

Abstract
Iron deficiency (ID) is one of the most prevalent nutritional deficiencies in the world. Iron deficiency in the late fetal and newborn period causes abnormal cognitive performance and emotional regulation, which can persist into adulthood despite iron repletion. Potential mechanisms contributing to these impairments include deficits in brain energy metabolism, neurotransmission, and myelination. Here, we comprehensively review the existing data that demonstrate diminished brain energetic capacity as a mechanistic driver of impaired neurobehavioral development due to early-life (fetal-neonatal) ID. We further discuss a novel hypothesis that permanent metabolic reprogramming, which occurs during the period of ID, leads to chronically impaired neuronal energetics and mitochondrial capacity in adulthood, thus limiting adult neuroplasticity and neurobehavioral function. We conclude that early-life ID impairs energy metabolism in a brain region- and age-dependent manner, with particularly strong evidence for hippocampal neurons. Additional studies, focusing on other brain regions and cell types, are needed.
Keywords: Iron, hippocampus, brain development, mitochondria, energy metabolism, iron deficiency
Introduction
Iron deficiency (ID) is one of the most common global nutritional health problems, affecting an estimated 2 billion people.1,2 Iron deficiency especially targets pregnant women, neonates, and young children with estimated prevalence rates of 15% to 42%3-5 and 80%6 in high- and low-resource countries, respectively. Early-life ID in humans affects learning and memory, gross and fine motor skills, and psychosocial behaviors both during the period of ID and after iron repletion.7-10 These findings are supported by numerous studies in nonhuman primate, pig, sheep, rodent, and cell culture models of early-life ID, which show iron-dependent impairments in the same neurobehavioral domains.7-10 Impaired brain energy metabolism, along with hypomyelination and impaired dopamine signaling, is consistently described as one of the mechanistic causes of the neurodevelopmental deficits associated with early-life ID. The evidence supporting mechanistic roles for myelination and dopamine signaling defects have been reviewed in detail, whereas the brain energy metabolism hypothesis has not received in-depth attention.11-14 Here, for the first time, we comprehensively review the existing data to support this hypothesis and identify gaps in our current knowledge of early-life ID effects on energy metabolism in the developing brain. It is important to be clear that the various proposed mechanisms (ie, disruptions in myelination, dopamine signaling, tissue hypoxia, epigenetic regulation of gene expression, and energy metabolism) are not mutually exclusive and all likely contribute to the neurobehavioral manifestation of early-life ID.
Both PubMed and Google Scholar were used to ensure that this review encompasses all relevant studies. The search terms included “iron and brain development,” “iron deficiency and brain,” “iron deficiency and brain and mitochondria,” “iron deficiency and brain and energy metabolism,” “iron deficiency and brain and ATP,” “iron deficiency and brain and glucose,” “iron deficiency and brain and cytochromes,” “iron deficiency and brain and oxidative phosphorylation,” iron deficiency and brain and glycolysis,” iron deficiency and brain and TCA cycle,” and various combinations of these keywords with more specific energy metabolism molecules (eg, NADH or succinate dehydrogenase). This review exclusively focuses on studies where ID occurs during the fetal and/or early postnatal period and includes data on brain energy metabolism. Studies where the ID was initiated at or after weaning were not considered other than to make relevant comparisons to studies of fetal-neonatal ID regarding timing of the insult. Studies that did not include measurements related to energy metabolism are not discussed.
Key aspects of energy metabolism/mitochondria during brain development
A detailed discussion of the role of energy metabolism in brain development is beyond the scope of this review and can be found in other excellent reviews.15-18 Here, we briefly summarize some key principles. It is important to keep in mind that much of our current knowledge comes from rodent models, where the energy requirements of the developing brain, ~2% of total body oxygen consumption,19 are far less than in humans. In contrast, the newborn human brain accounts for approximately 60% of total body oxygen and glucose utilization.19,20 This demonstrates the high-energy demands of human brain development due to rapid growth and increasing complexity during early life. Because of this, all nutrients are necessary for optimal brain maturation and the developing brain is particularly susceptible to disruption of metabolic homeostasis. The neurobehavioral manifestation of perturbations to nutritional/metabolic regulation of brain development depends on the severity, timing, and duration of the insult.10,17,21 This is because the brain is not a homogeneous organ. It has different regions and cell types, each with their own unique developmental trajectories and age- and cell-dependent nutritional and metabolic needs. Essentially all phases of brain development (eg, neurogenesis, migration, outgrowth/branching of axons and dendrites, synapse formation, myelination, neurotransmission) have high metabolic requirements.15-17 To provide the necessary energy, the developing brain employs cell type–specific mechanisms. For example, neural progenitors and astrocytes primarily rely on glycolysis, whereas postmitotic neurons predominantly utilize oxidative phosphorylation. As we will discuss below, with a few notable exceptions, there is still much to learn about how early-life ID affects brain mitochondria and energy metabolism pathways, especially in terms of timing and brain region and cell type specificity.
Key players in iron-dependent regulation of energy metabolism
Iron mainly participates in energy metabolism through its role as a catalytic component of mitochondrial proteins. Table 1 shows a list of iron-containing proteins known to be directly involved in regulating cellular energy metabolism. In eukaryotic cells, there are no known glycolytic proteins that contain iron. Among the tricarboxylic acid (TCA) cycle enzymes, aconitase and succinate dehydrogenase are iron-dependent. Similarly, cytochrome c and all 4 complexes of the mitochondrial electron transport chain (ETC) contain iron, as Fe/S clusters, heme groups, or both.
Table 1.
Iron-containing proteins involved in brain energy metabolism.
| Protein(s) | Iron group(s) | Metabolic pathway |
|---|---|---|
| Aconitase (IRP1) | 1 Fe-S | TCA cycle, iron homeostasis |
| Succinate dehydrogenase iron-sulfur and cytochrome b subunits (Complex II) | 1 Heme, 3 Fe-S | TCA cycle, ETC |
| NADH dehydrogenase iron-sulfur proteins 1-8 (Complex I) | 8 Fe-S | ETC |
| Ubiquinol: cytochrome c oxidoreductase (Complex III) | 3 Heme, 1 Fe-S | ETC |
| Cytochrome c | 1 Heme | ETC |
| Cytochrome c oxidase (Complex IV) | 2 Heme | ETC |
Abbreviations: ETC, electron transport chain; IRP, iron regulatory protein; TCA, tricarboxylic acid.
The ability of cells to take up iron is a key regulatory point in iron-dependent energy metabolism. Under conditions of iron perturbation, cellular iron uptake and homeostasis are predominately regulated through iron regulatory proteins (IRPs) binding to iron response elements (IREs) in the 5′ or 3′ UTR (untranslated region) of messenger RNA (mRNA). For example, to decrease iron storage and increase iron import during conditions of iron insufficiency, IRPs bind to the IRE in the 5′ UTR of ferritin mRNAs to prevent translation and to the 3′ UTR of transferrin receptor mRNA to block degradation and promote translation. Several mRNAs, encoding protein subunits that are involved in cellular control of energy metabolism, contain IREs in their 5′ UTRs and are regulated by cellular iron status through the IRP/IRE system (Table 2). There is also evidence in non-CNS (central nervous system) cells that IRP-dependent posttranscriptional regulation of transcription factors, such as HIF1A and HIF2A, directly modulates cellular energy metabolism pathway usage.22 The Hif2a (ie, Epas1) gene has an IRE in its 5′ UTR, allowing direct IRP-mediated regulation of HIF2A translation.23 The protein stability of both HIF1A and HIF2A is also regulated by prolyl hydroxylase proteins, which require iron for their activity.24
Table 2.
IRE-regulated genes involved in brain energy metabolism.
| Gene | IRE location | Protein; Metabolic Pathway |
|---|---|---|
| Mitochondrial aconitase 2 (Aco2) | 5′ UTR25 | Aconitase; TCA cycle |
| Succinate dehydrogenase subunit b (Sdhb) | 5′ UTR25 | Succinate dehydrogenase; TCA cycle and ETC |
| NADH: ubiquinone oxidoreductase subunit S1 (Ndufs1) | 5′ UTR26 | NADH dehydrogenase; ETC |
| Endothelial PAS domain protein 1 (Epas1) | 5′ UTR23 | Hypoxia inducible factor 2 alpha; energy metabolism gene regulation |
Abbreviations: ETC, electron transport chain; IRE, iron response elements; IRP, iron regulatory protein; TCA, tricarboxylic acid.
