

Abstract
Mutations in the myotubularin 1 (MTM1) gene can cause the fatal disease X-linked centronuclear myopathy (XLCNM), but the underlying mechanism is incompletely understood. In this report, using an Mtm1−/y disease model, we found that expression of the intragenic microRNA miR-199a-1 is up-regulated along with that of its host gene, dynamin 2 (Dnm2), in XLCNM skeletal muscle. To assess the role of miR-199a-1 in XLCNM, we crossed miR-199a-1−/− with Mtm1−/y mice and found that the resultant miR-199a-1-Mtm1 double-knockout mice display markers of improved health, as evidenced by lifespans prolonged by 30% and improved muscle strength and histology. Mechanistic analyses showed that miR-199a-1 directly targets nonmuscle myosin IIA (NM IIA) expression and, hence, inhibits muscle postnatal development as well as muscle maturation. Further analysis revealed that increased expression and phosphorylation of signal transducer and activator of transcription 3 (STAT3) up-regulates Dnm2/miR-199a-1 expression in XLCNM muscle. Our results suggest that miR-199a-1 has a critical role in XLCNM pathology and imply that this microRNA could be targeted in therapies to manage XLCNM.
Keywords: skeletal muscle, microRNA (miRNA), muscle physiology, cell differentiation, STAT3, dynamin 2 (Dnm2), epigenetic regulation, miR-199a-1, myotubularin 1 (Mtm1), X-linked centronuclear myopathy (XLCNM)
Introduction
Centronuclear myopathies (CNMs) are a series of fatal muscle diseases with muscular pathology characterized by atrophic muscle fibers, disordered sarcomere organization, and a large number of centronuclear myofibers (1). Among these diseases, X-linked CNM is associated with the most severe form of myopathies, and most patients die in the first year after birth due to respiratory failure of muscle weakness (2). Although these diseases have long been characterized, no clinical treatment is available as yet (1).
CNMs are caused by mutations in several genes, including myotubularin 1 (MTM1), dynamin 2 (DNM2), bridging integrator-1 (BIN1), ryanodine receptor (RYR1), and titin (TTN). Among these genes, MTM1 serves as a lipid phosphatase to dephosphorylate phosphatidylinositol 3-phosphate and phosphatidylinositol 3,5-bisphosphate, which are essential for membrane formation/trafficking, endocytosis, and endosome formation (3, 4). DNM2 is a membrane-associated protein that regulates membrane scission, endocytosis, and cytoskeletal remodeling (5–7). DNM2 is also suggested to function in transverse tubule (T-tubule) membrane invaginations specialized for calcium handling (8, 9). Mutation of DNM2 causes relatively mild forms of myopathy featuring nuclear centralization, muscle atrophy, and deformed T-tubule (10, 11). BIN1, a negative regulator of DNM2, is involved in membrane recycling and T-tubule formation. Mutation of BIN1 results in abnormal Ca2+ release with aberrant triad formation (12–14). Intriguingly, mutation of RYR1, the calcium channel in the sarcoplasmic reticulum, is also involved in CNM myopathy (15). Abnormal excitation-contraction coupling seems to be a common event in CNM diseases (16). Despite several important advances to date, the mechanistic regulation of CNM pathology remains to be determined.
The DNM2 protein is encoded by the 114-kb DNM2 gene, which contains 22 exons, and four intragenic miRNAs are located in different introns (17). DNM2 is ubiquitously expressed in different tissues under physiological conditions (18). In CNM, normal DNM2 protein is up-regulated in diseased muscles (19, 20). Overexpression of WT DNM2 causes CNM phenotypes (8, 21), such as centralized nuclei, muscle atrophy, and deformed T-tubules, whereas down-regulation of DNM2 expression inhibits CNM (19, 20). There are reports that down-regulated GTPase of DNM2 could protect against CNMs (22, 23). Alternatively, as the intragenic miRNAs of DNM2 are transcribed along with its host gene and regulated related biology process (24, 25), we proposed that miR-199a, a uniquely conserved intragenic miRNA of Dnm2 among species, is central to the myopathy process.
To address the model that miR-199a is a contributor to XLCNM, we used a mouse model with deletion of the Mtm1 gene (Mtm1−/y) as an XLCNM model and assessed the role of the intragenic miRNA miR-199a-1 in myopathy. We found that Dnm2 mRNA and miR-199a-1 were simultaneously elevated in Mtm1−/y skeletal muscle. Deletion of miR-199a-1 is suggested to attenuate the severity of myopathy in Mtm1−/y mice, as evidenced by increased muscle mass, improved muscle strength, and prolonged animal lifespan. Our analyses suggest that miR-199a-1 targeted nonmuscle myosin IIA (NM IIA) and thereby inhibited muscular postnatal development as well as muscle maturation. In addition, we found that the overexpression of DNM2/miR-199a-1 was regulated by ectopic Stat3 activation in XLCNM muscle. Our study revealed an important role for an intragenic microRNA in XLCNM pathological progression.
Results
Expression of Dnm2 and intragenic miRNAs are elevated in Mtm1−/y skeletal muscle
Using CRISPR technology, we established two mouse lines with a 5-bp (Mtm1Δ5/y) and 7-bp (Mtm1Δ7/y) deletion within the Mtm1 gene (Fig. 1A). Note that Mtm1 is encoded in the X chromosome and the Y chromosome does not encode a Mtm1 allele, and the mutant male mice were referred to as Mtm1−/y. These mutant mice showed a retarded body growth and smaller muscle sizes (Fig. 1, B and C), which were comparable with those reported previously (26). We used these lines as a XLCNM disease model.
Figure 1.

Overexpression of Dnm2/miR-199a-1 is present in XLCNM mice. A, truncated genomic sequence and protein of Mtm1 surrounding the targeted exon 3. The red marker reflects the mutant region, and the blue letters show the premature stop codon. aa, amino acid. B, comparisons of body and muscle size between 5-week-old Mtm1Δ5/y and WT mice. C, comparisons of body and muscle size between 6-week-old Mtm1Δ7/y and WT mice. D, Western blotting for Mtm1 and Dnm2 in lysates prepared from 3-week-old WT or Mtm1Δ5/y or Mtm1Δ7/y tibialis anterior (n = 4). E, the presence of intragenic miR-199a in the Dnm2 gene was conserved among different species. Hsa, human; Mml, Rhesus monkey; Ptr, chimpanzee; Ssc, pig; Bta, cattle; Cfa, dog; Mmu, mouse. F and G, quantitative PCR to detect the expression of Dnm2 mRNA and miR-199a-1 in D (n = 3–4). Graphs represent mean ± S.D. (error bars). *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
The Western blotting assay showed a higher level of Dnm2 in Mtm1−/y tibialis anterior (TA) muscle in contrast to the control (Fig. 1D). As miR-199a is the intragenic microRNA of the Dnm2 gene and evolutionarily conserved across species (Fig. 1E), we measured miR-199a-1 expression level in the mutant TA muscles. Interestingly, the miR-199a-1 level was also significantly elevated (Fig. 1, F and G). These results indicated a simultaneous elevation of Dnm2 and miR-199a-1 expression in the myopathy muscle.
