

Abstract
Purpose:
Currently, <50% of high-risk pediatric solid tumors like neuroblastoma can be cured, and many survivors experience serious or life-threatening toxicities, so more effective, less toxic therapy is needed. One approach is to target drugs to tumors using nanoparticles, which take advantage of the enhanced permeability of tumor vasculature.
Experimental Design:
SN38, the active metabolite of irinotecan (CPT-11), is a potent therapeutic agent that is readily encapsulated in polymeric nanoparticles. Tocopherol oxyacetate (TOA) is a hydrophobic mitocan that was linked to SN38 to significantly increase hydrophobicity and enhance nanoparticle retention. We treated neuroblastomas with SN38-TOA nanoparticles and compared the efficacy with the parent prodrug CPT-11 using a mouse xenograft model.
Results:
Nanoparticle treatment induced prolonged event-free survival (EFS) in most mice, compared with CPT-11. This was shown for both SH-SY5Y and IMR-32 neuroblastoma xenografts. Enhanced efficacy was likely due to increased and sustained drug levels of SN38 in the tumor compared with conventional CPT-11 delivery. Interestingly, when recurrent CPT-11-treated tumors were re-treated with SN38-TOA nanoparticles, the tumors transformed from undifferentiated neuroblastomas to maturing ganglioneuroblastomas. Furthermore, these tumors were infiltrated with Schwann cells of mouse origin, which may have contributed to the differentiated histology.
Conclusions:
Nanoparticle delivery of SN38-TOA produced increased drug delivery and prolonged EFS compared to conventional delivery of CPT-11. Also, lower total dose and drug entrapment in nanoparticles during circulation should decrease toxicity. We propose that nanoparticle-based delivery of a rationally designed prodrug is an attractive approach to enhance chemotherapeutic efficacy in pediatric and adult tumors.
Introduction
Neuroblastoma is a cancer of the sympathetic nervous system. It accounts for about 8% of all childhood cancers and 12% of childhood deaths from cancer (1). In some instances, neuroblastomas may regress spontaneously, especially in infants, or mature into benign ganglioneuromas in older patients. However, over half of all patients have advanced stages of disease that frequently recur or are resistant to very intensive, multimodality treatment, and the 5-year survival rate for high-risk neuroblastoma is only 40% to 50% (2). Furthermore, many survivors experience significant and even life-threatening short- and long-term toxicities, including second malignancies (3–5). Therefore, more effective and less toxic therapy is needed for these patients.
Generally, less than 1% of chemotherapeutic agents reach the tumor using conventional delivery of drugs by mouth or injection (6, 7). Therefore, one approach is to deliver more drug to the tumor using nanoparticles, taking advantage of the leaky capillaries that are found in many rapidly growing tumors. Nanoparticles can accumulate in tumors by the enhanced permeability and retention (EPR) effect (8), while bypassing most normal tissues. This approach can increase drug delivery to the tumor by many fold, so greater antitumor efficacy can be achieved while dramatically reducing patient toxicity. Thus, less total drug can be delivered with far greater effect, and the drug is entrapped in nanoparticles while it circulates, which also reduces systemic exposure.
Irinotecan (CPT-11), a derivative of camptothecin, is a commercially available topoisomerase I inhibitor used to treat a variety of pediatric and adult solid tumors. It is a water-soluble prodrug that is hydrolyzed by liver enzymes to the hydrophobic but active metabolite SN38. This chemotherapeutic is cell cycle (S-phase) dependent and is effective at killing rapidly proliferating cells. Previously, we formulated SN38 as a tocopherol succinate conjugate (SN38-TS) in polylactic acid-polyethylene glycol (PLA-PEG) nanoparticles to treat neuroblastomas in a xenograft model (9, 10). The tocopheryl moiety not only increased the hydrophobicity of the therapeutic agent, but also may have increased overall antitumor efficacy by acting as a mitocan (11–13). This therapy was both extremely effective and nontoxic.
To further improve the design of the prodrug, and to exclude side reactions potentially reducing recovery of pharmacologically active SN38, we reformulated our prodrug nanoparticles with SN38 hydrophobized with a tocopherol moiety attached via an oxyacetate linkage (SN38-TOA). The optimized formulation addresses essential requirements with regard to sterility, stability, and cryopreservation capacity for use in human patients and clinical trials. We tested this new formulation in our neuroblastoma xenograft preclinical model to determine its toxicity and efficacy in chemo-naive and CPT-11–pretreated animals, which in turn should indicate its potential utility to treat aggressive disease that cannot be managed effectively using standard chemotherapy.
Materials and Methods
Compounds
Irinotecan (CPT-11; 20 mg/mL; Camptosar, Pfizer) was obtained from the pharmacy at The Children’s Hospital of Philadelphia (CHOP, Philadelphia, PA) and was diluted in 0.9% normal saline before being administered intravenously.
Nanoparticle formulation
D-α-Tocopheryloxyacetic acid (TOA) was prepared as described previously (14). The SN38-TOA conjugate was synthesized with a 95% yield from SN38 (AK Scientific), and TOA was added by direct coupling according to a standard procedure using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) as an activator and N,N-dimethylaminopyridine tosylate (DPTS) as a catalyst (9). SN38-TOA–loaded nanoparticles were formulated by nanoprecipitation using poly(D,L-lactide)-block-poly(ethylene glycol; 5,000:5,000; JenKem Technology USA) and Pluronic F-68 (Sigma-Aldrich) as the particle-forming polymer and stabilizer, respectively. The particle size was analyzed by dynamic light scattering, and the drug loading was determined by UV-Vis spectrophotometry after SN38-TOA extraction in sec-butanol.
Cell lines
We used SH-SY5Y cells stably transfected with NTRK2 (clone BR6) and IMR-32 cells for all in vitro and in vivo studies (15–18). We tested the integrity and authenticity of these cell lines for endotoxins, mycoplasmas, bacterial, and other viral contaminations as well as genetic variations by multiplex PCR techniques. These tests were performed on an annual basis at the cell center services facility of University of Pennsylvania (Philadelphia, PA). Cells were grown in RPMI1640 medium (Gibco) containing 10% FBS (CellGro) and maintained in 150 cm3 culture flasks (Corning) in a humidified atmosphere of 95% air and 5% CO2. Transfected cells were maintained in media containing 0.3 mg/mL G418 sulfate (stock solution: 20 mg/mL; Corning). Cells were harvested using 0.02% tetrasodium EDTA in PBS.