Iron-dependent mitochondrial energy production is also controlled by the mitochondrial-specific iron pool and thus by proteins involved in mitochondrial iron delivery, uptake, and storage. Several mechanisms have been proposed for delivery of iron to mitochondria. In erythroid cells, there is evidence that endosomal iron, from transferrin-mediated uptake, is directly delivered to mitochondria through a “kiss-and-run” mechanism.27 Alternatively, there may be a chaperone protein which mediates the endosome-mitochondrion transfer of ferric iron. Iron released from ferritin storage through NCOA4-mediated ferritinophagy can also be delivered from the lysosome to mitochondria. However, the proteins mediating this mechanism of mitochondrial iron delivery remain to be determined. In HEK293 cells, divalent metal transporter 1 (DMT-1) was recently found to localize to the outer mitochondrial membrane where it could mediate mitochondrial iron uptake.28,29 Thus, DMT-1 may be the bridge between endosomal/lysosomal iron release and mitochondrial iron uptake. Mitoferrin 1 and 2 are inner mitochondrial membrane proteins that mediate iron translocation from the intermembrane space into the mitochondrial matrix, where Fe-S cluster and heme biosynthesis occur. Mitochondrial iron can also be stored in mitoferritin within the matrix. Very little is known about how developing brain cells regulate mitochondrial iron levels under normal or iron-deficient conditions.
Evidence for impaired neonatal brain energy metabolism due to early-life ID
Dysregulated expression of energy metabolism genes and proteins in the neonatal iron-deficient brain
In addition to its structural and enzymatic roles in mitochondrial protein function, iron also has a direct role in epigenetic regulation of gene transcription (recently reviewed by Barks et al10). Iron is required for DNA demethylation and hydroxymethylation through the iron-dependent family of TET hydroxymethylases and histone demethylation through the iron-dependent JmjC ARID-domain-containing histone demethylase family. As a result, early-life ID alters brain expression of a wide range of genes, including genes coding for proteins involved in energy metabolism, such as Hmox1, Hmox2, Glut1, Hk2, Pfkfb3, Pdk2, Ldha, Ndufc1, and Cox6a1.30-34 In particular, early-life rodent ID with anemia (IDA) differentially alters P15 hippocampal transcript abundance for genes encoding ETC complex protein subunits, as shown by Ingenuity Pathway Analysis (IPA) of microarray data (Figure 1A).30
Figure 1.

Electron transport chain gene expression abnormalities in hippocampus of P15 IDA and P65 formerly iron deficiency anemia (IDA) rat. The data presented are new pathway analyses from previously published P15 hippocampal microarray30 or P65 hippocampal RNA-Seq98 data sets from a rat model of fetal-neonatal nutritional IDA with early postnatal-onset (P7) of iron repletion. Hippocampal transcriptomes were analyzed by Ingenuity Pathway Analysis (IPA) and mapped altered gene expression, compared with iron-sufficient controls, onto functional pathways including the mitochondrial electron transport chain, ETC (eg, Cox and Nduf families). (A) Hippocampal ETC pathway gene profiles were altered in P15 IDA neonates, while the brain is iron-deficient.30 Several ETC genes remain altered in (B) P65 formerly iron-deficient adults, after iron repletion restored brain iron levels to normal.98 Green and red shading indicate down- and upregulation, respectively, of ETC complex gene expression. IMM, inner mitochondrial membrane; IMS, intermembrane space; MM, mitochondrial matrix.
Dallman et al.35 were the first to report the effects of early-life ID on energy metabolism pathways in the developing brain when they assessed the effect of the timing of IDA on brain cytochrome c levels. They showed that cytochrome c is 27% lower in the postnatal (P) day 21 rat whole brain following severe IDA that occurred during late pregnancy and nursing. In contrast, when IDA is restricted to the nursing period or is delayed until the postweaning period, brain cytochrome c levels are not altered.35-37 Regardless of the timing of IDA, rat brain cytochrome c is significantly spared compared with non-CNS tissues (ie, liver, skeletal muscle, intestinal mucosa, and heart), consistent with a hierarchal prioritization of available iron when negative iron balance occurs in early life. Nutritional ID beginning in early gestation does not change whole brain cytochrome c oxidase (ETC Complex IV) protein levels in P12 IDA rats.38 P15 cortical and hippocampal lactate dehydrogenase protein levels are not changed by fetal-neonatal IDA.39 To our knowledge, no other studies have assessed the effect of early-life ID on neonatal brain expression of proteins upstream of cytochrome c (ie, glycolytic, TCA cycle, and Complex I-III proteins).
The mechanistic target of rapamycin kinase (mTOR) pathway integrates cellular energy and growth signaling. Early-life ID disrupts mTOR signaling, as evidenced by dysregulation of both gene and protein expression levels throughout the pathway. Fetal-neonatal ID increases P15 hippocampal mRNA expression levels of Ddit4, Fkbp1a, Gltscr2, and Tsc1, upstream regulators of mTOR, which indicates an overall blunting of mTOR signaling.30 This is further supported by a decreased ratio of P-S6K (activated) to total S6K protein, a downstream phosphorylation target of mTOR, in the neonatal iron-deficient rodent hippocampus due to early-life IDA or phlebotomy-induced anemia (PIA).40,41 Hippocampal neuron-specific ID beginning in late gestation also dysregulates mTOR signaling in developing mice, underscoring the specific role of iron in this key metabolic regulatory pathway.42 However, in contrast to the IDA and PIA models, proteins at all levels of the mTOR pathway exhibited hyperphosphorylation at active amino acid sites, suggesting increased mTOR signaling. This apparent upregulation of mTOR signaling throughput may be an attempt to compensate for the lack of an important metabolic substrate (ie, iron). The discordance between IDA and neuron-specific ID effects on mTOR pathway signaling is likely due to differences in the timing, duration, and severity of brain ID in these unique models. Iron deficiency onset is at the end of gestation in the neuron-specific ID models compared with immediately after conception in the IDA, which results in the brain having experienced a shorter and less severe ID at the time of analysis. Additional factors in the IDA model that could contribute to these differences include systemic effects (eg, anemia causing brain hypoxia) and ID in non-neuronal cells.
Early-life ID disrupts neonatal brain energy metabolism enzyme activity
Rao, de Ungria and colleagues43,44 first demonstrated that early-life IDA impairs neonatal brain energy metabolism in a brain region-specific manner using an in situ cytochrome c oxidase activity assay. Cytochrome c oxidase activity is reduced by an average of 27% throughout the 20 brain regions analyzed. Hippocampal subregions, cingulum, orbital cortex, hypothalamic arcuate nucleus, and substantia nigra compacta are most severely affected (32%-42% reduced).43 Cytochrome c oxidase activity is not altered in several regions (eg, amygdala) despite a similar degree of tissue ID compared with the significantly affected regions. Neonatal hypoxic-ischemic injury, which occurs in 0.1% to 2% of all live births and impairs energy-dependent neurodevelopment,45,46 exacerbates the effect of early-life ID on cytochrome c oxidase activity in the hippocampus.44 Consistent with the brain region-specificity of this effect, cytochrome c oxidase activity is only reduced by 15% (nonsignificant trend) in whole brain lysates from P12 IDA rat pups.38 These studies highlight the need for brain region–specific and developmental timing–specific studies, informed by the unique metabolic activity trajectories of each brain region,17 to accurately determine the effects of early-life ID on brain energy metabolism. In the rodent hippocampus, for example, glucose utilization,47-49 iron uptake,50,51 mitochondrial enzyme expression,52 neurotrophic factor expression,53,54 dendritic arborization,55 synaptogenesis,56 calcium signaling,57,58 neurotransmitter receptor expression,59,60 and long-term potentiation (LTP)61,62 all reach peak activity around the second to third postnatal week.17
Neonatal brain-regional metabolite changes during ID
We have determined the effects of early-life brain ID on neonatal brain regional metabolites using in vivo 1H spectroscopy (MRS) in the fetal-neonatal nutritional model of IDA and the mouse PIA model.63-65 This technique permits noninvasive assessment of cerebral energy metabolism by simultaneously determining changes in the concentration of 18 metabolites indexing cellular processes such as energy status, neurotransmission, phospholipid synthesis, and neuronal and glial integrity.66 The metabolomic profile consists of the following 18 metabolites: alanine, glucose, lactate, creatine (Cr), and phosphocreatine (PCr, also known as creatine phosphate) (energy substrates and indicator of resting energy state); glycerophosphocholine (GPC), phosphocholine (PC), and phosphoethanolamine (PE) (phospholipid biosynthesis); N-acetylaspartate (NAA) (neuronal/mitochondrial integrity); aspartate, γ-aminobutyric acid (GABA), glutamate (Glu), glutamine (Gln), glycine, N-acetylaspartylglutamate (NAAG), and taurine (amino acids and neurotransmitters); myo-inositol (osmolyte and glial integrity); ascorbate and glutathione (antioxidants); and macromolecules.66,67 The measured concentrations are predominantly intracellular concentrations, although the exact cellular subtype cannot be determined. The noninvasiveness of the method also allows repeated longitudinal assessments in the same subject, which is advantageous for determining the effects throughout the period of ID and in adulthood after iron treatment.
