

Abstract
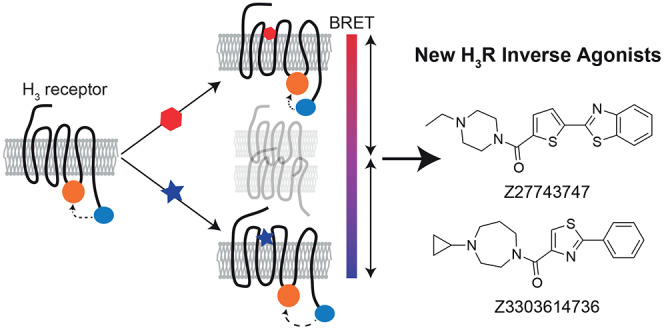
The histamine H3 receptor (H3R) represents a highly attractive drug target for the treatment of various central nervous system disorders, but the discovery of novel H3R targeting compounds relies on the assessment of highly amplified intracellular signaling events that do not only reflect H3R modulation and carry the risk of high false-positive and -negative screening rates. To address these limitations, we designed an intramolecular H3R biosensor based on the principle of bioluminescence resonance energy transfer (BRET) that reports the receptor’s real-time conformational dynamics and provides an advanced tool to screen for both H3R agonists and inverse agonists in a live cell screening-compatible assay format. This conformational G-protein-coupled receptor (GPCR) sensor allowed us to characterize the pharmacological properties of known and new H3 receptor ligands with unprecedented accuracy. Interestingly, we found that one newly developed H3 receptor ligand possesses even stronger inverse agonistic activity than reference H3R inverse agonists including the current gold standard pitolisant. Taken together, we describe here the design and validation of the first screening-compatible H3R conformational biosensor that will aid in the discovery of novel H3R ligands and can be employed to gain deeper insights into the (in-)activation mechanism of this highly attractive drug target.
Keywords: GPCR, BRET, conformational sensor, histamine receptor, inverse agonist, drug discovery
The group of histamine receptors belongs to the superfamily of G-protein-coupled receptors (GPCRs) and comprises four distinct subtypes, named H1, H2, H3, and H4 receptor (H1–4R), that are of highest interest for modern drug discovery.1 First, H1R and H2R antagonists have already been developed in the 1930s and 1970s, respectively, leading to their extensive medical use in numerous antiallergic and antiulcer pharmaceutics including loratadine (H1R) and ranitidine (H2R) that are listed in the WHO register of essential medicines.2,3
Likewise, the discovery of the subtype H3R has raised intense hopes for the development of similarly successful drugs.4 In humans, H3R is almost exclusively expressed in the central nervous system (CNS) and considered to work both as an autoreceptor in presynaptic membranes and as a postsynaptic heteroreceptor controlling the release of other neurotransmitters including acetylcholine, norepinephrine, and dopamine.5 Therefore, pharmacological modulation of the H3R represents an attractive approach to treat various central nervous system diseases such as Parkinson’s, Huntington’s, and Alzheimer’s diseases, as well as tic disorders.1,6−9 In addition, pitolisant, an H3R inverse agonist that reduces the basal signaling capacity of the receptor (known as receptor constitutive activity), has entered the market 4 years ago for the treatment of narcolepsia.10 Despite increasing efforts to develop novel H3R-modulating compounds,11−17 thus far, pitolisant represents the only H3R ligand that is approved by health authorities. This is partially due to the limited power of our current screening technologies that are mainly based on receptor downstream signaling events. For instance, measuring compound-induced β-arrestin translocation is one of the most common assays employed in GPCR drug discovery campaigns. However, this approach not only fails to detect G-protein-biased H3R agonists but also misses receptor inverse agonists like pitolisant. Identification of pitolisant with such a setup would instead require preincubation with selective H3R agonists but, in turn, hamper the discovery of H3 receptor agonists.
In contrast to these signaling-dependent assays, downstream-independent screening methods that detect ligands with distinct efficacies (e.g., agonists vs inverse agonists) and specific downstream signaling profiles (e.g., G-protein-biased vs β-arrestin-biased agonists) would allow for the simultaneous screening of compounds with differing pharmacological profiles and thus are highly desired to speed up the discovery of novel GPCR ligands.
Conformational GPCR biosensors based on fluorescence and bioluminescence resonance energy transfer (FRET and BRET, respectively) have proven valuable tools to determine the pharmacological profiles of GPCR ligands in a real-time and live cell assay system.18 Since the receptor’s conformational change follows directly upon ligand binding without any signal amplification, this approach represents the most undistorted way to assess ligand efficacy and potency; as a consequence, these conformational readouts reveal differences among distinct test compounds where downstream-dependent assays report indiscernible activities.19−21 Furthermore, single-cell studies employing FRET-based conformational biosensors of H1R and H3R have recently provided precious insights into the mechano-sensing mechanism and the activation kinetics of histamine receptors, respectively.22,23 Recently, the suitability of such sensors for high-throughput screening has been demonstrated by employing a novel BRET system composed of the small engineered luciferase NanoLuciferase (Nluc)24 and a red fluorescent HaloTag dye,25 as demonstrated for two class A (α2A-adrenergic and β2-adrenergic receptor) and one class B GPCRs (parathyroid hormone receptor 1).19
In this study, we set out to address the urgent need for a downstream-independent and very sensitive screening platform for the highly attractive drug target, the H3R. By fusing the BRET partners Nluc and HaloTag to this histamine receptor subtype, we generated a conformational biosensor that enabled the simultaneous detection of agonists and inverse agonists with screening-compatible assay sensitivity. Furthermore, using this biosensor, we characterized pharmacologically, in addition to several well-known H3R standard ligands, two novel H3R inverse agonists, which have recently been identified in a virtual screening campaign.26 We show that one of these ligands possesses even stronger inverse agonistic activity than pitolisant and reduces the H3R-mediated Gi-protein activity with nanomolar potencies.
