

Pathological cardiac hypertrophy is driven by neurohormonal activation of specific G protein-coupled receptors (GPCRs) in cardiomyocytes and is accompanied by large-scale changes in cardiomyocyte gene expression. These transcriptional changes require activity of positive transcription elongation factor b (P-TEFb), which is recruited to target genes by the bromodomain protein Brd4 or the super elongation complex (SEC). Here, we describe GPCR-specific regulation of these P-TEFb complexes and a novel mechanism for activating Brd4 in primary neonatal rat cardiomyocytes.
KEYWORDS: Brd4, G protein-coupled receptor, P-TEFb, cardiomyocyte hypertrophy, protein kinase A
ABSTRACT
Pathological cardiac hypertrophy is driven by neurohormonal activation of specific G protein-coupled receptors (GPCRs) in cardiomyocytes and is accompanied by large-scale changes in cardiomyocyte gene expression. These transcriptional changes require activity of positive transcription elongation factor b (P-TEFb), which is recruited to target genes by the bromodomain protein Brd4 or the super elongation complex (SEC). Here, we describe GPCR-specific regulation of these P-TEFb complexes and a novel mechanism for activating Brd4 in primary neonatal rat cardiomyocytes. The SEC was required for the hypertrophic response downstream of either the α1-adrenergic receptor (α1-AR) or the endothelin receptor (ETR). In contrast, Brd4 inhibition selectively impaired the α1-AR response. This was corroborated by the finding that the activation of α1-AR, but not ETR, increased Brd4 occupancy at promoters and superenhancers of hypertrophic genes. Transcriptome analysis demonstrated that the activation of both receptors initiated similar gene expression programs, but that Brd4 inhibition attenuated hypertrophic genes more robustly following α1-AR activation. Finally, we show that protein kinase A (PKA) is required for α1-AR stimulation of Brd4 chromatin occupancy. The differential role of the Brd4/P-TEFb complex in response to distinct GPCR pathways has potential clinical implications, as therapies targeting this complex are currently being explored for heart failure.
INTRODUCTION
The heart undergoes extensive remodeling in response to various mechanical and hormonal stressors during the progression to heart failure following myocardial infarction and/or sustained hypertension (1, 2). This includes hypertrophy of terminally differentiated cardiomyocytes in order to sustain cardiac output (3). While initially adaptive, prolonged cardiomyocyte hypertrophy leads to cardiomyocyte death, fibrosis, and progression to chronic heart failure (4). Cardiomyocyte hypertrophy is initiated in part by neurohormonal activation of G protein-coupled receptors (GPCRs), such as the α1-adrenergic receptor (α1-AR), endothelin-1 receptor (ETR), and β-adrenergic receptor (β-AR) families (5, 6). Upon ligand binding, GPCRs activate heterotrimeric G proteins comprised of a Gα subunit and the obligate heterodimer Gβγ. Both the α1-AR and ETR canonically activate Gαq signaling, whereas the β-AR activates Gαs signaling. Cardiac cell-specific overexpression of Gαq and Gαs isoforms in mice leads to cardiomyopathy phenotypes, including cardiomyocyte hypertrophy (7, 8). The Gα isoforms elicit distinct signaling pathways involving calcium release and cyclic AMP (cAMP) formation, respectively, which are capable of activating transcription factors and a gene expression program, culminating in cardiomyocyte hypertrophy (9, 10). These pathological gene expression changes also require the coordinated interplay between dynamic alterations in chromatin structure, various master transcription factors, and general transcriptional regulators, such as positive transcription elongation factor b (P-TEFb) (9, 11, 12).
P-TEFb, a heterodimer consisting of cyclin-dependent kinase 9 and cyclin T, positively regulates the release of RNA polymerase II (RNAPII) from a promoter-proximal paused state into productive elongation. P-TEFb phosphorylates multiple proteins in the RNAPII elongation complex, including the C-terminal repeat domain of RNAPII itself, DRB sensitivity inducing factor (DSIF), and negative elongation factor (NELF) (13). In cardiomyocytes, P-TEFb activity is regulated by Gαq signaling, as evidenced by cardiac cell-specific overexpression of Gαq in mice and ETR activation in primary neonatal rat cardiomyocytes (11). P-TEFb is a critical regulator for cardiomyocyte hypertrophy, with inhibition preventing the gene expression and cell size changes characteristic of cardiomyocyte hypertrophy (11). These transcriptional events require the recruitment of the active P-TEFb complex to chromatin. P-TEFb recruitment is predominantly regulated through interactions with the bromodomain and extraterminal (BET) protein Brd4 or through interactions with the super elongation complex (SEC) (14, 15).
Like other BET family members (Brd2, Brd3, and testis-specific BrdT), Brd4 contains two N-terminal bromodomains, which bind to acetylated lysines on histone proteins, leading to the recruitment of Brd4 to chromatin, as well as an extraterminal domain, which interacts with multiple transcriptional regulators (16). Brd4 and BrdT contain an additional domain that interacts with P-TEFb (17). The importance of Brd4 as a regulator of P-TEFb and transcription elongation in cardiomyocyte hypertrophy has been demonstrated using small-molecule inhibitors of the BET bromodomain/acetyllysine interaction, such as JQ1 (18–21). JQ1 treatment reduced stress-induced gene expression and cardiomyocyte hypertrophy in primary culture models and in mice subjected to pressure overload via transverse aortic constriction (TAC), a potent inducer of cardiac hypertrophy in vivo (18, 21). Brd4 inhibition was also able to partially reverse preestablished signs of heart failure in a mouse model of myocardial infarction and pressure overload (20). These effects are correlated with the loss of Brd4 from superenhancers and promoters of hypertrophic genes in cardiomyocytes, as well as reduced RNAPII elongation.
Multiple forms of SEC have been found in mammalian cells, comprised of P-TEFb, AF9, ENL, the three ELL family members (ELL1/2/3), EAF1/2, AFF1, and AFF4 (22). The SEC positively regulates the release of RNAPII from promoter-proximal pausing to productive elongation (23). Aberrant targeting and activity of the SEC underlies the development of various cancers and developmental diseases. For example, mixed-lineage leukemia 1 (MLL) is fused to various SEC subunits in certain types of acute leukemias (24), and a germ line Aff4 gain-of-function mutation leads to the developmental syndrome CHOPS (for cognitive impairment and coarse facies, heart defects, obesity, pulmonary involvement, and short stature and skeletal dysplasia) (25). Although RNAPII promoter-proximal pausing is dysregulated in cardiac hypertrophy, the role of the SEC in regulating the hypertrophic gene expression program in cardiomyocytes has not been investigated.
How diverse signaling pathways involved in cardiac remodeling cooperate to orchestrate the hypertrophic gene expression program in vivo remains poorly understood. Although neurohormonal signals induce similar hypertrophic responses in primary cardiomyocytes, distinct signaling pathways are initiated through the activation of their cognate GPCRs (26). Such effects are generally attributed to differential G protein coupling; however, receptor-specific differences in downstream effector protein activation of the same Gα subunit also have been demonstrated (27). The coordination between these signaling pathways, and the fact that each activates a unique combination of transcription factors, suggests that there are differences in how they regulate gene expression. However, comparisons of changes in gene expression have only been assessed for a limited repertoire of genes (28, 29). How different receptors alter global transcriptional regulation has not been systematically compared. Such differences may have important therapeutic implications for treating patients with heart disease stemming from varied clinical origins.
In this study, we focused on the differential impact of cardiomyocyte GPCR signaling pathways on mechanisms regulating transcription. We investigated the role of P-TEFb and its interacting partners, Brd4 and SEC, in cardiomyocyte hypertrophy caused by the activation of either of two GPCRs, the α1-AR or ETR. These receptors are canonically thought to elicit their hypertrophic responses through Gαq activation, with both receptors also able to activate additional Gα subunits, which has not been thoroughly assessed. We found that P-TEFb activity and the SEC are required for cardiomyocyte hypertrophy induced by the activation of either GPCR. However, only the α1-AR response was attenuated by Brd4 inhibition. Transcriptome analysis after Brd4 inhibition indicated the attenuation of α1-AR upregulated genes that were enriched for pathways involved in the pathophysiology of cardiomyocyte hypertrophy. Brd4 chromatin occupancy at promoters and superenhancers of hypertrophic genes was specifically induced by α1-AR activation, an effect that was dependent on the activity of protein kinase A (PKA). Lastly, we demonstrated that the hypertrophic response downstream of another receptor known to signal through PKA, the β-AR, was also attenuated by Brd4 inhibition. Our study suggests receptor-specific regulation of P-TEFb function and expands the currently known cellular repertoire of protein kinases capable of regulating Brd4 function. Further, our findings suggest that the clinical efficacy of BET inhibitors for heart failure depends on patients’ specific neurohormonal signaling patterns.
RESULTS
Evidence for receptor-specific P-TEFb regulation in cardiomyocyte hypertrophy.
We first revisited the requirement of P-TEFb activity for the hypertrophic response in primary neonatal rat cardiomyocytes (NRCMs). Previous experiments used the ATP analog 5,6-dichlorobenzimidazone-1-β-d-ribofuranoside (DRB), a cyclin-dependent kinase inhibitor that affects Cdk9, to implicate P-TEFb activity in the hypertrophic response (11). To confirm these results, we repeated this experiment using iCdk9, a Cdk9 inhibitor that is ∼1,000 times more potent and ∼100 times more selective than DRB (30). Following 24 h of treatment, agonists for the ETR (endothelin-1; ET-1) or α1-AR (phenylephrine; PE) increased cardiomyocyte surface area by 35 to 40% relative to that of the control, as assessed by analysis of α2-actinin immunostaining using high-content microscopy (Fig. 1). Simultaneous treatment with 0.2 μM iCdk9 completely abolished the increase in cardiomyocyte size elicited by either agonist, confirming a stringent requirement for P-TEFb activity in cardiomyocyte hypertrophy (Fig. 1).