Longitudinal assessment between P7 and P37 (ie, during the period of brain ID) in the dietary IDA model demonstrated elevated PCr concentration and PCr/Cr ratio in the hippocampus and striatum.63,64 Phosphocreatine is a readily available cytosolic energy store for buffering ATP synthesis when energy demands are increased or ATP synthesis is decreased.68 A decrease in PCr concentration is accompanied by a reciprocal increase in Cr concentration, a reaction mediated by the enzyme creatine kinase. Thus, PCr/Cr ratio is a measure of resting energy status. Conversely, Cr + PCr (total creatine) is a measure of energy store in a brain region. During normal development, Cr, PCr and Cr + PCr concentrations, and PCr/Cr ratio increase in the brain regions, paralleling the period of active growth, myelination, synaptogenesis, and acquisition of function in the region.17,66 Apparent increased resting energy status (elevated PCr and PCr/Cr ratio) in the context of IDA appears counterintuitive given that ID and hypoxia impair oxidative ATP production, creating a state of energy failure. Indeed, during acute energy failure (eg, due to acute hypoxia or hypoglycemia), PCr and PCr/Cr ratio is decreased,69,70 indicating increased utilization of energy storage. However, when energy substrate availability is chronically reduced (eg, chronic energy failure or hibernation), brain energy utilization is also often reduced, presumably as an adaptive mechanism to match energy supply and demand.71,72 Consistent with this possibility, PCr/Cr ratio is also increased in the hippocampus of rats exposed to chronic hypoxia during development.73 Thus, a more likely explanation for the increased PCr and PCr/Cr ratio in the neonatal IDA brain is reduced PCr consumption, secondary to an overall reduction in cerebral energy metabolism as an adaptation to ID-induced chronic subnormal energy production.
The effect of ID on PCr/Cr ratio also appears to be determined by the acuteness of onset and the duration of ID. Neonatal PIA causes a rapid onset and shorter duration IDA with a similar degree of anemia and brain ID as the dietary IDA model.65 Phlebotomy-induced anemia causes a lower PCr/Cr ratio and higher Cr concentration in the hippocampus,65 indicative of acute energy failure.70 Finally, it is noteworthy that increased PCr concentration in the setting of chronic ID does not confer neuroprotection during a superimposed acute energy failure (eg, hypoxia-ischemia), further corroborating that increased PCr/Cr ratio is an indicator of suppressed metabolism and neuronal activity.44,74
The concentrations of aspartate, GABA, glutamate, NAA, PE, taurine, and the Glu/Gln ratio were also higher in the early postnatal ID hippocampus.63 In the developing striatum, the concentrations of glucose, lactate, NAA, myo-inositol, taurine, and GPC + PC and Glu/Gln ratio were higher, and that of Gln lower in the IDA group.64
Glu/Gln ratio reflects glutamate-glutamine cycling between neurons and glia (ie, glutamatergic neurotransmission). Glu released from neurons is taken up by the adjacent astrocytes, converted to Gln, and transferred back to the neurons for reconversion to Glu. Glu uptake and conversion to Gln in astrocytes is an energy-demanding process. In addition, Glu and Gln synthesis is linked to the TCA cycle at the α-ketoglutarate step. Thus, increased Glu and Glu/Gln ratio, indicating suppression of energetically expensive glutamatergic neurotransmission,75 and increased levels of inhibitory neurotransmitter, GABA, are also supportive of decreased cerebral metabolism by reducing overall neuronal activity.
The increased glucose and lactate in the striatum suggests elevated glucose uptake and glycolysis to compensate for impaired iron-dependent mitochondrial oxidative phosphorylation as occurs in non-CNS cells.76 The increased Glut1 expression and cerebral vascular density in the neonatal iron-deficient brain support a mechanism for enhanced brain glucose uptake,33 which would also depend on systemic glucose availability. In postweaning models of ID, blood glucose concentrations and turnover are increased77; however, the effect of early-life ID on systemic glucose regulation has not been studied. Elevated taurine concentrations could also indicate an elevated cerebral glucose utilization rate.78 During normal brain development, glycolysis occurs predominantly in astrocytes and is highest during the period of peak brain functional maturation.79,80 Together, these findings point to increased blood-brain barrier glucose uptake and astrocytic glycolysis as an adaptive response to decreased oxidative energy production in the iron-deficient developing brain.
Iron is required for the function of thyroid peroxidase, the enzyme responsible for production of thyroid hormones in the thyroid gland. Thyroid hormone metabolites are critical regulators of mitochondrial energy metabolism in all tissues, including the developing brain. Triiodothyronine (T3) is the predominant active metabolite form and regulates transcription of nuclear- and mitochondrial-encoded genes involved in mitochondrial biogenesis and oxidative phosphorylation during brain development.81-83 Thyroid hormones also regulate expression and signaling for genes/proteins that control neuronal migration, differentiation, neurite outgrowth, myelination, and synaptic plasticity/function.84 Early-life ID with or without anemia reduces the concentration of T3 in the developing rat whole brain, cortex, and hippocampus, similar to mild hypothyroidism.38,85,86 This results in impaired expression of thyroid hormone-responsive genes in the neonatal cerebral cortex and hippocampus,85,86 an effect that is exacerbated when IDA is combined with a mild chemically induced hypothyroidism.39 Thus, fetal-neonatal IDA causes a mild hypothyroid state in the developing brain, consistent with impaired energy metabolism and reduction in energetically demanding maturation/growth processes.
Effects of early-life ID on cerebral energy metabolism in nonhuman primate infants
Nonhuman primate infants have closer similarity with human infants in brain energy requirements (~9% of total body oxygen consumption),19 genetic makeup, iron biology, neurodevelopmental trajectory, compared with rodents. Thus, investigation using the relatively noninvasive and sensitive MRS-based metabolomic assessment of cerebrospinal fluid (CSF) in rhesus macaque infants with IDA provides more clinically relevant outcome measures of ID effects on cerebral energy metabolism.87,88 In this animal model, ID and anemia occur spontaneously in a subset of the infants.89,90 Nonanemic ID becomes evident at 2 to 4 months, and anemia develops at 6 months. Anemia resolves over the next 4 to 6 months after weaning and the consumption of iron-containing food by the infant. However, the brain remains iron-deficient for 3 to 4 months after the hematology has normalized.91,92 Formerly anemic monkeys demonstrate impaired cognitive performance,93 similar to the effects seen in human infants.94
Six-month-old infant monkeys with IDA have evidence of disrupted energy metabolism in the intrathecal compartment.87 Compared with the iron-sufficient infants, the citrate/pyruvate (Cit/Pyr) and citrate/lactate (Cit/Lac) ratios were lower, and the pyruvate/glutamine (Pyr/Gln) ratio was higher in the iron-deficient infants. These results are consistent with suppressed TCA cycle activity and increased glycolysis in the iron-deficient rodent brain as described above. At 4 months of age, during the pre-anemic period, the Pyr/Gln ratio was already lower in the iron-deficient infant CSF.88 In addition, PCr/Cr ratio, a measure of tissue energy reserves, was lower and remained low after the resolution of anemia. Because CSF was obtained from the cisterna magna in the cervical region, the CSF is primarily drawn from the ventricles, and thus metabolite changes are likely more reflective of the changes in the brain parenchyma than CSF drawn from the lower lumbar region.
Early-life ID impairs energy production and mitochondrial structure/function in developing neurons
To directly measure the functional capacity of energy production pathways in developing iron-deficient neurons, we developed a mouse primary embryonic hippocampal neuron culture model of early-life ID.31 This nearly pure neuronal culture model closely mimics the degree of ID and the deleterious effects of dietary and neuronal-specific ID on neuronal structure and neurodevelopmental gene expression. Mitochondrial respiration is drastically blunted both at the beginning (ie, 11 days in vitro [DIV]) and peak (ie, 18 DIV) periods of rapid neuronal structural development.31,95 At 11 DIV, after 8 days of iron chelation, the oxygen consumption rate associated with basal respiration, maximal respiration, and ATP production are 30% to 60% lower, resulting in a 20% reduction in total intracellular ATP concentration.95 By 18 DIV, all measures of mitochondrial respiration are reduced by over 70%, compared with iron-sufficient neurons.31 In contrast to the in vivo neonatal iron-deficient brain, glycolytic rate and capacity are reduced in developing iron-deficient neuron cultures, suggesting a total dampening of the neuronal energetic capacity. The lack of compensatory increase in glycolysis is likely due to the presence of very few astrocytes, the primary glycolytic cells in the developing brain, in these cultures.
There is also evidence of impaired mitochondrial health and quality control in developing iron-deficient neurons.34,95 Mitochondria in the dendrites of 11 DIV iron-deficient hippocampal neurons move more slowly, primarily due to an increased rate of pausing.95 This effect is more pronounced in the anterograde direction, ultimately leading to decreased mitochondrial density in terminal dendrite segments of 18 DIV iron-deficient neurons. Iron deficiency also shifts the size distribution of dendritic mitochondria, with a significant increase in small, round mitochondria. This may be explained by a decreased rate of mitochondrial fusion, as ID decreases mRNA levels for mitofusin 1 and 2 and optic atrophy 1, which code for key mitochondrial fusion proteins, with little to no change in fission-associated genes. These deficits in mitochondrial motility and health are likely the result of impaired iron-dependent energy production.96
Evidence for persistently impaired energy metabolism due to early-life ID in the adult brain
Given the critical structural and catalytic role of iron in mitochondrial TCA cycle and ETC enzymes, it is not surprising that early-life ID causes extensive disruptions in brain energy metabolism and mitochondrial functions during the period of ID. The human adult brain also has high metabolic requirements (although not nearly as high as during development), accounting for approximately 20% of total body oxygen consumption and glucose utilization.19,20 The largest energy demands are in maintaining the ion gradients and vesicle cycling necessary for synaptic neurotransmission.97 In this section, we explore the evidence for a long-term disruption of brain energy metabolism, which could contribute to long-term neurobehavioral dysfunction, following recovery from early-life ID (Figure 2).