Ablation of miR-199a-1 improves the myopathy of Mtm1−/y mice
To assess the role of miR-199a-1 in XLCNM pathogenesis, we first established miR-199a-1 deletion lines of mice (Fig. 2A). The birth of mutant pups followed an expected Mendelian ratio. The mice were fertile and reached adulthood without any obvious abnormalities (Fig. 2, B–D). Western blotting assays showed no apparent alteration of Dnm2 protein expression after miR-199a-1 deletion (Fig. 2, E and F).
Figure 2.

miR-199a-1 KO mice do not exhibit a visible phenotypic defect. A, miR-199a-1 KO mice were generated with CRISPR-Cas9 technologies. The red and blue marker indicate the mutant region of miR-199a-1 and “seed sequence” of mature miR-199a-5p, respectively. B, whole-body weight of miR-199a-1 KO mice (n = 18–31). C, relative muscle weight of miR-199a-1 KO mice (n = 7–10). SOL, soleus. D, characteristic muscle biopsy from miR-199a-1 KO mice. Scale bar, 20 nm. Distribution of myofibers was grouped by cross-sectional area (n = 3–5). E and F, Western blotting measurements of Dnm2 protein in muscle lysates of 8-week-old miR-199a-1 KO mice. Protein levels relative to Gapdh were determined (n = 4–7). G, real-time PCR to detect miR-199a-5p level in TA muscle of the indicated 6-week-old mice (n = 6–8). H, Western blot analysis to measure Dnm2 protein in 6-week-old mice, with genotypes indicated. Protein levels relative to Gapdh were calculated (n = 4). Graphs represent mean ± S.D. (error bars). NS, no significant difference (two-tailed Student's t test).
We then crossed Mtm1−/y mice with miR-199a-1 knockout mice and obtained offspring with four genotypes: Mtm1Δ5/y; miR-199a-1I8/+ (Hetero-1), Mtm1Δ7/y; miR-199a-1Δ3/+ (Hetero-2), Mtm1Δ5/y; miR-199a-1I8/I8 (Homo-1), and Mtm1Δ7/y; miR-199a-1Δ3/Δ3 (Homo-2). Real-time PCR showed a significant reduction of miR-199a-5p RNA expression, which is the processed product of miR-199a-1 transcription in the muscle of Homo-1 mice and a moderate inhibition for Hetero-1 muscle, compared with Mtm1Δ5/y muscle (Fig. 2G). Western blotting showed a similar level of Dnm2 protein in the muscle of miR-199a-1I8/I8 and Homo-1 mice (Fig. 2H). Most Mtm1Δ5/y mice died ∼6–7 weeks after birth, and the average survival time was 43 ± 1.5 days (Fig. 3A). Surprisingly, most Homo-1 mice died ∼8–9 weeks after birth. The average survival time (57 ± 1.5 days) was significantly longer (∼30%) than that of Mtm1Δ5/y mice (p < 0.01). The Hetero-1 mice showed a moderately but significantly longer survival time than Mtm1Δ5/y (50 ± 2.1 days versus 43 ± 1.5 days, p < 0.05). This protective effect was also measured in Hetero-2 and Homo-2 mice (Fig. S1A). Thus, we concluded that reduction of intragenic miR-199a-1 expression inhibited the lethal phenotype of Mtm1−/y mice.
Figure 3.

Reduced miR-199a-1 expression partially improves pathology in XLCNM mice. A, lifespan curves of the indicated genotype mice (n = 17–30). B, whole-body weights of the indicated genotype mice were monitored from 3 to 8 weeks of age (n = 13–23). Results of WT mice are also presented in Fig. 2B. C, the relative muscle weight (muscle mass/body weight) of these mice was also measured (n = 5–8). The data of WT mice are also presented in Fig. 2C. D, muscle pathological sections of the indicated mice. Scale bar, 20 nm. E, the frequency of myofibers with central nuclei (n = 5). F, the myofiber ratio of the indicated genotype mice was grouped according to fiber area (n = 5). G, typical EDL contraction curve induced by electric stimulus. H, quantification of maximal forces in G (n = 4–7). All mice were sacrificed at 6 weeks of age. SOL, soleus. Graphs represent mean ± S.D. (error bars). NS, no significant difference; *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
Although the body weights among Mtm1−/y, Hetero-1/2 and Homo-1/2 mice showed no apparent difference (Fig. 3B and Fig. S1B), the ratio values of muscle mass/body weight of Homo-1 and Homo-2 mice were significantly larger than the controls (Fig. 3C and Fig. S1C). The ratio for the extensor digitorum longus (EDL) was even comparable with the WT muscle (0.036 ± 0.002% (WT) versus 0.035 ± 0.003% (Homo-1) versus 0.032 ± 0.002% (Homo-2), p > 0.05). Histological examination showed that the Homo-1/2 muscles had a decreased percentage of myofibers with central nuclei (10.95 ± 1.63% (Mtm1Δ5/y) versus 6.08 ± 0.95% (Homo-1), p < 0.05; 11.44 ± 1.42% (Mtm1Δ7/y) versus 6.27 ± 0.84% (Homo-2), p < 0.05) and more myofibers with larger area (Fig. 3 (D–F) and Fig. S1 (D–F)). These results indicated that miR-199a-1 deletion improved the pathological feature of XLCNM, particularly in the fast-twitch muscles.
We next measured the contractile response of the mutant EDL to electric stimulation (27). Mtm1Δ5/y muscle displayed a typical contraction action, but the maximal force was significantly less than that of WT muscle (3.44 ± 0.47 g versus 0.45 ± 0.06 g, p < 0.01). The Homo-1 muscle, in contrast to the Mtm1Δ5/y muscle, showed a significant increase in maximal force tension (0.99 ± 0.16 g versus 0.45 ± 0.07 g, p < 0.01) (Fig. 3G). Because the muscle masses were different among these muscles, we normalized the values and found that the maximal force of Homo-1 muscle was significantly higher than that of the Mtm1Δ5/y muscle (1.95 ± 0.25 N/cm2 versus 1.20 ± 0.14 N/cm2, p < 0.05), but still much lower than that of the WT control (1.95 ± 0.25 N/cm2 versus 3.73 ± 0.33 N/cm2, p < 0.01) (Fig. 3H). The phenotype was similar in Mtm1Δ7/y and Homo-2 muscle (Fig. S1, G and H). This result showed a functional improvement of the muscle after the deletion of miR-199a-1 in Mtm1−/y mice.
miR-199a-1 inhibits myoblast fusion by targeting nonmuscle myosin IIA
Whereas XLCNM muscle has no apparent defect of myogenesis during embryonic development (26), other reports suggested developmental defects for this muscle (28). We isolated single myofiber from 3-week-old Mtm1−/y mice and observed fewer nuclei in the muscle also (Fig. 4 (A and B) and Fig. S2 (A and B)). Measurement by Western blotting showed decreased expression of myosin II, a marker of myoblast fusion, in early postnatal muscle (Fig. 4 (C and D) and Fig. S2 (C and D)). This result indicated a defect of myoblast fusion in early postnatal Mtm1−/y muscle (29, 30).
Figure 4.