In vitro experiments
Sulforhodamine B (SRB) assays were performed to determine cell number and the effect of SN38-TOA nanoparticles on the survival and growth of the BR6 neuroblastoma cells. We plated 8 × 103 cells per well in 96-well plates and exposed cells to the formulation at different concentrations (1, 2, 4, 8, 16, 32 nmol/L), and harvested at 24, 48, 72, and 96 hours. The plates were processed for cell viability using standard SRB assay protocol (19) and read at 520 nm. All in vitro experiments were performed in quintuplicate and repeated at least three times.
Mice
We obtained 6-week-old Foxn1nu/Foxn1nu (JAXstock #007850) mice from The Jackson Laboratory. Mice were maintained under humidity- and temperature-controlled conditions in a light/dark cycle that was set at 12-hour intervals. The Institutional Animals Care and Use Committee of the CHOP Research Institute approved the mouse xenograft studies described in this report.
Flank xenograft experiments
Mice were injected subcutaneously in the right flank with 1 × 107 TrkB-expressing SY5Y (BR6) cells suspended in 0.1 mL of Matrigel (Corning). Tumors were measured manually twice per week in 2 dimensions (mm) using a caliper. The volume (cm3) was calculated as follows: [(0.523 × L × W2)/1,000)] where L >W. Body weights were obtained twice per week, and treatment doses adjusted if there was a >10% change in body weight.
All mice (n = 10/arm) were treated with either CPT-11 or SN38-TOA nanoparticles intravenously via tail vein injections twice per week for 4 weeks once tumor volumes reached 0.2 cm3. This schedule was based on a prior published study (10). SN38-TOA nanoparticles and CPT-11 were given at doses equivalent to 10 and 25 mg/kg drug, respectively. The nanoparticle drug dose was lower because CPT-11 is a prodrug that requires conversion to the active SN38 metabolite by the liver. Blank nanoparticles (vehicle only) and saline (no treatment) were used as negative controls. Mice were removed from study when tumor volumes reached 3.0 cm3. A subset of mice treated with CPT-11 was removed when the tumor volume reached 2.0 cm3 for a subsequent study. These mice (n = 4/arm) were re-treated with either SN38-TOA nanoparticles (10 mg/kg) or CPT-11 (25 mg/kg) twice per week for 4 weeks intravenously via tail vein and monitored in a similar fashion.
Histologic evaluation of tumors
Endpoint tumors were excised and fixed in 4% paraformaldehyde and delivered for processing and histochemical staining to the CHOP Pathology Core. Stains used included hematoxylin and eosin (H&E), Ki-67 for proliferation, SOX10 as a neural crest marker, p75/NGFR as a neural marker, S100 as a Schwann cell marker, and ab190710 (Abcam) as a human-specific nucleolar marker.
Pharmacokinetics analysis of SN38 and CPT-11 in mouse blood and tissues
Mice (n = 3/arm, per time point) with flank xenografts were given a single dose of either SN38-TOA nanoparticles (10 mg/kg) or CPT-11 (25 mg/kg) intravenously via tail vein. Blood was obtained via retro-orbital and terminal bleeds and collected into 2 mL collection tubes containing sodium heparin (BD). Tissues (tumor, lung, liver, spleen, kidney) were collected postsacrifice at 4, 24, and 72 hours after heart perfusion with cold saline. Plasma was separated from the blood by centrifugation for 15 minutes at 2,000 × g at 4°C and stored separately. All samples were stored in −80°C until analyzed by the CHOP Pharmacology Core. Total SN38 and CPT-11 levels were analyzed in mouse blood (1:1 diluted with water) and tissue homogenates by UPLC-MS/MS, as described previously (10, 20) with some modifications.
Tissues were homogenized using a Biologics Inc., Model 3000 ultrasonic homogenizer. We added 20:80 methanol:water with 1% formic acid to a known weight of tissue to obtain a ratio of 4 mL/g sample. Samples were homogenized on ice and frozen until analysis. SN38 prodrug–spiked mouse plasma and tissue homogenate were hydrolyzed using sodium hydroxide (1 mol/L, 15 μL) and incubated for 15 minutes at 37°C in a Thermo electron incubator. The reaction was stopped by adding 98% formic acid (10 μL). Analysis confirmed complete hydrolysis of SN38 prodrug under these conditions. Standards were prepared in CD-1 mouse plasma containing sodium heparin as an anticoagulant. A nine-point calibration curve was prepared at different concentrations by spiking a working stock. Plasma and tissue homogenate samples were extracted via acetonitrile precipitation in a 96-well format. Electrospray ionization in the positive ion mode was utilized for the tandem mass spectrometric detection of SN38-TOA (m/z 393.2→349.0 and m/z 393.2→249.1) and CPT-11 (m/z 587.3→124.2) using AB Sciex 4000 mass spectrometer. Separation was accomplished utilizing Kinetex PFP (50.4.1 mm id, 2.6 μm) column with Shimadzu LC 20AD HPLC system with a run time of 4.5 minutes. Assay was linear over the range of 1 to 1,000 ng/mL for both SN38 and CPT-11 in mouse plasma. The matrix factors of the mouse tissues (tumor, kidney, spleen, and liver) obtained using tissue homogenates spiked with 100 ng of SN38 prodrug per mL (n = 3) against mouse plasma calibration curves were applied in the drug assay calculations (10).
Statistical analysis
Cell growth inhibition was compared using two-way ANOVA and Bonferroni multiple comparisons tests. Event-free survival (EFS) curves were estimated using the Kaplan–Meier method and compared using a log-rank (Mantel–Cox) test.
Results
Effect of SN38-TOA nanoparticles on growth of neuroblastoma cells in vitro
We assessed the efficacy of SN38-TOA compared with free SN38 in vitro by performing a growth inhibition assay of neuroblastoma cells. Free SN38 had a mild inhibitory effect at the 1 nmol/L concentration (P = 0.0007). However, strong inhibition was observed at and above 2 nmol/L (P < 0.0001), and effectiveness increased as the concentration increased (Fig. 1, Supplementary Fig. S1). The effect of SN38-TOA nanoparticles on cell growth was delayed in comparison with free SN38, increasing toward 96 hours and presumably reflecting the gradual release of SN38 from the nanoparticles over time (Fig. 1, Supplementary Fig. S1), with no statistically significant difference in efficacy (P > 0.9999).
Figure 1.