Figure 2.
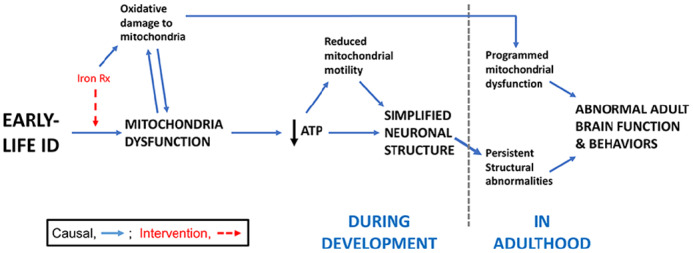
Summary conceptual model connecting energy metabolism deficits to long-term neurobehavioral dysfunction following early-life ID. Fetal and early postnatal ID, with or without anemia, acutely compromises mitochondrial function in the developing brain. This causes reduced cellular energy (ie, ATP) and impaired neuron structural development, ultimately blunting neurobehavioral function in adulthood. While reducing mitochondrial activity and metabolic load is potentially adaptive in the short run during the period of limited substrate availability, it can alter metabolic regulation across the lifespan. Mechanistically, inefficient mitochondrial energy metabolism (due to ID) and/or early-life iron repletion therapy could increase reactive oxygen species generation, causing oxidative damage to brain mitochondria. We hypothesize that permanent metabolic reprogramming, which occurs during the period of ID, leads to chronically impaired neuronal energetics and mitochondrial capacity in adulthood, thus limiting adult neurobehavioral function. Iron Rx = Iron repletion. ATP indicates adenosine triphosphate; ID, iron deficiency.
Transcriptomic data from the adult formerly iron-deficient hippocampus suggest that iron repletion only partially rescues the energy metabolism deficits observed in the neonatal brain of IDA rats (Figure 1B).98 Despite brain iron recovery, there is persistent dysregulation of genes involved in the ETC (eg, Ndufa5, Ndufs7, Ndufb8, and Cox6A1; see Figure 1B compared with Figure 1A), mitochondrial biogenesis/quality control (eg, Pgc1a, Opa1, Fis1, and Dnm1l), mTOR signaling (eg, Tsc1, Tsc2, Hif1a, Mlst8, and Fkbp1a), and thyroid hormone action (eg, Nrgn, Thrsp, and Pvalb).98 In contrast to persistently dysregulated mTOR pathway gene expression in the adult hippocampus, mTOR pathway protein phosphorylation states in the adolescent rodent hippocampus are normalized by early iron repletion in both dietary IDA and transgenic hippocampal neuron-specific ID models.99 Thus, further studies are needed to clarify whether activity of the mTOR pathway is permanently dysregulated by early-life ID. In a rodent proteomic study, Tran et al100 found persistently reduced protein levels of ATP synthase F1 subunit beta, cytochrome c oxidase subunit 4 isoform 1, cytochrome c oxidase subunit 6C, NDUFA4 mitochondrial complex–associated protein, and NADH: ubiquinone oxidoreductase subunit B5 at synapses from the formerly iron-deficient hippocampus. This suggests that permanently impaired energetic capacity may indeed directly contribute to the compromised neuronal structure and synaptic function that persists after recovery from early-life ID (Figure 2 and discussed below).
In a mouse model with less severe fetal-neonatal IDA, adult brain energy metabolism is not disrupted. Whole brain ATP levels and activities for lactate dehydrogenase, aconitase, succinate dehydrogenase, and cytochrome c oxidase are unaffected in mice with persistent moderate ID or those that received iron repletion at weaning.101 This suggests that degree of early-life ID is a critical determinant of whether brain energy metabolism deficits will persist after iron repletion.
The long-term effects of early-life ID on the metabolomic profiles of the brain regions have also been determined using in vivo 1H MRS in formerly iron-deficient adult rats.64,102 Assessment of formerly iron-deficient striatum on P37 demonstrated normalization of the metabolomic profile, likely because of the normalization of tissue ID in this brain region by P21.64,103 Conversely, assessment of the hippocampus at a later time point (P56) demonstrated persistent metabolomic alterations, despite normalization of tissue iron concentration by this time.102 The adult formerly iron-deficient hippocampal surface area is 12% smaller and is accompanied by lower concentrations of Cr, Cr + PCr, lactate, NAAG, and taurine. Hippocampal Gln concentration is higher and the Glu/Gln ratio is lower. As discussed above, lower Cr + PCr concentration is indicative of decreased energy stores. NAAG is a dipeptide consisting of NAA and Glu. Hydrolysis of NAAG by astrocyte-specific NAAG peptidase leads to release of Glu. Lower NAAG and Glu/Gln ratio, along with lower lactate, indicate decreased neuronal activity in the formerly iron-deficient hippocampus.104,105 However, in the setting of lower taurine, decreased lactate also could indicate decreased oxidative glucose metabolism.78 Consistent with this, Glut1 gene and protein expression is decreased in the formerly iron-deficient hippocampus, suggesting decreased brain glucose uptake.106 Overall, the neurochemical changes indicate a hypometabolic and hypoactive hippocampus, despite normalization of tissue iron concentration. The disparate metabolomic responses in the striatum and hippocampus that mediate different modalities of cognition (procedural memory by the striatum and spatial memory by the hippocampus) may explain the unbalanced development of memory systems in early-life ID.107,108 Studies assessing multiple brain regions in the same animal using 1H MRS provide further evidence that ID has disparate effects on the metabolomic profiles of the brain regions. In formerly iron-deficient adult rats with diet-induced early-life IDA, in vivo 1H MRS showed the neurochemical profile of the prefrontal cortex was normal, whereas that of the hippocampus remained abnormal.109 The metabolomic alterations were indicative of abnormal glutamatergic neurotransmission, hypomyelination, and abnormal phospholipid metabolism in the formerly iron-deficient hippocampus. Iron treatment at a dose 10-fold higher than the standard dose failed to correct these abnormalities. Correction of tissue ID in the prefrontal cortex prior to its peak period of development may have been responsible for the normalization of neurochemical profile in the region.
In vivo MRS studies in adult transgenic mice with ongoing nonanemic hippocampal neuron-specific ID due to conditional Slc11a2 (DMT-1) knockout beginning in late gestation also demonstrate abnormal energy metabolism in the hippocampus.110 Compared with wild-type controls, the concentration of PCr and lactate was lower in Slc11a2 knockout mice, suggesting a persistent hypometabolic state, similar to results in formerly iron-deficient adult rats discussed above.102 A subsequent study in this mouse model showed lower glucose and lactate concentrations and higher PCr/Cr ratio in the striatum, despite lack of tissue ID in the region.107 Collectively, these results indicate that altered energy metabolism due to ID in a brain region (in this case, the hippocampus) could lead to altered metabolism and function in a connected and non-iron-deficient brain region (in this case, the striatum) and impact its development. Consistent with this possibility, both hippocampus- and striatum-mediated behaviors were found to be abnormal in the Slc11a2 knockout mice.107,110 It is important to reiterate that hippocampal iron content in these transgenic mice remains low at the time of these analyses. However, adult Slc11a2 knockout mice have normal hippocampal Tfr1 expression, suggesting the tissue is functionally iron-sufficient and the metabolomic changes are predominately due to ID during development. However, further metabolomic studies are needed using a truly reversible model of neuron-specific ID99 to determine whether the persistent energy metabolism changes in the hippocampus following early-life dietary IDA are due to early-life neuronal ID, rather than tissue hypoxia due to anemia.
Two additional studies of ongoing ID, after early-life genetic disruption of iron homeostasis, showed impaired mitochondrial structure in the adult CNS.34,111 In adult mice with a dopaminergic neuron-specific Tfr1 knockout, enlarged mitochondria with abnormal cristae structure accumulate and aggregate in dopaminergic neurons of the substantia nigra pars compacta but not the ventral tegument area.34 Similarly, large mitochondria, with disrupted cristae and dysfunctional ETC enzyme activity, are also present in iron-deficient spinal cord motor neuron axons of adult Irp2 knockout mice.111 This phenotype is rescued by increasing cellular iron availability either with Tempol treatment to destabilize Fe/S clusters or genetic ferritin knockdown to decrease iron storage capacity.