Experimental Section
Plasmids
Wild-type human H3R DNA (GeneID 11255) in pcDNA3.1 vector was purchased from cDNA.org. Nluc was fused at position K445 to generate C-terminally tagged H3RNluc. Thereafter, a HaloTag was inserted between S307/G308 (full-length H3RNluc/Halo(618)) or between T229/F348 (Δicl3-H3RNluc/Halo(618)) within the third intracellular loop of H3RNluc. All cloning steps were performed using the established polymerase chain reaction (PCR) strategies, and restriction enzymes and constructs were verified by sequencing (Eurofins genomics). Plasmids encoding tagged and native G-protein subunits were kindly provided by A. Inoue (Tohoku University, Sendai, Japan).
Reagents
Histamine dihydrochloride, imetit dihydrobromide, clobenpropit dihydrobromide, thioperamide, and pitolisant (BF 2649 hydrochloride) were purchased from Tocris Bioscience (Wiesbaden-Nordenstadt, Germany). Poly-d-lysine and geneticin were obtained from Sigma-Aldrich (Taufkirchen, Germany). [3H]NAMH (specific activity: 79.7 Ci/mmol) was from PerkinElmer (Waltham, MA). Z27743747 and Z3303614736 were purchased from Enamine Ltd. (Kyiv, Ukraine). The Nluc substrate furimazine and the HaloTag fluorescent dye NanoBRET 618 were obtained from Promega (Madison, WI). UR-PI294, VUF4903, VUF4904, and VUF5207 were synthesized as described previously.27,28 White-wall, white-bottomed 96-well microtiter plates were from Brand (Wertheim, Germany) and Gibco (Waltham, MA).
Cell Culture
HEK293T and HEK293A cells were used for the transient expression of GPCR and G-protein biosensors and grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM glutamine, 10% fetal calf serum, 0.1 mg/mL streptomycin, and 100 units/mL penicillin at 37 °C with 5% CO2. To generate the cell line stably expressing Δicl3-H3RNluc/Halo(618), HEK293A cells grown in T75 flasks were transfected at a confluence of 50% with 1 μg of DNA using Lipofectamine 2000 (ThermoFisher Scientific, Waltham, MA). Stably transfected cells were selected with 2 mg/mL geneticin and maintained in supplemented DMEM containing 500 μg/mL geneticin.
Radioligand Binding
HEK293A cells stably expressing Δicl3-H3RNluc/Halo(618) or HEK293T cells transiently transfected with 2.5 μg of the wild-type H3R cDNA grown in 10 cm2 culture dishes were collected (48 h after transfection for the wild-type H3R) in cold phosphate-buffered saline (PBS) and centrifuged at 3000g for 15 min at 4 °C. The supernatant was discarded, and the cell pellet was stored at −20 °C until the day of the experiment. Prior to the experiment, cell pellets (200 μg/mL) were resuspended in binding buffer (50 mM Tris–HCl, pH 7.4) and disrupted using a Branson 250 sonifier (Boom B.V., Meppel, The Netherlands).
For saturation binding, 50 μL of cell pellets were incubated with increasing concentrations of [3H]NAMH for 2 h at 25 °C. Nonspecific binding was measured in the presence of 100 μM histamine. Equilibrium competition binding was assayed on 50 μL of cell homogenates with 2 nM [3H]NAMH in the absence and presence of increasing concentrations of unlabelled ligands for 2 h at 25 °C. To terminate incubation, homogenates were filtered over a 0.5% polyethyleneimine (PEI)-coated 96-well GF/C filter plate using a PerkinElmer 96-well Filtermate-harvester. After three rapid wash steps with ice-cold binding buffer, the GF/C filter plates were dried at 55 °C. Subsequently, 25 μL of Microscint-O scintillation liquid (PerkinElmer, Groningen, The Netherlands) was added per well and incubated for 2 h to quantify filter-bound radioactivity using a Microbeta Wallac Trilux scintillation counter (PerkinElmer).
Transient Transfection and Plating
The day before transient transfection, 1.5 × 106 HEK293T (for G-protein experiments) or HEK293A cells (for H3R conformational sensors) were seeded in T25 flasks. The next day, 1 μg of pcDNA3.1 plasmid encoding either of the two H3R biosensors was transfected using Lipofectamine 2000. For G-protein experiments, the cells were transfected with 200 ng of LargeBit-Gαi1, 1 μg of Gβ5-smallBit, 1 μg of Gγ2, and 400 ng of wild-type H3R plasmids. Twenty-four hours after transfection, the cells were resuspended in supplemented DMEM, mixed with 50 nM HaloTag NanoBret 618 if H3R conformational biosensors were transfected, and transferred to poly-d-lysine-precoated white 96-well plates at a density of 50 000 cells/well.
Recording of BRET Emission Spectra
HEK293T cells were transfected and labeled as described above. Luminescence emission spectra of H3R sensors were recorded in HBSS with 4 nm resolution upon addition of 1:1000 furimazine dilution using a CLARIOstar plate reader (BMG, Ortenberg, Germany). Spectra were normalized to the donor emission peak.
BRET and Luminescence Measurements
Cells transiently or stably expressing the H3R biosensors or H3R wild-type along with native and tagged G-protein subunits were washed with HBSS and incubated with 1/1000 dilution of furimazine stock solution. After incubation for 3 min at 37 °C, the basal BRET ratio (H3R biosensor) or absolute Nluc luminescence (G-protein) was measured. Subsequently, 10 μL of 10-fold ligand solution or vehicle control was applied per well and the stimulated BRET ratio or luminescence was recorded. All experiments were conducted at 37 °C with a Synergy Neo2 (Biotek, Winooski, VT) or a CLARIOstar plate reader. Nluc emission intensity was selected using a 460/40 nm filter (Neo2) or a 450/50 nm monochromator (CLARIOstar). For HaloTag NanoBRET 618, a 620/20 nm filter or a 620/30 nm monochromator was used. All experiments were conducted with an integration time of 0.3 s.