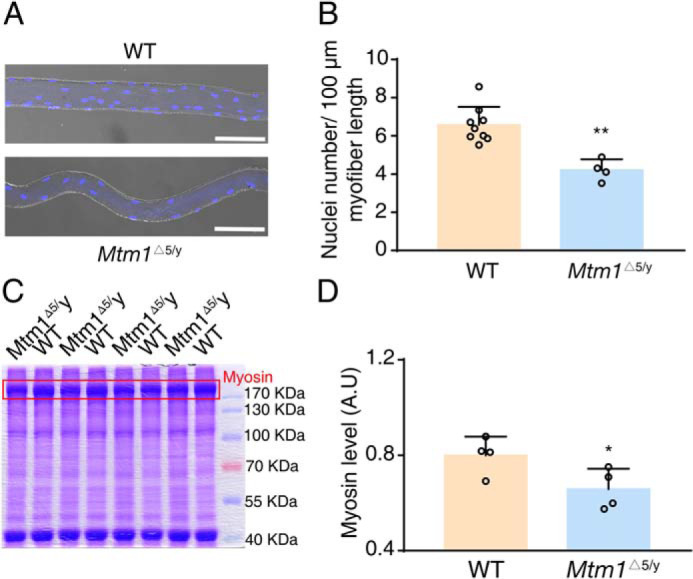
Deletion of Mtm1 inhibits myoblast fusion. A, immunofluorescence with 4′,6-diamidino-2-phenylindole staining for single myofiber from 3-week-old WT and Mtm1Δ5/y mice. Scale bar, 100 μm. B, quantification of nuclei number per 100-μm myofiber in A (n = 4–9). C, protein lysates from the TA muscle of 3-week-old mice were separated by an SDS-polyacrylamide gel and stained by Coomassie Brilliant Blue. The red box represents the myosin heavy chain band at ∼230 kDa. D, the expression of myosin in C was measured by gray level (n = 4). A.U., arbitrary units. Graphs represent mean ± S.D. (error bars). *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
To test whether miR-199a-1 participates in this fusion process, we first measured the dynamic expression of miR-199a-5p during the fusion of C2C12 myoblasts. Before induction, C2C12 myoblasts exhibited a lower level of miR-199a-5p, which peaked at 1 day after induction and sustained thereafter (Fig. 5A). We next examined the fusion ability of C2C12 myoblasts after treatment with an oligonucleotide mimic (199-M) or inhibitor (199-I) of miR-199a-5p (Fig. 5B). Upon transfection with 199-M, myogenic fusion was inhibited, as illustrated by the 50% reduction in Myog and MyHC protein expression levels. Transfection with 199-I resulted in a significant increase in MyHC expression and a slight enhancement of Myog expression (Fig. 5, C–E). Immunofluorescence analysis showed that 199-M transfection led to a significant decrease in Myog-positive cells (12.69 ± 3.18% (199-M) versus 23.66 ± 2.90% (199-C), p < 0.05) and multinuclear myotube area, whereas 199-I transfection led to an increase in Myog-positive cells (32.35 ± 2.37% (199-I) versus 23.66 ± 2.90% (199-C), p < 0.05) and a larger multinuclear myotube area (Fig. 5, F–H). These results suggested that miR-199a-1 expression during C2C12 differentiation was targeted during myoblast fusion.
Figure 5.

miR-199a-1 inhibits myoblast fusion. A, levels of miR-199a-5p were measured during myoblast differentiation. 5S rRNA were measured as controls (n = 3). B, levels of mature miR-199a-5p levels in C2C12 myoblasts treated with 199-M/I/C, respectively (n = 3). C–E, measurements for Myog and MyHC from lysates of differentiating C2C12 cells treated with 199-M/I/C, respectively. β-Actin was as internal controls (n = 10–11). F–H, immunofluorescence staining for Myog and MyHC in differentiating C2C12 cells treated with 199-M/I/C, respectively (n = 4–6). Scale bar, 200 μm. 199-M, miR-199a-5p mimic oligonucleotide; 199-I, miR-199a-5p inhibitor oligonucleotide; 199-C, scrambled oligonucleotide. Graphs represent mean ± S.D. (error bars). NS, no significant difference; *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
To identify the target gene of miR-199a-1, we used the bioinformatics tools TargetScan (RRID:SCR_010845), PicTar (RRID:SCR_003343), and DIANA TOOLS (RRID:SCR_018425) to predict candidate target genes. Given that deletion of miR-199a-1 led to improvement of myopathic muscle, the function of its target gene was expected to promote myoblast fusion or similar processes. Following this rationale, we identified four candidate target genes, MYH9, SULF1, RAD23B, and PPARGC1A, for miR-199a-5p. We transfected miR-199a-5p into C2C12 cells and measured the target proteins by Western blotting. Among these proteins, only NM IIA expression, the product of Myh9 translation, was inhibited by overexpression of miR-199a-5p (Fig. 6 (A and B) and Fig. S3A).
Figure 6.

NM IIA is the direct target of miR-199a-5p in muscle. A and B, measurements of NM IIA in C2C12 myoblasts treated with 199-M/I/C, respectively. Expression of NM IIA was normalized to that of β-actin (n = 4–5). C, the predicted structure of base-paired natural or mutant Myh9 3′-UTR/miR-199a-5p hybrid. The red and blue marker reflects the mutant region of Myh9 3′-UTR and “seed sequence” of mature miR-199a-5p, respectively. Hsa, human; Gga, chicken; Mmu, mouse. D, relative luciferase activity in cells transfected with the indicated vectors (n = 4–7). Blank, pGL3-Myh9-3′-UTR vector and blank expression vector; 199a-5p, pGL3-Myh9-3′-UTR vector and miR-199a-5p expression vector; Mut, pGL3-Myh9-3′-UTR-mut and miR-199a-5p expression vector. E and F, measurements of NM IIA and myogenic markers in C2C12 myoblasts treated with si-Myh9. Quantification was performed with β-actin as internal control (n = 3–5). G–I, immunofluorescence staining of Myog and MyHC in differentiated C2C12 cells treated with si-Myh9 (n = 6–10). Scale bar, 200 μm. J and K, regenerative TA muscles were separately injected with synthetic miR-199a-5p (Agomir), antisense oligonucleotides against miR-199a-5p (Antagomir), or randomized oligonucleotides (Scrambled). One week later, Western blotting was performed, and area distribution of regenerative myofibers was measured (n = 3). L, Western blotting with NM IIA antibody for lysates from 3-week-old TA muscle of the indicated genotype mice. Gapdh was as an internal control (n = 3). M, single myofiber was isolated from 6-week-old muscle and stained with sarcomere marker (α-actinin) and NM IIA. Scale bar, 5 μm. Graphs represent mean ± S.D. (error bars). NS, no significant difference; *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
To verify direct interactions of miR-199a-5p with Myh9 mRNA, we constructed luciferase reporters with the WT and mutant 3′-UTRs of Myh9 and the miR-199a-5p expression vector (Fig. 6C). Co-transfection showed that miR-199a-5p targeted the 982–988 region of the Myh9 3′-UTR (Fig. 6D). To further test whether Myh9 is the target of miR-199a-5p, we used a Myh9-specific siRNA (si-Myh9) to impede the expression of endogenous NM IIA in C2C12 myoblasts (∼25%; Fig. 6, E and F). Down-regulation of endogenous NM IIA caused a reduction of myogenic markers in Myh9 knockdown myotubes (Myog, ∼30%; MyHC, ∼10%) (Fig. 6, E and F). Immunofluorescence analysis also confirmed a fusion defect in si-Myh9 myoblasts, as illustrated by a decrease in Myog-positive cells (∼60%) and a smaller multinuclear myotube area (∼60%) (Fig. 6, G–I). In addition, the introduction of 199-agomir (miR-199a-5p analog) into regenerative muscle resulted in a striking reduction in NM IIA and Myog levels, along with smaller regenerative myofibers, whereas 199-antagomir (miR-199a-5p antagonist) led to the opposite effect (Fig. 6, J and K). Collectively, these results suggested that miR-199–5p inhibited myoblast fusion by directly targeting NM IIA.