Mechanisms underlying the long-term disruption of energy metabolism pathways following recovery from early-life ID are unknown but may include mitochondrial damage and reprogramming due to ID- or iron treatment-induced oxidative stress (Figure 2). This hypothesis is consistent with findings in other highly metabolic cells (eg, pancreatic beta cells), where fetal nutrient restriction permanently reprograms mitochondrial and cytosolic energy metabolism by causing mitochondrial damage, with consequent loss of structural and functional integrity in adulthood.112-114 Disrupted epigenetic regulation of gene expression and brain hypoxia (due to early-life anemia) are additional mechanisms that could contribute to persistent impairment of brain energy metabolism pathways. Ultimately, any persistent brain energy metabolism deficits following recovery from early-life ID will contribute to the long-term brain functional impairments (Figure 2).
Functional consequences of early-life ID consistent with impaired brain energy metabolism
Many excellent reviews have detailed the structural, functional, and neurobehavioral outcomes of early-life ID in humans and animal models.7-13 Of particular importance are the deleterious consequences that are present in adolescence and adulthood, despite early-life iron repletion. Briefly, early-life ID or IDA in humans results in persistent deficits in learning/memory, speed of processing, motor skills, language, attention, and increased risk of depression, anxiety, attention-deficit hyperactivity disorder, and schizophrenia. Together, human and animal studies have associated persistently impaired speed of processing with hypomyelination and permanent deficits in psychosocial and motor domains with altered regulation of monoamine neurotransmission. In this section, we briefly discuss some of the key impairments with specific attention to relationships with impaired brain energy metabolism.
One fundamental consequence of reduced energy availability and compromised mitochondrial capacity during a critical period of neuronal differentiation is simplified neuronal architecture (Figure 2). Studies at multiple levels of investigation in the dendritic arbor of hippocampal CA1 neurons indicate that ID distorts dendrites and simplifies their branching patterns.31,99,110,115,116 The alterations are evident during the period of ID and remain permanently altered in adulthood despite repletion of iron status, resulting in a 12% reduction in adult hippocampal volume.99,102,110,115,116 The functional capacity of hippocampal neurons is directly related to dendrite complexity.117
Beyond morphology, disruptions in energy metabolism alter neurophysiologic capacity, defined as the ability to generate action potentials. Long-term potentiation, a fundamental read-out of hippocampal learning, relies on adequate ATP generation to support this high-energy event.97 Normal glutamate metabolism is essential as the hippocampus is largely a glutamate-driven neurotransmitter system. The balance between excitatory and inhibitory neurotransmitters (eg, glutamate and GABA) also dictates the field potential. Finally, membrane fluidity is critical for a synaptic vesicle to merge with the extracellular presynaptic membrane to release the neurotransmitters. As discussed above, ID during development alters neuronal glutamate, GABA, and fatty acid metabolism. The functional consequence is that ID retards the maturation of LTP during development. Likely because metabolism is programmed during development for the lifespan, developmental ID also results in loss of LTP capacity in adulthood, despite brain iron recovery.118,119
Compromised anatomy and physiologic capacity due to the metabolic deficits induced by developmental ID results in acute and long-term behavioral deficits. The hippocampus is critical for the processing of recognition memory events. Fetal-neonatal ID in humans results in poorer recognition memory at birth.120,121 Recognition memory deficits directly related to the degree of fetal ID remain until at least 3.5 years of age.122 Iron deficiency during toddlerhood results in poorer memory function at 10 and 19 years of age and likely beyond, despite iron repletion.123,124 Preclinical models demonstrate that the cellular metabolism43,63,102,109 and altered dendritic structure99,115,116 are directly associated with reduced spatial memory function99,108 and novel object recognition125; both hippocampally mediated tasks.
The anatomic, electrophysiologic, and functional fidelity of primary structures such as the hippocampus during development is also essential to the functional capacity of more complex circuits that depend on connections from the primary structures (eg, ventral tegmental area loop, prefrontal cortex).126 Preclinical models of IDA and isolated hippocampal neuronal ID demonstrate that developmental alterations to a primary structure such as the hippocampus result in extra-hippocampal electrophysiologic and neurobehavioral effects in adulthood.107,119,127 The range of neurobehavioral effects include poorer implicit memory function,107 reduced pre-pulse inhibition control,119 and increased attentional distraction.127 The latter 2 abnormalities parallel the literature in humans that shows an increased risk of schizophrenia and attentional issues in formerly developmentally iron-deficient subjects.123,128
Conclusions
Taken together, the data compiled on early-life ID and neonatal brain energy metabolism indicate a key role for these critical cellular pathways in mediating the short-term effects of early-life ID on hippocampal-mediated learning and memory function. Deficits in hippocampal energy metabolism also likely contribute to long-term hippocampal dysfunction through impaired neuronal structural maturation or persistently impaired energetic capacity (Figure 2). Additional data are needed to support the latter hypothesis. Whether disruption of energy metabolism in other brain regions or cell types mechanistically contributes to the neurobehavioral manifestations of early-life ID (eg, oligodendrocyte energy metabolism and hypomyelination) is less clear and warrants further investigation.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The writing of this manuscript was supported by National Institutes of Health Grants R01 HD29241 (MKG), R01 HD094809 (MKG), R01 NS099178 (PVT), and R01 HD089989 (RR).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: TWB, RR and MKG reviewed the literature and wrote and edited the manuscript. PVT reviewed the literature, performed the new IPA data analyses for Figure 1, and edited the manuscript. All authors reviewed and gave approval of the intellectual content of the final manuscript.
ORCID iD: Thomas W Bastian  https://orcid.org/0000-0001-5486-9471
https://orcid.org/0000-0001-5486-9471
References
- 1. McLean E, Cogswell M, Egli I, Wojdyla D, de Benoist B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993-2005. Public Health Nutr. 2009;12:444-454. doi: 10.1017/S1368980008002401. [DOI] [PubMed] [Google Scholar]
- 2. Yip R. Iron deficiency: contemporary scientific issues and international programmatic approaches. J Nutr. 1994;124:1479S-1490S. [DOI] [PubMed] [Google Scholar]
- 3. Cogswell ME, Looker AC, Pfeiffer CM, et al. Assessment of iron deficiency in US preschool children and nonpregnant females of childbearing age: National Health and Nutrition Examination Survey 2003-2006. Am J Clin Nutr. 2009;89:1334-1342. doi: 10.3945/ajcn.2008.27151. [DOI] [PubMed] [Google Scholar]
- 4. Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999-2006. Am J Clin Nutr. 2011;93:1312-1320. doi: 10.3945/ajcn.110.007195. [DOI] [PubMed] [Google Scholar]
- 5. Auerbach M, Abernathy J, Juul S, Short V, Derman R. Prevalence of iron deficiency in first trimester, nonanemic pregnant women [published online ahead of print June 3, 2019]. J Matern Fetal Neonatal Med. doi: 10.1080/14767058.2019.1619690. [DOI] [PubMed] [Google Scholar]
- 6. Walker SP, Wachs TD, Gardner JM, et al. Child development: risk factors for adverse outcomes in developing countries. Lancet. 2007;369:145-157. doi: 10.1016/S0140-6736(07)60076-2. [DOI] [PubMed] [Google Scholar]
- 7. Lozoff B, Beard JL, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev. 2006;64:S34-S43; discussion S72-S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Georgieff MK. Long-term brain and behavioral consequences of early iron deficiency. Nutr Rev. 2011;69:S43-S48. doi: 10.1111/j.1753-4887.2011.00432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Markova V, Holm C, Pinborg A, Thomsen L, Moos T. Impairment of the developing human brain in iron deficiency: correlations to findings in experimental animals and prospects for early intervention therapy. Pharmaceuticals. 2019;12:120. doi: 10.3390/ph12030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barks A, Hall AM, Tran PV, Georgieff MK. Iron as a model nutrient for understanding the nutritional origins of neuropsychiatric disease. Pediatr Res. 2019;85:176-182. doi: 10.1038/s41390-018-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badaracco ME, Siri MVR, Pasquini JM. Oligodendrogenesis: the role of iron. Biofactors. 2010;36:98-102. doi: 10.1002/biof.90. [DOI] [PubMed] [Google Scholar]