Data Analysis and Statistics
BRET ratios were defined as acceptor emission/donor emission. Three individual luminescence or BRET values were averaged before and after ligand addition (lumbasal and lumstim; ratiobasal and ratiostim, respectively). To quantify ligand-induced changes, Δluminescence (Δlum) and ΔBRET were calculated for each well as a percent over basal ([(lumstim – lumbasal)/lumbasal] × 100; [(ratiostim – ratiobasal)/ratiobasal] × 100). Subsequently, the average Δlum/ΔBRET of vehicle control was subtracted. The Z-factors were calculated based on the following equation
where σ and μ are the standard deviations (SDs) and average ΔBRET values of 10 μM histamine and vehicle control, respectively. For the Z-factor experiments using pitolisant as a positive control, the denominator in the equation was replaced by “(μ[vehicle] – μ[compound])”.
Signal-to-noise (S/N) ratios in the Δicl3-H3RNluc/Halo(618) assay were calculated according to the following equation
S/N ratios in the split Nluc-based Gi1 assay were calculated according to the following equation
Data were analyzed using Prism 5.0 software (GraphPad, San Diego, CA). The data from BRET and luminescence concentration–response experiments were fitted using a four-parameter fit. Data from radioligand saturation binding experiments were fitted using a one-site fitting model. Competition-binding curves were fitted to a one-site binding model to obtain the IC50 value. Equilibrium dissociation constants (Ki) of unlabeled ligands were subsequently calculated using the Cheng–Prusoff equation
where [L] is the concentration of the labeled ligand and KD is the equilibrium dissociation constant of the labeled ligand. Statistical differences were assessed by one-way ANOVA followed by Bonferroni multiple comparison or extra-sum-of-squares F-test. Differences were considered significant for values of p < 0.05.
Results and Discussion
Design and Comparison of Two H3R Conformational Biosensor Versions
The development of ligand-sensitive conformational GPCR biosensors often requires the testing of different (i) resonance energy partner combinations, (ii) insertion sites for the FRET and BRET tags, or (iii) major modifications of the original receptor sequence.
We have previously shown that the combination of Nluc and HaloTag(618) yields the most sensitive conformational sensors for three model GPCRs19 and therefore employed this BRET pair to create two distinct variants of conformational H3RNluc/Halo biosensors and monitor their conformational dynamics in a 96-well microtiter format (Scheme 1a,b). To minimize the manipulation of the natural H3 receptor, we first generated a full-length H3R sensor version by placing HaloTag between S307 and G308 in the third intracellular loop (icl3) and fusing NanoLuc to the C-terminal amino acid K445 (full-length H3RNluc/Halo(618)). In some previously published conformational GPCR sensors, a long icl3, as it is also present in H3R (142 amino acids), has been associated with issues for sensor engineering and was truncated to yield a ligand-sensitive biosensor.29−31 Thus, in a second approach, we took advantage of a previously validated FRET-based H3R conformational sensor design that enabled the assessment of H3R activation kinetics in a single-cell assay format.23 In compliance with this sensor design, we fused Nluc to K445 and placed HaloTag between amino acids T229 and F348 to generate an analogous BRET H3R conformational sensor (Δicl3-H3RNluc/Halo(618)) and monitor ligand–H3R interaction in a 96-well microtiter format (Scheme 1c).23
Scheme 1. Principle of the Sensor and Assay Design.

(a) BRET partners HaloTag and Nluc are fused to the third intracellular loop and C-terminus of the histamine H3 receptor, respectively. The proximity of the BRET patners allows for energy transfer from the donor Nluc to HaloTag. Upon binding of full agonists or full inverse agonists, the receptor undergoes conformational changes to the fully active (top right) or inactive (bottom right) state, affecting the efficiency of energy transfer. (b) Assay protocol. (c) Insertion sites of Nluc and HaloTag in two H3R biosensor versions.
These two sensor constructs were transiently expressed in HEK293 cells to record the luminescence emission spectra upon addition of the Nluc substrate furimazine (Figure 1a,b). Both variants displayed the typical Nluc emission maximum around 450 nm and an additional peak at 620 nm. The latter is specific for the BRET acceptor fluorophore HaloTag(618) and indicates resonance energy transfer in the basal ligand-free states of both biosensors. Subsequently, we assessed the ability of these sensor variants to detect the GPCR conformational dynamics upon the addition of the well-characterized H3R ligands histamine (endogenous full agonist), imetit (synthetic full agonist), clobenpropit (inverse agonist), and pitolisant (inverse agonist) (Chart 1).
Figure 1.
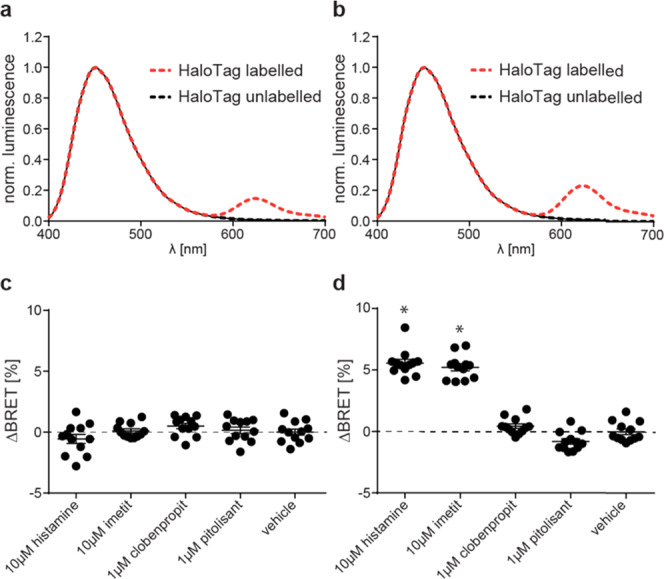
Comparison of two H3RNluc/Halo(618) biosensor versions. (a, b) Representative luminescence emission spectra of full-length H3RNluc/Halo(618) (a) and Δicl3-H3RNluc/Halo(618) (b). (c, d) BRET changes reported by full-length H3RNluc/Halo(618) (c) and Δicl3-H3RNluc/Halo(618) (d) upon addition of H3R reference ligands. All experiments were conducted in HEK293 cells transiently transfected with the indicated biosensor. Data in (c) and (d) show pooled data from three independent experiments. *p < 0.05 vs vehicle control.