FIG 1.

Inhibition of the P-TEFb kinase subunit Cdk9 prevents cardiomyocyte hypertrophy in response to α1-AR or ETR activation. (A) NRCMs were treated with PE or ET-1 for 24 h, as indicated. Cardiomyocytes were stained with Hoechst dye and identified by staining for the cardiomyocyte-specific marker α2-actinin. (B) Fold change in cardiomyocyte surface area following 24 h of treatment over the surface area of cardiomyocytes from the same biological replicate at 0 h. Data are presented as means ± standard errors of the means (SEM), with each point representing a biological replicate. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed (***, P < 0.001; ****, P < 0.0001).
To further characterize P-TEFb function in the hypertrophic response, we assessed the roles of the SEC and Brd4, two key P-TEFb-interacting proteins (31). We first treated NRCMs undergoing hypertrophy with KL-2, a small molecule previously shown to prevent the interaction between the cyclin T component of P-TEFb and the SEC scaffolding subunit Aff4 in cell lines (32). KL-2 treatment blocked the increase in cell size in response to both ETR and α1-AR activation, similar to the effect of Cdk9 inhibition (Fig. 2A and B). To determine the effect on the expression of hypertrophic genes, we monitored changes in mRNA levels for established hypertrophy marker genes Nppa, Nppb, and Serpine1 genes using reverse transcription-quantitative PCR (RT-qPCR). Whereas mRNA levels for these genes were robustly increased in response to the activation of either receptor, the induction of Nppb and Nppa was blocked by KL-2 cotreatment (Fig. 2C). Serpine1 induction was unaffected by KL-2 treatment, indicating a gene-specific aspect to SEC function. To disrupt SEC function using an independent method, we reduced Aff4 levels using short interfering RNA (siRNA) and verified knockdown by RT-qPCR (Fig. 2D). Similar to KL-2 treatment, knockdown of Aff4 blocked the increase in cell size following the activation of either receptor (Fig. 2E).
FIG 2.

Disruption of SEC-P-TEFb interaction blocks the hypertrophic response following activation of either receptor. (A) NRCMs were treated for 24 h, as indicated. Cardiomyocytes were stained with Hoechst dye and identified by staining of the cardiomyocyte-specific marker α2-actinin. (B) Fold change in surface area of identified cardiomyocytes over siRNA control-transfected cardiomyocytes from the same biological replicate at 0 h. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed. (C) Expression of three genes previously identified as upregulated in hypertrophic cardiomyocytes, the Nppb, Nppa, and Serpine1 genes, was determined by RT-qPCR. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed. (D) Aff4 knockdown in cardiomyocytes 72 h after transfection with Aff4-targeted siRNA was validated by RT-qPCR. An unpaired t test was performed. (E) Fold change in surface area of identified cardiomyocytes over the surface area of siRNA control-transfected cardiomyocytes from the same biological replicate at 0 h. Cardiomyocytes were transfected 72 h prior to treatment with 50 nM the specified siRNA. Data are presented as means ± SEM, with each point representing a biological replicate. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
We next tested the role of Brd4 using the pan-BET small-molecule inhibitor JQ1. JQ1 targets the BET family bromodomains, acting to competitively inhibit their interaction with acetylated lysine residues (33). Interestingly, BET inhibition attenuated the hypertrophic response in a receptor-specific manner: the response to α1-AR activation was decreased, whereas there was no effect on the ETR-mediated increase in cell size (Fig. 3A and B). We also observed that JQ1 treatment more strongly reduced the expression of hypertrophic marker genes in cells stimulated with the α1-AR agonist compared to cells stimulated with an ETR agonist (Fig. 3C). One possible explanation for the receptor-specific effect of JQ1 is that the dose of ET-1 used to drive hypertrophy was sufficiently high to overcome JQ1 inhibition. To address this, we tested the effect of JQ1 on the ET-1-driven hypertrophic response over a wide range of ET-1 doses. The ET-1 response was insensitive to JQ1 at all doses tested (Fig. 4). Thus, receptor-specific differences in JQ1 sensitivity likely reflect intrinsic differences in the respective GPCR signaling outcomes. These data suggest that the SEC is generally required for P-TEFb function in the hypertrophic response, whereas the P-TEFb/Brd4 complex mediates receptor-specific functions.
FIG 3.

Effects of BET inhibitor JQ1 on cardiomyocyte hypertrophy are specific to the receptor driving the response. (A) NRCMs treated for 24 h were fixed and stained with Hoechst dye and identified by staining for the cardiomyocyte-specific marker α2-actinin. (B) Fold change in surface area of cardiomyocytes over surface area of cardiomyocytes at 0 h from the same biological replicate. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed. (C) Expression of genes previously demonstrated to be upregulated in hypertrophic cardiomyocytes was determined by RT-qPCR. Data are presented as means ± SEM, with each point representing a biological replicate. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
FIG 4.
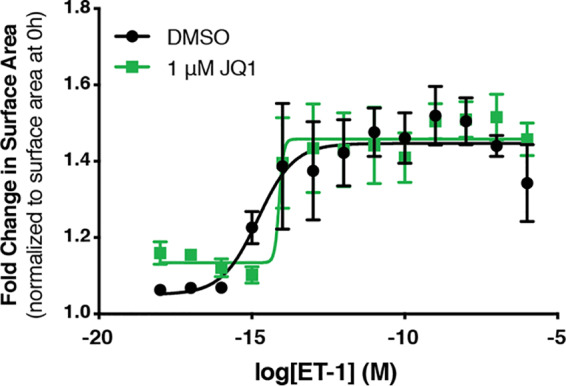
ETR-mediated hypertrophy is insensitive to BET inhibition independent of ET-1 concentration. Dose-response curves were generated to assess the effect of JQ1 on cardiomyocyte surface area at a range of ET-1 concentrations. Cardiomyocytes were treated for 24 h, as indicated, followed by fixation and staining with Hoechst dye for α2-actinin to identify NCRMs. Fold change in surface area over cardiomyocytes from the same biological replicate at 0 h was determined. Data are presented as means ± SEM for 3 or 4 independent experiments. Dose-response curves were plotted using sigmoidal dose-response (variable slope) curves by nonlinear regression.
As JQ1 inhibits all members of the BET family of bromodomain proteins, we confirmed that the effects on hypertrophy were mediated by Brd4 and not Brd2 and/or Brd3. Brd2, Brd3, and Brd4 were individually depleted in NRCMs using siRNA (Fig. 5A). Hypertrophic responses then were induced through the activation of the α1-AR or ETR. Brd2 and Brd3 knockdown reduced the basal size of NRCMs relative to that of control siRNA (Fig. 5B), but the response following activation of either receptor was comparable to that observed with control siRNA (Fig. 5C). In contrast, Brd4 knockdown recapitulated the effects observed with JQ1, in that it attenuated the response to α1-AR activation but did not affect ETR-mediated hypertrophy (Fig. 5B and C). These data argue that Brd4 inhibition accounts for the receptor-specific effects of JQ1 on cardiomyocyte hypertrophy.
FIG 5.

Role of individual BET family members expressed in cardiomyocytes assessed after siRNA-mediated knockdown. (A) Brd2, Brd3, and Brd4 knockdown efficiency in NRCMs 72 h after transfection with targeted siRNA was determined by RT-qPCR. An unpaired t test was performed. (B) Fold change in surface area over cardiomyocytes transfected with control siRNA from the same biological replicate at 0 h. Following 72 h of knockdown with the indicated siRNA, cardiomyocytes were treated for 24 h, as indicated. Cardiomyocytes were fixed and identified by staining for the cardiomyocyte-specific marker α2-actinin. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed. (C) Change in cardiomyocyte size from panel B is presented relative to respective vehicle/siRNA treatment to normalize for the difference in basal size. Data are presented as means ± SEM, with each point representing a biological replicate. Two-way ANOVA followed by post hoc t tests with Bonferroni correction was performed (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To gain further insight into how Brd4 function is differentially affected by distinct GPCR signaling pathways, we assessed gene-specific Brd4 localization to chromatin using chromatin immunoprecipitation coupled to qPCR (ChIP-qPCR). ChIP was performed using an antibody recognizing endogenous Brd4 and was quantified by qPCR using primer pairs near the transcription start sites of the Nppb, Nppa, and Serpine1 genes. Occupancy at previously defined superenhancers for Nppb and Serpine1 in cardiomyocytes was also assessed (19). At all genomic loci tested, 24 h of α1-AR activation increased Brd4 chromatin occupancy compared to that of vehicle treatment (Fig. 6A). We observed no change in Brd4 occupancy compared to that of vehicle following ETR treatment, despite the fact that mRNA levels for the same genes were similarly induced by PE and ET-1 (Fig. 2C and 3C). The effects on Brd4 occupancy were not simply a reflection of altered Brd4 protein levels, as immunoblots performed on cell extracts from NRCMs treated with PE or ET-1 revealed slight decreases in expression compared to those of vehicle controls (Fig. 6B and C). This suggests that signaling through α1-AR, but not ETR, triggers recruitment of Brd4, making gene expression changes downstream of this receptor more sensitive to Brd4 inhibition by JQ1.
FIG 6.

