

Abstract
Introduction
N6-methyladenosine (m6A) is one of the most common post-transcriptional modifications in RNA, which has been related to several biological processes. The accurate prediction of m6A sites from RNA sequences is one of the challenging tasks in computational biology. Several computational methods utilizing machine-learning algorithms have been proposed that accelerate in silico screening of m6A sites, thereby drastically reducing the experimental time and labor costs involved.
Methodology
In this study, we proposed a novel computational predictor termed ERT-m6Apred, for the accurate prediction of m6A sites. To identify the feature encodings with more discriminative capability, we applied a two-step feature selection technique on seven different feature encodings and identified the corresponding optimal feature set.
Results
Subsequently, performance comparison of the corresponding optimal feature set-based extremely randomized tree model revealed that Pseudo k-tuple composition encoding, which includes 14 physicochemical properties significantly outperformed other encodings. Moreover, ERT-m6Apred achieved an accuracy of 78.84% during cross-validation analysis, which is comparatively better than recently reported predictors.
Conclusion
In summary, ERT-m6Apred predicts Saccharomyces cerevisiae m6A sites with higher accuracy, thus facilitating biological hypothesis generation and experimental validations.
Keywords: Extremely randomized tree, feature optimization, N6-methyladenosine sites, cross-validation, RNA sequences, Saccharomyces cerevisiae
1. INTRODUCTION
Post-transcriptional modifications in RNA are the variations that occur on a newly transcribed primary RNA transcript. To date, approximately 150 kinds of RNA modifications have been determined [1, 2]. The most abundant RNA modification is N6-methyladenosine (m6A), which is prevalent among viruses, plants, insects, mammals, and eukaryotes such as yeast [3-7]. m6A denotes the methylation at N-6 position of adenosine nucleotide catalyzed by a methyltransferase complex and this reaction is reversible by demethylases (ALKBH5 and FTO). m6A modification has been involved in a series of biological processes, such as mRNA exporting, nascent mRNA synthesis, splicing events, nuclear translation, and translocation [8-10]. Importantly, unusual modifications of m6A have been associated with several diseases, including prostate cancer, thyroid tumor, leukemia, etc. [11-13]. Therefore, accurate identification of m6A modification sites would be of great benefit for cell biologists to better understand the disease mechanism.
Several experimental approaches, including high performance liquid chromatography [14], next-generation sequencing technologies [15, 16], and two-dimensional thin layer chromatography [17] have been widely applied in the identification of m6A sites. Particularly, next-generation sequencing is not available for large-scale genomic sequences’ m6A identification. Overall, these experimental approaches are time-consuming and cost-ineffective, when applied on large-scale genome analysis. Therefore, the development of an accurate and efficient computational method for m6A identification is necessary to complement experimental approaches.
Previous decade has witnessed tremendous growth in the development of various machine-learning (ML)-based methods to predict m6A sites from RNA sequences in different species, such as Homo sapiens, Saccharomyces cerevisiae, Mus musculus, and Arabidopsis thaliana. In this study, we focused on S. cerevisiae because it has been widely recognized as an attractive model organism. To date, 14 prediction models have been developed for this species to predict m6A sites. Chen et al., [18] proposed the first predictor, where they constructed a reliable benchmark dataset of 1307 positive samples (m6A sites) and an equal number of negative samples (non-m6A sites) for S. cerevisiae based on the experimental data [19]. They developed a predictor called “iRNA-Methyl” by employing SVM and pseudo-nucleotide composition features, that achieved an accuracy of 65.59%. Notably, iRNA-Methyl benchmark dataset acted as a base for the development of other methods, including m6Apred [20], pRNAm-PC [21], RNA-MethylPred [22], TargetM6A [23], M6A-HPCS [24], RAM-ESVM [25], RAM-NPPS [26], M6APred-EL [27], iRNA(m6A)-PseDNC [28], iMethyl-STTNC [29], BERMP [30], M6AMRFS [31], and iRNA-Freq [32]. Researchers employed different approaches and feature encoding schemes to improve the predictive performance. A detailed description of the performance of each method and their approaches have been provided in the recent review [33]. Although substantial growth has been made, it still remains a challenging task to extract appropriately useful features to accurately differentiate m6A sites from non-m6A sites.
In the current study, we proposed a novel computational approach called ERT-m6Apred. Firstly, 14 physicochemical properties (PCPs) were incorporated into Pseudo k-tupler composition (PseKNC) and were considered as the features to differentiate m6A from non-m6A samples. To optimize the feature space, we combined F-score and sequential forward search (SFS) using extremely randomized tree (ERT) for enhancing the ability of the feature representation. Our benchmark result shows that the proposed method ERT-m6Apred can achieve improved performance when compared to the existing methods.