Chart 1. Chemical Structures of Histamine H3 Receptor Agonists and Inverse Agonists Applied in This Study.

We incubated HEK293 cells transiently expressing either of the two sensor versions with these reference ligands or vehicle control and recorded the resulting change in BRET over baseline in 96-well microtiter plates. These experiments revealed that the full-length H3RNluc/Halo(618) does not detect the conformational dynamics of the H3R since no significant BRET change was observed (Figure 1c). In contrast, the BRET ratio of Δicl3-H3RNluc/Halo(618) expressing cells increased upon stimulation with the two full agonists histamine and imetit and decreased slightly after the addition of the inverse agonist pitolisant (Figure 1d).
Validation of Δicl3-H3RNluc/Halo
The Δicl3-H3RNluc/Halo(618) biosensor proved capable of detecting the ligand-induced receptor conformational changes in transiently transfected HEK293 cells (Figure 1d). To evaluate the ligand-binding properties of Δicl3-H3RNluc/Halo(618), we next conducted classical radioligand saturation binding experiments. Here, binding of the radiolabeled H3R agonist N-α-methylhistamine ([3H]NAMH) saturated with a pKD of 8.7 ± 0.2 (mean ± SD), similar to what we observed with cells expressing wild-type H3R (pKD = 8.9 ± 0.1) (Figure 2a and Supporting Table 1). In addition, radioligand displacement experiments at Δicl3-H3RNluc/Halo(618) with a panel of H3R reference ligands yielded pKi values that were in general accordance with the previously published data (Figure 2b, Supporting Table 1 and Figure 1). Importantly, Δicl3-H3RNluc/Halo(618) maintains the distinct binding affinities for the enantiomers (R)-α-methylhistamine (RAMH) and (S)-α-methylhistamine (SAMH) known from wild-type H3R. Taken together, these binding experiments reveal that Δicl3-H3RNluc/Halo(618), similar to its original FRET analogue,23 possesses wild-type binding properties, which represents a key requirement for its utility in ligand screening experiments.
Figure 2.
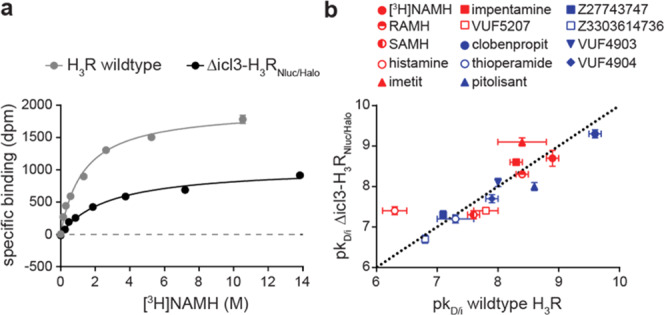
Ligand-binding properties of Δicl3-H3RNluc/Halo(618). (a) Saturation binding of the radiolabeled H3R agonist [3H]NAMH to the wild-type H3R and Δicl3-H3RNlucHalo(618). (b) Correlation of ligand-binding affinities to the wild-type H3R (values extracted from literature except for [3H]NAMH; see also Supporting Table 1) and Δicl3-H3RNlucHalo(618) (red: agonists; blue: inverse agonists). Dotted line indicates line of unity. Experiments were conducted using membranes from transiently (a) or stably expressing (b) HEK293 cells. Data show the mean ± standard error of mean (SEM) of one representative (a) or at least three independent experiments (b).
To improve the assay’s dynamic range, we next generated a HEK293 cell line stably expressing Δicl3-H3RNluc/Halo(618) and conducted live cell BRET experiments to demonstrate its suitability to study the pharmacology of H3R ligands. Therefore, we first stimulated these sensor cells with 10 μM of six reference H3R ligands (histamine, imetit, UR-PI294, thioperamide, clobenpropit, and pitolisant) that possess distinct intrinsic efficacies ranging from full agonism to inverse agonism (Chart 1 and Supporting Table 1). The BRET time-courses plateaued within several minutes after ligand addition and remained stable for at least 40 min (Figure 3a). The stability of the BRET response illustrates the broad time window for signal detection and underlines the applicability of this assay for automated screening conditions where stacked ligand-treated microtiter plates are read consecutively. Furthermore, all reference ligands yielded saturating BRET concentration–response curves and the EC50-based order of potency resembled the pKi-based order of affinity from our previous binding experiments (Figure 3b and Supporting Table 1). For few ligands, however, we obtained distinct affinities/potencies using the radioligand displacement assay in cell lysates or the BRET-based conformational readout in intact cells (e.g., for clobenpropit). Similar deviations have been reported recently for H3R ligand affinities assessed in either radioligand displacement or NanoBRET binding experiments on the very same Nluc-tagged H3R construct.32 Therefore, we suppose that differences in assay conditions (e.g., buffer composition or receptor environment in intact cells vs cell lysates) account for the few deviations observed in our experiments.
Figure 3.

Validation of the Δicl3-H3RNluc/Halo(618) biosensor. (a) ΔBRET time-course of six reference ligands (legend in b). (b) BRET concentration–response curves of six reference ligands. (c) Concentration–response curves of impentamine and three structural analogues. (d) Z-factors of Δicl3-H3RNluc/Halo(618) to assess the screening windows for H3R agonists and inverse agonists. (e) BRET signals of one representative 96-well plate treated with 10 μM histamine or vehicle. (f) BRET signals of one representative 96-well plate treated with 10 μM pitolisant or vehicle. All experiments were conducted in HEK293 cells stably expressing the Δicl3-H3RNluc/Halo(618) biosensor. Data in (a)–(d) show the mean ± SEM of at least three independent experiments.
