK activity even when BRAF is WT (7), and also have been associated with increased MYC expression (51) or beta-catenin signaling (52). Although there are no previous reports of ASH1L functioning upstream of CCAT1, this pathway is worthy of further study in malignancies and in normal physiology.
Published data suggest that the abundance of ASH1L protein in FTC is primarily regulated by posttranslational processes, rather than by the level of the mRNA (25). We found a similar phenomenon in ATC, though the mechanism appears to be different. MiR-200b is strongly down-regulated in ATC relative to PTC and FTC (26, 27), and our data support the hypothesis that miR-200b-3p binds to the 3′ UTR of ASH1L and inhibits its translation. These results suggest that increasing the level of a miR-200b-3p mimic in ATC should decrease ASH1L protein and may be a novel therapeutic approach. Alternatively, inhibiting ASH1L methyltransferase activity, increasing the degradation of ASH1L protein, and decreasing CCAT1 activity also could be novel avenues of therapy.
Materials and methods
Human tumor samples
Primary human tumor samples were collected at the time of surgical thyroidectomy by the Tissue Procurement Service of the University of Michigan Department of Pathology and were stored at −80°C. The histology of each case was reviewed by Dr. Thomas Giordano. Institutional review board approval was obtained. This study abided by the Declaration of Helsinki principles.
Cell culture
The human ATC cell lines BHT-101 and SW1736 and the FTC cell lines FTC-133 and TT2609-C02 were obtained from Dr. Rebecca Schweppe (University of Colorado, Denver). BHT-101 cells were maintained in Dulbecco's modified Eagle medium (catalog no. 12430; Gibco, Gaithersburg, MD) plus 10% fetal bovine serum (FBS) (catalog no. F2442; Sigma-Aldrich, St. Louis, MO) and 1 mm sodium pyruvate. SW1736 cells were maintained in RPMI 1640 (catalog no. 11875; Gibco) plus 10% FBS. FTC-133 cells were maintained in DMEM–Ham's F-12 (catalog no. 11330; Gibco) plus 10% FBS. TT2609-C02 cells were maintained in RPMI 1640 plus 10% FBS, 1 mm sodium pyruvate, and 1× insulin-transferrin-selenium-ethanolamine (catalog no. 51500056; Thermo Fisher Scientific, Waltham, MA). The human ATC cell line JEM493 was a gift from Dr. J. A. Copland III (Mayo Clinic, Jacksonville, FL) and was maintained in RPMI 1640 plus 5% FBS, 1% nonessential amino acids, 1 mm sodium pyruvate, and 10 mm HEPES. The human FTC cell lines FTC-236 and FTC-238 were obtained from Dr. Orlo Clark (University of California, San Francisco) and were maintained in DMEM-Ham's F-12 plus 10% FBS, amphotericin B (250 ng/ml), bovine insulin (10 mg/ml; catalog no. 10516; Sigma-Aldrich), and bovine TSH (1 U/ml; catalog no. T-8931; Sigma-Aldrich). The human ATC cell line 8505C (Sigma-Aldrich) was maintained in Eagle minimal essential medium (catalog no. M7278; Sigma-Aldrich) plus 10% FBS and nonessential amino acids. All media contained penicillin-streptomycin, and all cell lines were incubated at 37°C in 5% CO2. The authenticity of all cell lines was confirmed by short-tandem-repeat profiling.
Cell lysis and immunoblotting
Total cellular extracts were prepared in HEPES buffer containing protease inhibitor mixture (catalog no. 11836170001; Sigma-Aldrich) as described previously (53). Cell lysates were then gently resuspended and incubated at 4°C with gentle rocking for 40 min to 1 h, followed by microcentrifugation at 13,000 rpm for 10 min at 4°C. The supernatants were transferred to new tubes, and protein concentrations were determined. Nuclear and cytoplasmic extracts were prepared using NE-PER (catalog no. 78833; Thermo Fisher Scientific). Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes, and immunoblotting was performed using antibodies to ASH1L (catalog no. A301-749A; Bethyl Laboratories, Montgomery, TX) and lamin B1 (catalog no. 12987-1AP; Proteintech, Rosemont, IL). The primary and secondary antibodies were diluted with signal enhancer HIKAR solution 1 and solution 2, respectively, as described previously (53). Detection by enhanced chemiluminescence was carried out with a SuperSignal West Dura kit (Thermo Fisher Scientific) and a Bio-Rad Fluor-S Max multi-imager.