2. MATERIALS AND METHODS
2.1. Dataset
Chen et al., [18] constructed a benchmark dataset containing 2614 sequences from S. cerevisiae. Of those, 1307 sequences are positive samples (m6A sites) and 1307 sequences are negative samples (non-m6A sites). Notably, both negative and positive samples are 51-base pair (bp) long with the adenosine in the center position. Furthermore, sequence similarity of this dataset is lower than 85%. This dataset has been widely used in the development of various prediction models [8, 21, 34]. For fair comparison with the existing methods, we also employed the same benchmark dataset for model development. The benchmark dataset can be downloaded from previous work: http://server.malab.cn/M6APred-EL/.
2.2. Feature Encoding Scheme
2.2.1. Pseudo k-tupler Composition (PseKNC)
In this work, we utilized Type-II PseKNC to represent RNA samples, which has been widely applied in previous methods [34-36]. This encoding can efficiently capture short-range and long-range information of RNA sequences. For a given sequence, it is denoted by:
where,
where denotes the counted rank of the correlations along an RNA sequence, is the normalized frequency of the uth k-tuple nucleotide in RNA segment, is the weight factor, and is defined as:
The correlation function is defined as:
where = 14 physicochemical properties (PCPs), which includes 'Tilt', 'Roll', 'Rise', 'Shift', 'Slide', 'Twist', 'GC content', 'Adenine content', Uracil content', 'Stacking energy', 'Bending stiffness', 'Electron_interaction', 'Enthalpy', and 'Entropy.' We note that the above PCPs are based on DNA, which were modified accordingly to RNA sequence (replacing one base pair from Uracil to Thymine). L is the sequence length. is the numerical value of the vth physicochemical index of dinucleotide at position a and denotes the corresponding value of the dinucleotide at position a+b. Based on our preliminary analysis of these three parameters (), we set =1.0, = 0.8, and k = 6 to calculate the Type-II PseKNC that generates 4098-dimensional feature vector.
2.2.2. Feature Optimization
In general, the feature vectors with higher dimensions (>100) have noisy and irrelevant information [37, 38]. Therefore, it is necessary to exclude that information and improve classification accuracy, which is regarded as one of the key steps in ML-based model development. A two-step feature selection scheme was applied that has been widely used in computational biology [39-42]. Firstly, ranking the original features by F-score algorithm and generate a ranked feature list (from highest to the lowest ranked features) [43]. Secondly, two features were selected at each time from the ranked list, and added consecutively to ERT and developed their corresponding models by 10-fold cross-validation (CV). Eventually, the feature set equivalent to the model with the highest accuracy was considered as optimal features.
The F-score of the kth feature is defined as:
where , , and denote an average of the kth feature in the combined (both negative and positive), negative, and positive datasets, respectively. and represent the number of negative and positive samples, respectively. and represent the kth feature of lth negative instance and kth feature of lth positive instance, respectively.
2.2.3. Extremely Randomized Tree
Guertz et al. proposed ERT method in 2006 [44], which has been widely applied in various fields [45-51]. A brief description of how we implemented ERT in this study has been described previously [52, 53]. Grid search approach was implemented to optimize three ERT parameters, which include the number of randomly selected features (mtry), number of trees (ntree), and minimum number of samples required to split an internal node (nsplit). The three parameters search ranges were: 312 with a step size of 1, 401000 with a step size of 20, and 415 with a step size of 1.
2.2.4. Cross-validation
We carried out 10-fold CV test to assess model performance. In 10-fold CV, the benchmark dataset was randomly separated into 10 subgroups with roughly equal size [54-64], with each subgroup containing the same number of m6A and non-m6A samples [65, 66]. One of the subgroups was considered as the validation set to assess the trained model and the remaining subgroups were used to train the model. This step was repeated 10 times by considering each one of the subgroups used at least once as a validation [67-69]. The performance of 10 validation sets was averaged and provided an assessment of the global performance.
2.2.5. Performance Assessment
The commonly used four sets of metrics in the fields of computational biology and bioinformatics [57, 70-77] were utilized to quantitatively evaluate the performance of the proposed method. These metrics included Matthews correlation coefficient (MCC), accuracy (ACC), sensitivity (SN), and specificity (SP), which were computed as follows:
Where TP and TN respectively represent the number of m6A samples and the number of non-m6A samples correctly predicted. FP and FN represent the numbers of m6A or non-m6A samples wrongly predicted, respectively.