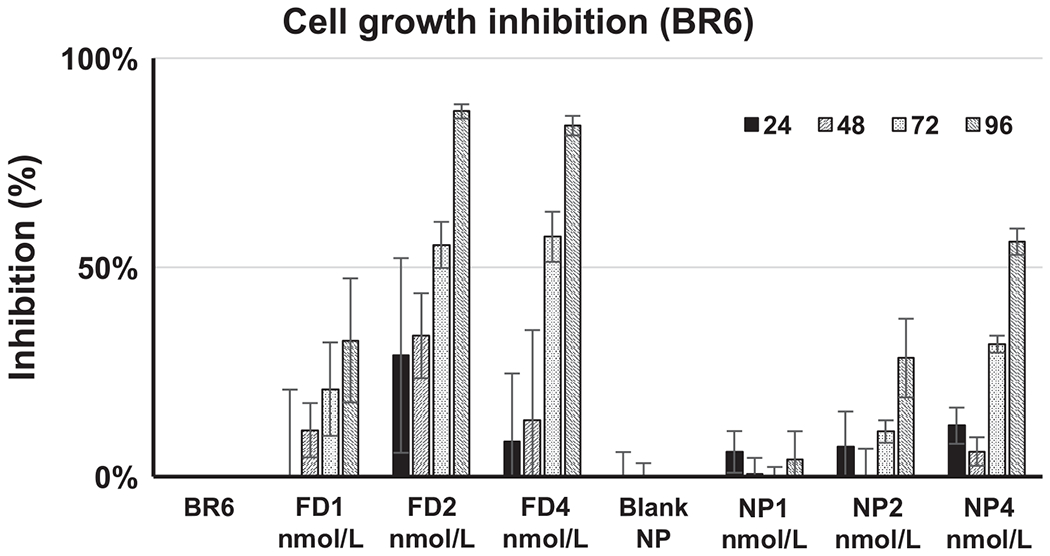
SRB assay assessing efficacy of free SN38 and SN38-TOA nanoparticle. Growth inhibition by free SN38 is statistically significant for 2 and 4 nmol/L at 72 hours (P > 0.0001) and 96 hours (P > 0.0001), and 1 nmol/L at 96 hours (P = 0.0007). Growth inhibition by SN38-TOA is statistically significant for 4 nmol/L at 72 hours (P = 0.0002), and 2 and 4 nmol/L at 96 hours (P < 0.0001). FD, free drug; NP, nanoparticles.
Effect of SN38-TOA nanoparticles on growth of neuroblastoma flank xenografts
We performed mouse flank xenograft experiments (n = 10/arm) with BR6 neuroblastoma cells to examine the effect of SN38-TOA in vivo. Thirty days after treatment initiation, all of the mice that received either blank nanoparticle (vehicle only) or saline (no treatment) reached the endpoint tumor volume (3.0 cm3). CPT-11 was able to extend survival time by approximately 45 days (Fig. 2A). Tumor growth was suppressed by CPT-11 during the course of the treatment, but many tumors began to grow immediately after cessation of treatment, reaching a volume of over 3.0 cm3 in 15 days. Mice that received SN38-TOA nanoparticles had nearly undetectable tumors for at least 60 days, with 100% survival at 110 days, and 60% survival 180 days post-initiation of treatment (P < 0.0001; Fig. 2B). Of the remaining 6 surviving mice, half did not have detectable tumors, and half had stable tumors that were less than 0.5 cm3.
Figure 2.
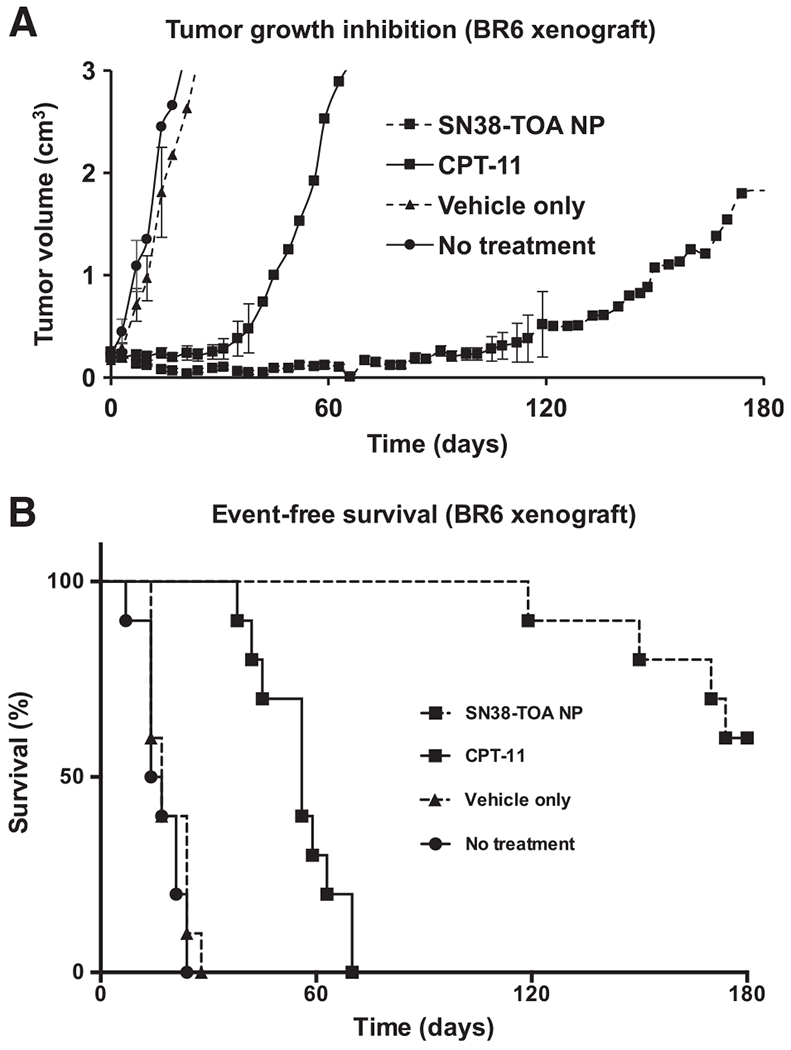
Efficacy of SN38-TOA nanoparticles (NP) on BR6 flank xenografts. A, Tumor growth inhibition by SN38-TOA. Trk-B-expressing SH-SY5Y (BR6) cells were implanted subcutaneously in the flank of each mouse. All mice (n = 10/arm) were treated intravenously via tail vein injections twice per week for 4 weeks when tumors reached a volume of 0.2 cm3. SN38-TOA nanoparticles and CPT-11 were given at doses equivalent to 10 and 25 mg/kg drug, respectively. B, Event-free survival graph. Mice were removed from the study when the tumor volumes reached 3.0 cm3. SN38-TOA nanoparticles are more effective than CPT-11 (P < 0.0001) at inhibiting tumor growth and extending survivability.