RNA isolation, reverse transcription, and RT-qPCR
Total RNA was isolated with an RNeasy minikit (catalog no. 74104; Qiagen, Germantown, MD) according to the manufacturer's instructions. Four micrograms of total RNA was reverse transcribed using a SuperScript III first-strand synthesis system (catalog no. 18080051; Thermo Fisher Scientific), and RT-qPCR was performed as described previously (54). All primer sequences are listed in Table S4.
CRISPR-Cas9 KO of ASH1L
For KO of ASH1L by CRISPR-Cas9, four small guide RNA sequences targeting the human ASH1L gene (NCBI reference sequence NC_000001.1) were designed (sgRNA1, 81528:81547; sgRNA2, 82120:82139; sgRNA3, 192199:192218; and sgRNA4, 192317:192336; numbers indicate the location in ASH1L genomic DNA) via Deskgen web tools (55), followed by ligation into pX459 (catalog no. 62988; Addgene, Cambridge, MA) using BbsI restriction enzyme sites. Transfection of pX459-sgRNA was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Clonal cell lines were isolated, and ASH1L gene mutations were confirmed by Sanger sequencing of PCR-amplified genomic DNA. Deconvolution of the sequencing profiles was performed using the Tide web tool from Deskgen.
Cell proliferation assay
Measurement of the cell proliferation rate was performed by using an in vitro MTT assay according to the instructions of the manufacturer (catalog no. V13154; Invitrogen). Since the MTT assay is an indirect measure of cell number, cell proliferation also was assessed by direct cell counting after trypsinization and cell suspension, using a Luna-FL automated fluorescence cell counter with acridine orange/propidium iodide staining.
Thyroid tumor xenograft model
Five- to six-week-old NOD.CB17-Prkdcscid/J (NOD SCID; stock number 001303) female mice were purchased from the Jackson Laboratory, Bar Harbor, ME. Six mice per group were used to inject 5 × 106 shControl BHT-101 or shAsh1L cells in 50% Matrigel (catalog no. 354234; Corning Life Sciences, New York) subcutaneously into the flanks of the mice. Similarly, 5 × 106 WT BHT-101 cells or ASH1L KO cells mixed with 50% Matrigel were injected subcutaneously into the flanks of the mice. Tumor volumes were estimated weekly using digital calipers following the formula (L × W2), where L is length and W is width of the tumor. The University of Michigan Committee on the Use and Care of Animals approved all in vivo studies.
RNA-Seq assay
Total RNA was extracted using an RNeasy minikit (catalog no. 74104; Qiagen) with DNase 1 on-column digestion following the manufacturer's protocol. Samples for analysis included three independent cultures of BHT-101 cells and one culture each of 4 independent BHT-101 cell lines with CRISPR-Cas9-mediated deletion of ASH1L protein expression. Library construction (TruSeq RNA library preparation kit; Illumina, San Diego, CA) and sequencing on an Illumina NextSeq 500 using 75-nt single-end reads were done per the manufacturers' protocols, performed by the University of Michigan Advanced Genomics Core. The samples were barcoded and run in four lanes; an average of 75 million reads were sequenced per sample.
RNA-Seq data analysis
Quality control of raw RNA-Seq data was performed using FastQC (v0.11.5) (RRID:SCR_014583), RseQC (v2.6.4) (56), and MultiQC (v1.7) (57). The adapters were trimmed by TrimGalore (v0.4.1) (RRID:SCR_011847), and the trimmed reads were aligned to the GRCh38.p10 human genome (GRCh38_GencodeV26) (RRID:SCR_014966) using STAR (v2.5.3a) (58) with default parameters. Gene expression levels were quantified by HTseq (v0.11.2) (59) with the following options: “–mode=union,” “–stranded=reverse,” “-f bam,” “-a 30,” and “-t exon.” The differential expression analysis was conducted with the Bioconductor package edgeR (v3.20.9) (60) using “tagwise” and “robust” options for dispersion estimation and the likelihood ratio test “glmLRT” for the comparison between ASH1L KO and WT groups. FDR was controlled using the Benjamini-Hochberg method (61). The genes with FDR of <0.05 and absolute FC of >2 in the same direction across the four KO samples were selected as the differentially expressed genes.