Brd4 chromatin occupancy increases in response to α1-AR but not ETR activation. (A) Cardiomyocytes were treated for 24 h, as indicated. Following treatment, cross-linked chromatin was immunoprecipitated with an anti-Brd4 antibody, followed by DNA purification and quantification by qPCR using primers at the indicated loci. Each immunoprecipitation was normalized to the percent input for exogenous S. pombe spike-in DNA at the cdc2+ loci. Data were analyzed by one-way ANOVA, followed by Dunnett’s post hoc comparison. (B) A Western blot of whole-cell lysates to assess changes in Brd4 protein expression following the indicated treatment for 24 h. (C) Densitometry-based quantification of Brd4 normalized to Hsp90 expression. Data are presented as means ± SEM, with each point representing a biological replicate. Data were analyzed by one-way ANOVA, followed by Dunnett’s post hoc comparison (*, P < 0.05; **, P < 0.01).
RNA-seq reveals differences in GPCR-dependent signaling between hypertrophic agonists.
To comprehensively profile receptor-specific effects on cardiomyocyte hypertrophy, we performed transcriptome sequencing (RNA-seq) on NRCMs following the activation of either receptor for 24 h in the presence or absence of JQ1 (see Table S1 in the supplemental material). When comparing agonist versus vehicle conditions, we observed robust gene expression changes [log2(fold change) of >±1 and P < 0.05] for hundreds of genes following ETR (209 upregulated, 192 downregulated) or α1-AR (269 upregulated, 279 downregulated) activation. The genes regulated by either receptor overlapped significantly, although there were more genes uniquely regulated by α1-AR activation than by ET-1 activation (Fig. 7A and B). The combined effect of receptor agonists and JQ1 on differential expression was visualized by performing K-means clustering (Fig. 7C and D). This analysis identified three major gene clusters that were similarly regulated by agonist and JQ1: one in which genes were repressed by agonist in the presence or absence of JQ1 (cluster 1), one in which genes were activated by agonist and attenuated by JQ1 (cluster 2), and one in which genes were activated by agonist in the presence or absence of JQ1 (cluster 3). The observation that the primary effect of JQ1 was to dampen the expression of genes regulated by receptor activation aligns with the known roles of Brd4 in recruiting P-TEFb to regulate pause-release and activate transcription (15) and is consistent with the effect of JQ1 on cardiac stress-induced genes previously characterized (20).
FIG 7.

Transcriptome analysis of gene expression programs mediated by receptor activation and the effect of Brd4 inhibition. (A and B) Venn diagrams of significantly upregulated genes [log2(fold change) of >1 for agonist/vehicle versus vehicle/vehicle; P < 0.05] (A) or downregulated genes [log2(fold change) of <−1 for agonist/vehicle versus vehicle/vehicle; P < 0.05] (B) following 24 h of receptor activation. (C and D) Heat maps were generated for genes differentially regulated following activation of the specified receptor for 24 h. Each row was normalized, with the color representing the z score for the specific row. K-means clustering was performed to identify subsets of genes with distinct patterns following Brd4 inhibition. (E) Venn diagram of genes upregulated by respective receptor activation [log2(fold change) of >1 for agonist/vehicle versus vehicle/vehicle; P < 0.05] and attenuated by Brd4 inhibition from the activated state [log2(fold change) of <−0.5 for agonist/JQ1 versus agonist/vehicle; P < 0.05]. (F) Number of genes uniquely upregulated by either agonist or both and whether expression was attenuated by JQ1. Genes were first categorized as uniquely upregulated by α1-AR or ETR or upregulated by both receptors [log2(fold change) of >1 for agonist/vehicle versus vehicle/vehicle; P < 0.05]. Within each category, genes were further characterized as JQ1 sensitive if they were attenuated by Brd4 inhibition from the activated state [log2(fold change) of <−0.5 for agonist/JQ1 versus agonist/vehicle; P < 0.05]. In the category of genes upregulated by both receptors, genes were categorized if they were attenuated by Brd4 inhibition when upregulated by either receptor (upregulated by both receptors/attenuated by JQ1) or if the effect of Brd4 inhibition was unique to a specific receptor. (G) Gene ontology enrichment for genes attenuated by JQ1 following receptor activation (from panel E) performed with DAVID. The false discovery rate (FDR) indicates whether the pathway was significantly enriched in the gene list. (H) Changes in transcription factor activity predicted by Ingenuity Pathway Analysis (IPA). The z score represents the predicted change in transcription factor activity between the two treatment groups, and the P value indicates whether the transcription factor’s targets are significantly enriched in the gene set. The purple dots indicate the change in activity following agonist treatment alone [log2(fold change) of > 1 for agonist/vehicle versus vehicle/vehicle; P < 0.05]. The green dots indicate the JQ1-dependent decrease in activity from the activated state [log2(fold change) of <−0.5 for agonist/JQ1 versus agonist/vehicle; P < 0.05].
We focused on groups of genes for which increased expression caused by the activation of either receptor was attenuated by JQ1 [log2(fold change) < −0.5 compared to agonist alone; P < 0.05]. JQ1 attenuated the expression of 107 ETR-induced genes and 155 α1-AR-induced genes (termed receptor+/JQ1−) (Fig. 7E). Roughly equal proportions of genes induced by the activation of either receptor were JQ1 sensitive, irrespective of whether they were induced by one receptor or both (Fig. 7F). Gene ontology term analysis of α1-AR+/JQ1− and ETR+/JQ1− gene sets revealed that the terms inflammatory response, defense response, cell adhesion, cardiac muscle tissue growth, and heart growth, which correspond to the pathophysiology of cardiomyocyte hypertrophy and align with those previously identified to be affected by JQ1, were significantly enriched among the α1-AR+/JQ1− genes (Fig. 7G) (20). In contrast, none of these terms were significantly enriched among ETR+/JQ1− genes, consistent with the selective effect of JQ1 on the α1-AR response.
Previous studies have identified multiple transcription factors that are required to activate prohypertrophic genes in cardiomyocytes (9, 34). Some of these transcription factors have been associated with Brd4 activity in cardiomyocytes, through either motif enrichment in genomic loci with high Brd4 occupancy or various gene set enrichment methods for JQ1-sensitive genes. The pathway-specific effect of Brd4 inhibition suggests that specific transcription factors are dependent on Brd4 to activate transcription. We initially focused on those transcription factors that were previously implicated, including NF-κB, GATA4, and the AP-1 subunits Jun and Fos (18–20). To determine the effect of JQ1 on these transcription factors, we used Ingenuity Pathway Analysis software (IPA; Qiagen) to predict changes in their activity (Fig. 7H). IPA upstream regulator analysis provides a z score, to indicate the predicted change in activity between the two treatment groups, and a Fisher exact test P value, to indicate whether particular upstream regulator’s target genes are significantly enriched in the gene expression program. Interestingly, IPA predicted increased activity of these transcription factors following the activation of either receptor (positive z score, agonist/vehicle versus vehicle/vehicle), but activity was specifically attenuated by JQ1 following α1-AR activation (negative z score, agonist/JQ1 versus agonist/vehicle) (Fig. 7H). This suggests that receptor-specific activation mechanisms for these transcription factors dictate their dependence on Brd4 activity. When we expanded the analysis to include all activated transcription factors, a more ubiquitous effect of Brd4 inhibition was observed. We identified 78 transcription factors with enhanced activity following α1-AR activation, of which the activity of 39 (50%) was attenuated by cotreatment with JQ1. In contrast, ETR activation was predicted to enhance the activity of 50 transcription factors, and only eight (16%) were attenuated by JQ1. Thus, although specific transcription factors may function to recruit Brd4 to specific loci, the more ubiquitous effect of Brd4 inhibition on α1-AR-mediated transcription factor activity suggests that α1-AR signaling increases the pool of active Brd4.
Signaling pathway regulating Brd4 recruitment to chromatin involves PKA.
We hypothesized that a distinct signaling pathway activated by the α1-AR determines differential recruitment of Brd4 and inhibitory effect of JQ1 on transcription factor activity. Brd4 is activated following the phosphorylation of its phosphorylation-dependent interaction domain (PDID). An in vitro kinase assay demonstrated that PKA was able to phosphorylate this region, although the functional significance was not determined (35). We have previously demonstrated that α1-AR, but not ETR, activation led to Gαs-dependent activation of cAMP/PKA signaling in HEK 293 cells (36). Thus, we hypothesized that PKA, a protein kinase activated by cAMP, regulates the specific effects of α1-AR signaling on Brd4 in cardiomyocytes.
We confirmed that α1-AR and not ETR signaling activated PKA in NRCMs. Cardiomyocytes were transduced with a nuclearly localized Förster resonance energy transfer (FRET)-based PKA biosensor (AKAR4-NLS) to monitor PKA activity (37). When phosphorylated, the biosensor undergoes a conformational change that moves the two fluorophores into closer proximity, leading to an increased FRET ratio. Nuclear localization of the AKAR biosensor was confirmed by fluorescence microscopy (Fig. 8A). We then generated a dose-response relationship for PKA activity following stimulation with ET-1 and PE. Alprenolol was included in the PE experiments to prevent off-target effects on the β-AR at high concentrations. We observed a dose-dependent increase in FRET, indicating an increase in PKA activity, following 15 min of α1-AR activation. In contrast, no change in activity was observed following 15 min of ETR activation (Fig. 8B). This demonstrated that the α1-AR uniquely activates PKA in the nucleus of cardiomyocytes, similar to what we detected in HEK 293 cells (36).
FIG 8.