- 12. Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: the role of iron. Glia. 2009;57:467-478. doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- 13. Lozoff B. Early iron deficiency has brain and behavior effects consistent with dopaminergic dysfunction. J Nutr. 2011;141:740S-746S. doi: 10.3945/jn.110.131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J, Wessling-Resnick M. Iron and mechanisms of emotional behavior. J Nutr Biochem. 2014;25:1101-1107. doi: 10.1016/j.jnutbio.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rangaraju V, Lewis TL, Hirabayashi Y, et al. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J Neurosci. 2019;39:8200-8208. doi: 10.1523/JNEUROSCI.1157-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Princz A, Kounakis K, Tavernarakis N. Mitochondrial contributions to neuronal development and function. Biol Chem. 2018;399:723-739. doi: 10.1515/hsz-2017-0333. [DOI] [PubMed] [Google Scholar]
- 17. Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. 2004;73:397-445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 18. Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014;13:217-232. doi: 10.1016/S1474-4422(13)70261-8. [DOI] [PubMed] [Google Scholar]
- 19. Kuzawa CW. Adipose tissue in human infancy and childhood: an evolutionary perspective. Am J Phys Anthropol. 1998;107:177-209. [DOI] [PubMed] [Google Scholar]
- 20. Kuzawa CW, Chugani HT, Grossman LI, et al. Metabolic costs and evolutionary implications of human brain development. Proc Natl Acad Sci. 2014;111:13010-13015. doi: 10.1073/pnas.1323099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cusick SE, Georgieff MK. The role of nutrition in brain development: the golden opportunity of the “first 1000 days.” J Pediatr. 2016;175:16-21. doi: 10.1016/j.jpeds.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Liu Y, Shang L, et al. Iron regulatory protein 2 modulates the switch from aerobic glycolysis to oxidative phosphorylation in mouse embryonic fibroblasts. Proc Natl Acad Sci. 2019;116:9871-9876. doi: 10.1073/pnas.1820051116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420-426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 24. Bailey P, Nathan J. Metabolic regulation of hypoxia-inducible transcription factors: the role of small molecule metabolites and iron. Biomedicines. 2018;6:60. doi: 10.3390/biomedicines6020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gray NK, Pantopoulos K, Dandekar T, Ackrell BA, Hentze MW. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. Proc Natl Acad Sci. 1996;93:4925-4930. doi: 10.1073/pnas.93.10.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin E, Graziano JH, Freyer GA. Regulation of the 75-kDa subunit of mitochondrial complex I by iron. J Biol Chem. 2001;276:27685-27692. doi: 10.1074/jbc.M100941200. [DOI] [PubMed] [Google Scholar]
- 27. Zhang A-S, Sheftel AD, Ponka P. Intracellular kinetics of iron in reticulocytes: evidence for endosome involvement in iron targeting to mitochondria. Blood. 2005;105:368-375. doi: 10.1182/blood-2004-06-2226. [DOI] [PubMed] [Google Scholar]
- 28. Wolff NA, Ghio AJ, Garrick LM, et al. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 2014;28:2134-2145. doi: 10.1096/fj.13-240564. [DOI] [PubMed] [Google Scholar]
- 29. Wolff NA, Garrick MD, Zhao L, Garrick LM, Ghio AJ, Thévenod F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep. 2018;8:211. doi: 10.1038/s41598-017-18584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus. 2007;17:679-691. doi: 10.1002/hipo.20307. [DOI] [PubMed] [Google Scholar]
- 31. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci. 2016;38:264-276. doi: 10.1159/000448514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clardy SL, Wang X, Zhao W, et al. Acute and chronic effects of developmental iron deficiency on mRNA expression patterns in the brain. J Neural Transm Suppl. 2006;71:173-196. [DOI] [PubMed] [Google Scholar]
- 33. Bastian TW, Santarriaga S, Nguyen TA, Prohaska JR, Georgieff MK, Anderson GW. Fetal and neonatal iron deficiency but not copper deficiency increases vascular complexity in the developing rat brain. Nutr Neurosci. 2015;18:365-375. doi: 10.1179/1476830515Y.0000000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matak P, Matak A, Moustafa S, et al. Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice. Proc Natl Acad Sci. 2016;113:3428-3435. doi: 10.1073/pnas.1519473113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dallman PR. Iron restriction in the nursing rat: early effects upon tissue heme proteins, hemoglobin and liver iron. J Nutr. 1969;97:475-480. [DOI] [PubMed] [Google Scholar]
- 36. Weinberg J, Dallman PR, Levine S. Iron deficiency during early development in the rat: behavioral and physiological consequences. Pharmacol Biochem Behav. 1980;12:493-502. doi: 10.1016/0091-3057(80)90179-3. [DOI] [PubMed] [Google Scholar]
- 37. Mackler B, Person R, Miller LR, Inamdar AR, Finch CA. Iron deficiency in the rat: biochemical studies of brain metabolism. Pediatr Res. 1978;12:217-220. doi: 10.1203/00006450-197803000-00011. [DOI] [PubMed] [Google Scholar]
- 38. Bastian TW, Prohaska JR, Georgieff MK, Anderson GW. Perinatal iron and copper deficiencies alter neonatal rat circulating and brain thyroid hormone concentrations. Endocrinology. 2010;151:4055-4065. doi: 10.1210/en.2010-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bastian TW, Prohaska JR, Georgieff MK, Anderson GW. Fetal and neonatal iron deficiency exacerbates mild thyroid hormone insufficiency effects on male thyroid hormone levels and brain thyroid hormone-responsive gene expression. Endocrinology. 2014;155:1157-1167. doi: 10.1210/en.2013-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tran PV, Fretham SJ, Wobken J, Miller BS, Georgieff M. Gestational-neonatal iron deficiency suppresses and iron treatment re-activates IGF signaling in developing rat hippocampus. Am J Physiol Endocrinol Metab. 2011;302:E316-E324. doi: 10.1152/ajpendo.00369.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wallin DJ, Zamora TG, Alexander M, Ennis KM, Tran PV, Georgieff MK. Neonatal mouse hippocampus: phlebotomy-induced anemia diminishes and treatment with erythropoietin partially rescues mammalian target of rapamycin signaling. Pediatr Res. 2017;82:501-508. doi: 10.1038/pr.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fretham SJ, Carlson ES, Georgieff MK. Neuronal-specific iron deficiency dysregulates mammalian target of rapamycin signaling during hippocampal development in nonanemic genetic mouse models. J Nutr. 2013;143:260-266. doi: 10.3945/jn.112.168617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Ungria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res. 2000;48:169-176. [DOI] [PubMed] [Google Scholar]
- 44. Rao R, de Ungria M, Sullivan D, et al. Perinatal brain iron deficiency increases the vulnerability of rat hippocampus to hypoxic ischemic insult. J Nutr. 1999;129:199-206. [DOI] [PubMed] [Google Scholar]
- 45. Millar LJ, Shi L, Hoerder-Suabedissen A, Molnár Z. Neonatal hypoxia ischaemia: mechanisms, models, and therapeutic challenges. Front Cell Neurosci. 2017;11:78. doi: 10.3389/fncel.2017.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greco P, Nencini G, Piva I, et al. Pathophysiology of hypoxic–ischemic encephalopathy: a review of the past and a view on the future. Acta Neurol Belg. 2020;120:277-288. doi: 10.1007/s13760-020-01308-3. [DOI] [PubMed] [Google Scholar]
- 47. Meibach RC, Ross DA, Cox RD, Glick SD. The ontogeny of hippocampal energy metabolism. Brain Res. 1981;204:431-435. doi: 10.1016/0006-8993(81)90603-X. [DOI] [PubMed] [Google Scholar]
- 48. Nehlig A, de Vasconcelos AP, Boyet S. Quantitative autoradiographic measurement of local cerebral glucose utilization in freely moving rats during postnatal development. J Neurosci. 1988;8:2321-2333. doi: 10.1523/JNEUROSCI.08-07-02321.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vannucci SJ. Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J Neurochem. 1994;62:240-246. doi: 10.1046/j.1471-4159.1994.62010240.x. [DOI] [PubMed] [Google Scholar]
- 50. Siddappa AJ, Rao RB, Wobken JD, Leibold EA, Connor JR, Georgieff MK. Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J Neurosci Res. 2002;68:761-775. doi: 10.1002/jnr.10246. [DOI] [PubMed] [Google Scholar]
- 51. Taylor EM, Morgan EH. Developmental changes in transferrin and iron uptake by the brain in the rat. Brain Res Dev Brain Res. 1990;55:35-42. [DOI] [PubMed] [Google Scholar]
- 52. Mellgren SI. Distribution of succinate-, alpha-glycerophosphate-, NADH-, and NADPH dehydrogenases (tetrazolium reductases) in the hippocampal region of the rat during postnatal development. Z Zellforsch Mikrosk Anat. 1973;141:347-373. doi: 10.1007/BF00307411. [DOI] [PubMed] [Google Scholar]