Gene set enrichment testing
Gene set enrichment testing was performed using the RNA-Enrich functionality in LRpath (RRID:SCR_018572) (62) with the “directional test” option. The p values of genes from the differential expression analysis were modified by taking the concordance of the four KO samples into account, and the genes were ranked by the modified p values. The following criteria were applied to modify the raw p value for each gene: (i) if the gene showed consistent down- or up-regulation by at least an FC of 2 in all four KO samples (n = 4), the original p value was kept; (ii) if the gene showed consistent down- or up-regulation (FC > 2) in three KO samples (n = 3), the p value was multiplied by 1.5; (iii) if the gene showed consistent down- or up-regulation (FC > 2) in two KO samples (n = 2), the p value was multiplied by 2; (iv) if the gene was down- or up-regulated (FC > 2) in only one KO sample (n = 1), the p value was multiplied by 2.5; and (v) if the gene failed to show down- or up-regulation with an FC of >2 in any KO samples (n = 0), the p value was multiplied by 3. The genes with less concordance in the expression change across the four KO samples compared with controls were intended to be ranked down in the list. Gene IDs, modified p values, FC values, and average counts per million from the differential expression analysis were used as input in the RNA-Enrich testing. GO biological processes, KEGG pathways, and four categories of custom gene sets (cancer gene neighborhood, c4.cgn.v7.0.entrez.gmt.txt; oncogenic gene sets, c6.oncogenic.all.v7.0.entrez.gmt.txt; immunologic gene sets, c7.immunologic.all.v7.0.entrez.gmt.txt; and hallmark gene sets, hallmark.all.v7.0.entrez.gmt.txt) from MSigDB (40) were used for the test. Only gene sets with <1,000 genes were considered for this analysis, and those with FDR of <0.05 were considered significantly up- or down-regulated by ASH1L KO.
shRNA knockdown of ASH1L and CCAT1
Knockdown of ASH1L in ATC cell lines was performed by infection with a lentivirus expressing either of two shRNAs targeting ASH1L or an shControl. Two different Mission lentivirus-based plasmids of shRNAs (clone numbers TRCN0000016168 and TRCN0000016172) against human ASH1L and the shControl vector TRC2 pLKO.5-puro nonmammalian shRNA (SHC202) were obtained from Sigma-Aldrich. 293T cells were cotransfected with the shRNA and packaging plasmids psPAX2 and pMD2 by the calcium phosphate method to produce the lentivirus as described previously (54). shRNAs targeting human CCAT1 were Sigma Mission custom vectors in pLKO.5-puro, and TRC2 pLKO.5-puro nonmammalian shRNA (SHC202) was the shControl vector. The CCAT1 shRNA targeting sequences are shCCAT1-1 (CAGGAGGGTGCTTGACAATAA) and shCCAT1-2 (CCCAGCCACCCACAATTCTTT). The transfection of shCCAT1 and production of lentivirus in 293T cells were performed as described above.
ChIP-Seq assay
ChIP was performed using SimpleChIP kit no. 9003 (Cell Signaling Technology, Danvers, MA) following the manufacturer's protocol. Briefly, 3 × 107 cells were cross-linked with 1% formaldehyde at room temperature for 10 min, quenched with 0.125 M glycine, and resuspended in SDS lysis buffer. Chromatin fragments were obtained by partial digestion with micrococcal nuclease, followed by ChIP using H3K36me2 antibody catalog no. ab9049 from Abcam (Cambridge, MA). The ChIP and input DNA samples were used for next-generation library construction (ThruPLEX library preparation kit; Takara Bio, Mountain View, CA) and DNA sequencing on an Illumina HiSeq 4000 per the manufacturers' protocols using 50-nt single-end reads, performed by the University of Michigan Advanced Genomics Core. The WT and 4 KO samples from three biological replicate experiments were barcoded and run together in five lanes; an average of 68 million reads were sequenced per input sample, and 81 million reads per ChIP sample.