3. RESULTS AND DISCUSSION
3.1. Exploration of Different Feature Encodings in m6A Site Prediction
To identify the appropriate feature encoding in m6A site prediction seven feature encodings were explored, including PseKNC, k-mer (concatenation of mono-, di, tri-, tetra-, and penta-nucleotide composition), binary profile (BPF), electron-ion interaction pseudo potential (PseEIIP), numerical representation of nucleotides (NUM), ring-function-hydrogen-chemical (RFHC) properties, and a combination of dinucleotide binary encoding, and local position-specific dinucleotide frequency (DPE_LPF). A detailed description of the other six feature encodings (except PseKNC) has been provided in previous studies [68, 78, 79] and we modified according to RNA sequence. Seven prediction models were developed and the performances are shown in Fig. (1). As shown in Fig. (1), we observed that four encodings (BPF, RFHC, DPE_LPF, NUM) achieved a similar performance, whose ACC in the range of 73.8-75.5%; (ii) the next two encodings (Kmer and PseKNC) achieved the same performance (ACC of 71.8%), which is significantly lower than the above four encodings, and EIIP achieved the worst performance. Finally, none of the featured encodings achieved significant performance over others. It should be noted that all these encodings (except NUM and EIIP) have greater than 100-dimensional feature vector. Hence, we applied SFS to see whether any encoding has an advantage over others in terms of performance.
Fig. (1).
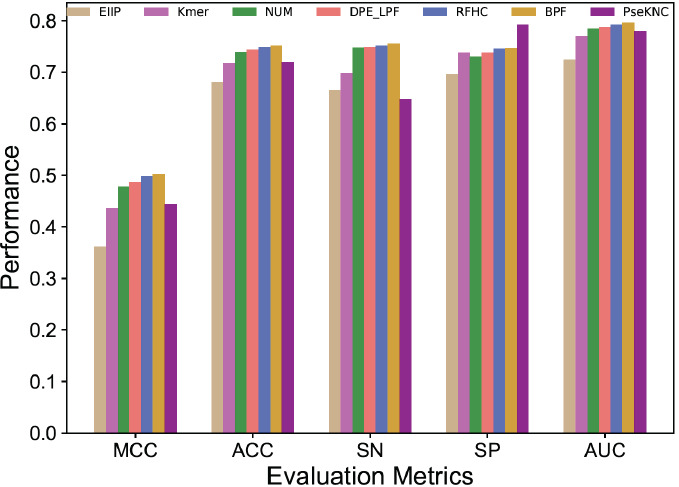
Performance comparison of seven different feature encodings in terms of the Matthews Correlation Coefficient (MCC), Accuracy (ACC), Sensitivity (SN), Specificity (SP) and the Area Under the Curve (AUC). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3.2. Optimal Feature Selection of Each Encoding and their Performance Comparison
Fig. (2A) shows the ACC curves with gradual increment of features for seven encodings. The result shows that all
Fig. (2).
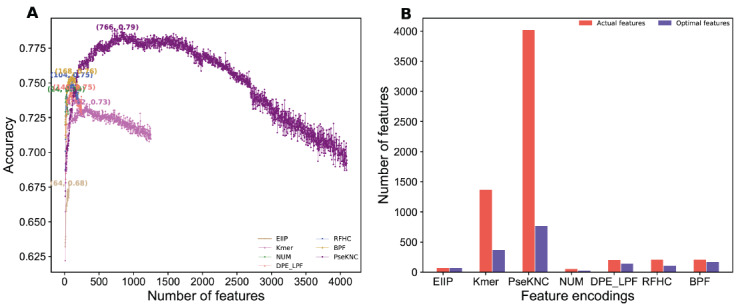
(A) Sequential forward search for distinguishing between m6A and non-m6A sites for seven different encodings. (B) Comparison of the original and optimal features dimension. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
encodings follow the same pattern, where ACC curve gradually improved, reached its highest point and then deteriorated upon the increment of features. Remarkably, two-step feature selection procedure significantly improved the prediction performance in two encodings (Kmer and PseKNC), slight improvement in four encodings (BPF, DPE_LPF, RFHC and NUM), and no improvement in EIIP encoding when compared to their control (using all features). Next, we examined the highest ACC achieved by each encoding corresponding optimal features. Result shows that Kmer, PseKNC, BPF, RFHC, LPDF, and NUM encodings respectively considered 26.60%, 18.7%, 82.36% 50.98%, 70.0%, and 47.1% as the optimal features when compared to their original dimension (Fig. 2B). It appears that all features are equally important in case of EIIP encoding, because of its best performance with all features.
Next, we compared the optimal feature-based performances. Fig. (3) shows that PseKNC achieved the ACC of 78.84%, which is 3.17-10.74% higher than other encodings, thus indicating that PseKNC has more discriminative capability to identify m6A sites. Hence, we selected PseKNC-based model namely, ERT-m6Apred, as the final one.
Fig. (3).