α1-AR activation leads to increased nuclear PKA signaling. (A) Cardiomyocytes were transduced with AAV9-AKAR4-NLS virus at an MOI of 5,000 and imaged 72 h later. Nuclei were visualized by staining live cells with Draq5. (B) Dose-response curves for PKA activation following activation of the ETR or α1-AR were generated. Alprenolol was included to prevent off-target β-AR activation by high concentrations of PE. Data are presented means ± SEM for three biological replicates. Dose-response curves were plotted using sigmoidal dose-response (variable slope) curves by nonlinear regression.
To determine if PKA activity downstream of the α1-AR regulates Brd4 function in cardiomyocytes, we performed ChIP-qPCR after inhibition or activation of PKA. To inhibit PKA, we used the competitive inhibitor KT5720 (38). As we anticipated that long-term PKA inhibition could cause other changes in cellular physiology that would complicate the interpretation of the experiments, we used a treatment time of 1.5 h. We quantified Brd4 localization using primer pairs near the transcription start site of c-Fos and Ctgf as well as previously defined superenhancer regions of Ctgf (19). The activation of the α1-AR for 1.5 h enhanced Brd4 occupancy near the transcription start sites of both genes and along the previously defined Ctgf superenhancers, whereas ETR stimulation had no effect (Fig. 9A and B). These data suggest that the specific effect of α1-AR signaling on Brd4 chromatin occupancy is maintained at the shorter treatment time. Pretreatment with KT5720 abrogated the increase in Brd4 occupancy following α1-AR activation, consistent with a requirement for PKA activity for chromatin recruitment of Brd4 downstream of α1-AR signaling (Fig. 9A and B). PKA inhibition also increased the basal occupancy of Brd4 at these sites, perhaps reflecting a repressive function for PKA in unstimulated NRCMs. We also stimulated the activation of PKA in NRCMs by increasing intracellular cAMP levels with forskolin and 3-isobutyl-1-methylxanthine (IBMX), an adenylyl cyclase activator and phosphodiesterase inhibitor, respectively (39). Sustained PKA activation (1.5 h) increased Brd4 chromatin association at two of the four genomic loci assessed, reinforcing the key role of PKA in Brd4 activation (Fig. 9C).
FIG 9.

PKA signaling regulates recruitment of Brd4 to chromatin. (A and B) Effect of PKA inhibition with the small-molecule inhibitor KT5720 on receptor-mediated increases in Brd4 occupancy. Cardiomyocytes were pretreated for 30 min with the PKA inhibitor prior to receptor activation for 1.5 h with the indicated agonists. Two-way ANOVA followed by post hoc t test comparisons with Bonferroni correction was performed. (C) PKA was activated for 1.5 h by increasing intracellular cAMP levels with forskolin and IBMX, an adenylyl cyclase activator and phosphodiesterase inhibitor, respectively. Following the indicated treatment, cardiomyocytes were fixed and ChIP was performed with an anti-Brd4 antibody. ChIP was quantified by qPCR using primers at the indicated loci. An unpaired t test was performed. (D) After 24 h of the indicated treatments, NRCMs were fixed and stained with Hoechst dye and for the cardiomyocyte-specific marker α2-actinin. (E) Fold change in surface area after 24 h of the indicated treatment over cardiomyocytes fixed at 0 h from the same biological replicate. Two-way ANOVA followed by post hoc t test comparisons with Bonferroni correction was performed. Data are presented as means ± SEM, with each point representing a separate biological replicate (*, P < 0.05; ***, P < 0.001; ****, P < 0.0001).
To test whether a regulatory link between PKA and Brd4 could be detected in response to other GPCRs coupled to Gαs, we examined the role of Brd4 downstream of the β-AR, the activation of which is strongly prohypertrophic in cardiomyocytes (40, 41). The primary signaling pathway downstream of this receptor in cardiomyocytes (and other cell types as well) involves adenylyl cyclase activation, cAMP production, and increased protein kinase A activity (41). Thus, we predicted that hypertrophy mediated by the β-AR also would be attenuated by the inhibition of Brd4 with JQ1. Following 24 h of treatment with the agonist isoproterenol, we observed an ∼25% increase in surface area that was completely blocked by cotreatment with JQ1 (Fig. 9D and E). This demonstrates that the observed connection between PKA and Brd4 is not unique to α1-AR signaling and may reflect a general Gαs-coupled GPCR-dependent pathway for Brd4 activation. Taken together, our results point to PKA and Brd4 as central players underlying receptor-specific gene regulatory mechanisms in hypertrophic cardiomyocytes (Fig. 10).
FIG 10.

Model of P-TEFb complex activation following activation of the α1-AR or ETR. Both the ETR and α1-AR activate a signaling cascade that increases active P-TEFb and requires subsequent recruitment through the SEC. The α1-AR activation also leads to Brd4-dependent recruitment due to the activation of a PKA signaling pathway.
DISCUSSION
In this study, we showed that the activation of distinct GPCRs in cardiomyocytes can result in hypertrophic responses and gene expression programs that are qualitatively similar but that operate through different transcriptional regulatory mechanisms. We also identified a novel mechanism regulating Brd4 in the development of cardiomyocyte hypertrophy that expands our understanding of how specific signaling pathways regulate the recruitment of general transcription regulators and that may have relevance in other physiological contexts.
Chromatin occupancy of Brd4 undergoes extensive redistribution in order to positively regulate the expression of the cardiomyocyte hypertrophic gene program. These changes lead to enhanced Brd4 occupancy on specific superenhancers and promoter regions. Previous Brd4 ChIP-seq experiments have used primary cardiomyocytes treated with PE (in accord with our results) or cardiac tissue isolated from mice subjected to transverse aortic constriction (TAC) (19, 20). The increased genomic loading of Brd4 in heart failure models has been attributed to increases in Brd4 expression (19), similar to proposed mechanisms in various forms of cancer (42–44). In heart failure, increased Brd4 protein expression is thought to occur due to the decreased expression of the Brd4 targeting microRNA miR-9 (21, 45). These reports assessed whole cardiac tissue not enriched for cardiomyocytes or in vitro cardiomyocyte studies, which reported conflicting evidence regarding changes in Brd4 expression (18, 21). Following 24 h of activation of the α1-AR or ETR in cardiomyocytes, we did not observe a significant change in Brd4 protein expression (Fig. 6B). Instead, the increased Brd4 recruitment following α1-AR activation was dependent on the cAMP/PKA signaling pathway.
The phosphorylation of Brd4 is critical for its activation and also mediates alterations in its interactome. Brd4 hyperphosphorylation correlates with its oncogenic potential, and increased phosphorylation levels lead to the development of BET inhibitor resistance in certain types of cancer (46, 47). At present, casein kinase 2 (CK2) and CK1δ are the only protein kinases demonstrated to directly phosphorylate Brd4 in vivo, although others have been shown to regulate Brd4 activity (35, 46, 48, 49). The CK2 and CK1δ phosphorylation sites reside within the PDID domain, where phosphorylation results in a conformational change that unmasks the second bromodomain and enables interactions with acetyllysine residues (35). An in vitro kinase screen of the PDID domain identified PKA as a potential Brd4 kinase, aligning with the PKA regulatory effect on Brd4 we observed (35). This suggests that in cardiomyocytes, enhanced PKA activity could increase PDID phosphorylation and drive the conformational change required for Brd4 to interact with chromatin. Further work remains to identify the putative phosphorylated sites and elucidate their functional role(s). Conversely, we also observed an increase in basal Brd4 occupancy following PKA inhibition (Fig. 9A). Such increased Brd4 occupancy may be related to PKA’s known role in regulating histone deacetylases (50). The balance between these two opposing processes regulated by PKA is likely linked to the highly localized nature of PKA signaling through interactions with A kinase-anchoring proteins (AKAPs) (51).
RNA-seq analyses revealed that the transcriptional programs triggered by the activation of α1-AR or ETR were highly overlapping, consistent with the similar hypertrophic response downstream of either receptor. The mechanistic difference between the two pathways instead was linked to the fact that specific groups of genes relevant to the hypertrophic response were differentially sensitive to JQ1 downstream of α1-AR activation compared to that of ETR. Previous reports have identified inflammatory pathways enriched in JQ1-attenuated genes using both in vitro and in vivo models of cardiomyocyte hypertrophy (20). We observed JQ1-selective attenuation of these inflammatory pathways following α1-AR activation (Fig. 7G). We argue that this difference stems from a greater role for Brd4 downstream of α1-AR, consistent with our Brd4 ChIP results. Such inflammatory responses are characteristic of heart failure, and the production of several cytokines is increased during cardiac remodeling (52, 53). Importantly, inflammation is a driver of cardiomyocyte hypertrophy, and the inhibition of those pathways prevents the progression of heart failure (54, 55). Although specific pathways were not enriched among genes attenuated by JQ1 following ETR activation, a large number of genes did exhibit JQ1 sensitivity. We suspect the observed effects are due to functions of Brd2 and/or Brd3 in regulating these genes, although further work is required to confirm this.