Of note, the three agonists histamine, imetit, and UR-PI294 elevated the BRET ratio with distinct maximal effects (Emax). To the best of our knowledge, this is the first data indicating that imetit behaves as a strong partial rather than a full agonist at the receptor level. In contrast to the increase in BRET evoked by agonists, the H3R inverse agonists thioperamide, clobenpropit, and pitolisant induced a reduction in BRET, in line with their distinct downstream effects. However, surprisingly, thioperamide, clobenpropit, and pitolisant plateaued at different BRET signals (Emax of pitolisant > clobenpropit > thioperamide), similar to the differences we observed for agonists, i.e., imetit and histamine. This suggests that also these ligands possess distinct inverse efficacies, with pitolisant being the strongest inverse agonist.
Next, we aimed to explore whether Δicl3-H3RNluc/Halo(618) can be utilized to conduct structure-activity-relationship (SAR) studies with H3R ligands. We selected a panel of three chemical analogues of the H3R agonist impentamine that present distinct chemical substitutions at the amine group (Chart 1). When characterized at the second messenger cyclic adenosine monophosphate (cAMP) level, these ligands displayed varying efficacies ranging from strong partial agonism for impentamine to inverse agonism for VUF4903 (Supporting Table 1).28 The conformational H3R sensor Δicl3-H3RNluc/Halo(618) detected these differential ligand activities and reported partial agonistic responses for the parent compound impentamine and its analogue VUF5207, as well as inverse agonistic responses with distinct Emax values for VUF4904 and VUF4903 (Figure 3c and Supporting Table 1). Overall, these results demonstrate that Δicl3-H3RNluc/Halo(618) provides a valuable tool to conduct SAR studies of H3R ligands at the level of receptor conformation.
Last, we aimed to evaluate the sensitivity and reproducibility of this sensor cell line to screen simultaneously H3R agonists and inverse agonists in a single high-throughput campaign (i.e., without the requirement to preincubate the cells with competing H3R ligands). Therefore, we measured the Z-factors for agonists and inverse agonists by applying either histamine or pitolisant as a positive control, respectively.33 The resulting average Z-factors were well above 0.5 (histamine: 0.58 ± 0.02; pitolisant: 0.72 ± 0.02; mean ± SEM) and showed low interday variability (histamine: 4.6%; pitolisant: 5.6%; coefficient of variation), highlighting the substantial agonist and inverse agonist screening windows and the robustness of this conformational readout (Figure 3d–f and Supporting Figure 2). These results underline the applicability of this conformational biosensor to screen for and characterize novel H3R ligands with varying intrinsic activities using the same experimental setup.
Pharmacological Characterization of Two Novel H3R Inverse Agonists
To further demonstrate the ability of the conformational biosensor to characterize new H3R targeting compounds, we used it to assess the pharmacological properties of two new H3R ligands, Z27743747 and Z3303614736 (Chart 1). These compounds were discovered in a recent in-silico docking screen and validated for binding H3R with nanomolar affinities.26 However, further information on the pharmacological properties of Z27743747 and Z3303614736 is lacking, and this is hampering a reliable evaluation of their potential as novel H3R modulating lead structures. We first incubated the stable H3R biosensor cell line with 10 μM of either compound and in parallel conducted the same experiments using our previously described conformational biosensor of the β2-adrenergic receptor (β2ARNluc/Halo).19 Both ligands evoked fast and stable negative BRET changes in H3RNluc/Halo expressing cells but, as control, no BRET signals were observed in β2ARNluc/Halo (Figure 4a,b). Furthermore, application of increasing compound concentrations yielded sigmoidal concentration–response curves with nanomolar EC50-values for H3RNluc/Halo but not β2ARNluc/Halo (Figure 4c,d and Supporting Table 1). Of note, Z27743747 evoked an even stronger BRET response than the reference inverse agonist pitolisant (Supporting Table 1 and Figure 3), indicating that this ligand presents a promising chemical scaffold for the development of novel H3R ligands with even greater intrinsic inverse agonistic activity.
Figure 4.
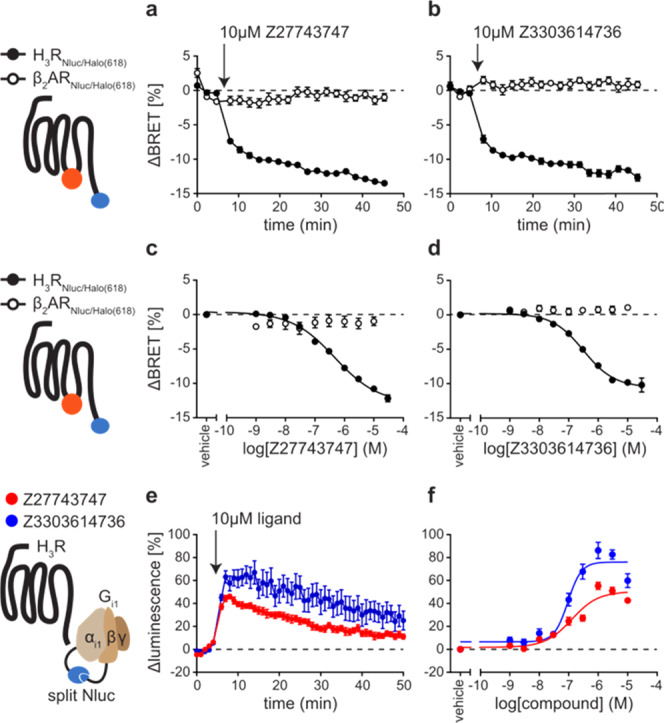
Characterization of new H3R inverse agonists. (a, b) ΔBRET time-course of Δicl3-H3RNluc/Halo(618) and β2ARNluc/Halo(618) upon addition of Z27743747 (a) and Z3303614736 (b). (c, d) Concentration–response curves of Z27743747 (c) and Z3303614736 (d) applied to cells expressing Δicl3-H3RNluc/Halo(618) or β2ARNluc/Halo(618). (e, f) ΔLuminescence time-course (e) and concentration–response (f) of the split Nluc-based G-protein sensor upon treatment with Z27743747 or Z3303614736. Experiments in (a)–(d) were conducted in HEK293 cells stably expressing either of the two conformational GPCR biosensors. Experiments in (e) and (f) were conducted in HEK293 cells transiently transfected with H3R wild-type and the split Nluc-based Gi1 sensor and pre-stimulated with 300 nM histamine (for 5 min before time point 0). Data show the mean ± SEM of at least three independent experiments.