Biodistribution of SN38-TOA nanoparticles
To measure SN38 levels in blood, tumor, and other tissues from mice (n = 3/arm, per time point) given a single dose of either CPT-11 (25 mg/kg) or SN38-TOA nanoparticles (10 mg/kg), we performed biodistribution studies. SN38 levels were measured at 4, 24, and 72 hours in blood, tumor, liver, spleen, lung, and kidney (Table 1). At 4 hours, the level of SN38 in blood was over 400 times higher in mice treated with SN38-TOA nanoparticles compared with those treated with CPT-11, suggesting that this nanoparticle formulation results in prolonged retention, which allows for both continued accumulation in the tumor and decreased systemic exposure. The level of SN38 in the tumor was 3-fold higher at 4 hours in the nanoparticle-treated mice (Fig. 3). More importantly, intratumoral SN38 remained high in the nanoparticle-treated mice for a protracted period of time (about 50% of the 4-hour level at 24 hours, and 25% of the 4-hour level at 72 hours). In contrast, SN38 levels were virtually undetectable at 24 and 72 hours in tumors from mice treated with CPT-11 (Table 1, Fig. 3). As expected, there was substantial accumulation of SN38 in the liver and spleen, presumably reflecting sequestration of nanoparticles in macrophages. However, no hepatic or other toxicity was seen in the nanoparticle-treated mice.
Table 1.
Levels of SN38 in blood, tumor and normal organs after 1 dose of CPT-11 or SN38-TOA NPs
| SN38 | Blood (ng/ml) |
Tumor (ng/g) |
Liver (ng/g) |
Spleen (ng/g) |
Lung (ng/g) |
Kidney (ng/g) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| levels | CPT-11 | SN38-TOA NP | CPT-11 | SN38-TOA NP | CPT-11 | SN38-TOA NP | CPT-11 | SN38-TOA NP | CPT-11 | SN38-TOA NP | CPT-11 | SN38-TOA NP |
| 4 hrs | 8 (± 5) | 4537(± 978) | 147 (± 26) | 383 (± 18) | 1245 (± 101) | 169965 (± 13524) | 56 (± 8) | 122691 (± 26283) | 38 (± 16) | 1710 (± 1328) | 421 (± 81) | 1289 (± 378) |
| 24 hrs | <1 | 221 (± 8) | <10 | 193 (± 17) | 201 (± 1) | 126266 (± 8573) | 5(± 5) | 1262239 (± 18376) | 5(± 4) | 1976 (± 497) | 13 (± 4) | 1171 (± 335) |
| 72 hrs | <1 | 25 (± 10) | <10 | 102 (± 18) | 6 (± 6) | 106890 (± 13106) | 4(± 5) | 110360 (± 31374) | <2 | 1445 (± 413) | 6 (± 11) | 664 (± 424) |
Figure 3.
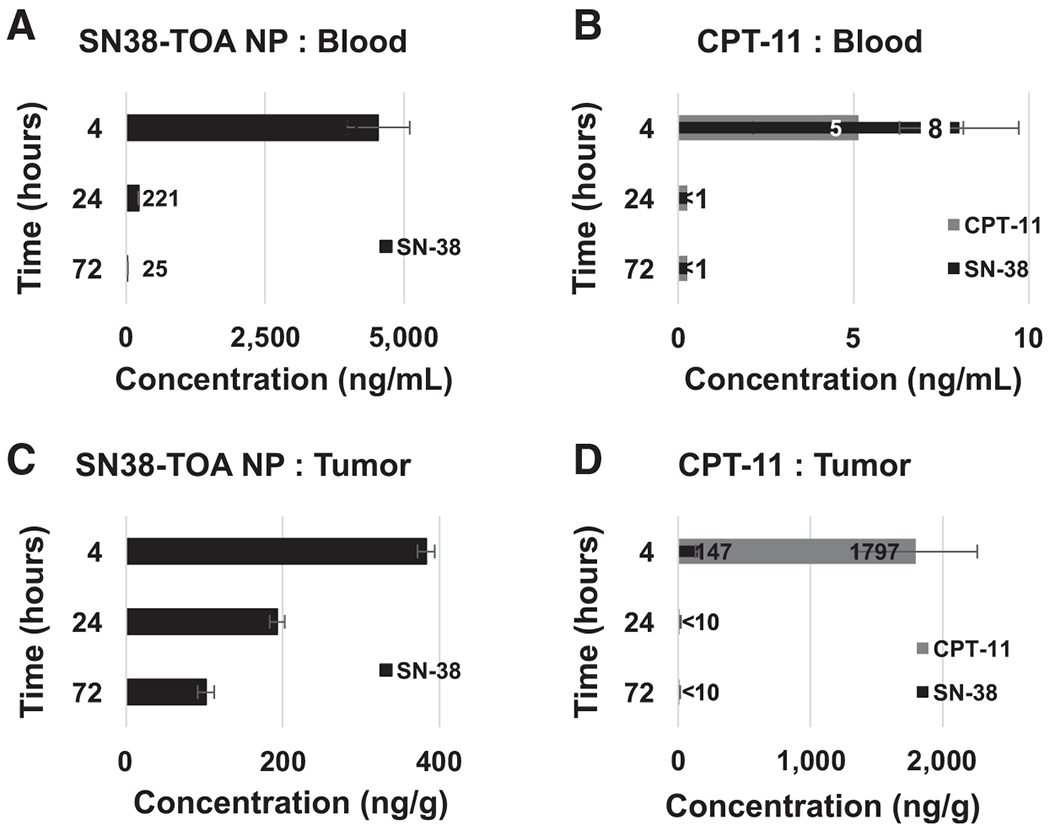
Biodistribution profile of SN38-TOA nanoparticles (NP). Mice (n = 3/arm, per time point) were given a single dose of SN38-TOA nanoparticles (10 mg/kg) or CPT-11 (25 mg/kg) intravenously via tail vein. A and B, Blood: SN38-TOA nanoparticle treated (A) and CPT-11 treated (B). C and D, Tumor, SN38-TOA nanoparticle treated (C) and CPT-11 treated (D) were collected post sacrifice at 4, 24, and 72 hours. SN38 levels in SN38-TOA nanoparticle-treated mice were much higher compared with CPT-11–treated mice at all time points. Values are shown ±SE.