ChIP-Seq data analysis
Quality control of H3K36me2 ChIP-Seq data was performed using FastQC (v0.11.5) (RRID:SCR_014583) and MultiQC (v1.7) (57). The adapters were trimmed by TrimGalore (v0.4.1) (RRID:SCR_011847), and the trimmed reads were aligned to GRCh38.p10 human genome (GRCh38_GencodeV26) (RRID:SCR_014966) using bowtie2 (v2.2.9) (63) with default parameters. DeepTools (64) was used for a second round of quality control on the postalignment data to ensure that the libraries had high IP quality and only high-quality reads were used for this analysis. The uniquely mapped reads were extracted from the alignment files and used for peak calling. MACS2 was used to call H3K36me2 peaks with the following parameters: -f BAM -g hs –bdg –SPMR –keep-dup 1 –broad –broad-cutoff 0.05. The peaks with q values of <0.05 were considered significant. The identified peaks were annotated to known genes using the annotatr Bioconductor package (v1.4.1) (65). The BEDTools (v2.29.2) (66) intersect function was used to identify peaks unique to WT cells and absent from ASH1L KO cells. For data visualization, the read alignment bam files were converted to bigWig files by DeepTools using the “RPKM normalization” option and then imported into the IGV browser (v2.4.10) (67).
Forced expression of miR-200b
hsa-miR-200b (catalog no. HmiR0001-MR03; GeneCopoeia, Rockville, MD) in the lentiviral vector pEZX-MR03 and the scrambled control vector (catalog no. CmiR0001-MR03) were cotransfected with packaging plasmids psPAX2 and pMD2 in 293T cells, followed by infection of BHT-101 and JEM493 cells with the harvested lentivirus particles. Stably infected cells were selected with puromycin (1 µg/ml). For analysis of miRNA expression, RNA was isolated using a miRNeasy minikit (catalog no. 217004; Qiagen), followed by reverse transcription with a TaqMan miRNA reverse transcription kit (catalog no. 4366596; Thermo Fisher) and qPCR using a TaqMan miRNA assay (catalog no. A25576 for miR-200b-3p and no. 4427975 for RNU38B) as a normalization control for small RNA.
Data availability
The ChIP-Seq and RNA-Seq data have been deposited in Gene Expression Omnibus, accession numbers GSE147074 and GSE147076, respectively. Data presented as not shown are available upon request from Ronald Koenig (rkoenig@umich.edu) or Bin Xu (bxu@umich.edu), University of Michigan. All other data are presented in this article.
Supplementary Material
Acknowledgments
We thank Dr. Brian Emmer (University of Michigan) for advice and technical support in planning ASH1L gene editing and Michelle Vinco (University of Michigan) for preparation of human tumor samples.
This article contains supporting information.
Author contributions—B. X. and R. J. K. conceptualization; B. X., T. Q., and M. A. S. data curation; B. X., T. Q., M. A. S., and R. J. K. formal analysis; B. X., T. Q., J. Y., T. J. G., M. A. S., and R. J. K. investigation; B. X., T. Q., J. Y., M. A. S., and R. J. K. methodology; B. X., T. Q., M. A. S., and R. J. K. writing-original draft; B. X., T. Q., J. Y., T. J. G., M. A. S., and R. J. K. writing-review and editing; T. J. G. funding acquisition; R. J. K. supervision; R. J. K. validation; R. J. K. project administration.
Funding and additional information—Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA046592 (to T. J. G.) by the use of the Rogel Cancer Center Tissue and Molecular Pathology Shared Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ATC
- anaplastic thyroid cancer
- ASH1L
- ASH1 like histone lysine methyltransferase
- B2M
- beta-2 microglobulin
- CCAT1
- colon cancer associated transcript 1
- DTC
- differentiated thyroid cancer
- FBS
- fetal bovine serum
- FC
- fold change
- FDR
- false discovery rate
- FTC
- follicular thyroid cancer
- GO
- Gene Ontology
- gRNA
- guide RNA
- KO
- knockout
- lncRNA
- long noncoding RNA
- MAPK
- mitogen-activated protein kinase
- miRNA
- microRNA
- MTT
- (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide)
- nt
- nucleotide
- PTC
- papillary thyroid cancer
- RT-qPCR
- real-time quantitative PCR
- TSH
- thyroid-stimulating hormone.