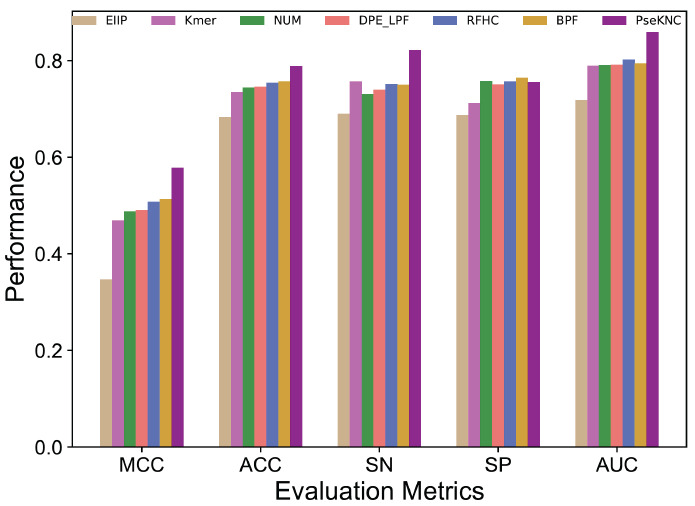
Performance comparison of seven different feature encodings based on optimal feature set encodings in terms of Accuracy (ACC), the Matthews Correlation Coefficient (MCC), Sensitivity (SN), Specificity (SP) and the Area Under the Curve (AUC). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3.3. Comparison with the Existing Methods
To see whether our proposed method is better than the existing predictors, we compared our method with the four recent methods, namely M6AMRFS, BERMP, iMethyl-STTNC and iRNA-Freq. Notably, all these methods were trained and validated on the same benchmark dataset as employed in this study, indicating a fair comparison. As shown in Table 1, our method ERT-m6Apred achieved an ACC of 78.84% and MCC of 0.578. Explicitly, ACC and MCC were 1.7-9.0% and 2.8-20% higher than the existing methods, thus indicating that our predictor ERT-m6Apred is more accurate and balanced than the existing predictors in m6A site prediction.
Table 1. Performance comparison of the proposed predictor with the existing methods.
| Methods | MCC | ACC | SN | SP |
|---|---|---|---|---|
| M6A-ERT | 0.578 | 0.788 | 0.822 | 0.755 |
| M6AMRFS | 0.485 | 0.743 | 0.752 | 0.733 |
| BERMP | 0.430 | 0.713 | 0.730 | 0.696 |
| iMethyl-STTNC | 0.380 | 0.698 | 0.703 | 0.682 |
| iRNA-Freq | 0.550 | 0.771 | 0.836 | 0.719 |
First column represents the method name. The second, third, fourth, and fifth respectively represents Matthews Correlation Coefficient (MCC), Accuracy (ACC), Sensitivity (SN) and Specificity (SP).
CONCLUSION
In this study, we reported a novel predictor for m6A site prediction from RNA sequence. To the best of our knowledge, this is the first instance where ERT and PseKNC were employed in m6A site prediction. Comparative analysis of CV results shows that the proposed predictor is better than existing predictors. Although our proposed method showed improved performance over other methods, it still has room for improvement. Recently, several novel computational approaches have been proposed in computational biology [68, 78, 80-85] to identify function from the sequence. Hence, developing a novel prediction model by utilizing such approaches may improve the prediction performance.
ACKNOWLEDGEMENTS
Declared none.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in M6APred-EL database at http://server.malab.cn/M6APred-EL/ [27].
FUNDING
This work has been supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Sci- ence and Technology [2018R1D1A1B07049572].