Retreatment of recurrent tumors in CPT-11–treated mice
Tumors in mice initially treated with CPT-11 were re-treated with either SN38-TOA nanoparticles (n = 4) or CPT-11 (n = 4) when their recurrent tumor volumes reached 2.0 cm3 (Fig. 4A). Retreatment with CPT-11 was only able to slow the rate of tumor progression modestly, and most of the mice were removed from the study before receiving the full retreatment course. Mice retreated with SN38-TOA nanoparticles responded immediately, evidenced by tumor regression after initiation of treatment. Overall, the tumors decreased in volume by over 50% after 14 days. After cessation of treatment, the tumors began to regrow, but in a very slow and protracted manner. Interestingly, 100% of the mice were surviving with slowly growing tumors at 120 days postretreatment (P = 0.0067; Fig. 4A), so we investigated the histology of these tumors.
Figure 4.
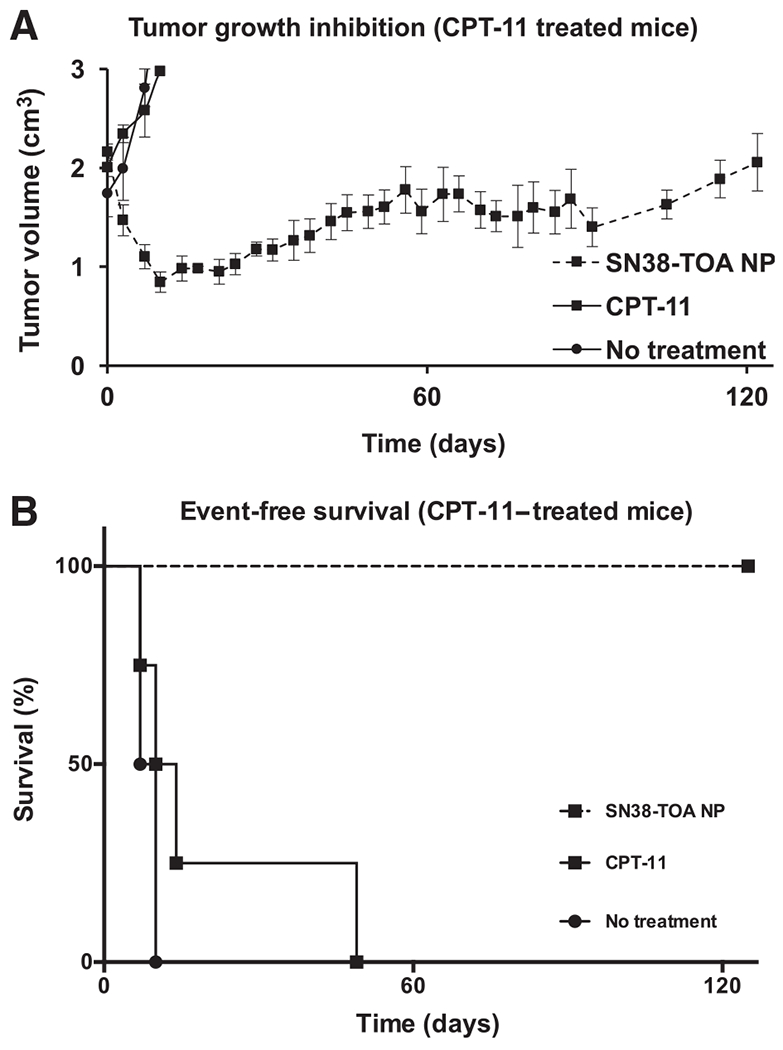
SN38-TOA nanoparticle (NP) treatment of CPT-11–treated tumors. A, Kinetic pattern of tumor growth inhibition. Tumors recurred in mice previously treated with CPT-11. When these tumors reached a volume of 2.0 cm3, they (n = 4/arm) received a second treatment course of either SN38-TOA nanoparticles (10 mg/kg drug) or CPT-11 (25 mg/kg drug) twice per week for 4 weeks. B, Event-free survival curves of CPT-11–re-treated mice. CPT-11 was ineffective, while tumors immediately responded to SN38-TOA nanoparticles (P = 0.0067) and took 100 days to return to their original size.
Histologic evaluation of tumors
Tumors that were untreated, treated initially with CPT-11 or SN38-TOA nanoparticles, or treated and re-treated with CPT-11 had similar histology. They all appeared as undifferentiated small, round, blue cells with focal necrosis, no neuropil (Fig. 5A and B), and high proliferative index (>30%), as demonstrated by Ki-67 index (Supplementary Fig. S2A). However, tumors retreated with SN38-TOA nanoparticles had a dramatically different appearance, both grossly and microscopically. Tumors were composed mostly of cells that were large and differentiated, resembling ganglion cells (Fig. 5C). There was abundant neuropil, and the proliferative index was extremely low (<1%), as demonstrated by Ki-67 index (Supplementary Fig. S2B). There were also fields of Schwann cells (Fig. 5D) positive for SOX-10 staining around the ganglionic cells (Fig. 5E), and these cells were negative for a human nucleolar-specific antibody (Fig. 5F), suggesting that these were infiltrating mouse cells.
Figure 5.