Further evidence for the effect of JQ1 on the inflammatory response is the negative effect on inflammatory transcription factors, such as NF-κB and AP-1 (Fig. 7H) (34, 56, 57). We focused on these transcription factors for two reasons: their gene expression signatures were previously identified in JQ1-sensitive, TAC-induced genes, and a causal role in regulating Brd4 recruitment in cardiomyocytes has been identified (18, 19). Importantly, these transcription factors are also directly implicated in driving pathological cardiomyocyte hypertrophy in various in vitro and in vivo models (58–62). Despite the fact that similar transcription factors were activated downstream of α1-AR and ETR, JQ1 only attenuated the α1-AR response. The receptor-specific attenuation of transcription factor activity may be due to distinct signaling mechanisms required for transcription factor activation, creating a differential dependence on Brd4 activity (63–66). However, the greater number of transcription factors attenuated by JQ1 following α1-AR activation suggests that it leads to an active form of Brd4 that more readily binds to chromatin and promotes the activity of transcription factors. Although we predict that PKA activates Brd4 directly, indirect activation through a downstream factor is also possible. Notably, a direct role for PKA activity in regulating P-TEFb also has been observed. For example, PKA-dependent phosphorylation of Cdk9 promotes its association with the viral transactivator Tat, and phosphorylation of hexamethylene bisacetamide inducible protein 1 (HEXIM1) by PKA releases P-TEFb from the inhibitory 7SK sRNP complex (67, 68). Thus, P-TEFb itself could be an important PKA target, although how these phosphorylation events impact its interaction with Brd4 is not known. Further investigation is required to determine the effects of JQ1 on Brd4’s interactome and phosphorylation status following α1-AR or ETR activation.
The receptor-specific activation of Brd4 we identified may have implications for the clinical use of Brd4 inhibitors in cardiovascular disease. Efficacy will be dependent on the specific neurohormonal signaling pathways altered in a particular patient. Specifically, we would expect a negative correlation with a patient’s ET-1 levels. Therefore, more extensive characterization of signaling molecules in patients might be important predictors of drug efficacy. Importantly, we expect that Brd4 inhibition will be less effective in severe and/or late stages of heart failure as PKA activity is reduced (41) and levels of endothelin-1 or its precursors (69–71) increases. The chronic infusion of these neurohormones in mice is required to determine how the receptor-specific effects of Brd4 inhibition affect cardiac remodeling. Furthermore, although JQ1 has been demonstrated to reverse established heart failure in mouse TAC and myocardial infarction mouse models (20), we expect JQ1 efficacy would decrease as heart failure progresses.
Our finding that JQ1 sensitivity is dependent on the activation of PKA signaling raises the question of whether Brd4 inhibition is also an effective therapeutic for other pathologies associated with enhanced PKA signaling. For example, the chronic activation of PKA is a hallmark of dopamine-dependent neuronal pathologies, such as cocaine addiction and L-DOPA-induced dyskinesia (LID) (72, 73). Recent studies have implicated Brd4 in regulating the neuronal transcriptional programs and behavioral effects driven by dopamine signaling in these contexts. The systemic administration of JQ1 reduces reward-seeking behavior in addiction models and prevents LID development in Parkinson’s models (74, 75). The correlation between PKA and Brd4 activity in these cases suggests that the regulation of Brd4 chromatin occupancy by PKA is a common regulatory mechanism for other GPCRs and cell types. Furthermore, certain adrenocortical adenomas are driven by enhanced basal PKA activity due to activating mutations in Gαs or the catalytic subunit of PKA (76, 77). We expect these adenomas would be highly sensitive to Brd4 inhibition, although further work is required to establish the requirement of Brd4 in the progression of these cancers.
The dysregulated activation and recruitment of P-TEFb is an underlying cause of several diseases and developmental disorders (78). While the recruitment of P-TEFb is regulated either by Brd4 or the SEC, little is known about the functional relationship between these two complexes. It has been suggested that these complexes work together to target P-TEFb to different substrates, whereas others have shown that Brd4 assists in recruiting the SEC (79, 80). Our results indicate the cooperative nature of these two complexes is dependent on the signaling pathway employed to activate transcriptional responses. Following ETR activation, the SEC alone is sufficient to elicit a gene expression program required for cardiomyocyte hypertrophy, whereas the activation of the Gαs/cAMP/PKA pathway by the α1-AR leads to an additional dependence on Brd4. This suggests that the SEC form of P-TEFb has a more general transcriptional regulatory role than that associated with Brd4, which may have a more restricted signal-responsive role. Further genome-wide investigation is required to assess whether these complexes have distinct or overlapping functions in cardiomyocytes. Furthermore, expanding our understanding of how signaling pathways activate these complexes is an important step to improve therapeutic approaches in diseases with dysregulated P-TEFb.
MATERIALS AND METHODS
Primary neonatal rat cardiomyocyte isolation, tissue culture, transfection, and treatments.
Unless otherwise stated, all reagents were obtained from Sigma. Primary rat cardiomyocytes were isolated from 1- to 3-day-old Sprague-Dawley rats (Charles River Laboratories, St-Constant, Quebec, Canada) as previously described, with minor modifications (81). Following isolation, cardiomyocytes were seeded at a density of 40,000 cells/cm2 on tissue culture dishes coated with 0.1% gelatin and 10 μg/ml fibronectin in Dulbecco’s modified Eagle medium (DMEM) low glucose (Wisent) supplemented with 7% (vol/vol) fetal bovine serum (FBS; Wisent), 1% (vol/vol) penicillin-streptomycin (P/S), and 10 μM cytosine-β-d-arabinoside (MP Biomedicals). After 24 h, plates were washed twice with DMEM low glucose, and medium was changed to cardiomyocyte maintenance medium (DMEM low glucose, 1% [vol/vol] insulin-selenium-transferrin [Wisent], and 1% [vol/vol] P/S) with 10 μM cytosine-β-d-arabinoside. Twenty-four hours later, medium was replaced with fresh cardiomyocyte maintenance medium, and experiments were initiated 24 h later. Cardiomyocytes were maintained at 37°C with 5% CO2, and typical cultures contained >90% cardiomyocytes. Cardiomyocytes were treated with endothelin-1 (Bachem), phenylephrine, iCdk9 (Novartis), KL-2 (ProbeChem Biochemicals), JQ1, alprenolol, KT5720, forskolin, 3-isobutyl-1-methylxanthine (IBMX), or isoproterenol.
For small interfering RNA (siRNA) transfection (siGENOME SMARTPool; Horizon Discover), cardiomyocytes were pelleted at 400 × g for 5 min at 4°C after isolation, resuspended in DMEM low glucose supplemented with 2.5% (vol/vol) FBS, and plated at a density of 60,000 cells/cm2. Cardiomyocytes were transfected with 50 nM siRNA for the specified target gene with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. After 5 h of incubation, the medium was replaced with DMEM low glucose supplemented with 7% (vol/vol) FBS, 1% (vol/vol) P/S, and 10 μM cytosine-β-d-arabinoside and cultured as previously described.
Immunofluorescence and measurement of cell area.
Cardiomyocytes were plated in 96-well plates and cultured as described above. Following the indicated treatment, cells were fixed with methanol for 5 min at –20°C, permeabilized with 0.2% (vol/vol) Triton X-100 in phosphate-buffered saline (PBS) for 5 min at room temperature, and blocked with 10% horse serum (Wisent) in PBS for 1 h at room temperature. Primary anti-α2-actinin antibody (A7811; 1/200; Sigma) in 10% horse serum–PBS was incubated with cardiomyocytes overnight at 4°C. The following day, the fixed cardiomyocytes were incubated with anti-mouse Alexa Fluor 488-conjugated secondary antibody (A-11029; 1/500; Invitrogen) in 10% horse serum–PBS for 1 h at room temperature and 10 min with Hoechst dye (1 μg/μl; Invitrogen) in PBS at room temperature. Stained cardiomyocytes were imaged with an Operetta high-content screening system (PerkinElmer) with ×20 magnification and analyzed with the Columbus image analysis system (PerkinElmer). Hoechst dye was excited with a 360- to 400-nm filter and emissions were detected at 410 to 480 nm; Alexa Fluor 488 was excited with a 460- to 490-nm filter and emissions were detected at 500 to 550 nm. The average from two technical replicates was taken for all treatments.
AKAR4-NLS AAV transduction and FRET experiments.
The AKAR4-NLS construct was a gift from Jin Zhang (UCSD), and pAAV-CAG-GFP was a gift from Karel Svoboda (plasmid number 28014; Addgene) (82, 83). To generate the AKAR4-NLS biosensor for adeno-associated virus (AAV) production, the AKAR4-NLS biosensor was excised and cloned into the pAAV backbone using BamHI and EcoRI (New England Biolabs) at the 5′ and 3′ end, respectively. For AAV production, HEK 293T cells were maintained in DMEM high glucose supplemented with 10% (vol/vol) FBS and 1% (vol/vol) P/S in a controlled environment of 37°C and 5% CO2. Adeno-associated viruses were produced as previously described (84).
Twenty-four hours after plating cardiomyocytes, medium was changed to cardiomyocyte maintenance medium with AAV9-packaged AKAR4-NLS biosensor at a multiplicity of infection (MOI) of 5,000. Following 24 h of transduction, medium was changed to cardiomyocyte maintenance medium and changed every 24 h until the experiment was performed. After 48 h of incubation, cardiomyocyte maintenance medium was removed and cells were washed with Krebs solution (146 mM NaCl, 4.2 mM KCl, 0.5 mM MgCl2, 1 mM CaCl2, 10 mM HEPES [pH 7.4], 1 g/liter glucose) and incubated for 1 h at 37°C with 5% CO2 in Krebs solution prior to FRET readings. All cardiomyocyte FRET experiments were performed using the Opera Phenix high-content screening system (PerkinElmer) with the confocal setting at ×40 magnification at 37°C and 5% CO2 and analyzed with the Columbus Image Analysis System (PerkinElmer). Each well was excited with 425-nm light and emissions detected at 434 to 515 nm for cyan fluorescent protein (CFP) and 500 to 550 nm for yellow fluorescent protein (YFP). Basal FRET images were obtained prior to the addition of agonist, and stimulated FRET images were obtained 15 min after the addition of agonist to the indicated final concentration. For experiments requiring a β-AR antagonist, 1 μM alprenolol was added to cardiomyocytes 30 min prior to obtaining basal FRET images. The FRET ratio was calculated as YFP emission/CFP emission. For all experiments, ΔFRET refers to the following formula: (stimulated agonist FRET ratio – basal agonist FRET ratio) – (stimulated vehicle FRET ratio – basal FRET ratio). The average from three technical replicates was taken for all treatments.