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Maden B. The numerous modified nucleotides in eukaryotic ribosomal RNA. Progress in nucleic acid research and molecular biology. Vol. 39. Elsevier; 1990. pp. 241–303. [DOI] [PubMed] [Google Scholar]
- 2.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue Y., Liu J., He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29(13):1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei C.M., Gershowitz A., Moss B. 5′-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15(2):397–401. doi: 10.1021/bi00647a024. [DOI] [PubMed] [Google Scholar]
- 5.Zhong S., Li H., Bodi Z., Button J., Vespa L., Herzog M., Fray R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20(5):1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodi Z., Button J.D., Grierson D., Fray R.G. Yeast targets for mRNA methylation. Nucleic Acids Res. 2010;38(16):5327–5335. doi: 10.1093/nar/gkq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clancy M.J., Shambaugh M.E., Timpte C.S., Bokar J.A. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30(20):4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu N., Pan T. N6-methyladenosine-encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016;23(2):98–102. doi: 10.1038/nsmb.3162. [DOI] [PubMed] [Google Scholar]
- 9.Edupuganti R.R., Geiger S., Lindeboom R.G.H., Shi H., Hsu P.J., Lu Z., Wang S-Y., Baltissen M.P.A., Jansen P.W.T.C., Rossa M., Müller M., Stunnenberg H.G., He C., Carell T., Vermeulen M.N. 6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017;24(10):870–878. doi: 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slobodin B., Han R., Calderone V., Vrielink J. A. O., Loayza-Puch F., Elkon R., Agami R. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell. 2017;169(2):326–337 e12.. doi: 10.1016/j.cell.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akilzhanova A., Nurkina Z., Momynaliev K., Ramanculov E., Zhumadilov Z., Rakhypbekov T., Hayashida N., Nakashima M., Takamura N. Genetic profile and determinants of homocysteine levels in Kazakhstan patients with breast cancer. Anticancer Res. 2013;33(9):4049–4059. [PubMed] [Google Scholar]
- 12.Machiela M.J., Lindström S., Allen N.E., Haiman C.A., Albanes D., Barricarte A., Berndt S.I., Bueno-de-Mesquita H.B., Chanock S., Gaziano J.M., Gapstur S.M., Giovannucci E., Henderson B.E., Jacobs E.J., Kolonel L.N., Krogh V., Ma J., Stampfer M.J., Stevens V.L., Stram D.O., Tjønneland A., Travis R., Willett W.C., Hunter D.J., Le Marchand L., Kraft P. Association of type 2 diabetes susceptibility variants with advanced prostate cancer risk in the Breast and Prostate Cancer Cohort Consortium. Am. J. Epidemiol. 2012;176(12):1121–1129. doi: 10.1093/aje/kws191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heiliger K-J., Hess J., Vitagliano D., Salerno P., Braselmann H., Salvatore G., Ugolini C., Summerer I., Bogdanova T., Unger K., Thomas G., Santoro M., Zitzelsberger H. Novel candidate genes of thyroid tumourigenesis identified in Trk-T1 transgenic mice. Endocr. Relat. Cancer. 2012;19(3):409–421. doi: 10.1530/ERC-11-0387. [DOI] [PubMed] [Google Scholar]
- 14.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C-M., Li C.J., Vågbø C.B., Shi Y., Wang W-L., Song S-H., Lu Z., Bosmans R.P., Dai Q., Hao Y.J., Yang X., Zhao W.M., Tong W.M., Wang X.J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H.E., Klungland A., Yang Y.G., He C. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 16.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keith G. Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie. 1995;77(1-2):142–144. doi: 10.1016/0300-9084(96)88118-1. [DOI] [PubMed] [Google Scholar]
- 18.Chen W., Feng P., Ding H., Lin H., Chou K.C. iRNA-Methyl: identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz S., Agarwala S.D., Mumbach M.R., Jovanovic M., Mertins P., Shishkin A., Tabach Y., Mikkelsen T.S., Satija R., Ruvkun G., Carr S.A., Lander E.S., Fink G.R., Regev A. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155(6):1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W., Tran H., Liang Z., Lin H., Zhang L. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015;5:13859. doi: 10.1038/srep13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z., Xiao X., Yu D-J., Jia J., Qiu W-R., Chou K-C. pRNAm-PC: Predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem. 2016;497:60–67. doi: 10.1016/j.ab.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Jia C-Z., Zhang J-J., Gu W-Z. RNA-MethylPred: a high-accuracy predictor to identify N6-methyladenosine in RNA. Anal. Biochem. 2016;510:72–75. doi: 10.1016/j.ab.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 23.Li G-Q., Liu Z., Shen H-B., Yu D-J. TargetM6A: Identifying N6-methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans. Nanobioscience. 2016;15(7):674–682. doi: 10.1109/TNB.2016.2599115. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M., Sun J-W., Liu Z., Ren M-W., Shen H-B., Yu D-J. Improving N(6)-methyladenosine site prediction with heuristic selection of nucleotide physical-chemical properties. Anal. Biochem. 2016;508:104–113. doi: 10.1016/j.ab.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Chen W., Xing P., Zou Q., Detecting N. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017;7:40242. doi: 10.1038/srep40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing P., Su R., Guo F., Wei L., Identifying N. Identifying N6-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017;7:46757. doi: 10.1038/srep46757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei L., Chen H., Su R. M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther. Nucleic Acids. 2018;12:635–644. doi: 10.1016/j.omtn.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W., Ding H., Zhou X., Lin H., Chou K-C. iRNA(m6A)-PseDNC: identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018;561-562:59–65. doi: 10.1016/j.ab.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Akbar S., Hayat M. iMethyl-STTNC: Identification of N6-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. J. Theor. Biol. 2018;455:205–211. doi: 10.1016/j.jtbi.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y., He N., Chen Y., Chen Z., Li L. BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018;14(12):1669–1677. doi: 10.7150/ijbs.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiang X., Chen H., Ye X., Su R., Wei L. M6AMRFS: robust prediction of N6-methyladenosine sites with sequence-based features in multiple species. Front. Genet. 2018;9:495. doi: 10.3389/fgene.2018.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhuang Y.Y., Liu H.J., Song X., Ju Y., Peng H. A linear regression predictor for identifying N6-methyladenosine sites using frequent gapped K-mer pattern. Mol. Ther. Nucleic Acids. 2019;18:673–680. doi: 10.1016/j.omtn.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu X., He J., Zhao S., Tao W., Xiong Y., Bi S. A comprehensive comparison and analysis of computational predictors for RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Brief. Funct. Genomics. 2019;18(6):367–376. doi: 10.1093/bfgp/elz018. [DOI] [PubMed] [Google Scholar]
- 34.Feng P., Yang H., Ding H., Lin H., Chen W., Chou K.C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. 2019;111(1):96–102. doi: 10.1016/j.ygeno.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Dao F.Y., Lv H., Wang F., Feng C.Q., Ding H., Chen W., Lin H. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. 2019;35(12):2075–2083. doi: 10.1093/bioinformatics/bty943. [DOI] [PubMed] [Google Scholar]
- 36.Lou C., Zhao J., Shi R., Wang Q., Zhou W., Wang Y., Wang G., Huang L., Feng X., Zhou F. sefOri: selecting the best-engineered squence features to predict DNA replication origins. Bioinformatics. 2019;36(1):49–55. doi: 10.1093/bioinformatics/btz506. [DOI] [PubMed] [Google Scholar]
- 37.Tan J.X., Li S.H., Zhang Z.M., Chen C.X., Chen W., Tang H., Lin H. Identification of hormone binding proteins based on machine learning methods. Math. Biosci. Eng. 2019;16(4):2466–2480. doi: 10.3934/mbe.2019123. [DOI] [PubMed] [Google Scholar]
- 38.Yang H., Yang W., Dao F.Y., Lv H., Ding H., Chen W., Lin H. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 2019;•••:bbz123. doi: 10.1093/bib/bbz123. [DOI] [PubMed] [Google Scholar]
- 39.Xu Z-C., Feng P-M., Yang H., Qiu W-R., Chen W., Lin H. iRNAD: a computational tool for identifying D modification sites in RNA sequence. Bioinformatics. 2019;35(23):4922–4929. doi: 10.1093/bioinformatics/btz358. [DOI] [PubMed] [Google Scholar]
- 40.Manavalan B., Lee J. SVMQA: support-vector-machine-based protein single-model quality assessment. Bioinformatics. 2017;33(16):2496–2503. doi: 10.1093/bioinformatics/btx222. [DOI] [PubMed] [Google Scholar]
- 41.Lai H.Y., Zhang Z.Y., Su Z.D., Su W., Ding H., Chen W., Lin H. iProEP: A computational predictor for predicting promoter. Mol. Ther. Nucleic Acids. 2019;17:337–346. doi: 10.1016/j.omtn.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng C.Q., Zhang Z.Y., Zhu X.J., Lin Y., Chen W., Tang H., Lin H. iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. 2019;35(9):1469–1477. doi: 10.1093/bioinformatics/bty827. [DOI] [PubMed] [Google Scholar]
- 43.Ding H., Li D. Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino Acids. 2015;47(2):329–333. doi: 10.1007/s00726-014-1862-4. [DOI] [PubMed] [Google Scholar]
- 44.Geurts P., Ernst D., Wehenkel L. Extremely randomized trees. Mach. Learn. 2006;63(1):3–42. doi: 10.1007/s10994-006-6226-1. [DOI] [Google Scholar]
- 45.Paul A., Furmanchuk A., Liao W.K., Choudhary A., Agrawal A. Property prediction of organic donor molecules for photovoltaic applications using extremely randomized trees. Mol. Inform. 2019;38(11-12):e1900038. doi: 10.1002/minf.201900038. [DOI] [PubMed] [Google Scholar]