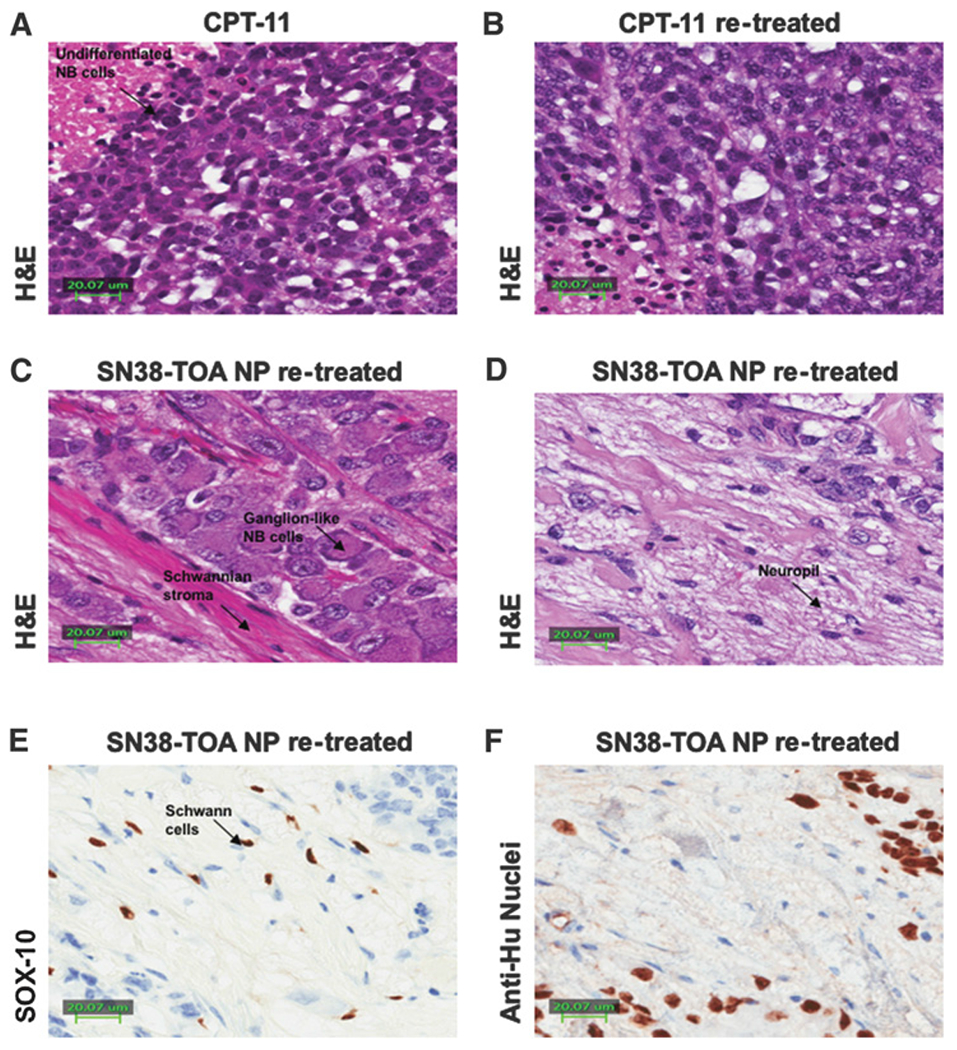
Histology of tumor samples. A and B, H&E staining of a CPT-11–treated tumor (A) and a CPT-11–re-treated tumor (B) shows undifferentiated neuroblastoma with areas of necrosis. There is no evidence of Schwannian differentiation, and no ganglion cells. C, H&E of a SN38-TOA nanoparticle (NP)–treated tumor shows ganglion cells embedded in Schwannian stroma with a background of neuropil. D–F, A SN38-TOA nanoparticle–treated tumor with H&E staining (D) shows neuroblastoma cells, ganglion cells embedded in Schwannian stroma, Schwann cells, and neuropil; SOX-10 (E) staining shows positive spindle cell nuclei, consistent with Schwann cells, while neuroblastoma cells and ganglion cells are negative. F, Anti-Hu nucleolar staining shows positive undifferentiated neuroblastoma cells, and negative Schwannian spindle cells consistent with murine origin.
Confirmation of SN38-TOA nanoparticle efficacy in the IMR-32 neuroblastoma line
To demonstrate that the therapeutic response to SN38-TOA nanoparticles could be extended to MYCN-amplified tumors, which are a very aggressive subset of high-risk neuroblastomas, we also tested the nanoparticles against xenografts established using the IMR-32 neuroblastoma line. Cells were implanted subcutaneously in the flank of each mouse. Mice (n = 4/arm) were treated intravenously via tail vein injections with SN38-TOA nanoparticles (10 mg/kg) twice per week for 4 weeks when tumors reached a volume of 1.5 cm3, substantially larger than the BR6-treated tumors described above. Mice were removed from the study when the tumor volumes reached 3.0 cm3. SN38-TOA nanoparticles caused the tumors to completely regress in all of the mice, and so far, there have been no recurrences after more than 180 days (P = 0.0101; Fig. 6).
Figure 6.
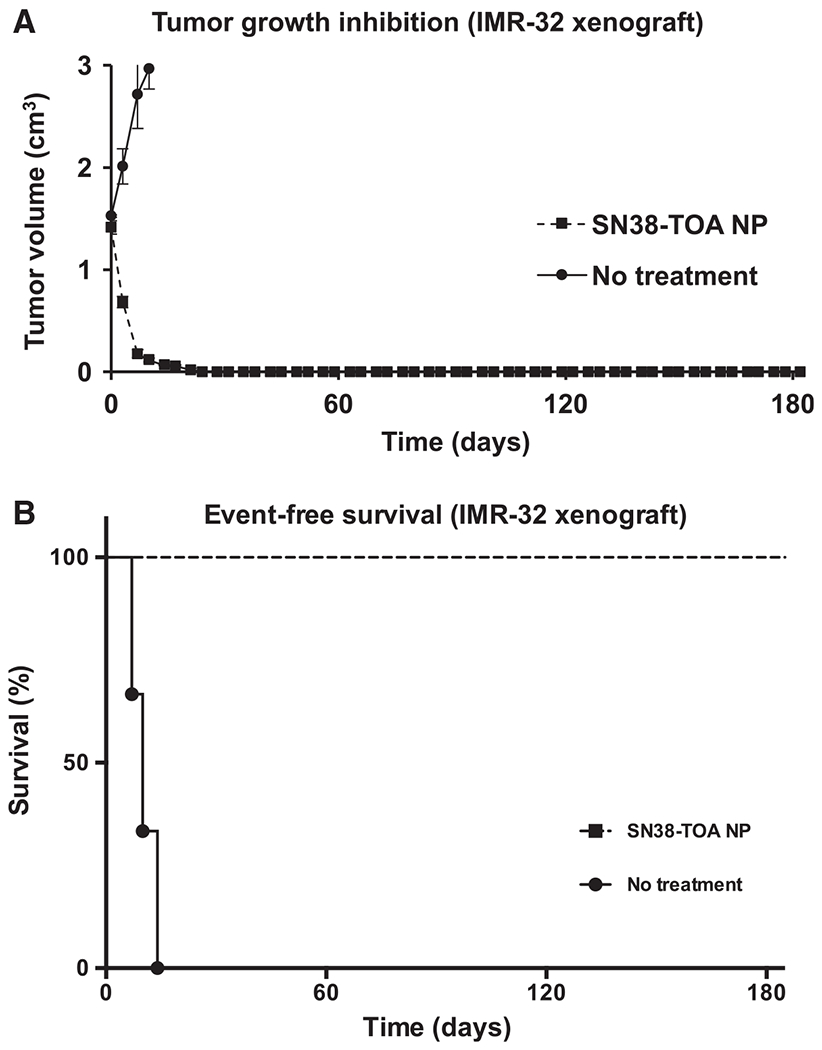
Efficacy of SN38-TOA nanoparticles (NP) on IMR-32 flank xenografts. A, Tumor growth inhibition by SN38-TOA. IMR-32 cells were implanted subcutaneously in the flank of each mouse. Mice (n = 4/arm) were treated intravenously via tail vein injections with SN38-TOA nanoparticles (10 mg/kg) twice per week for 4 weeks when tumors reached a volume of 1.5 cm3. B, Event-free survival curves of CPT-11–re-treated mice. Mice were removed from the study when the tumor volumes reached 3.0 cm3. SN38-TOA nanoparticles caused the tumors to completely regress in all of the mice (P = 0.0101).