To assess whether NM IIA was involved in the XLCNM, we first detected its expression in Mtm1−/y muscle. As we expected, a striking reduction in NM IIA was measured in these mice (Fig. 6L and Fig. S3B). As NM IIA participated in myofibrillogenesis that might reflect myotube fusion and muscle maturation (31), we examined the muscle fibers by staining for α-actinin. The WT fibers showed clear and well-organized sarcomeres, whereas Mtm1Δ5/y sarcomeres showed misalignment of Z-lines, as reported previously (19). NM IIA was abundantly co-localized with α-actinin in WT muscle, but its abundance was greatly decreased in the Mtm1Δ5/y sarcomeres. However, in Homo-1 sarcomeres, there appeared to be an improvement of the Z-line arrangement, together with an increase in the amount of NM IIA (Fig. 6M). These results indicated that the impaired maturation of the mutant muscles was attributable to the down-regulation of NM IIA by miR-199a-1.
Stat3 signaling contributes to the ectopic expression of the Dnm2 gene and miR-199a-1
To investigate the regulatory mechanism of overexpression of Dnm2/miR-199a-1 in XLCNM muscle, we analyzed the regulatory region of the Dnm2 gene with JASPAR (RRID:SCR_003030). We found conserved signal transducer and activator of transcription 3 (Stat3) binding sites upstream of the Dnm2 gene. Given the important role of Stat3 during myoblast fusion (32), we hypothesized that the abundant Stat3 cis-elements located within the region might contribute to the up-regulation of Dnm2/miR-199a-1 (Fig. 7A). To address the potential Stat3 regulation of the Dnm2/miR-199a-1 locus, we first measured Stat3 signaling in XLCNM muscle and found a significant elevation of the levels and phosphorylation of Stat3 (Pho-Stat3) compared with that of WT littermates (Fig. 7B). We then made a series of luciferase reporters containing Dnm2 promotor regions with either intact or mutant Stat3-binding sites (Fig. 7C). Upon transfecting HEK 293T cells with the reporters, we found that luciferase activity was significantly reduced in the Stat3-binding site mutant groups compared with the intact group (∼50%; Fig. 7C). Furthermore, we treated C2C12 myoblasts with Stat3 siRNA, measured the expression of Dnm2/miR-199a-1, and found that miR-199a-1 level was decreased by ∼50% with reduced Dnm2 expression (Fig. 7, D–F). Considering that phosphorylation is essential for Stat3 activation, we applied the specific inhibitor (5,15-DPP) of Pho-Stat3 to mice injured by BaCl2 and found a predicted reduction in Dnm2/miR-199a-1 level and an increase in NM IIA expression (Fig. 7, G–I). These results indicate that Stat3 activation drives Dnm2/miR-199a-1 expression in muscle.
Figure 7.

Overexpression of Stat3 in muscle activates Dnm2/miR-199a-1 signaling. A, schematics of Dnm2 promoter regulatory regions with predicted Stat3-binding sites. The number below the graph represents the binding site positions. B1/B2, binding site. TSS, transcriptional start site. B, Western blotting for Stat3 and phosphorylated Stat3 (Pho-Stat3) with muscle lysates of 3-week-old mice. C, top, schematic of the luciferase reporter constructs. Bottom, relative luciferase activity of cells transfected with Dnm2-WT/B1/B2 luciferase reporters, respectively (n = 6). D and E, Western blotting measurements for Stat3 and Dnm2 in C2C12 myoblasts treated with si-Stat3. Gapdh was used as a normalization control (n = 3). F, transcript levels of Dnm2 and miR-199a-1 in D (n = 4). G and H, Western blotting for protein with lysates from the acute injury model treated with 5,15-DPP. Gapdh was used as an internal control (n = 4). I, transcript levels of Dnm2 and miR-199a-1 in the samples from G (n = 3–5). Graphs represent mean ± S.D. (error bars). NS, no significant difference; *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
Collectively, aberrant activation of Stat3 in XLCNM muscle induced expression of the Dnm2 gene and intragenic miR-199a-1, which inhibited myoblast fusion and muscle maturation during early postnatal development via direct down-regulation of NM IIA (Fig. 8).
Figure 8.
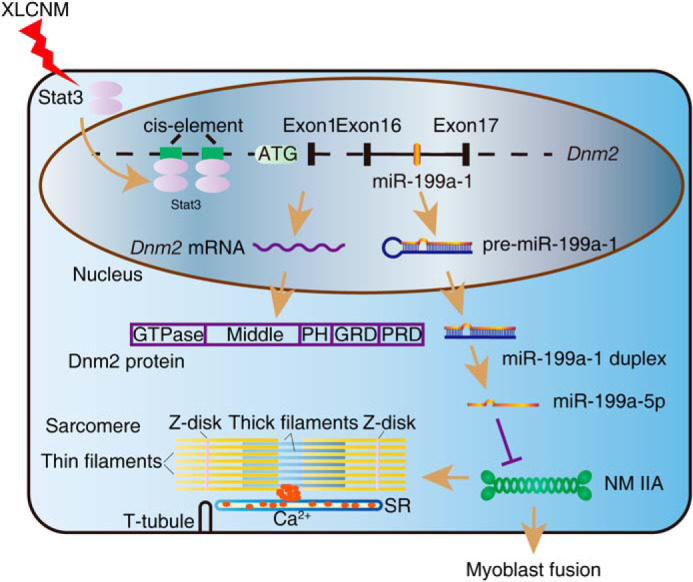
Up-regulated miR-199a-1 contributes to pathogenesis of XLCNM. During muscle development, abnormally activated Stat3 expression in XLCNM results in enhanced transcription of Dnm2 gene and intragenic miR-199a-1, which inhibits myoblast fusion and muscle maturity via regulation of NM IIA expression, leading to defect in myofiber area and muscle strength.