To verify these data on a receptor downstream level, we employed a split luciferase complementation-based readout reflecting the G-protein activity.34 We co-expressed Gαi1 and Gβ5 subunits tagged with complementary Nluc fragments along with native Gγ2 and wild-type H3R in HEK293 cells and pre-stimulated with 300 nM histamine to induce the dissociation of the heterotrimeric G-protein complex.
Upon the addition of 10 μM Z27743747 or Z3303614736, the luminescence intensity increased and peaked within several minutes, corresponding to a reassembly of the heterotrimeric G-protein complex (Figure 4e). Moreover, these signals were concentration-dependent and resulted in very similar IC50 values as measured with the H3R conformational biosensor (Z27743747: 6.90 ± 0.11; Z3303614736: 7.03 ± 0.07; pIC50 ± SD) (Figure 4f and Supporting Table 1). Of note, only Z27743747, but not Z3303614736, was also able to induce an increase in G-protein luminescence when applied to basal H3R (i.e., without 300 nM histamine preincubation) demonstrating its stronger inverse activity compared to that of Z3303614736 (Supporting Figure 4a). However, the signal-to-noise ratio (S/N) of this luminescence increase was substantially lower as compared to (i) that of histamine pre-stimulated H3R-Gi condition and (ii) the S/N ratio assessed with Δicl3-H3RNluc/Halo(618) (also without agonist pre-stimulation) (Supporting Figure 4b).
Taken together, these results demonstrate that Z27743747 and Z3303614736 are new strong inverse agonists of H3R that inactivate the receptor with nanomolar potencies. Additionally, comparison of the different assay systems (Δicl3-H3RNluc/Halo(618) vs H3R wild-type plus Gi1 sensor) underlines the accuracy and outstanding sensitivity of the new conformational H3R biosensor to identify and characterize novel H3R inverse agonist.
Conclusions
The histamine H3 receptor is involved in multiple CNS disorders and rated among the most attractive and most targeted GPCRs by compounds in clinical trials.35 Despite the extensive interest in regulating this receptor with new pharmaceutics, pitolisant represents so far the only approved H3R-targeting drug partially due to the limitations of current H3R-suited screening assays. In this work, we describe a new screening-compatible biosensor to aid in the future identification and pharmacological assessment of H3R-modulating compounds.
Novel H3R Conformational Biosensor Allows for the Synchronous Screening of Agonists and Inverse Agonists
This new biosensor is composed of the BRET partners Nluc and a red fluorescent HaloTag dye fused to H3R to translate the ligand-induced conformational dynamics of this receptor into optical signals.
We show that this sensor maintains wild-type-like ligand-binding properties and facilitates the assessment of ligand potency and efficacy at the most proximal level, i.e., directly after ligand binding. Consequently, we were able to detect differences in ligand efficacies between the two H3R agonists histamine and imetit as well as between the inverse agonists thioperamide, clobenpropit, and pitolisant (Supporting Table 1). These distinct intrinsic activities could not be observed in previous studies using the FRET-based conformational H3R sensor23 or downstream-dependent H3R assays,36 which might be a consequence of differential sensitivities of FRET- vs BRET-based sensors37 or, with respect to histamine and imetit, be due to the impact of signal amplification in downstream-dependent assays. Supporting this hypothesis, similar discrepancies between ligand efficacies assessed with FRET vs BRET conformational sensors (UK 14 304 response at α2AARCFP/Flash vs α2AARNluc/Halo(618))19,38 and downstream vs receptor conformation assays have been reported for the α2A-adrenergic (norepinephrine vs UK 14 304) and β2-adrenergic receptor (epinephrine vs norepinephrine).19−21 Our findings with the H3R conformational biosensor underline the superior resolution provided by conformational GPCR sensors for the determination of ligand efficacies.
Furthermore, we demonstrate that the H3R biosensor can be employed to explore structure–activity relationships and provides excellent sensitivity and robustness for the simultaneous screening of H3R agonists and inverse agonists. This feature is of great value in GPCR-targeted ligand screening campaigns since, in contrast to downstream-dependent assays, no preincubation with (or subsequent addition of) an H3R agonists is required to identify inverse agonists or vice versa.
To the best of our knowledge, such a screening-compatible and uniform assay format is not provided by any other GPCR screening technology available today.
Characterization of Novel H3R Ligands with Marked Inverse Agonistic Efficacy
In the last part of this study, we employed the novel H3R conformational sensor to assess the pharmacological properties of two new H3R ligands that were recently discovered through virtual screening.26 Both compounds bind H3R with nanomolar affinity, but their intrinsic efficacies have not been determined so far. Interestingly, our results with the H3R conformational biosensor revealed pronounced inverse agonistic responses of the ligands Z27743747 and Z3303614736. In particular, Z27743747 induced even higher BRET amplitudes than the validated inverse agonists thioperamide, clobenpropit, and pitolisant. This suggests that Z27743747 presents a stronger inverse agonistic activity than the current gold standard pitolisant (Supporting Table 1 and Figure 3) by stabilizing a distinct conformation of H3R that is characterized by no or very little residual receptor signaling capacity.
Therefore, Z27743747 and Z3303614736 represent two promising H3R lead structures with novel chemical scaffolds that can help in designing superior H3R ligands with advantageous pharmacodynamic properties.
Taken together, this work describes the successful generation of the first screening applicable H3R conformational sensor. This optical tool will aid in the development of novel receptor ligands and may be employed to gain new insights into the molecular mechanisms underlying H3R conformational dynamics. The successful implementation of this sensor design for the H3R opens up the possibility to expand this technique to other class A GPCRs, e.g., other members of the histamine receptor family, to assess the pharmacological properties of GPCR ligands directly at the level of receptor conformation.
Acknowledgments
We thank Kristina Lorenz (University of Wuerzburg, Germany) and Anna Krook (Karolinska Institutet, Sweden) for access to the Synergy Neo2 and ClarioStar plate reader, respectively. Furthermore, we thank Asuka Inoue (Tohoku University, Japan) for providing plasmids encoding the G-protein sensor.