References
- 1. Siegel R. L., Miller K. D., and Jemal A. (2020) Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2. Kebebew E., Greenspan F. S., Clark O. H., Woeber K. A., and McMillan A. (2005) Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer 103, 1330–1335 10.1002/cncr.20936 [DOI] [PubMed] [Google Scholar]
- 3. Kitamura Y., Shimizu K., Nagahama M., Sugino K., Ozaki O., Mimura T., Ito K., Ito K., and Tanaka S. (1999) Immediate causes of death in thyroid carcinoma: clinicopathological analysis of 161 fatal cases. J. Clin. Endocrinol. Metab. 84, 4043–4049 10.1210/jcem.84.11.6115 [DOI] [PubMed] [Google Scholar]
- 4. Nikiforova M. N., Kimura E. T., Gandhi M., Biddinger P. W., Knauf J. A., Basolo F., Zhu Z., Giannini R., Salvatore G., Fusco A., Santoro M., Fagin J. A., and Nikiforov Y. E. (2003) BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 88, 5399–5404 10.1210/jc.2003-030838 [DOI] [PubMed] [Google Scholar]
- 5. Bonhomme B., Godbert Y., Perot G., Al Ghuzlan A., Bardet S., Belleannee G., Crinière L., Do Cao C., Fouilloux G., Guyetant S., Kelly A., Leboulleux S., Buffet C., Leteurtre E., Michels J. J., et al. (2017) Molecular pathology of anaplastic thyroid carcinomas: a retrospective study of 144 cases. Thyroid 27, 682–692 10.1089/thy.2016.0254 [DOI] [PubMed] [Google Scholar]
- 6. Kunstman J. W., Juhlin C. C., Goh G., Brown T. C., Stenman A., Healy J. M., Rubinstein J. C., Choi M., Kiss N., Nelson-Williams C., Mane S., Rimm D. L., Prasad M. L., Höög A., Zedenius J., et al. (2015) Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet. 24, 2318–2329 10.1093/hmg/ddu749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Landa I., Ibrahimpasic T., Boucai L., Sinha R., Knauf J. A., Shah R. H., Dogan S., Ricarte-Filho J. C., Krishnamoorthy G. P., Xu B., Schultz N., Berger M. F., Sander C., Taylor B. S., Ghossein R., et al. (2016) Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Invest. 126, 1052–1066 10.1172/JCI85271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molinaro E., Romei C., Biagini A., Sabini E., Agate L., Mazzeo S., Materazzi G., Sellari-Franceschini S., Ribechini A., Torregrossa L., Basolo F., Vitti P., and Elisei R. (2017) Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 13, 644–660 10.1038/nrendo.2017.76 [DOI] [PubMed] [Google Scholar]
- 9. Donghi R., Longoni A., Pilotti S., Michieli P., Della Porta G., and Pierotti M. A. (1993) Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Invest. 91, 1753–1760 10.1172/JCI116385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakamura T., Yana I., Kobayashi T., Shin E., Karakawa K., Fujita S., Miya A., Mori T., Nishisho I., and Takai S. (1992) p53 gene mutations associated with anaplastic transformation of human thyroid carcinomas. Jpn. J. Cancer Res. 83, 1293–1298 10.1111/j.1349-7006.1992.tb02761.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pallante P., Battista S., Pierantoni G. M., and Fusco A. (2014) Deregulation of microRNA expression in thyroid neoplasias. Nat. Rev. Endocrinol. 10, 88–101 10.1038/nrendo.2013.223 [DOI] [PubMed] [Google Scholar]
- 12. Schuettengruber B., Martinez A. M., Iovino N., and Cavalli G. (2011) Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 12, 799–814 10.1038/nrm3230 [DOI] [PubMed] [Google Scholar]
- 13. Nakamura T., Blechman J., Tada S., Rozovskaia T., Itoyama T., Bullrich F., Mazo A., Croce C. M., Geiger B., and Canaani E. (2000) huASH1 protein, a putative transcription factor encoded by a human homologue of the Drosophila ash1 gene, localizes to both nuclei and cell-cell tight junctions. Proc. Natl. Acad. Sci. U. S. A. 97, 7284–7289 10.1073/pnas.97.13.7284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. An S., Yeo K. J., Jeon Y. H., and Song J. J. (2011) Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J. Biol. Chem. 286, 8369–8374 10.1074/jbc.M110.203380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanaka Y., Katagiri Z., Kawahashi K., Kioussis D., and Kitajima S. (2007) Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene 397, 161–168 10.1016/j.gene.2007.04.027 [DOI] [PubMed] [Google Scholar]