Following FRET experiments, cardiomyocytes were stained with 5 μM Draq5 at room temperature for 5 min. Images were obtained on the Opera Phenix high-content screening system (PerkinElmer) using the confocal setting at ×40 magnification. Draq5 was imaged using 640-nm excitation and emission detected at 650 to 760 nm, YFP using 425-nm excitation and emission detected at 434 to 515 nm, and CFP using 425-nm excitation and emission detected at 500 to 550 nm.
RT-qPCR.
Following the indicated treatments of cardiomyocytes, cells were lysed in TRI Reagent and RNA was extracted by following the manufacturer’s protocol. Reverse transcription was performed with random hexamer primers using a Moloney murine leukemia virus (MMLV)-RT platform (Promega) according to the manufacturer’s protocol. Subsequent qPCR analysis was performed with BrightGreen 2× qPCR master mix (Applied Biological Materials, Inc.) on a Bio-Rad 1000 series thermal cycling CFX96 optical reaction module. Threshold cycle (CT) values were normalized to U6 snRNA, and fold change over the respective control was calculated using the 2−ΔΔCT method. Primer sequences were the following: Nppb, 5′-CAATCCACGATGCAGAAGCTG-3′ and 5′-TTTTGTAGGGCCTTGGTCCTTT-3′; Nppa, 5′-CCTGGACTGGGGAAGTCAAC-3′ and 5′-ATCTATCGGAGGGGTCCCAG-3′; Serpine1, 5′-TCCTCGGTGCTGGCTATGCT-3′ and 5′-TGGAGAGCTTTCGGAGGGCA-3′; and U6 snRNA, 5′-TGGAACGATACAGAGAAGATTAG-3′ and 5′-GAATTTGCGTGTCATCCTTG-3′.
RNA-seq analysis.
RNA was isolated with the RNeasy minikit (Qiagen) according to the manufacturer’s instructions. Libraries were prepared using the NEBNext rRNA-depleted (HMR) stranded library kit and single-read 50-bp sequencing completed on the Illumina HiSeq 4000 at the McGill University and Génome Québec Innovation Centre, Montréal, Canada. Reads were trimmed with Trim Galore (0.6.0) (85, 86) using the following settings: –phred33 –length 36 -q 5 –stringency 1 -e 0.1. Following processing, reads were aligned to the Ensembl rat reference genome (Rattus_norvegicus.Rnor_6.0.94) (87) with STAR (2.7.1a) (88). Transcripts were assembled with StringTie (1.3.4d) (89) and imported into R (3.6.1) with tximport (1.12.3) (90). Differential gene expression was assessed with DESeq2 (1.24.0) (91) with the independent hypothesis weighting (IHW) library for multiple testing adjustment (92). Heat maps and K-means clustering were completed with pheatmap, and the removeBatchEffect function from limma (3.40.6) (93) was used prior to data visualization. Pathway analysis was completed with Ingenuity Pathway Analysis (IPA; Qiagen Inc.; https://www.qiagenbio-informatics.com/products/ingenuity-pathway-analysis) (94). The data are available at the NCBI Gene Expression Omnibus (GEO) under the accession GSE147402. Our code to analyze the RNA-seq data is available at https://github.com/tannylab/Cardiomyocyte-RNA-seq.git.
Chromatin immunoprecipitation-qPCR.
Preparation and immunoprecipitation of cardiomyocyte chromatin were performed as previously described, with minor modifications (95). Following the indicated treatments, cardiomyocytes were cross-linked with 1% formaldehyde in DMEM low glucose for 10 min at room temperature with slight agitation. Crosslinking was quenched by the addition of glycine to 125 mM final concentration and incubated for 5 min at room temperature with slight agitation. Cardiomyocytes were placed on ice following fixation, washed once with cold PBS, scraped into PBS with 1 mM phenylmethylsulfonyl fluoride (PMSF), and pelleted at 800 × g for 5 min at 4°C. The pellet was resuspended in lysis buffer (10 mM Tris-HCl [pH 8.0], 10 mM EDTA, 0.5 mM EGTA, 0.25% Triton X-100, 1 mM PMSF, 1× protease inhibitor cocktail) and incubated for 10 min at 4°C on a nutator. Nuclei were pelleted at 800 × g for 5 min at 4°C and resuspended in nucleus lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS, 1 mM PMSF, 1× protease inhibitor cocktail). Nuclei were incubated for 15 min on ice, followed by sonication with a BioRuptor (Diagenode) (18 cycles, 30 s on/off, high power). Insoluble cellular debris was removed by centrifugation at 14,000 × g for 10 min at 4°C. A small aliquot was taken for quantification and the remaining sample stored at −80°C until use. The aliquot was incubated at 65°C overnight to reverse cross-links, treated with RNase A (50 μg/ml) for 15 min at 37°C, and then treated with proteinase K (200 μg/ml) for 1.5 h at 42°C. Protein was removed by phenol-chloroform extraction and DNA was precipitated at −80°C with 0.3 M sodium acetate (pH 5.2), 2.5 volumes of 100% ethanol, and 20 μg of glycogen. Samples were centrifuged for 20 min at 16,000 × g, and the pellet was washed with 70% ethanol, resuspended with double-distilled water, and quantified using a NanoDrop spectrophotometer (Thermo Fisher) to determine the concentration of chromatin for each sample.
For immunoprecipitations, 10 μg of chromatin was diluted 9-fold with dilution buffer (16.7 mM Tris-HCl [pH 8.0], 1.2 mM EDTA, 167 mM NaCl, 0.01% SDS, 1.1% Triton X-100, 1× protease inhibitor cocktail). Schizosaccharomyces pombe chromatin, prepared as previously described (96), was spiked in to each sample for normalization. A rabbit anti-Brd4 antibody (A301-985A; 5 μg; Bethyl) or rabbit IgG antibody (12-370; 5 μg; Millipore), as well as anti-S. pombe H2B antibody (ab188271; Abcam), was added to respective IPs, and 1% input sample was taken for subsequent analysis. Each IP was incubated at 4°C overnight on a nutator, followed by the addition of 15 μl protein G Dynabeads (Invitrogen) in dilution buffer for 4 h. Beads were washed twice with low-salt buffer (20 mM Tris-HCl [pH 8.0], 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100), twice with high-salt buffer (20 mM Tris-HCl [pH 8.0], 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100), once with LiCl buffer (10 mM Tris [pH 8.0], 1 mM EDTA, 0.25 M LiCl, 1% NP-40, 1% deoxycholate), and once with TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) at 4°C. Beads were resuspended in elution buffer (200 mM NaCl, 1% [wt/vol] SDS) and heated at 65°C for 20 min to elute chromatin. The eluted chromatin was incubated at 65°C overnight to reverse cross-links and then incubated with proteinase K (200 μg/ml) for 2 h at 37°C. DNA was purified and quantified as described above.
Localization was assessed by qPCR with primers for specific genomic loci; a primer pair amplifying S. pombe cdc2+ was used for normalization. All qPCRs were performed using a Bio-Rad 1000 series thermal cycling CFX96 optical reaction module and iQ SYBR green supermix (Bio-Rad). For each primer pair in a given experimental condition, percent input for IgG control IP was subtracted from the percent input for the Brd4 IP, followed by normalization to the percent input of S. pombe cdc2+. Primer sequences were the following: Nppb SE (chromosome 5 [chr5] 164778453 to 164778528, 5′-AGGTGGCACCCCCTCTTCTAC-3′ and 5′-TTGGGGGAGTCTCAGCAGCTT-3′), Nppb TSS (chr5 164796330 to 164796402, 5′-TTTCCTTAATCTGTCGCCGC-3′ and 5′-GGATTGTTCTGGAGACTGGC-3′), Nppa TSS (chr5 164808403 to 164808456, 5′-GTGACGGACAAAGGCTGAGA-3′ and 5′-ATGTTTGCTGTCTCGGCTCA-3′), Serpine1 SE #1 (chr12 22636488 to 22636538, 5′-TCCCCCGCTAACTCGAACGC-3′ and 5′-TTGTTTGGAGAGCCACCAGGC-3′), Serpine1 SE #2 (chr12 22634466 to 22634539, 5′-TTGAGTGGCAGACAGCCGACA-3′ and 5′-GGCGGCCTCCAACATTCCTC-3′), Serpine1 TSS (chr12 22640931 to 22641011, 5′-AGCCCCACCCACCTTCTAACTC-3′ and 5′-TACTGGGAGGGAGGGAAGGAGA-3′), Ctgf SE #1 (chr1 21871291 to 21871393, 5′-AGCCCTGGAATGCTGTTT-3′ and 5′-ACCGCATGATATCTCCTAAACC-3′), Ctgf SE #2 (chr1 21984665 to 21984753, 5′-AGTGAGTCAGGGAGGAAGAA-3′ and 5′-CTCCTGCAGCCTGTGATTAG-3′), Ctgf TSS (chr1 21854660 to 21854725, 5′-CAGACCCACTCCAGCTCCGA-3′ and 5′-GTGGCTCCTGGGGTTGTCCA-3′), Fos TSS (chr6 109300463 to 109300526, 5′-GACTGGATAGAGCCGGCGGA-3′ and 5′-CAGAGCAGAGCTGGGTGGGA-3′), and S. pombe cdc2+ (II 1500254 to 1500328, 5′-ATCATTCTCGCATCTCTATTA-3′ and 5′-ATTCTCCATTGCAAACCACTA-3′).