Discussion
Mutation of Mtm1 causes multiple skeletal muscle pathologies, including growth retardation, skeletal muscle wasting, and other severe complications (2). As observed in previous studies, dysfunction of the Mtm1 gene is the leading cause of XLCNM (33). In this report, we found that, during the pathological process, the DNM2 gene, along with its intragenic miR-199a-1, was secondarily up-regulated in XLCNM muscle. Importantly, inhibition of miR-199a-1 expression attenuated the severity of the myopathy and, hence, prolonged lifespan significantly. As miR-199a-1 was able to apparently inhibit the process of myoblast fusion both in vitro and in vivo, we suggested that, during the process of myopathy, the elevated miR-199a-1 functioned as an inhibitor of myoblast fusion and myofiber maturation. In other words, the myopathy phenotype of XLCNM is partially attributable to the ectopic expression of miR-199a-1. Based on the collective results, we suggest a working model for XLCNM pathological progression, in which the Mtm1 mutation causes activation of Stat3 signaling and hence promotes Dnm2/miR-199a-1 expression, which inhibits myoblast fusion and muscle maturation. It was reported that up-regulation of miR-199a was present in muscle of eight major muscular disorders (34). The pathogenesis of miR-199a may therefore extend to other myopathies or muscle injuries. It is noted that even in XLCNM, the severer form of myopathy mice, inhibition of miR-199a-1 is still able to prolong lifespan by ∼30% as reported here. This implies that targeting miR-199a-1 might shows more efficacy in the case of other myopathy with relative mild phenotypes.
miR-199a has been demonstrated to be involved in several biological processes (35–37) in which several genes are directly targeted. Based on our observations, nonmuscle myosin IIA serves as a new target of miR-199a-1 in the process of muscular development. During myofibrillogenesis, NM IIA is predominantly expressed at nascent premyofibrils and then disappears in mature myofibrils, showing a featured turnover of muscle and nonmuscle myosin molecules (31). As the reduction in NM IIA impaired sarcomere organization in XLCNM muscle, we suggest that the miR-199a-1–regulated NM IIA might function in muscle myosin turnover during myofibril maturation also. In addition, as the nonmuscle tissues are able to express NM IIA along with DNM2, it is reasonable to predict that the down-regulation of NM IIA by miR-199a-1 might also contribute to the pathology of nonmuscle tissues in XLCNM patients, who showed a high percentage of nonmuscular comorbidity with ear and kidney (2). The similar phenotypes observed in patients with NM IIA mutations also support this speculation (38, 39).
Recent studies propose that up-regulation of MTM1 protein or activity, which inhibits phosphatidylinositol 3-kinase accumulation and reduces DNM2 expression, is effective for XLCNM treatment in animals (40–45). Based on our results, combinations of these strategies with inhibition of miR-199a-1 would obtain a better therapeutic outcome. As STAT3 potently regulates miR-199a-1 and DNM2, application of STAT3 inhibitor, a drug that has been approved by the Food and Drug Administration for myelofibrosis (ruxolitinib), would be an optimal strategy for XLCNM therapy (45).
Experimental procedures
Antibodies
The primary antibodies used were anti-Myog (SC-576, Santa Cruz Biotechnology, Inc.), MyHC (MAB4470, R&D Systems), ACTB (β-actin; A5441, Sigma-Aldrich), Nonmuscle myosin IIA (NM IIA; catalog no. 3403, Cell Signaling Technology), STAT3 (catalog no. 9139, Cell Signaling Technology), phospho-STAT3 (catalog no. 9145, Cell Signaling Technology), ACTN2 (α-actinin; ab9465, Abcam), DNM2 (ab151555, Abcam), Gapdh (SC-32233, Santa Cruz Biotechnology), and MTM1 (ab1350, Abcam). Alexa Fluor–conjugated secondary antibodies were obtained from Invitrogen (Alexa Fluor 546, A-11003 and A-11010; Alexa Fluor 488, A-11008 and A-11001). Secondary antibodies conjugated with horseradish peroxidase were purchased from Thermo Fisher Scientific (catalog nos. 31460 and 31430).
Genetic mouse models
The Mtm1– and Mir199a-1–deficient mice were constructed with CRISPR-Cas9 technology, as described previously (46). Briefly, a sgRNA (5′-GTAACTCCCCTGGGAGCCGA-3′) targeting the third exon of Mtm1 was cloned into the pUC57-U6-sgRNA vector with the BbsI cleavage site. Then the sgRNA was transcribed and purified in vitro, whereas the Cas9 mRNA was purchased from the Nanjing Biomedical Research Institute of Nanjing University. Next, the sgRNA and Cas9 mRNA were co-injected into the pronucleus and cytoplasm of C57BL/6 zygotes, respectively. Then the manipulated embryos were implanted into the oviducts of pseudopregnant female mice to generate chimeras. Mice with a 5-/7-bp deletion of Mtm1 in the germline were backcrossed for at least 6 generations into the C57BL/6 background for subsequent research. The generation of Mir199a-1 KO mice was similar to that of Mtm1-deficient mice with an sgRNA for miR-199a-5p (GAACAGGTAGTCTGAACATC).
Intramuscular injection operation
The acute muscle injury model was generated by injection of BaCl2 (50 μl of 1.2% (w/v) in saline) into the TA of one hind limb as described previously (47); another TA with saline (50 μl) treatment was used as a control. For the miR-199a-5p oligonucleotide operation, the acute injury model was first induced in two hind limbs of adult C57BL/6 mice by BaCl2. On the second day, the regenerative muscle of one hind limb was injected with agomir or antagomir (1 nmol, RiboBio Ltd. Co., Guangzhou, China). Meanwhile, the introduction of Scrambled (1 nmol; RiboBio) into another regenerative TA was used as a control. The injections were consecutively performed at intervals of 1 day until the TA muscle was isolated and subjected to experimental measurements, within a week. In addition, 5,15-DPP (2.5 nmol; D4071, Sigma–Aldrich), a specific Stat3 inhibitor, was introduced into acutely injured TA in a similar manner as the miR-199a-5p oligonucleotides, and an equal volume of DMSO was used as a control.
Cell culture
All cell lines were purchased from MuCyte Ltd. Co. (Nanjing, China). C2C12 myoblasts and HEK 293T cells were maintained in Dulbecco's modified Eagle's medium (12100046, Gibco) supplemented with 10% fetal bovine serum (growth medium (GM)) at 37 °C and 5% CO2. Myoblasts were induced to differentiate after the cells reached ∼90% confluence by replacing the GM with Dulbecco's modified Eagle's medium containing 2% horse serum (differentiation medium or DM).
Plasmid construction and luciferase assays
To construct miR-199a-5p expression vectors, a region including miR-199a-1 was amplified from C57BL/6 mouse genomic DNA by PCR. The PCR fragment was digested with BglII and SalI and then inserted into pIRES2-EGFP (BD Biosciences Clontech) using the same cutting sites. For the pGL3-Myh9-3′-UTR vector, a 1.2-kb fragment of the Myh9 3′-UTR including putative miR-199a-5p–binding sites was amplified from C57BL/6 mouse cDNA by PCR. The PCR product was digested with SpeI and cloned into the firefly luciferase gene downstream of the pGL3-promoter vector (Promega), which was digested with XbaI. The mutation vector of the miR-199a-5p–binding site of the Myh9 3′-UTR (pGL3-Myh9-3′-UTR-mut) was constructed using a MutanBEST Kit (D401, TaKaRa) with PCR. Similarly, 2 kb upstream of the Dnm2 gene, including the WT or B1/B2 mutant Stat3-binding site, was inserted into the multiple-cloning site of the pGL3-Basic vector (Promega). The primers are listed in Table S1.