- 16. Bell O., Wirbelauer C., Hild M., Scharf A. N., Schwaiger M., MacAlpine D. M., Zilbermann F., van Leeuwen F., Bell S. P., Imhof A., Garza D., Peters A. H., and Schübeler D. (2007) Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 26, 4974–4984 10.1038/sj.emboj.7601926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stessman H. A., Xiong B., Coe B. P., Wang T., Hoekzema K., Fenckova M., Kvarnung M., Gerdts J., Trinh S., Cosemans N., Vives L., Lin J., Turner T. N., Santen G., Ruivenkamp C., et al. (2017) Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526 10.1038/ng.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shen W., Krautscheid P., Rutz A. M., Bayrak-Toydemir P., and Dugan S. L. (2019) De novo loss-of-function variants of ASH1L are associated with an emergent neurodevelopmental disorder. Eur. J. Med. Genet. 62, 55–60 10.1016/j.ejmg.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 19. Jones M., Chase J., Brinkmeier M., Xu J., Weinberg D. N., Schira J., Friedman A., Malek S., Grembecka J., Cierpicki T., Dou Y., Camper S. A., and Maillard I. (2015) Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells. J. Clin. Invest. 125, 2007–2020 10.1172/JCI78124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu L., Li Q., Wong S. H., Huang M., Klein B. J., Shen J., Ikenouye L., Onishi M., Schneidawind D., Buechele C., Hansen L., Duque-Afonso J., Zhu F., Martin G. M., Gozani O., et al. (2016) ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 6, 770–783 10.1158/2159-8290.CD-16-0058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trissal M. C., Wong T. N., Yao J. C., Ramaswamy R., Kuo I., Baty J., Sun Y., Jih G., Parikh N., Berrien-Elliott M. M., Fehniger T. A., Ley T. J., Maillard I., Reddy P. R., and Link D. C. (2018) MIR142 loss-of-function mutations derepress ASH1L to increase HOXA gene expression and promote leukemogenesis. Cancer Res. 78, 3510–3521 10.1158/0008-5472.CAN-17-3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu L., Kimball S., Liu H., Holowatyj A., and Yang Z. Q. (2015) Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 6, 2466–2482 10.18632/oncotarget.2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujimoto A., Furuta M., Totoki Y., Tsunoda T., Kato M., Shiraishi Y., Tanaka H., Taniguchi H., Kawakami Y., Ueno M., Gotoh K., Ariizumi S., Wardell C. P., Hayami S., Nakamura T., et al. (2016) Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509 10.1038/ng.3547 [DOI] [PubMed] [Google Scholar]
- 24. Skawran B., Steinemann D., Weigmann A., Flemming P., Becker T., Flik J., Kreipe H., Schlegelberger B., and Wilkens L. (2008) Gene expression profiling in hepatocellular carcinoma: upregulation of genes in amplified chromosome regions. Mod. Pathol. 21, 505–516 10.1038/modpathol.3800998 [DOI] [PubMed] [Google Scholar]
- 25. Colamaio M., Puca F., Ragozzino E., Gemei M., Decaussin-Petrucci M., Aiello C., Bastos A. U., Federico A., Chiappetta G., Del Vecchio L., Torregrossa L., Battista S., and Fusco A. (2015) miR-142-3p down-regulation contributes to thyroid follicular tumorigenesis by targeting ASH1L and MLL1. J. Clin. Endocrinol. Metab. 100, E59–E69 10.1210/jc.2014-2280 [DOI] [PubMed] [Google Scholar]
- 26. Braun J., Hoang-Vu C., Dralle H., and Hüttelmaier S. (2010) Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene 29, 4237–4244 10.1038/onc.2010.169 [DOI] [PubMed] [Google Scholar]
- 27. Hébrant A., Floor S., Saiselet M., Antoniou A., Desbuleux A., Snyers B., La C., de Saint Aubain N., Leteurtre E., Andry G., and Maenhaut C. (2014) miRNA expression in anaplastic thyroid carcinomas. PLoS One 9, e103871 10.1371/journal.pone.0103871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghafouri-Fard S., and Taheri M. (2019) Colon cancer-associated transcripts 1 and 2: roles and functions in human cancers. J. Cell. Physiol. 234, 14581–14600 10.1002/jcp.28176 [DOI] [PubMed] [Google Scholar]
- 29. Guo X., and Hua Y. (2017) CCAT1: an oncogenic long noncoding RNA in human cancers. J. Cancer Res. Clin. Oncol. 143, 555–562 10.1007/s00432-016-2268-3 [DOI] [PubMed] [Google Scholar]