Protein extraction and Western blotting.
Treated cardiomyocytes were lysed in radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 0.5% sodium deoxycholate) and protein was quantified by Bradford assay. Proteins were denatured at 65°C for 15 min in Laemmli buffer, and protein expression was assessed by Western blotting. Western blots were probed with anti-Brd4 (A301-985A; 1:1,000; Bethyl) or anti-Hsp90 (AC88; 1:1,000; Enzo Life Sciences) in 5% milk overnight at 4°C. The following day, blots were visualized with peroxidase-conjugated secondary antibodies and an Amersham Imager 600.
Statistical analysis.
All statistical analyses were performed using GraphPad Prism 8 software. Two-way analysis of variance (ANOVA) was performed, followed by post hoc t tests with Bonferroni correction (Fig. 1B, 2B, C, and E, 3B and C, 5B and C, and 9A and C). An unpaired t test was completed for the validation of gene knockdown by siRNA (Fig. 2D and 5A) and for Brd4 ChIP with forskolin and IBMX (Fig. 9B). One-way analysis of variance followed by Dunnett’s post hoc comparison was performed for Brd4 ChIP (Fig. 6A) and Brd4 protein expression (Fig. 6C) following 24 h of receptor activation. For DAVID GO term enrichment (Fig. 7G), the false discovery rate (FDR) was calculated by Fisher’s exact test completed within DAVID. For the upstream regulator prediction (Fig. 7H), the P value was obtained by Fisher’s exact test completed within the Ingenuity Pathway Analysis software.
Data availability.
The data determined here are available at the NCBI Gene Expression Omnibus (GEO) under accession number GSE147402.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Heart and Stroke Foundation of Canada (G-15-0008938) to T.E.H. and J.C.T., a grant from the Canadian Institute of Health Science (Canadian Institutes for Health Research [CIHR]) (MOP 130-362) to J.C.T., and a grant from CIHR (PJT 159687) to T.E.H. R.M. was supported by a studentship from the McGill-CIHR Drug Development Training Program and McGill Faculty of Medicine.
We thank Jin Zhang (UCSD), Karel Svoboda, and Novartis Institutes for Biomedical Research (Emeryville, CA) for providing materials instrumental to this study. We thank the McGill Imaging and Molecular Biology Platform for assistance with our high-content microscopy experiments. Lastly, we thank all the members of T. E. Hébert’s and J. C. Tanny’s laboratories for discussions and critical reading of the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Sutton MG, Sharpe N. 2000. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 2.Nadruz W. 2015. Myocardial remodeling in hypertension. J Hum Hypertens 29:1–6. doi: 10.1038/jhh.2014.36. [DOI] [PubMed] [Google Scholar]
- 3.Gerdes AM. 2002. Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail 8:S264–S268. doi: 10.1054/jcaf.2002.129280. [DOI] [PubMed] [Google Scholar]
- 4.Hill JA, Olson EN. 2008. Cardiac plasticity. N Engl J Med 358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 5.Heineke J, Molkentin JD. 2006. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 6.van Berlo JH, Maillet M, Molkentin JD. 2013. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Investig 123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW II. 1997. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A 94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwase M, Bishop SP, Uechi M, Vatner DE, Shannon RP, Kudej RK, Wight DC, Wagner TE, Ishikawa Y, Homcy CJ, Vatner SF. 1996. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS α overexpression. Circ Res 78:517–524. doi: 10.1161/01.RES.78.4.517. [DOI] [PubMed] [Google Scholar]
- 9.Akazawa H, Komuro I. 2003. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res 92:1079–1088. doi: 10.1161/01.RES.0000072977.86706.23. [DOI] [PubMed] [Google Scholar]
- 10.Ho MK, Su Y, Yeung WW, Wong YH. 2009. Regulation of transcription factors by heterotrimeric G proteins. Curr Mol Pharmacol 2:19–31. doi: 10.2174/1874467210902010019. [DOI] [PubMed] [Google Scholar]
- 11.Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD. 2002. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med 8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 12.Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Vigano V, Stirparo GG, Latronico MV, Hasenfuss G, Chen J, Condorelli G. 2013. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A 110:20164–20169. doi: 10.1073/pnas.1315155110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Q, Li T, Price DH. 2012. RNA polymerase II elongation control. Annu Rev Biochem 81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. 2010. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 16.Shi J, Vakoc CR. 2014. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. 2007. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A 104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, Haldar SM. 2013. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, Ambardekar AV, Sucharov CC, Bradner JE, Haldar SM, McKinsey TA. 2016. Signal-dependent recruitment of BRD4 to cardiomyocyte super-enhancers is suppressed by a microRNA. Cell Rep 16:1366–1378. doi: 10.1016/j.celrep.2016.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM. 2017. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med 9:eaah5084. doi: 10.1126/scitranslmed.aah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, Bradner JE, McKinsey TA. 2013. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol 63:175–179. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo Z, Lin C, Shilatifard A. 2012. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol 13:543–547. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- 23.Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, Shilatifard A. 2011. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 25:1486–1498. doi: 10.1101/gad.2059211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KS, Allis CD, Roeder RG. 2011. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A 108:15751–15756. doi: 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, Zackai EH, Deardorff MA, Clark D, Allen JL, Dorsett D, Misulovin Z, Komata M, Bando M, Kaur M, Katou Y, Shirahige K, Krantz ID. 2015. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet 47:338–344. doi: 10.1038/ng.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Gareri C, Rockman HA. 2018. G-protein-coupled receptors in heart disease. Circ Res 123:716–735. doi: 10.1161/CIRCRESAHA.118.311403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clerk A, Sugden PH. 1997. Regulation of phospholipases C and D in rat ventricular myocytes: stimulation by endothelin-1, bradykinin and phenylephrine. J Mol Cell Cardiol 29:1593–1604. doi: 10.1006/jmcc.1997.0395. [DOI] [PubMed] [Google Scholar]
- 28.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW II. 2006. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem 281:1442–1448. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 29.Hong HM, Song EJ, Oh E, Kabir MH, Lee C, Yoo YS. 2011. Endothelin-1- and isoproterenol-induced differential protein expression and signaling pathway in HL-1 cardiomyocytes. Proteomics 11:283–297. doi: 10.1002/pmic.201000018. [DOI] [PubMed] [Google Scholar]
- 30.Lu H, Xue Y, Xue Y, Yu GK, Arias C, Lin J, Fong S, Faure M, Weisburd B, Ji X, Mercier A, Sutton J, Luo K, Gao Z, Zhou Q. 2015. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 4:e06535. doi: 10.7554/eLife.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Liu M, Chen LF, Chen R. 2018. P-TEFb: finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 9:88–94. doi: 10.1080/21541264.2017.1281864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang K, Smith ER, Aoi Y, Stoltz KL, Katagi H, Woodfin AR, Rendleman EJ, Marshall SA, Murray DC, Wang L, Ozark PA, Mishra RK, Hashizume R, Schiltz GE, Shilatifard A. 2018. Targeting processive transcription elongation via SEC disruption for MYC-induced cancer therapy. Cell 175:766–779. doi: 10.1016/j.cell.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. 2010. Selective inhibition of BET bromodomains. Nature 468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon JW, Shaw JA, Kirshenbaum LA. 2011. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res 108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 35.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. 2013. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin RD, Sun Y, Bourque K, Audet N, Inoue A, Tanny JC, Hébert TE. 2018. Receptor- and cellular compartment-specific activation of the cAMP/PKA pathway by α1-adrenergic and ETA endothelin receptors. Cell Signal 44:43–50. doi: 10.1016/j.cellsig.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Depry C, Allen MD, Zhang J. 2011. Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst 7:52–58. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- 38.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. 1987. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 39.Boularan C, Gales C. 2015. Cardiac cAMP: production, hydrolysis, modulation and detection. Front Pharmacol 6:203. doi: 10.3389/fphar.2015.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J. 2001. Β-adrenergic cardiac hypertrophy is mediated primarily by the β(1)-subtype in the rat heart. J Mol Cell Cardiol 33:561–573. doi: 10.1006/jmcc.2000.1332. [DOI] [PubMed] [Google Scholar]