HEK 293T cells were seeded in plates and cultured overnight. The transfection procedures were performed using LipoMax or Lipofectamine 2000 according to the manufacturer's protocol (32012, Sudgen; 11668019, Invitrogen). After 24 h, the cells were collected and analyzed using the Dual-Luciferase reporter system (E1910, Promega). The Renilla luciferase reporter plasmid (pRL-TK, E2241, Promega) was used as an internal control. Relative luciferase activity was calculated as the ratio of firefly luciferase to Renilla luciferase.
Oligonucleotide transfection
C2C12 myoblasts were transfected with 100 nm negative control oligonucleotides (199-C), miR-199a-5p inhibitor (199-I), or miR-199a-5p mimic (199-M) (RiboBio) in GM, respectively. After 24 h, the myoblasts were switched to DM, and immunoblots and immunofluorescence analysis were performed at 3 days after culture in DM. The siRNA oligonucleotides for Myh9 and Stat3 were obtained from Invitrogen and RiboBio, respectively. In total, 20 nm oligonucleotides were transfected into C2C12 myoblasts using LipoMax according to the manufacturer's instructions. Next, the myoblasts were cultured in DM until immunoblot or immunofluorescence analysis on the third day.
RT-PCR
Total RNA was extracted from cells or tissues using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Mature miR-199a-5p expression was detected using previously described procedures with PCR (48). Briefly, the mRNA was extracted and linked to poly(A) tail with Escherichia coli poly(A) polymerase (New England BioLabs Inc., M0276). Then the reverse transcription was performed with specific RT primer (5′-GCTGTCAACGATACGCTACGTAACGGCATGACAGTGTTTTTTTTTTTTTTTTTTTTTTA-3′), which recognizes poly(A) to generate an mRNA cDNA library. Next, expression of miR-199a-5p was tested by real-time PCR with specific primer (forward, 5′-CCCAGTGTTCAGACTACCTGTTC-3′; reverse, 5′-GCTGTCAACGATACGCTACGTAACG-3′). The 5S rRNA was detected as an internal reference. Reverse transcription was performed using a PrimeScript RT reagent kit (DRR037A, TaKaRa). Real-time PCR was performed using SYBR Premix Ex Taq (TaKaRa) and the ABI Prism Step One system. The primers are listed in Table S1.
Western blot analysis
Cells or muscles were harvested at the indicated times. The medium was removed, and the cells were washed with D-Hanks' balanced salt solution. Ice-cold sample buffer (2% SDS, 10 mm DTT, 10% glycerol, a trace amount of bromphenol blue, and 50 mm Tris-HCl, pH 7.4) was used to lyse the cells and dissolve total protein. The samples were homogenized, incubated at 85 °C for 5 min, and stored at room temperature for 60 min. Then the cell lysates were centrifuged at 12,000 rpm for 10 min. The resultant samples were subjected to SDS-PAGE. The primary antibody dilutions were as follows: MYOG, 1:1000; MyHC, 1:1000; ACTB, 1:10,000; NM 2A, 1:1000; STAT3, 1:1000; phospho-STAT3, 1:1000; DNM2, 1:1000; GAPDH, 1:2000; MTM1, 1:1000. Horseradish peroxidase–conjugated secondary antibody dilutions were 1:5000. For protein pattern analysis, the polyacrylamide gel was incubated in Coomassie Brilliant Blue for 2 h and developed after treatment with destaining solution (10% acetic acid, 5% alcohol, 85% H2O).
Histology test
Fresh TA or gastrocnemius (GAS) muscle was isolated and immediately frozen in precooled isopentane. Sections of 10-μm thickness were made and stained with hematoxylin and eosin. Next, the stained sections were analyzed with Image-Pro Plus software. Cross-sectional area was calculated from 3–5 mice/group with over 200 fibers for each mouse. The percentage of myofibers with paracentral or central nuclei was measured from 3–5 mice/genotype with over 200 fibers for each mouse.
Immunofluorescence analysis
Cells for immunofluorescence analysis were plated on 0.1% gelatin-coated glass coverslips and cultured until the indicated time. Then the cells were washed with D-Hanks', fixed in 4% paraformaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and stained with the appropriate antibodies. The slides were costained with 4′,6-diamidino-2-phenylindole (Sigma) to mark nuclei. For NM 2A location analysis, the gastrocnemius of the indicated mice was isolated and fixed in 4% paraformaldehyde overnight. Then a single muscle fiber was isolated and manipulated using a protocol similar to that described for the cells above. The primary antibody dilutions were as follows: MYOG, 1:100; MyHC, 1:100; NM 2A, 1:100; ACTN2, 1:100. Alexa Fluor–conjugated secondary antibodies were used at a dilution of 1:250. For myonucleus number analysis in myofiber, the EDL muscles of the indicated 3-week-old mice were isolated and underwent a procedure similar to that above.
Muscle force measurement
The EDL muscles from the indicated mice were isolated and then mounted on force-displacement transducers (MLT0202, ADInstruments), which were connected to a recording device (PowerLab, ADInstruments). The muscles were incubated in 37 °C Krebs–Ringer buffer (NaCl, 118.07 mm; KCl, 4.69 mm; CaCl2, 2.52 mm; MgSO4, 1.16 mm; NaH2PO4 1.01 mm; NaHCO3, 25 mm; glucose, 11.1 mm). The maximal force was generated by a 10-V electric stimulus with a frequency of 100 Hz for 250 ms. Meanwhile, the physiological muscle optimal length (L0) and muscle weight (W) were measured. Assuming that the density of muscle was 1.06 g·cm−3 (ρ), the cross-sectional area (S) was equal to the ratio of W and (L0 × ρ), and the maximal force was calculated as the maximal specific force/S.
Statistics
All of the data are presented as the mean ± S.D. (n ≥ 3). The differences between two groups were determined using two-tailed Student's t test analysis with GraphPad Prism version 7. For survival experiments, the log-rank (Mantel–Cox) test was performed. p < 0.05 was considered significantly different.
Study approval
All animal experiments were approved by the Animal Care and Use Committee of the Model Animal Research Center at Nanjing University, which is a member of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Data availability
All data are contained within the article.
Supplementary Material
Acknowledgments
We thank the Core Facilities of the Model Animal Research Center of Nanjing University for the excellent imaging technology.
This article contains supporting information.
Author contributions—X. C., Y.-Q. G., Y.-Y. Z., J. L., J. S., and M.-S. Z. data curation; X. C., Y.-Q. G., Y.-Y. Z., W. W., P. W., and W. Z. software; X. C., Y.-Q. G., W. W., J. L., T. T., J. S., and L. W. validation; X. C., Y.-Q. G., Y.-Y. Z., W. Z., L. W., Y. L., and X. Z. investigation; X. C., Y.-Q. G., P. W., T. T., Y. L., Y. Z., X. Z., and H.-Q. C. visualization; X. C., Y.-Q. G., Y.-Y. Z., P. W., J. L., T. T., J. S., L. W., Y. Z., and X. Z. methodology; X. C., H.-Q. C., and M.-S. Z. writing-original draft; X. C. and M.-S. Z. writing-review and editing; Y.-Q. G., W. Z., Z. G., and M.-S. Z. formal analysis; Y.-Y. Z. and M.-S. Z. resources; Z. G., H.-Q. C., and M.-S. Z. conceptualization; H.-Q. C. and M.-S. Z. supervision; H.-Q. C. and M.-S. Z. project administration; M.-S. Z. funding acquisition.