- 46.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. AtbPpred: A robust sequence-based prediction of anti-tubercular peptides using extremely randomized trees. Comput. Struct. Biotechnol. J. 2019;17:972–981. doi: 10.1016/j.csbj.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nattee C., Khamsemanan N., Lawtrakul L., Toochinda P., Hannongbua S. A novel prediction approach for antimalarial activities of Trimethoprim, Pyrimethamine, and Cycloguanil analogues using extremely randomized trees. J. Mol. Graph. Model. 2017;71:13–27. doi: 10.1016/j.jmgm.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Soltaninejad M., Yang G., Lambrou T., Allinson N., Jones T.L., Barrick T.R., Howe F.A., Ye X. Automated brain tumour detection and segmentation using superpixel-based extremely randomized trees in FLAIR MRI. Int. J. CARS. 2017;12(2):183–203. doi: 10.1007/s11548-016-1483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xia B., Zhang H., Li Q., Li T. PETS: a stable and accurate predictor of protein-protein interacting sites based on extremely-randomized trees. IEEE Trans. Nanobioscience. 2015;14(8):882–893. doi: 10.1109/TNB.2015.2491303. [DOI] [PubMed] [Google Scholar]
- 50.Scalzo F., Hamilton R., Asgari S., Kim S., Hu X. Intracranial hypertension prediction using extremely randomized decision trees. Med. Eng. Phys. 2012;34(8):1058–1065. doi: 10.1016/j.medengphy.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marée R., Geurts P., Wehenkel L. Random subwindows and extremely randomized trees for image classification in cell biology. BMC Cell Biol. 2007;8(Suppl. 1):S2. doi: 10.1186/1471-2121-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manavalan B., Subramaniyam S., Shin T.H., Kim M.O., Lee G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018;17(8):2715–2726. doi: 10.1021/acs.jproteome.8b00148. [DOI] [PubMed] [Google Scholar]
- 53.Manavalan B., Shin T.H., Kim M.O., Lee G. AIPpred: sequence-based prediction of anti-inflammatory peptides using random forest. Front. Pharmacol. 2018;9:276. doi: 10.3389/fphar.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charoenkwan P., Kanthawong S., Schaduangrat N., Yana J., Shoombuatong W. PVPred-SCM: improved prediction and analysis of phage virion proteins using a scoring card method. Cells. 2020;9(2):E353. doi: 10.3390/cells9020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hasan M.M., Manavalan B., Khatun M.S., Kurata H. i4mCROSE,a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 2019;S0141-8130(19):38547–38542.. doi: 10.1016/j.ijbiomac.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Hasan M.M., Manavalan B., Khatun M.S., Kurata H. Prediction of S-nitrosylation sites by integrating support vector machines and random forest. Mol Omics. 2019;15(6):451–458. doi: 10.1039/C9MO00098D. [DOI] [PubMed] [Google Scholar]
- 57.Su R., Hu J., Zou Q., Manavalan B., Wei L. Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief. Bioinform. 2019;••• doi: 10.1093/bib/bby124. [DOI] [PubMed] [Google Scholar]
- 58.Wei L., Su R., Luan S., Liao Z., Manavalan B., Zou Q., Shi X. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics. 2019;35(23):4930–4937. doi: 10.1093/bioinformatics/btz408. [DOI] [PubMed] [Google Scholar]
- 59.Shoombuatong W., Schaduangrat N., Pratiwi R., Nantasenamat C. THPep: A machine learning-based approach for predicting tumor homing peptides. Comput. Biol. Chem. 2019;80:441–451. doi: 10.1016/j.compbiolchem.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Schaduangrat N., Nantasenamat C., Prachayasittikul V., Shoombuatong W. ACPred: a computational tool for the prediction and analysis of anticancer peptides. Molecules. 2019;24(10):E1973. doi: 10.3390/molecules24101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laengsri V., Shoombuatong W., Adirojananon W., Nantasenamat C., Prachayasittikul V., Nuchnoi P. ThalPred: a web-based prediction tool for discriminating thalassemia trait and iron deficiency anemia. BMC Med. Inform. Decis. Mak. 2019;19(1):212. doi: 10.1186/s12911-019-0929-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laengsri V., Nantasenamat C., Schaduangrat N., Nuchnoi P., Prachayasittikul V., Shoombuatong W. TargetAntiAngio: a sequence-based tool for the prediction and analysis of anti-angiogenic peptides. Int. J. Mol. Sci. 2019;20(12):E2950. doi: 10.3390/ijms20122950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Charoenkwan P., Schaduangrat N., Nantasenamat C., Piacham T., Shoombuatong W. iQSP: a sequence-based tool for the prediction and analysis of quorum sensing peptides via Chou’s 5-steps rule and informative physicochemical properties. Int. J. Mol. Sci. 2019;21(1):E75. doi: 10.3390/ijms21010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uchida T., Furukawa M., Kikawada T., Yamazaki K., Gohara K. Intracellular trehalose via transporter TRET1 as a method to cryoprotect CHO-K1 cells. Cryobiology. 2017;77:50–57. doi: 10.1016/j.cryobiol.