Discussion
There is a compelling need to develop more effective, less toxic therapy for children and adults with high-risk cancers. Clinically used and experimental strategies include cellular or humoral immunotherapy that selectively targets specific antigens, as well as drugs that selectively target a particular gene, protein, or pathway that represents an oncogenic driver and tumor vulnerability. Each of these approaches can benefit from enhancing its on-target effect while limiting systemic exposure and toxicity, resulting in safer and more effective therapies. Improving tumor-specific delivery of conventional as well as targeted agents using nanoparticles permits enhanced accumulation and retention in the tumor, and it offers a strategy for making a therapeutically effective response achievable at well-tolerated drug doses.
We chose SN38 as the anticancer agent to deliver in nanoparticles for several reasons. It has poor solubility, which makes it unsuitable for conventional delivery but enhances its retention in nanoparticles. Also, SN38 is a very potent and active agent, whereas CPT-11 is a prodrug that requires activation by the liver, which is an inefficient process. SN38 is an S-phase–specific agent that is both selective for rapidly proliferating cells and is highly effective at killing them, while sparing normal, quiescent cells. Thus, it should have limited toxicity in other organs where nanoparticles typically accumulate, such as the liver and spleen. In addition, a reduced amount of CPT-11 and SN38 was administered in these studies. During each experiment, no weight loss was observed, and there was a steady increase in body weight over the course of the treatment. On the basis of these observations, it was decided that gastrointestinal toxicity was minimal, and no specific pharmacokinetic study of the gastrointestinal tract was performed, as luminal drug would account for any toxicity.
We reversibly hydrophobized SN38 with tocopheryl oxyacetate using a linkage that is readily hydrolyzed in a nonenzymatic manner in an aqueous environment. In addition, tocopheryl oxyacetate is a mitocan that targets the mitochondria by the generation of reactive oxygen species, leading to apoptosis, so it is essentially a codrug that acts on tumor cells by an independent mechanism (11–13).
We are using polymeric nanoparticles of 50 to 70 nm that have been size-optimized to be long-circulating, as well as for tumor penetration, retention and payload delivery. Our nanoparticles composed of PEGylated polylactic acid chains are highly biocompatible and fully biodegradable, so there is no accumulation of toxic elements with repeated exposure. This matrix is suitable for entrapment and delivery of a variety of cell cycle–specific hydrophobic agents to solid tumors, taking advantage of the leaky blood vessels commonly found in rapidly growing tumors.
As expected, the entrapped SN38 in nanoparticles was not freely available to cells in vitro, so free SN38 is more potent (Fig. 1). However, the advantage of nanoparticle drug delivery was readily evident in our in vivo xenograft studies. There was dramatic tumor shrinkage and prolongation of EFS in mice with BR6 xenografts compared with the CPT-11–treated mice, with half the mice surviving disease free for over 180 days (Fig. 2). This effect was even more dramatic in the IMR-32 xenografts, with 100% of the mice surviving disease free at 180 days (Fig. 6). This is especially important, given the negative prognostic impact of MYCN amplification in neuroblastomas (21, 22). Thus, SN38-TOA nanoparticles clearly outperformed CPT-11, even though the dose of drug in the nanoparticles was significantly less (10 vs. 25 mg/kg).
The improved outcome with nanoparticle prodrug delivery is likely due to the much higher drug levels we were able to achieve in the tumor, especially at later time points (Fig. 3, Table 1). The nanoparticles are long-circulating, allowing continued accumulation over time in tumors due to the EPR effect. Thus, we were able to have greater efficacy while administering less total drug compared with conventional delivery of CPT-11. The entrapment of SN38 in nanoparticles may contribute to further reducing systemic exposure and toxicity. Thus, the delivery of a reduced amount of SN38 as a reversibly hydrophobized prodrug encased in nanoparticles achieved increased antitumor efficacy, while theoretically minimizing the potential for toxicity.
Others have used PEGylated SN38 (EZN-2208) to treat neuroblastoma xenografts, and it was also shown to be superior to CPT-11 in terms of drug penetration and tumor control (23–25). However, the results from a phase I clinical trial in neuroblastomas were disappointing (26). Another group used nano-liposomal CPT-11 (MM-398) to treat pediatric solid tumors as xenografts (27). There was substantial efficacy and prolongation of survival in Ewing family tumor xenografts, but the results were not as good with neuroblastoma and with rhabdomyosarcoma xenografts. This agent is in clinical trials in adult solid tumors, and it demonstrates moderate antitumor activity, with a manageable side effect profile (28, 29). These results support the advantages of nanoformulations of SN38/CPT-11 and warrant further exploration of other approaches.
The complementary role of TOA as a mitocan may be adding to the anticancer effect of these nanoparticles, but this is difficult to model accurately in vitro. Nevertheless, we saw not only an enhanced anticancer effect, but also differentiation of the neuroblastoma xenograft cells in tumors that were pretreated with CPT-11 alone. Two groups were treated with SN38-TOA nanoparticles: small untreated tumors (0.2 cm3), and large pretreated tumors (2.0 cm3). Only the re-treated group presented a dramatically different histology. This was also observed previously when large untreated tumors were treated with SN38-TS (10). In both instances, exposing large tumors to high amounts of SN38 resulted in differentiated tumors. The role of each component is unclear, as a larger concentration of SN38 was also delivered. However, in addition to neuroblastomacells differentiated into ganglionic cells, we also saw fields of Schwannian stroma surrounding the differentiated neural cells. We confirmed that they were Schwann cells by SOX-10 staining, but they were also identified as mouse in origin, due to the absence of staining with a human-specific antibody (Fig. 5F). Although this model is clearly different than the spontaneous development of ganglioneuromas in humans, it lends support to the hypothesis that the Schwann cells in these tumors are normal host-derived, infiltrating cells, rather than an alternate differentiation path of malignant sympathoadrenal progenitor cells (30). It is possible that the TOA component of this nanoparticle codrug may have contributed to the differentiated state of these retreated neuroblastomas.