Funding and additional information—This study was supported by National Natural Science Foundation of China Grants 31330034 and 31671548.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CNM
- centronuclear myopathy
- XLCNM
- X-linked centronuclear myopathy
- DNM2
- dynamin 2
- NM IIA
- non-muscle myosin IIA
- MTM1
- myotubularin 1
- T-tubule
- transverse tubule
- TA
- tibialis anterior
- GAS
- gastrocnemius
- EDL
- extensor digitorum longus
- Hetero-1
- Mtm1Δ5/y; Mir199a-1I8/+
- Hetero-2
- Mtm1Δ7/y; Mir199a-1Δ3/+
- Homo-1
- Mtm1Δ5/y; Mir199a-1I8/I8
- Homo-2
- Mtm1Δ7/y; Mir199a-1Δ3/Δ3
- g
- gravitational force unit
- N
- newtons
- 5,15-DPP
- 5,15-diphenylporphyrin
- sgRNA
- single guide RNA
- GM
- growth medium.
References
- 1. Tasfaout H., Cowling B. S., and Laporte J. (2018) Centronuclear myopathies under attack: a plethora of therapeutic targets. J. Neuromuscul. Dis. 5, 387–406 10.3233/JND-180309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amburgey K., Tsuchiya E., de Chastonay S., Glueck M., Alverez R., Nguyen C. T., Rutkowski A., Hornyak J., Beggs A. H., and Dowling J. J. (2017) A natural history study of X-linked myotubular myopathy. Neurology 89, 1355–1364 10.1212/WNL.0000000000004415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cowling B. S., Toussaint A., Muller J., and Laporte J. (2012) Defective membrane remodeling in neuromuscular diseases: insights from animal models. PLoS Genet. 8, e1002595 10.1371/journal.pgen.1002595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ketel K., Krauss M., Nicot A. S., Puchkov D., Wieffer M., Müller R., Subramanian D., Schultz C., Laporte J., and Haucke V. (2016) A phosphoinositide conversion mechanism for exit from endosomes. Nature 529, 408–412 10.1038/nature16516 [DOI] [PubMed] [Google Scholar]
- 5. Kreitzer G., Marmorstein A., Okamoto P., Vallee R., and Rodriguez-Boulan E. (2000) Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat. Cell Biol. 2, 125–127 10.1038/35000081 [DOI] [PubMed] [Google Scholar]
- 6. Warnock D. E., Baba T., and Schmid S. L. (1997) Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol. Biol. Cell 8, 2553–2562 10.1091/mbc.8.12.2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gu C., Yaddanapudi S., Weins A., Osborn T., Reiser J., Pollak M., Hartwig J., and Sever S. (2010) Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 29, 3593–3606 10.1038/emboj.2010.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cowling B. S., Toussaint A., Amoasii L., Koebel P., Ferry A., Davignon L., Nishino I., Mandel J. L., and Laporte J. (2011) Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 178, 2224–2235 10.1016/j.ajpath.2011.01.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chin Y. H., Lee A., Kan H. W., Laiman J., Chuang M. C., Hsieh S. T., and Liu Y. W. (2015) Dynamin-2 mutations associated with centronuclear myopathy are hypermorphic and lead to T-tubule fragmentation. Hum. Mol. Genet. 24, 5542–5554 10.1093/hmg/ddv285 [DOI] [PubMed] [Google Scholar]
- 10. Durieux A. C., Vignaud A., Prudhon B., Viou M. T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Lainé J., Romero N. B., Guicheney P., and Bitoun M. (2010) A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 19, 4820–4836 10.1093/hmg/ddq413 [DOI] [PubMed] [Google Scholar]
- 11. Kutchukian C., Szentesi P., Allard B., Trochet D., Beuvin M., Berthier C., Tourneur Y., Guicheney P., Csernoch L., Bitoun M., and Jacquemond V. (2017) Impaired excitation-contraction coupling in muscle fibres from the dynamin2(R465W) mouse model of centronuclear myopathy. J. Physiol. 595, 7369–7382 10.1113/JP274990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee E., Marcucci M., Daniell L., Pypaert M., Weisz O. A., Ochoa G. C., Farsad K., Wenk M. R., and De Camilli P. (2002) Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196 10.1126/science.1071362 [DOI] [PubMed] [Google Scholar]
- 13. Nicot A. S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J. M., Biancalana V., Oldfors A., Mandel J. L., and Laporte J. (2007) Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 39, 1134–1139 10.1038/ng2086 [DOI] [PubMed] [Google Scholar]
- 14. Fugier C., Klein A. F., Hammer C., Vassilopoulos S., Ivarsson Y., Toussaint A., Tosch V., Vignaud A., Ferry A., Messaddeq N., Kokunai Y., Tsuburaya R., de la Grange P., Dembele D., Francois V., et al. (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 17, 720–725 10.1038/nm.2374 [DOI] [PubMed] [Google Scholar]
- 15. Wilmshurst J. M., Lillis S., Zhou H., Pillay K., Henderson H., Kress W., Müller C. R., Ndondo A., Cloke V., Cullup T., Bertini E., Boennemann C., Straub V., Quinlivan R., Dowling J. J., et al. (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 68, 717–726 10.1002/ana.22119 [DOI] [PubMed] [Google Scholar]
- 16. Jungbluth H., Treves S., Zorzato F., Sarkozy A., Ochala J., Sewry C., Phadke R., Gautel M., and Muntoni F. (2018) Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol. 14, 151–167 10.1038/nrneurol.2017.191 [DOI] [PubMed] [Google Scholar]
- 17. Durieux A. C., Prudhon B., Guicheney P., and Bitoun M. (2010) Dynamin 2 and human diseases. J. Mol. Med. 88, 339–350 10.1007/s00109-009-0587-4 [DOI] [PubMed] [Google Scholar]
- 18. Cao H., Garcia F., and McNiven M. A. (1998) Differential distribution of dynamin isoforms in mammalian cells. Mol. Biol. Cell 9, 2595–2609 10.1091/mbc.9.9.2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cowling B. S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N. B., and Laporte J. (2014) Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 124, 1350–1363 10.1172/JCI71206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cowling B. S., Prokic I., Tasfaout H., Rabai A., Humbert F., Rinaldi B., Nicot A. S., Kretz C., Friant S., Roux A., and Laporte J. (2017) Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J. Clin. Invest. 127, 4477–4487 10.1172/JCI90542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu N., Bezprozvannaya S., Shelton J. M., Frisard M. I., Hulver M. W., McMillan R. P., Wu Y., Voelker K. A., Grange R. W., Richardson J. A., Bassel-Duby R., and Olson E. N. (2011) Mice lacking microRNA 133a develop dynamin 2–dependent centronuclear myopathy. J. Clin. Invest. 121, 3258–3268 10.1172/JCI46267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kenniston J. A., and Lemmon M. A. (2010) Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 29, 3054–3067 10.1038/emboj.2010.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L., Barylko B., Byers C., Ross J. A., Jameson D. M., and Albanesi J. P. (2010) Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J. Biol. Chem. 285, 22753–22757 10.1074/jbc.C110.130013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joshi H. P., Subramanian I. V., Schnettler E. K., Ghosh G., Rupaimoole R., Evans C., Saluja M., Jing Y., Cristina I., Roy S., Zeng Y., Shah V. H., Sood A. K., and Ramakrishnan S. (2014) Dynamin 2 along with microRNA-199a reciprocally regulate hypoxia-inducible factors and ovarian cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 111, 5331–5336 10.1073/pnas.1317242111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tay Y., Tan S. M., Karreth F. A., Lieberman J., and Pandolfi P. P. (2014) Characterization of dual PTEN and p53-targeting microRNAs identifies microRNA-638/Dnm2 as a two-hit oncogenic locus. Cell Rep. 8, 714–722 10.1016/j.celrep.2014.06.