- 53. Tran PV, Carlson ES, Fretham SJ, Georgieff MK. Early-life iron deficiency anemia alters neurotrophic factor expression and hippocampal neuron differentiation in male rats. J Nutr. 2008;138:2495-2501. doi: 10.3945/jn.108.091553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luesse H-G, Roskoden T, Linke R, Otten U, Heese K, Schwegler H. Modulation of mRNA expression of the neurotrophins of the nerve growth factor family and their receptors in the septum and hippocampus of rats after transient postnatal thyroxine treatment. Exp Brain Res. 1998;119:1-8. doi: 10.1007/s002210050313. [DOI] [PubMed] [Google Scholar]
- 55. Pokorný J, Yamamoto T. Postnatal ontogenesis of hippocampal CA1 area in rats. I. Development of dendritic arborisation in pyramidal neurons. Brain Res Bull. 1981;7:113-120. [DOI] [PubMed] [Google Scholar]
- 56. Pokorný J, Yamamoto T. Postnatal ontogenesis of hippocampal CA1 area in rats. II. Development of ultrastructure in stratum lacunosum and moleculare. Brain Res Bull. 1981;7:121-130. doi: 10.1016/0361-9230(81)90076-9. [DOI] [PubMed] [Google Scholar]
- 57. Krnjević K, Cherubini E, Ben-Ari Y. Anoxia on slow inward currents of immature hippocampal neurons. J Neurophysiol. 1989;62:896-906. doi: 10.1152/jn.1989.62.4.896. [DOI] [PubMed] [Google Scholar]
- 58. Jones OT, Bernstein GM, Jones EJ, et al. N-Type calcium channels in the developing rat hippocampus: subunit, complex, and regional expression. J Neurosci. 1997;17:6152-6164. doi: 10.1523/JNEUROSCI.17-16-06152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hattori H, Wasterlain CG. Excitatory amino acids in the developing brain: ontogeny, plasticity, and excitotoxicity. Pediatr Neurol. 1990;6:219-228. doi: 10.1016/0887-8994(90)90111-d. [DOI] [PubMed] [Google Scholar]
- 60. Insel TR, Miller LP, Gelhard RE. The ontogeny of excitatory amino acid receptors in rat forebrain—I. N-methyl-D-aspartate and quisqualate receptors. Neuroscience. 1990;35:31-43. doi: 10.1016/0306-4522(90)90117-m. [DOI] [PubMed] [Google Scholar]
- 61. Bekenstein JW, Lothman EW. An in vivo study of the ontogeny of long-term potentiation (LTP) in the CA1 region and in the dentate gyrus of the rat hippocampal formation. Brain Res Dev Brain Res. 1991;63:245-251. doi: 10.1016/0165-3806(91)90084-V. [DOI] [PubMed] [Google Scholar]
- 62. Harris KM, Teyler TJ. Developmental onset of long-term potentiation in area CA1 of the rat hippocampus. J Physiol. 1984;346:27-48. doi: 10.1113/jphysiol.1984.sp015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK. Perinatal iron deficiency alters the neurochemical profile of the developing rat hippocampus. J Nutr. 2003;133:3215-3221. [DOI] [PubMed] [Google Scholar]
- 64. Ward KL, Tkac I, Jing Y, et al. Gestational and lactational iron deficiency alters the developing striatal metabolome and associated behaviors in young rats. J Nutr. 2007;137:1043-1049. doi: 10.1093/jn/137.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wallin DJ, Tkac I, Stucker S, et al. Phlebotomy-induced anemia alters hippocampal neurochemistry in neonatal mice. Pediatr Res. 2015;77:765-771. doi: 10.1038/pr.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tkáč I, Rao R, Georgieff MK, Gruetter R. Developmental and regional changes in the neurochemical profile of the rat brain determined by in vivo 1 H NMR spectroscopy. Magn Reson Med. 2003;50:24-32. doi: 10.1002/mrm.10497. [DOI] [PubMed] [Google Scholar]
- 67. Maliszewski-Hall AM, Alexander M, Tkáč I, Öz G, Rao R. Differential effects of intrauterine growth restriction on the regional neurochemical profile of the developing rat brain. Neurochem Res. 2017;42:133-140. doi: 10.1007/s11064-015-1609-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wallimann T, Tokarska-Schlattner M, Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids. 2011;40:1271-1296. doi: 10.1007/s00726-011-0877-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jensen F, Tsuji M, Offutt M, Firkusny I, Holtzman D. Profound, reversible energy loss in the hypoxic immature rat brain. Brain Res Dev Brain Res. 1993;73:99-105. doi: 10.1016/0165-3806(93)90051-B. [DOI] [PubMed] [Google Scholar]
- 70. Rao R, Ennis K, Long JD, Ugurbil K, Gruetter R, Tkac I. Neurochemical changes in the developing rat hippocampus during prolonged hypoglycemia: hippocampal neurochemistry in hypoglycemia. J Neurochem. 2010;114:728-738. doi: 10.1111/j.1471-4159.2010.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Henry P-G, Russeth KP, Tkac I, Drewes LR, Andrews MT, Gruetter R. Brain energy metabolism and neurotransmission at near-freezing temperatures: in vivo 1 H MRS study of a hibernating mammal. J Neurochem. 2007;101:1505-1515. doi: 10.1111/j.1471-4159.2007.04514.x. [DOI] [PubMed] [Google Scholar]
- 72. Staples JF, Buck LT. Matching cellular metabolic supply and demand in energy-stressed animals. Comp Biochem Physiol A Mol Integr Physiol. 2009;153:95-105. doi: 10.1016/j.cbpa.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 73. Raman L, Tkac I, Ennis K, Georgieff MK, Gruetter R, Rao R. In vivo effect of chronic hypoxia on the neurochemical profile of the developing rat hippocampus. Brain Res Dev Brain Res. 2005;156:202-209. doi: 10.1016/j.devbrainres.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 74. Rao R, Tkac I, Townsend EL, Ennis K, Gruetter R, Georgieff MK. Perinatal iron deficiency predisposes the developing rat hippocampus to greater injury from mild to moderate hypoxia-ischemia. J Cereb Blood Flow Metab. 2007;27:729-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133-1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 76. Oexle H, Gnaiger E. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta. 1999;1413:99-107. [DOI] [PubMed] [Google Scholar]
- 77. Brooks GA, Henderson SA, Dallman PR. Increased glucose dependence in resting, iron-deficient rats. Am J Physiol. 1987;253:E461-E466. doi: 10.1152/ajpendo.1987.253.4.E461. [DOI] [PubMed] [Google Scholar]
- 78. van Gelder NM. Brain taurine content as a function of cerebral metabolic rate: osmotic regulation of glucose derived water production. Neurochem Res. 1989;14:495-497. doi: 10.1007/BF00964908. [DOI] [PubMed] [Google Scholar]
- 79. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metabolism. 2011;14:724-738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 80. Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metabolism. 2014;19:49-57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vega-Núñez E, Menéndez-Hurtado A, Garesse R, Santos A, Perez-Castillo A. Thyroid hormone-regulated brain mitochondrial genes revealed by differential cDNA cloning. J Clin Invest. 1995;96:893-899. doi: 10.1172/JCI118136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Martinez B, Del Hoyo P, Martin MA, Arenas J, Perez-Castillo A, Santos A. Thyroid hormone regulates oxidative phosphorylation in the cerebral cortex and striatum of neonatal rats: T3 regulates brain mitochondrial activity. J Neurochem. 2001;78:1054-1063. doi: 10.1046/j.1471-4159.2001.00487.x. [DOI] [PubMed] [Google Scholar]
- 83. Sinha RA, Pathak A, Mohan V, et al. Evidence of a bigenomic regulation of mitochondrial gene expression by thyroid hormone during rat brain development. Biochem Biophys Res Commun. 2010;397:548-552. doi: 10.1016/j.bbrc.2010.05.154. [DOI] [PubMed] [Google Scholar]
- 84. Bernal J. Thyroid hormones in brain development and function. In: Feingold KR, Anawalt B, Boyce A, et al. , eds. Endotext [Internet]. MDText.com, Inc; 2015:1-62. [Google Scholar]
- 85. Bastian TW, Anderson JA, Fretham SJ, Prohaska JR, Georgieff MK, Anderson GW. Fetal and neonatal iron deficiency reduces thyroid hormone-responsive gene mRNA levels in the neonatal rat hippocampus and cerebral cortex. Endocrinology. 2012;153:5668-5680. doi: 10.1210/en.2012-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hu X, Wang R, Shan Z, et al. Perinatal iron deficiency-induced hypothyroxinemia impairs early brain development regardless of normal iron levels in the neonatal brain. Thyroid. 2016;26:891-900. doi: 10.1089/thy.2015.0293. [DOI] [PubMed] [Google Scholar]
- 87. Rao R, Ennis K, Oz G, Lubach GR, Georgieff MK, Coe CL. Metabolomic analysis of cerebrospinal fluid indicates iron deficiency compromises cerebral energy metabolism in the infant monkey. Neurochem Res. 2013;38:573-580. doi: 10.1007/s11064-012-0950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rao R, Ennis K, Lubach GR, Lock EF, Georgieff MK, Coe CL. Metabolomic analysis of CSF indicates brain metabolic impairment precedes hematological indices of anemia in the iron-deficient infant monkey. Nutr Neurosci. 2018;21:40-48. doi: 10.1080/1028415X.2016.1217119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Coe CL, Lubach GR, Busbridge M, Chapman RS. Optimal iron fortification of maternal diet during pregnancy and nursing for investigating and preventing iron deficiency in young rhesus monkeys. Res Vet Sci. 2013;94:549-554. doi: 10.1016/j.rvsc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lubach GR, Coe CL. Preconception maternal iron status is a risk factor for iron deficiency in infant rhesus monkeys (Macaca mulatta). J Nutr. 2006;136:2345-2349. doi: 10.1093/jn/136.9.2345. [DOI] [PubMed] [Google Scholar]