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Qiao Y., Xiong Y., Gao H., Zhu X., Chen P. Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinformatics. 2018;19(1):14. doi: 10.1186/s12859-018-2009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He J., Fang T., Zhang Z., Huang B., Zhu X., Xiong Y., Pse U.I., Pse U.I. Pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics. 2018;19(1):306. doi: 10.1186/s12859-018-2321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cao R., Freitas C., Chan L., Sun M., Jiang H., Chen Z. ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules. 2017;22(10):1732. doi: 10.3390/molecules22101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Basith S., Manavalan B., Shin T.H., Lee G. SDM6A: A web-based integrative machine-learning framework for predicting 6mA sites in the rice genome. Mol. Ther. Nucleic Acids. 2019;18:131–141. doi: 10.1016/j.omtn.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Basith S., Manavalan B., Shin T.H., Lee G. iGHBP: Computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput. Struct. Biotechnol. J. 2018;16:412–420. doi: 10.1016/j.csbj.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boopathi V., Subramaniyam S., Malik A., Lee G., Manavalan B., Yang D-C. mACPpred: a support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019;20(8):1964. doi: 10.3390/ijms20081964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manavalan B., Shin T.H., Lee G. DHSpred: support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget. 2017;9(2):1944–1956. doi: 10.18632/oncotarget.23099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manavalan B., Shin T.H., Lee G. PVP-SVM: sequence-based prediction of phage virion proteins using a support vector machine. Front. Microbiol. 2018;9:476. doi: 10.3389/fmicb.2018.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu Q., Xiong Y., Dai H., Kumari K.M., Xu Q., Ou H-Y., Wei D-Q. PDC-SGB: prediction of effective drug combinations using a stochastic gradient boosting algorithm. J. Theor. Biol. 2017;417:1–7. doi: 10.1016/j.jtbi.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 74.Xiong Y., Wang Q., Yang J., Zhu X., Wei D.Q. PredT4SE-stack: prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 2018;9:2571. doi: 10.3389/fmicb.2018.02571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang W., Zhu X.J., Huang J., Ding H., Lin H. A brief survey of machine learning methods in protein sub-Golgi localization. Curr. Bioinform. 2019;14:234–240. doi: 10.2174/1574893613666181113131415. [DOI] [Google Scholar]
- 76.Ding H., Yang W., Tang H., Feng P.M., Huang J., Chen W., Lin H. PHYPred: a tool for identifying bacteriophage enzymes and hydrolases. Virol. Sin. 2016;31(4):350–352. doi: 10.1007/s12250-016-3740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Basith S., Manavalan B., Hwan Shin T., Lee G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 2020;••• doi: 10.1002/med.21658. [DOI] [PubMed] [Google Scholar]
- 78.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol. Ther. Nucleic Acids. 2019;16:733–744. doi: 10.1016/j.omtn.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei L., Luan S., Nagai L.A.E., Su R., Zou Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics. 2019;35(8):1326–1333. doi: 10.1093/bioinformatics/bty824. [DOI] [PubMed] [Google Scholar]
- 80.Wang J., Li J., Yang B., Xie R., Marquez-Lago T.T., Leier A., Hayashida M., Akutsu T., Zhang Y., Chou K.C., Selkrig J., Zhou T., Song J., Lithgow T. Bastion3: a two-layer ensemble predictor of type III secreted effectors. Bioinformatics. 2019;35(12):2017–2028. doi: 10.1093/bioinformatics/bty914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y., Yu S., Xie R., Li J., Leier A., Marquez-Lago T.T., Akutsu T., Smith A.I., Ge Z., Wang J. PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins. Bioinformatics. 2019;1:9. doi: 10.1093/bioinformatics/btz629. [DOI] [PubMed] [Google Scholar]
- 82.Li F., Chen J., Leier A., Marquez-Lago T., Liu Q., Wang Y., Revote J., Smith A.I., Akutsu T., Webb G.I., Kurgan L., Song J. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics. 2019;•••:btz721. doi: 10.1093/bioinformatics/btz721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics. 2019;35(16):2757–2765. doi: 10.1093/bioinformatics/bty1047. [DOI] [PubMed] [Google Scholar]
- 84.Yu B., Qiu W., Chen C., Ma A., Jiang J., Zhou H., Ma Q. SubMito-XGBoost: predicting protein submitochondrial localization by fusing multiple feature information and eXtreme gradient boosting. Bioinformatics. 2019;•••:btz734. doi: 10.1093/bioinformatics/btz734. [DOI] [PubMed] [Google Scholar]
- 85.Wei L., Zhou C., Chen H., Song J., Su R. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics. 2018;34(23):4007–4016. doi: 10.1093/bioinformatics/bty451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of the article is available in M6APred-EL database at http://server.malab.cn/M6APred-EL/ [27].