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J. F., and Mandel J. L. (2002) The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 99, 15060–15065 10.1073/pnas.212498399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Price F. D., von Maltzahn J., Bentzinger C. F., Dumont N. A., Yin H., Chang N. C., Wilson D. H., Frenette J., and Rudnicki M. A. (2014) Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181 10.1038/nm.3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bachmann C., Jungbluth H., Muntoni F., Manzur A. Y., Zorzato F., and Treves S. (2017) Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Hum. Mol. Genet. 26, 320–332 10.1093/hmg/ddw388 [DOI] [PubMed] [Google Scholar]
- 29. White R. B., Biérinx A. S., Gnocchi V. F., and Zammit P. S. (2010) Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 10, 21 10.1186/1471-213X-10-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shin J. Y., Méndez-López I., Hong M., Wang Y., Tanji K., Wu W., Shugol L., Krauss R. S., Dauer W. T., and Worman H. J. (2017) Lamina-associated polypeptide 1 is dispensable for embryonic myogenesis but required for postnatal skeletal muscle growth. Hum. Mol. Genet. 26, 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanger J. W., Wang J., Fan Y., White J., Mi-Mi L., Dube D. K., Sanger J. M., and Pruyne D. (2017) Assembly and maintenance of myofibrils in striated muscle. Handb. Exp. Pharmacol. 235, 39–75 10.1007/164_2016_53 [DOI] [PubMed] [Google Scholar]
- 32. Sun L., Ma K., Wang H., Xiao F., Gao Y., Zhang W., Wang K., Gao X., Ip N., and Wu Z. (2007) JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 179, 129–138 10.1083/jcb.200703184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Biancalana V., Beggs A. H., Das S., Jungbluth H., Kress W., Nishino I., North K., Romero N. B., and Laporte J. (2012) Clinical utility gene card for: centronuclear and myotubular myopathies. Eur. J. Hum. Genet. 20, 10.1038/ejhg.2012.91 10.1038/ejhg.2012.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eisenberg I., Eran A., Nishino I., Moggio M., Lamperti C., Amato A. A., Lidov H. G., Kang P. B., North K. N., Mitrani-Rosenbaum S., Flanigan K. M., Neely L. A., Whitney D., Beggs A. H., Kohane I. S., and Kunkel L. M. (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. 104, 17016–17021 10.1073/pnas.0708115104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hou J., Lin L., Zhou W., Wang Z., Ding G., Dong Q., Qin L., Wu X., Zheng Y., Yang Y., Tian W., Zhang Q., Wang C., Zhang Q., Zhuang S.-M., et al. (2011) Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 19, 232–243 10.1016/j.ccr.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 36. Rane S., He M., Sayed D., Vashistha H., Malhotra A., Sadoshima J., Vatner D. E., Vatner S. F., and Abdellatif M. (2009) Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 104, 879–886 10.1161/CIRCRESAHA.108.193102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alexander M. S., Kawahara G., Motohashi N., Casar J. C., Eisenberg I., Myers J. A., Gasperini M. J., Estrella E. A., Kho A. T., Mitsuhashi S., Shapiro F., Kang P. B., and Kunkel L. M. (2013) MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ. 20, 1194–1208 10.1038/cdd.2013.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verver E. J., Topsakal V., Kunst H. P., Huygen P. L., Heller P. G., Pujol-Moix N., Savoia A., Benazzo M., Fierro T., Grolman W., Gresele P., and Pecci A. (2016) Nonmuscle myosin heavy chain IIA mutation predicts severity and progression of sensorineural hearing loss in patients with MYH9-related disease. Ear Hear. 37, 112–120 10.1097/AUD.0000000000000198 [DOI] [PubMed] [Google Scholar]
- 39. Sekine T., Konno M., Sasaki S., Moritani S., Miura T., Wong W. S., Nishio H., Nishiguchi T., Ohuchi M. Y., Tsuchiya S., Matsuyama T., Kanegane H., Ida K., Miura K., Harita Y., et al. (2010) Patients with Epstein-Fechtner syndromes owing to MYH9 R702 mutations develop progressive proteinuric renal disease. Kidney Int. 78, 207–214 10.1038/ki.2010.21 [DOI] [PubMed] [Google Scholar]
- 40. Buj-Bello A., Fougerousse F., Schwab Y., Messaddeq N., Spehner D., Pierson C. R., Durand M., Kretz C., Danos O., Douar A. M., Beggs A. H., Schultz P., Montus M., Denèfle P., and Mandel J. L. (2008) AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 17, 2132–2143 10.1093/hmg/ddn112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Childers M. K., Joubert R., Poulard K., Moal C., Grange R. W., Doering J. A., Lawlor M. W., Rider B. E., Jamet T., Danièle N., Martin S., Rivière C., Soker T., Hammer C., Van Wittenberghe L., et al. (2014) Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci. Transl. Med. 6, 220ra10 10.1126/scitranslmed.3007523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kutchukian C., Lo Scrudato M., Tourneur Y., Poulard K., Vignaud A., Berthier C., Allard B., Lawlor M. W., Buj-Bello A., and Jacquemond V. (2016) Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 113, 14432–14437 10.1073/pnas.1604099113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabha N., Volpatti J. R., Gonorazky H., Reifler A., Davidson A. E., Li X., Eltayeb N. M., Dall'Armi C., Di Paolo G., Brooks S. V., Buj-Bello A., Feldman E. L., and Dowling J. J. (2016) PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Invest. 126, 3613–3625 10.1172/JCI86841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tasfaout H., Buono S., Guo S., Kretz C., Messaddeq N., Booten S., Greenlee S., Monia B. P., Cowling B. S., and Laporte J. (2017) Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 8, 15661 10.1038/ncomms15661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tasfaout H., Lionello V. M., Kretz C., Koebel P., Messaddeq N., Bitz D., Laporte J., and Cowling B. S. (2018) Single intramuscular injection of AAV-shRNA reduces DNM2 and prevents myotubular myopathy in mice. Mol. Ther. 26, 1082–1092 10.1016/j.ymthe.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ran F. A., Hsu P. D., Lin C. Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., and Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ge Y., Sun Y., and Chen J. (2011) IGF-II is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol. 192, 69–81 10.1083/jcb.201007165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fu H., Tie Y., Xu C., Zhang Z., Zhu J., Shi Y., Jiang H., Sun Z., and Zheng X. (2005) Identification of human fetal liver miRNAs by a novel method. FEBS Lett. 579, 3849–3854 10.1016/j.febslet.2005.05.064 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.