- 41.de Lucia C, Eguchi A, Koch WJ. 2018. New insights in cardiac β-adrenergic signaling during heart failure and aging. Front Pharmacol 9:904. doi: 10.3389/fphar.2018.00904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan Y, Wang L, Du Y, Liu X, Chen Z, Weng X, Guo J, Chen H, Wang M, Wang X. 2018. Inhibition of BRD4 suppresses tumor growth in prostate cancer via the enhancement of FOXO1 expression. Int J Oncol 53:2503–2517. doi: 10.3892/ijo.2018.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu L, Chen Z, Lin X, Tian L, Su Q, An P, Li W, Wu Y, Du J, Shan H, Chiang CM, Wang H. 2020. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ 27:255–268. doi: 10.1038/s41418-019-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, Chen T, Shimizu K, Beca F, Blattner M, Vasudevan D, Buckley DL, Qi J, Buser L, Liu P, Inuzuka H, Beck AH, Wang L, Wild PJ, Garraway LA, Rubin MA, Barbieri CE, Wong KK, Muthuswamy SK, Huang J, Chen Y, Bradner JE, Wei W. 2017. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 23:1063–1071. doi: 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stratton MS, McKinsey TA. 2015. Acetyl-lysine erasers and readers in the control of pulmonary hypertension and right ventricular hypertrophy. Biochem Cell Biol 93:149–157. doi: 10.1139/bcb-2014-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R, Cao XJ, Kulej K, Liu W, Ma T, MacDonald M, Chiang CM, Garcia BA, You J. 2017. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc Natl Acad Sci U S A 114:E5352–E5361. doi: 10.1073/pnas.1703071114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Brown J, D'Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu S-Y, Chiang C-M, Anders L, Young RA, Winer E, Letai A, Barry WT, Carroll JS, Long H, Brown M, Liu XS, Meyer CA, Bradner JE, Polyak K. 2016. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529:413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, Qi J, Lemieux ME, Srivastava D, Lam MPY, Haldar SM, Lin CY, McKinsey TA. 2019. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res 125:662–677. doi: 10.1161/CIRCRESAHA.119.315125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penas C, Maloof ME, Stathias V, Long J, Tan SK, Mier J, Fang Y, Valdes C, Rodriguez-Blanco J, Chiang CM, Robbins DJ, Liebl DJ, Lee JK, Hatten ME, Clarke J, Ayad NG. 2019. Time series modeling of cell cycle exit identifies Brd4 dependent regulation of cerebellar neurogenesis. Nat Commun 10:3028. doi: 10.1038/s41467-019-10799-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG. 2010. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 107:15467–15472. doi: 10.1073/pnas.1000462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehmann LH, Worst BC, Stanmore DA, Backs J. 2014. Histone deacetylase signaling in cardioprotection. Cell Mol Life Sci 71:1673–1690. doi: 10.1007/s00018-013-1516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuusisto J, Karja V, Sipola P, Kholova I, Peuhkurinen K, Jaaskelainen P, Naukkarinen A, Yla-Herttuala S, Punnonen K, Laakso M. 2012. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart 98:1007–1013. doi: 10.1136/heartjnl-2011-300960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erten Y, Tulmac M, Derici U, Pasaoglu H, Altok Reis K, Bali M, Arinsoy T, Cengel A, Sindel S. 2005. An association between inflammatory state and left ventricular hypertrophy in hemodialysis patients. Ren Fail 27:581–589. doi: 10.1080/08860220500200072. [DOI] [PubMed] [Google Scholar]
- 54.Huang S, Frangogiannis NG. 2018. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol 175:1377–1400. doi: 10.1111/bph.14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, Popov AF, Dohmen PM, Choi YH, Wahlers T, Weymann A. 2016. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res 22:75–79. doi: 10.12659/MSMBR.900437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogatsky I, Adelman K. 2014. Preparing the first responders: building the inflammatory transcriptome from the ground up. Mol Cell 54:245–254. doi: 10.1016/j.molcel.2014.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fiordelisi A, Iaccarino G, Morisco C, Coscioni E, Sorriento D. 2019. NFκB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int J Mol Sci 20:E1599. doi: 10.3390/ijms20071599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. 2005. Requirement of nuclear factor-κB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 59.Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. 2001. Activation of NF-κB is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A 98:6668–6673. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. 2006. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 61.Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. 2001. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem 276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 62.Omura T, Yoshiyama M, Yoshida K, Nakamura Y, Kim S, Iwao H, Takeuchi K, Yoshikawa J. 2002. Dominant negative mutant of c-Jun inhibits cardiomyocyte hypertrophy induced by endothelin 1 and phenylephrine. Hypertension 39:81–86. doi: 10.1161/hy0102.100783. [DOI] [PubMed] [Google Scholar]
- 63.Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. 2005. NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. 2009. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol Cell Biol 29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garg R, Caino MC, Kazanietz MG. 2013. Regulation of transcriptional networks by PKC isozymes: identification of c-Rel as a key transcription factor for PKC-regulated genes. PLoS One 8:e67319. doi: 10.1371/journal.pone.0067319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katanasaka Y, Suzuki H, Sunagawa Y, Hasegawa K, Morimoto T. 2016. Regulation of cardiac transcription factor GATA4 by post-translational modification in cardiomyocyte hypertrophy and heart failure. Int Heart J 57:672–675. doi: 10.1536/ihj.16-404. [DOI] [PubMed] [Google Scholar]
- 67.Sun Y, Liu Z, Cao X, Lu Y, Mi Z, He C, Liu J, Zheng Z, Li MJ, Li T, Xu D, Wu M, Cao Y, Li Y, Yang B, Mei C, Zhang L, Chen Y. 2019. Activation of P-TEFb by cAMP-PKA signaling in autosomal dominant polycystic kidney disease. Sci Adv 5:eaaw3593. doi: 10.1126/sciadv.aaw3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garber ME, Mayall TP, Suess EM, Meisenhelder J, Thompson NE, Jones KA. 2000. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol Cell Biol 20:6958–6969. doi: 10.1128/mcb.20.18.6958-6969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang CL, Xie S, Qiao X, An YM, Zhang Y, Li L, Guo XB, Zhang FC, Wu LL. 2017. Plasma endothelin-1-related peptides as the prognostic biomarkers for heart failure: a PRISMA-compliant meta-analysis. Medicine (Baltimore) 96:e9342. doi: 10.1097/MD.0000000000009342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei CM, Lerman A, Rodeheffer RJ, McGregor CG, Brandt RR, Wright S, Heublein DM, Kao PC, Edwards WD, Burnett JC Jr.. 1994. Endothelin in human congestive heart failure. Circulation 89:1580–1586. doi: 10.1161/01.cir.89.4.1580. [DOI] [PubMed] [Google Scholar]
- 71.Rodeheffer RJ, Lerman A, Heublein DM, Burnett JC Jr.. 1992. Increased plasma concentrations of endothelin in congestive heart failure in humans. Mayo Clin Proc 67:719–724. doi: 10.1016/S0025-6196(12)60795-2. [DOI] [PubMed] [Google Scholar]
- 72.Feyder M, Bonito-Oliva A, Fisone G. 2011. L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci 5:71. doi: 10.3389/fnbeh.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lynch WJ, Kiraly DD, Caldarone BJ, Picciotto MR, Taylor JR. 2007. Effect of cocaine self-administration on striatal PKA-regulated signaling in male and female rats. Psychopharmacology (Berlin) 191:263–271. doi: 10.1007/s00213-006-0656-0. [DOI] [PubMed] [Google Scholar]
- 74.A Figge D, Standaert DG. 2017. Dysregulation of BET proteins in levodopa-induced dyskinesia. Neurobiol Dis 102:125–132. doi: 10.1016/j.nbd.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sartor GC, Powell SK, Brothers SP, Wahlestedt C. 2015. Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J Neurosci 35:15062–15072. doi: 10.1523/JNEUROSCI.0826-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weigand I, Ronchi CL, Rizk-Rabin M, Dalmazi GD, Wild V, Bathon K, Rubin B, Calebiro D, Beuschlein F, Bertherat J, Fassnacht M, Sbiera S. 2017. Differential expression of the protein kinase A subunits in normal adrenal glands and adrenocortical adenomas. Sci Rep 7:49. doi: 10.1038/s41598-017-00125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C, Beuschlein F, Reincke M, Lorenz K, Allolio B, Kisker C, Fassnacht M, Lohse MJ. 2014. PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nat Commun 5:5680. doi: 10.1038/ncomms6680. [DOI] [PubMed] [Google Scholar]
- 78.Lee TI, Young RA. 2013. Transcriptional regulation and its misregulation in disease. Cell 152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu X, Zhu X, Li Y, Liu M, Yu B, Wang Y, Rao M, Yang H, Zhou K, Wang Y, Chen Y, Chen M, Zhuang S, Chen LF, Liu R, Chen R. 2016. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res 44:6853–6867. doi: 10.1093/nar/gkw571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. 2011. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. 1998. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Investig 101:812–818. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang JH, Polanowska-Grabowska RK, Smith JS, Shields CWT, Saucerman JJ. 2014. PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to β-adrenergic signaling. J Mol Cell Cardiol 66:83–93. doi: 10.1016/j.yjmcc.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mao T, Kusefoglu D, Hooks BM, Huber D, Petreanu L, Svoboda K. 2011. Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 72:111–123. doi: 10.1016/j.neuron.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burger C, Nash KR. 2016. Small-scale recombinant adeno-associated virus purification. Methods Mol Biol 1382:95–106. doi: 10.1007/978-1-4939-3271-9_7. [DOI] [PubMed] [Google Scholar]
- 85.Krueger F. Trim galore! http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed 10 March 2019.
- 86.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 87.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P. 2018. Ensembl 2018. Nucleic Acids Res 46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soneson C, Love MI, Robinson MD. 2015. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ignatiadis N, Klaus B, Zaugg JB, Huber W. 2016. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods 13:577–580. doi: 10.1038/nmeth.3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kramer A, Green J, Pollard J Jr, Tugendreich S. 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolli P, Vardabasso C, Bernstein E, Chaudhry HW. 2013. Chromatin immunoprecipitation of adult murine cardiomyocytes. Curr Protoc Cell Biol Chapter 17:Unit17.14. doi: 10.1002/0471143030.cb1714s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mbogning J, Tanny JC. 2017. Chromatin immunoprecipitation of histone modifications in fission yeast. Methods Mol Biol 1528:199–210. doi: 10.1007/978-1-4939-6630-1_12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data determined here are available at the NCBI Gene Expression Omnibus (GEO) under accession number GSE147402.