- 30. Ivanova A. V., Goparaju C. M., Ivanov S. V., Nonaka D., Cruz C., Beck A., Lonardo F., Wali A., and Pass H. I. (2009) Protumorigenic role of HAPLN1 and its IgV domain in malignant pleural mesothelioma. Clin. Cancer Res. 15, 2602–2611 10.1158/1078-0432.CCR-08-2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu X. M., and Long H. (2018) Nicotinamide N-methyltransferase as a potential marker for cancer. Neoplasma 65, 656–663 10.4149/neo_2018_171024N680 [DOI] [PubMed] [Google Scholar]
- 32. Xia S., Ji R., Xu Y., Ni X., Dong Y., and Zhan W. (2017) Twisted gastrulation BMP signaling modulator 1 regulates papillary thyroid cancer cell motility and proliferation. J. Cancer 8, 2816–2827 10.7150/jca.18482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mebarki S., Désert R., Sulpice L., Sicard M., Desille M., Canal F., Dubois-Pot Schneider H., Bergeat D., Turlin B., Bellaud P., Lavergne E., Le Guével R., Corlu A., Perret C., Coulouarn C., Clément B., and Musso O. (2016) De novo HAPLN1 expression hallmarks Wnt-induced stem cell and fibrogenic networks leading to aggressive human hepatocellular carcinomas. Oncotarget 7, 39026–39043 10.18632/oncotarget.9346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ulanovskaya O. A., Zuhl A. M., and Cravatt B. F. (2013) NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 9, 300–306 10.1038/nchembio.1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feichtinger J., Aldeailej I., Anderson R., Almutairi M., Almatrafi A., Alsiwiehri N., Griffiths K., Stuart N., Wakeman J. A., Larcombe L., and McFarlane R. J. (2012) Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget 3, 843–853 10.18632/oncotarget.580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang Z., Zhuang Q., Hu G., and Geng S. (2019) MORC4 is a novel breast cancer oncogene regulated by miR-193b-3p. J. Cell. Biochem. 120, 4634–4643 10.1002/jcb.27751 [DOI] [PubMed] [Google Scholar]
- 37. Hua F., Shang S., Yang Y. W., Zhang H. Z., Xu T. L., Yu J. J., Zhou D. D., Cui B., Li K., Lv X. X., Zhang X. W., Liu S. S., Yu J. M., Wang F., Zhang C., Huang B., and Hu Z. W. (2019) TRIB3 interacts with beta-catenin and TCF4 to increase stem cell features of colorectal cancer stem cells and tumorigenesis. Gastroenterology 156, 708–721.e15 10.1053/j.gastro.2018.10.031 [DOI] [PubMed] [Google Scholar]
- 38. Sato T., Omura M., Saito J., Hirasawa A., Kakuta Y., Wakabayashi Y., and Nishikawa T. (2000) Neutrophilia associated with anaplastic carcinoma of the thyroid: production of macrophage colony-stimulating factor (M-CSF) and interleukin-6. Thyroid 10, 1113–1118 10.1089/thy.2000.10.1113 [DOI] [PubMed] [Google Scholar]
- 39. Kumari N., Dwarakanath B. S., Das A., and Bhatt A. N. (2016) Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 37, 11553–11572 10.1007/s13277-016-5098-7 [DOI] [PubMed] [Google Scholar]
- 40. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agarwal V., Bell G. W., Nam J. W., and Bartel D. P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y., and Wang X. (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, D127–D131 10.1093/nar/gkz757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gregory G. D., Vakoc C. R., Rozovskaia T., Zheng X., Patel S., Nakamura T., Canaani E., and Blobel G. A. (2007) Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol. Cell Biol. 27, 8466–8479 10.1128/MCB.00993-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan W., Xu M., Huang C., Liu N., Chen S., and Zhu B. (2011) H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 10.1074/jbc.M110.194027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jih G., Chase J., Zhou Y., Friedman A., Feng X., Nakayama M., Koseki H., Nishioka K., Huang G., and Maillard I. (2018) The trithorax-group protein ASH1L regulates hematopoietic stem cell homeostasis independently of its histone methyltransferase activity. Blood 132, 1270 10.1182/blood-2018-99-117367 [DOI] [Google Scholar]