- 91. Coe CL, Lubach GR, Bianco L, Beard JL. A history of iron deficiency anemia during infancy alters brain monoamine activity later in juvenile monkeys. Dev Psychobiol. 2009;51:301-309. doi: 10.1002/dev.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Geguchadze RN, Coe CL, Lubach GR, Clardy TW, Beard JL, Connor JR. CSF proteomic analysis reveals persistent iron deficiency-induced alterations in non-human primate infants. J Neurochem. 2008;105:127-136. doi: 10.1111/j.1471-4159.2007.05113.x. [DOI] [PubMed] [Google Scholar]
- 93. Lubach GR, Coe CL. Selective impairment of cognitive performance in the young monkey following recovery from iron deficiency. J Dev Behav Pediatr. 2008;29:11-17. doi: 10.1097/DBP.0b013e31815f24a9. [DOI] [PubMed] [Google Scholar]
- 94. Lozoff B, Smith JB, Kaciroti N, Clark KM, Guevara S, Jimenez E. Functional significance of early-life iron deficiency: outcomes at 25 years. J Pediatr. 2013;163:1260-1266. doi: 10.1016/j.jpeds.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bastian TW, von Hohenberg WC, Georgieff MK, Lanier LM. Chronic energy depletion due to iron deficiency impairs dendritic mitochondrial motility during hippocampal neuron development. J Neuroscience. 2019;39:802-813. doi: 10.1523/JNEUROSCI.1504-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bastian TW. Potential mechanisms driving mitochondrial motility impairments in developing iron-deficient neurons. J Exp Neurosci. 2019;13:117906951985835. doi: 10.1177/1179069519858351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762-777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 98. Tran PV, Kennedy BC, Pisansky MT, et al. Prenatal choline supplementation diminishes early-life iron deficiency–induced reprogramming of molecular networks associated with behavioral abnormalities in the adult rat hippocampus. J Nutr. 2016;146:484-493. doi: 10.3945/jn.115.227561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus. 2012;22:1691-1702. doi: 10.1002/hipo.22004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tran PV, Dakoji S, Reise KH, Storey KK, Georgieff MK. Fetal iron deficiency alters the proteome of adult rat hippocampal synaptosomes. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1297-R1306. doi: 10.1152/ajpregu.00292.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kwik-Uribe CL, Gietzen D, German JB, Golub MS, Keen CL. Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism and myelin composition. J Nutr. 2000;130:2821-2830. doi: 10.1093/jn/130.11.2821. [DOI] [PubMed] [Google Scholar]
- 102. Rao R, Tkac I, Schmidt AT, Georgieff MK. Fetal and neonatal iron deficiency causes volume loss and alters the neurochemical profile of the adult rat hippocampus. Nutr Neurosci. 2011;14:59-65. doi: 10.1179/1476830511Y.0000000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Unger EL, Hurst AR, Georgieff MK, et al. Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J Nutr. 2012;142:2040-2049. doi: 10.3945/jn.112.162198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mangia S, Tkáč I, Gruetter R, Van de Moortele P-F, Maraviglia B, Uğurbil K. Sustained neuronal activation raises oxidative metabolism to a new steady-state level: evidence from 1 H NMR spectroscopy in the human visual cortex. J Cereb Blood Flow Metab. 2007;27:1055-1063. doi: 10.1038/sj.jcbfm.9600401. [DOI] [PubMed] [Google Scholar]
- 105. Rao R, Nashawaty M, Fatima S, Ennis K, Tkac I. Neonatal hyperglycemia alters the neurochemical profile, dendritic arborization and gene expression in the developing rat hippocampus. NMR Biomed. 2018;31:e3910. doi: 10.1002/nbm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ennis K, Felt B, Georgieff MK, Rao R. Early-life iron deficiency alters glucose transporter-1 expression in the adult rodent hippocampus. J Nutr. 2019;149:1660-1666. doi: 10.1093/jn/nxz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Carlson ES, Fretham SJ, Unger E, et al. Hippocampus specific iron deficiency alters competition and cooperation between developing memory systems. J Neurodev Disord. 2010;2:133-143. doi: 10.1007/s11689-010-9049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schmidt AT, Waldow KJ, Grove WM, Salinas JA, Georgieff MK. Dissociating the long-term effects of fetal/neonatal iron deficiency on three types of learning in the rat. Behav Neurosci. 2007;121:475-482. doi: 10.1037/0735-7044.121.3.475. [DOI] [PubMed] [Google Scholar]
- 109. Rao R, Tkac I, Unger EL, et al. Iron supplementation dose for perinatal iron deficiency differentially alters the neurochemistry of the frontal cortex and hippocampus in adult rats. Pediatr Res. 2013;73:31-37. doi: 10.1038/pr.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Carlson ES, Tkac I, Magid R, et al. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. 2009;139:672-679. doi: 10.3945/jn.108.096354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jeong SY, Crooks DR, Wilson-Ollivierre H, et al. Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice. PLoS ONE. 2011;6:e25404. doi: 10.1371/journal.pone.0025404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Simmons RA, Suponitsky-Kroyter I, Selak MA. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to beta-cell failure. J Biol Chem. 2005;280:28785-28791. doi: 10.1074/jbc.M505695200. [DOI] [PubMed] [Google Scholar]
- 113. Peterside IE, Selak MA, Simmons RA. Impaired oxidative phosphorylation in hepatic mitochondria in growth-retarded rats. Am J Physiol Endocrinol Metab. 2003;285:E1258-E1266. doi: 10.1152/ajpendo.00437.2002. [DOI] [PubMed] [Google Scholar]
- 114. Selak MA, Storey BT, Peterside I, Simmons RA. Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. Am J Physiol Endocrinol Metab. 2003;285:E130-E137. doi: 10.1152/ajpendo.00322.2002. [DOI] [PubMed] [Google Scholar]
- 115. Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci. 2003;25:412-420. doi: 10.1159/000075667. [DOI] [PubMed] [Google Scholar]
- 116. Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci. 2010;32:238-248. doi: 10.1159/000314341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lefebvre JL, Sanes JR, Kay JN. Development of dendritic form and function. Annu Rev Cell Dev Biol. 2015;31:741-777. doi: 10.1146/annurev-cellbio-100913-013020. [DOI] [PubMed] [Google Scholar]
- 118. Jorgenson LA, Sun M, O’Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus. 2005;15:1094-1102. doi: 10.1002/hipo.20128. [DOI] [PubMed] [Google Scholar]
- 119. Pisansky MT, Wickham RJ, Su J, et al. Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus. 2013;23:952-962. doi: 10.1002/hipo.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, Deregnier R-A. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res. 2004;55:1034-1041. doi: 10.1203/01.pdr.0000127021.38207.62. [DOI] [PubMed] [Google Scholar]
- 121. Geng F, Mai X, Zhan J, et al. Impact of fetal-neonatal iron deficiency on recognition memory at 2 months of age. J Pediatr. 2015;167:1226-1232. doi: 10.1016/j.jpeds.2015.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Riggins T, Miller NC, Bauer PJ, Georgieff MK, Nelson CA. Consequences of low neonatal iron status due to maternal diabetes mellitus on explicit memory performance in childhood. Dev Neuropsychol. 2009;34:762-779. doi: 10.1080/87565640903265145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lukowski AF, Koss M, Burden MJ, et al. Iron deficiency in infancy and neurocognitive functioning at 19 years: evidence of long-term deficits in executive function and recognition memory. Nutr Neurosci. 2010;13:54-70. doi: 10.1179/147683010X12611460763689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Congdon EL, Westerlund A, Algarin CR, et al. Iron deficiency in infancy is associated with altered neural correlates of recognition memory at 10 years. J Pediatr. 2012;160:1027-1033. doi: 10.1016/j.jpeds.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kennedy BC, Dimova JG, Siddappa AJ, Tran PV, Gewirtz JC, Georgieff MK. Prenatal choline supplementation ameliorates the long-term neurobehavioral effects of fetal-neonatal iron deficiency in rats. J Nutr. 2014;144:1858-1865. doi: 10.3945/jn.114.198739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wachs TD, Georgieff M, Cusick S, McEwen BS. Issues in the timing of integrated early interventions: contributions from nutrition, neuroscience, and psychological research: timing of integrated early interventions. Ann N Y Acad Sci. 2014;1308:89-106. doi: 10.1111/nyas.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schmidt AT, Ladwig EK, Wobken JD, Grove WM, Georgieff MK. Delayed alternation performance in rats following recovery from early iron deficiency. Physiol Behav. 2010;101:503-508. doi: 10.1016/j.physbeh.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Insel BJ, Schaefer CA, McKeague IW, Susser ES, Brown AS. Maternal iron deficiency and the risk of schizophrenia in offspring. Arch Gen Psychiatry. 2008;65:1136-1144. doi: 10.1001/archpsyc.65.10.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]