Glossary
Abbreviations Used
- GPCR
G-protein-coupled receptor
- H3R
histamine H3 receptor
- CNS
central nervous system
- FRET
fluorescence resonance energy transfer
- BRET
bioluminescence resonance energy transfer
- icl3
intracellular loop 3
- [3H]NAMH
N-α-methylhistamine
- SAR
structure–activity relationship
- S/N ratio
signal-to-noise ratio
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssensors.0c00397.
Summary of H3R ligand affinities/potencies and efficacies assessed with different assays (Table S1); competition binding of selected unlabeled H3R ligands with [3H]NAMH to cells stably expressing Δicl3-H3RNlucHalo(618) (Figure S1); BRET signals and Z-factors of Δicl3-H3RNluc/Halo(618) assessed in four independent 96-well plates using histamine (Figure S2); concentration–response curves of H3R inverse agonists assessed with Δicl3-H3RNluc/Halo(618) (Figure S3); and comparison of assay sensitivity without H3R agonist pre-stimulation (Figure S4) (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Institutes of Health (NIH, 0255-8521-4609), SFB/TR166, and the Bundesministerium für Bildung und Forschung (BMBF, Federal Ministry of Education and Research–OptiMAR) to M.J.L. and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, 427840891) to H.S. The work at Karolinska Institutet was supported by grants from Karolinska Institutet, the Swedish Research Council (2017-04676), and the Swedish Cancer Society (CAN2017/561). The work at Vrije Universiteit was supported by a CSC Chinese scholarship grant to X.M. (File No. 201703250074).
The authors declare the following competing financial interest(s): The authors declare that the University of Wuerzburg holds a patent on this technology: WO2004057333 A1.
Supplementary Material
References
- Panula P.; Chazot P. L.; Cowart M.; Gutzmer R.; Leurs R.; Liu W. L. S.; Stark H.; Thurmond R. L.; Haas H. L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. 10.1124/pr.114.010249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw J.; Brittain R. T.; Clitherow J. W.; Daly M. J.; Jack D.; Price B. J.; Stables R. Ranitidine (AH 19065): a new potent, selective histamine H2-receptor antagonist [proceedings]. Br. J. Pharmacol. 1979, 66, 464. [PMC free article] [PubMed] [Google Scholar]
- Barnett A.; Iorio L. C.; Kreutner W.; Tozzi S.; Ahn H. S.; Gulbenkian A. Evaluation of the CNS properties of SCH 29851, a potential non-sedating antihistamine. Agents Actions 1984, 14, 590–597. 10.1007/BF01978891. [DOI] [PubMed] [Google Scholar]
- Arrang J.-M.; Garbarg M.; Schwartz J.-C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- Nieto-Alamilla G.; Márquez-Gómez R.; García-Gálvez A.-M.; Morales-Figueroa G.-E.; Arias-Montano J.-A. The Histamine H3 Receptor: Structure, Pharmacology, and Function. Mol. Pharmacol. 2016, 90, 649–673. 10.1124/mol.116.104752. [DOI] [PubMed] [Google Scholar]
- Ferrada C.; Ferré S.; Casadó V.; Cortés A.; Justinova Z.; Barnes C.; Canela E. I.; Goldberg S. R.; Leurs R.; Lluis C.; Franco R. Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 2008, 55, 190–197. 10.1016/j.neuropharm.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ruiz M.; Moreno E.; Moreno-Delgado D.; Navarro G.; Mallol J.; Cortés A.; Lluís C.; Canela E. I.; Casadó V.; McCormick P. J.; Franco R. Heteroreceptor Complexes Formed by Dopamine D1, Histamine H3, and N-Methyl-D-Aspartate Glutamate Receptors as Targets to Prevent Neuronal Death in Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 4537–4550. 10.1007/s12035-016-9995-y. [DOI] [PubMed] [Google Scholar]
- Rapanelli M. The magnificent two: histamine and the H3 receptor as key modulators of striatal circuitry. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 73, 36–40. 10.1016/j.pnpbp.2016.10.002. [DOI] [PubMed] [Google Scholar]
- Rapanelli M.; Frick L.; Pogorelov V.; Ohtsu H.; Bito H.; Pittenger C. Histamine H3R receptor activation in the dorsal striatum triggers stereotypies in a mouse model of tic disorders. Transl. Psychiatry 2017, 7, e1013 10.1038/tp.2016.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed Y. Y. Pitolisant: First Global Approval. Drugs 2016, 76, 1313–1318. 10.1007/s40265-016-0620-1. [DOI] [PubMed] [Google Scholar]
- Wágner G.; Mocking T. A. M.; Arimont M.; Provensi G.; Rani B.; Silva-Marques B.; Latacz G.; Da Costa Pereira D.; Karatzidou C.; Vischer H. F.; Wijtmans M.; Kiec-Kononowicz K.; de Esch I. J. P.; Leurs R. 4-(3-Aminoazetidin-1-yl)pyrimidin-2-amines as High-Affinity Non-imidazole Histamine H3 Receptor Agonists with in Vivo Central Nervous System Activity. J. Med. Chem. 2019, 62, 10848–10866. 10.1021/acs.jmedchem.9b01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wágner G.; Mocking T. A. M.; Kooistra A. J.; Slynko I.; Ábrányi-Balogh P.; Keserű G. M.; Wijtmans M.; Vischer H. F.; de Esch I. J. P.; Leurs R. Covalent Inhibition of the Histamine H3 Receptor. Molecules 2019, 24, 4541 10.3390/molecules24244541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauwert N. J.; Mocking T. A. M.; Da Costa Pereira D.; Lion K.; Huppelschoten Y.; Vischer H. F.; De Esch I. J. P.; Wijtmans M.; Leurs R. A Photoswitchable Agonist for the Histamine H3 Receptor, a Prototypic Family A G-Protein-Coupled Receptor. Angew. Chem., Int. Ed. 2019, 58, 4531–4535. 10.1002/anie.201813110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras F. H.; Pockes S.; Huang G.; Wehle S.; Strasser A.; Wittmann H. J.; Nimczick M.; Sotriffer C. A.; Decker M. Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. 10.1021/cn4002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari N.; Dastmalchi S.; Zarei O.; Arias-Montaño J.-A.; Reiner D.; Ustun-Alkan F.; Stark H.; Hamzeh-Mivehroud M. In silico and in vitro studies of two non-imidazole multiple targeting agents at histamine H3 receptors and cholinesterase enzymes. Chem. Biol. Drug Des. 2020, 95, 279–290. 10.1111/cbdd.13642. [DOI] [PubMed] [Google Scholar]