- 46. Ryder M., Ghossein R. A., Ricarte-Filho J. C., Knauf J. A., and Fagin J. A. (2008) Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 15, 1069–1074 10.1677/ERC-08-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caillou B., Talbot M., Weyemi U., Pioche-Durieu C., Al Ghuzlan A., Bidart J. M., Chouaib S., Schlumberger M., and Dupuy C. (2011) Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS One 6, e22567 10.1371/journal.pone.0022567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tang T., Guo C., Xia T., Zhang R., Zen K., Pan Y., and Jin L. (2019) LncCCAT1 promotes breast cancer stem cell function through activating WNT/β-catenin signaling. Theranostics 9, 7384–7402 10.7150/thno.37892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang C., Chen F., Fan Z., Yao C., and Xiao L. (2019) lncRNA CCAT1/miR-490-3p/MAPK1/c-Myc positive feedback loop drives progression of acute myeloid leukemia. J. Biochem. 167, 379–388 10.1093/jb/mvz107 [DOI] [PubMed] [Google Scholar]
- 50. Chen S., Liu Y., Wang Y., and Xue Z. (2019) LncRNA CCAT1 promotes colorectal cancer tumorigenesis via a miR-181b-5p/TUSC3 axis. Onco Targets Ther. 12, 9215–9225 10.2147/OTT.S216718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Enomoto K., Zhu X., Park S., Zhao L., Zhu Y. J., Willingham M. C., Qi J., Copland J. A., Meltzer P., and Cheng S. Y. (2017) Targeting MYC as a therapeutic intervention for anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 102, 2268–2280 10.1210/jc.2016-3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garcia-Rostan G., Tallini G., Herrero A., D'Aquila T. G., Carcangiu M. L., and Rimm D. L. (1999) Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 59, 1811–1815 [PubMed] [Google Scholar]
- 53. Xu B., O'Donnell M., O'Donnell J., Yu J., Zhang Y., Sartor M. A., and Koenig R. J. (2016) Adipogenic differentiation of thyroid cancer cells through the Pax8-PPARγ fusion protein is regulated by thyroid transcription factor 1 (TTF-1). J. Biol. Chem. 291, 19274–19286 10.1074/jbc.M116.740324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu B., Gerin I., Miao H., Vu-Phan D., Johnson C. N., Xu R., Chen X. W., Cawthorn W. P., MacDougald O. A., and Koenig R. J. (2010) Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS One 5, e14199 10.1371/journal.pone.0014199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hough S. H., Ajetunmobi A., Brody L., Humphryes-Kirilov N., and Perello E. (2016) Desktop genetics. Per. Med. 13, 517–521 10.2217/pme-2016-0068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L., Wang S., and Li W. (2012) RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 10.1093/bioinformatics/bts356 [DOI] [PubMed] [Google Scholar]
- 57. Ewels P., Magnusson M., Lundin S., and Käller M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T. R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anders S., Pyl P. T., and Huber W. (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robinson M. D., McCarthy D. J., and Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 62. Lee C., Patil S., and Sartor M. A. (2016) RNA-Enrich: a cut-off free functional enrichment testing method for RNA-seq with improved detection power. Bioinformatics 32, 1100–1102 10.1093/bioinformatics/btv694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ramírez F., Dündar F., Diehl S., Grüning B. A., and Manke T. (2014) deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 10.1093/nar/gku365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cavalcante R. G., and Sartor M. A. (2017) annotatr: genomic regions in context. Bioinformatics 33, 2381–2383 10.1093/bioinformatics/btx183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Quinlan A. R., and Hall I. M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thorvaldsdottir H., Robinson J. T., and Mesirov J. P. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-Seq and RNA-Seq data have been deposited in Gene Expression Omnibus, accession numbers GSE147074 and GSE147076, respectively. Data presented as not shown are available upon request from Ronald Koenig (rkoenig@umich.edu) or Bin Xu (bxu@umich.edu), University of Michigan. All other data are presented in this article.