In conclusion, we have shown that nanoparticle drug delivery of SN38-TOA had a dramatically greater tumor response and prolonged control compared with treatment with comparable or higher levels of the parent drug, CPT-11. This was due presumably to the higher and sustained drug levels in the tumor using nanoparticle prodrug delivery. These nanoparticles were able to achieve prolonged EFS in 50% to 100% of the mice, as defined by tumor remission and control for at least 180 days. Furthermore, these anticancer effects were achieved using a lower total drug dose, while drug entrapment in nanoparticles should further reduce systemic exposure and toxicity. Although this nanoparticle prodrug formulation was tested in a model of neuroblastoma in these experiments, it should be effective in treating many other aggressive solid tumors in children and adults.
Supplementary Material
Acknowledgments
These results were supported in part by grants from Alex’s Lemonade Stand, St. Baldrick’s Foundation, CURE Childhood Cancer, and Solving Kids’ Cancer.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
F. Nguyen and I. Alferiev are co-first authors of this article.
M. Chorny and G.M. Brodeur are co-last authors of this article.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Brodeur GM, Hogarty MD, Bagatell R, Mosse YP, Maris JM. Neuroblastoma In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. 7th ed. Philadelphia, PA: Wolters, Kluwer; 2016. p 772–97. [Google Scholar]
- 2.Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol 2009;27:1007–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Applebaum MA, Vaksman Z, Lee SM, Hungate EA, Henderson TO, London WB, et al. Neuroblastoma survivors are at increased risk for second malignancies: A report from the International Neuroblastoma Risk Group Project. Eur J Cancer 2017;72:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hobbie WL, Moshang T Carlson CA, Goldmuntz E, Sacks N, Goldfarb SB, et al. Late effects in survivors of tandem peripheral blood stem cell transplant for high-risk neuroblastoma. Pediatric Blood Cancer 2008;51: 679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreno L, Vaidya SJ, Pinkerton CR, Lewis IJ, Imeson J, Machin D, et al. European Neuroblastoma Study G, Children’s C, Leukaemia G. Long-term follow-up of children with high-risk neuroblastoma: the ENSG5 trial experience. Pediatr Blood Cancer 2013;60:1135–40. [DOI] [PubMed] [Google Scholar]
- 6.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A 2011;108:1850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res 2002;8:878–84. [PubMed] [Google Scholar]
- 8.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 2000;65:271–84. [DOI] [PubMed] [Google Scholar]
- 9.Alferiev IS, Iyer R, Croucher JL, Adamo RF, Zhang K, Mangino JL, et al. Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials 2015;51:22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer R, Croucher JL, Chorny M, Mangino JL, Alferiev IS, Levy RJ, et al. Nanoparticle delivery of an SN38 conjugate is more effective than irinotecan in a mouse model of neuroblastoma. Cancer Lett 2015; 360:205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong LF, Freeman R, Liu J, Zobalova R, Marin-Hernandez A, Stantic M, et al. Suppression of tumor growth in vivo by the mitocan alphatocopheryl succinate requires respiratory complex II. Clin Cancer Res 2009;15:1593–600. [DOI] [PubMed] [Google Scholar]
- 12.Neuzil J, Wang XF, Dong LF, Low P, Ralph SJ. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett 2006;580: 5125–9. [DOI] [PubMed] [Google Scholar]
- 13.Prochazka L, Dong LF, Valis K, Freeman R, Ralph SJ, Turanek J, et al. alpha-Tocopheryl succinate causes mitochondrial permeabilization by preferential formation of Bak channels. Apoptosis 2010;15: 782–94. [DOI] [PubMed] [Google Scholar]
- 14.Lawson KA, Anderson K, Menchaca M, Atkinson J, Sun L, Knight V, et al. Novel vitamin E analogue decreases syngeneic mouse mammary tumor burden and reduces lung metastasis. Mol Cancer Ther 2003;2: 437–44. [PubMed] [Google Scholar]
- 15.Brodeur GM, Green AA, Hayes FA, Williams KJ, Williams DL, Tsiatis AA. Cytogenetic features of human neuroblastomas and cell lines. Cancer Res 1981;41(11 Pt 1):4678–86. [PubMed] [Google Scholar]
- 16.Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res 2002;62:6462–6. [PubMed] [Google Scholar]
- 17.Ross RA, Spengler BA, Biedler JL. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 1983;71:741–7. [PubMed] [Google Scholar]
- 18.Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res 1970; 30:2110–8. [PubMed] [Google Scholar]
- 19.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1:1112–6. [DOI] [PubMed] [Google Scholar]
- 20.Goldwirt L, Lemaitre F, Zahr N, Farinotti R, Fernandez C. A new UPLC-MS/MS method for the determination of irinotecan and 7-ethyl-10-hydroxycamptothecin (SN-38) in mice: application to plasma and brain pharmacokinetics. J Pharm Biomed Anal 2012;66: 325–33. [DOI] [PubMed] [Google Scholar]
- 21.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003;3:203–16. [DOI] [PubMed] [Google Scholar]
- 22.Campbell K, Gastier-Foster JM, Mann M, Naranjo AH, Van Ryn C, Bagatell R, et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer 2017;123:4224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pastorino F, Loi M, Sapra P, Becherini P, Cilli M, Emionite L, et al. Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin Cancer Res 2010;16:4809–21. [DOI] [PubMed] [Google Scholar]
- 24.Sapra P, Zhao H, Mehlig M, Malaby J, Kraft P, Longley C, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res 2008;14: 1888–96. [DOI] [PubMed] [Google Scholar]
- 25.Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem 2008;19:849–59. [DOI] [PubMed] [Google Scholar]
- 26.Norris RE, Shusterman S, Gore L, Muscal JA, Macy ME, Fox E, et al. Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr Blood Cancer 2014;61:1792–7. [DOI] [PubMed] [Google Scholar]
- 27.Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, et al. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin Cancer Res 2015;21: 1139–50. [DOI] [PubMed] [Google Scholar]
- 28.Ko AH, Tempero MA, Shan YS, Su WC, Lin YL, Dito E, et al. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM-398) for patients with gemcitabine-refractory metastatic pancreatic cancer. Br J Cancer 2013;109:920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy AC, Park SR, Cunningham D, Kang YK, Chao Y, Chen LT, et al. A randomized phase II study of PEP02 (MM-398), irinotecan or docetaxel as a second-line therapy in patients with locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma. Ann Oncol 2013;24:1567–73. [DOI] [PubMed] [Google Scholar]
- 30.Ambros IM, Zellner A, Roald B, Amann G, Ladenstein R, Printz D, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med 1996;334:1505–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.