- Lutsenko K.; Hagenow S.; Affini A.; Reiner D.; Stark H. Rasagiline derivatives combined with histamine H3 receptor properties. Bioorg. Med. Chem. Lett. 2019, 29, 126612 10.1016/j.bmcl.2019.08.016. [DOI] [PubMed] [Google Scholar]
- Łażewska D.; Kieć-Kononowicz K. Progress in the development of histamine H3 receptor antagonists/inverse agonists: a patent review (2013-2017). Expert Opin. Ther. Pat. 2018, 28, 175–196. 10.1080/13543776.2018.1424135. [DOI] [PubMed] [Google Scholar]
- Lohse M. J.; Nuber S.; Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol. Rev. 2012, 64, 299–336. 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- Schihada H.; Vandenabeele S.; Zabel U.; Frank M.; Lohse M. J.; Maiellaro I. A universal bioluminescence resonance energy transfer sensor design enables high-sensitivity screening of GPCR activation dynamics. Commun. Biol. 2018, 1, 105 10.1038/s42003-018-0072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner S.; Ambrosio M.; Hoffmann C.; Lohse M. J. Differential signaling of the endogenous agonists at the beta2-adrenergic receptor. J. Biol. Chem. 2010, 285, 36188–36198. 10.1074/jbc.M110.175604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J.; Horst R.; Katritch V.; Stevens R. C.; Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogmus S.; Storch U.; Danner L.; Becker J.; Winter M.; Ziegler N.; Wirth A.; Offermanns S.; Hoffmann C.; Gudermann T.; y Mederos M. M. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 2019, 10, 5784 10.1038/s41467-019-13722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zeng H.; Pediani J. D.; Ward R. J.; Chen L.-Y.; Wu N.; Ma L.; Tang M.; Yang Y.; An S.; Guo X. -X.; Hao Q.; Xu T. R. Visualization of the activation of the histamine H3 receptor (H3R) using novel fluorescence resonance energy transfer biosensors and their potential application to the study of H3R pharmacology. FEBS J. 2018, 285, 2319–2336. 10.1111/febs.14484. [DOI] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 2012, 7, 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 2008, 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Schaller D.; Hagenow S.; Stark H.; Wolber G. Ligand-guided homology modeling drives identification of novel histamine H3 receptor ligands. PLoS One 2019, 14, e0218820 10.1371/journal.pone.0218820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igel P.; Schnell D.; Bernhardt G.; Seifert R.; Buschauer A. Tritium-labeled N(1)-[3-(1H-imidazol-4-yl)propyl]-N(2)-propionylguanidine ([(3)H]UR-PI294), a high-affinity histamine H(3) and H(4) receptor radioligand. ChemMedChem 2009, 4, 225–231. 10.1002/cmdc.200800349. [DOI] [PubMed] [Google Scholar]
- Wieland K.; Bongers G.; Yamamoto Y.; Hashimoto T.; Yamatodani A.; Menge W. M. B. P.; Timmerman H.; Lovenberg T. W.; Leurs R. Constitutive activity of histamine h(3) receptors stably expressed in SK-N-MC cells: display of agonism and inverse agonism by H(3) antagonists. J. Pharmacol. Exp. Ther. 2001, 299, 908–914. [PubMed] [Google Scholar]
- Vilardaga J.-P.; Bünemann M.; Krasel C.; Castro M.; Lohse M. J. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat. Biotechnol. 2003, 21, 807–812. 10.1038/nbt838. [DOI] [PubMed] [Google Scholar]
- Hoffmann C.; Gaietta G.; Bünemann M.; Adams S. R.; Oberdorff-Maass S.; Behr B.; Vilardaga J.-P.; Tsien R. Y.; Ellisman M. H.; Lohse M. J. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat. Methods 2005, 2, 171–176. 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- Kauk M.; Hoffmann C. Intramolecular and Intermolecular FRET Sensors for GPCRs - Monitoring Conformational Changes and Beyond. Trends Pharmacol. Sci. 2018, 39, 123–135. 10.1016/j.tips.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Mocking T. A. M.; Verweij E. W. E.; Vischer H. F.; Leurs R. Homogeneous, Real-Time NanoBRET Binding Assays for the Histamine H3 and H4 Receptors on Living Cells. Mol. Pharmacol. 2018, 94, 1371–1381. 10.1124/mol.118.113373. [DOI] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Inoue A.; Raimondi F.; Kadji F. M. N.; Singh G.; Kishi T.; Uwamizu A.; Ono Y.; Shinjo Y.; Ishida S.; Arang N.; Kawakami K.; Gutkind J. S.; Aoki J.; Russell R. B. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947. 10.1016/j.cell.2019.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schioth H. B.; Gloriam D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H. D.; van Rijn R. M.; Ling P.; Bakker R. A.; Thurmond R. L.; Leurs R. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J. Pharmacol. Exp. Ther. 2005, 314, 1310–1321. 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- Boute N.; Jockers R.; Issad T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 2002, 23, 351–354. 10.1016/S0165-6147(02)02062-X. [DOI] [PubMed] [Google Scholar]
- Nikolaev V. O.; Hoffmann C.; Bünemann M.; Lohse M. J.; Vilardaga J.-P. Molecular basis of partial agonism at the neurotransmitter alpha2A-adrenergic receptor and Gi-protein heterotrimer. J. Biol. Chem. 2006, 281, 24506–24511. 10.1074/jbc.M603266200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.