

Abstract
Background
Dickeya solani is an important plant pathogenic bacterium causing severe losses in European potato production. This species draws a lot of attention due to its remarkable virulence, great devastating potential and easier spread in contrast to other Dickeya spp. In view of a high need for extensive studies on economically important soft rot Pectobacteriaceae, we performed a comparative genomics analysis on D. solani strains to search for genetic foundations that would explain the differences in the observed virulence levels within the D. solani population.
Results
High quality assemblies of 8 de novo sequenced D. solani genomes have been obtained. Whole-sequence comparison, ANIb, ANIm, Tetra and pangenome-oriented analyses performed on these genomes and the sequences of 14 additional strains revealed an exceptionally high level of homogeneity among the studied genetic material of D. solani strains. With the use of 22 genomes, the pangenome of D. solani, comprising 84.7% core, 7.2% accessory and 8.1% unique genes, has been almost completely determined, suggesting the presence of a nearly closed pangenome structure. Attribution of the genes included in the D. solani pangenome fractions to functional COG categories showed that higher percentages of accessory and unique pangenome parts in contrast to the core section are encountered in phage/mobile elements- and transcription- associated groups with the genome of RNS 05.1.2A strain having the most significant impact. Also, the first D. solani large-scale genome-wide phylogeny computed on concatenated core gene alignments is herein reported.
Conclusions
The almost closed status of D. solani pangenome achieved in this work points to the fact that the unique gene pool of this species should no longer expand. Such a feature is characteristic of taxa whose representatives either occupy isolated ecological niches or lack efficient mechanisms for gene exchange and recombination, which seems rational concerning a strictly pathogenic species with clonal population structure. Finally, no obvious correlations between the geographical origin of D. solani strains and their phylogeny were found, which might reflect the specificity of the international seed potato market.
Keywords: Soft rot, Blackleg, Pectinolytic bacteria, Erwinia chrysanthemi, Pectobacteriaceae, Next-generation sequencing, Whole genome sequencing, Pacific biosciences, Clusters of orthologous groups, Average nucleotide identity
Background
Dickeya spp. together with Pectobacterium spp. belong to the family Pectobacteriaceae [1] and are causative agents of economically important soft rot and blackleg diseases affecting various crops, vegetables and ornamentals worldwide [2]. These bacterial phytopathogens decay host tissue due to the production of a broad range of plant cell wall degrading enzymes (PCWDEs) i.e. pectinases (pectate and pectin lyases, polygalacturonases, pectin-methyl and acetyl esterases), cellulases and proteases, which are secreted via type I or II secretion systems [3, 4]. Because of the activities of PCWDEs, these necrotrophic bacteria get access to valuable sources of nutrients accumulated within the plant cell. Other worth mentioning virulence factors of Pectobacteriaceae include biofilm formation [5], motility [6], siderophores production [7], lipopolysaccharide [8] or synthesis of bacteriocins [7]. Such a molecular or adaptive repertoire takes part in progression of the incited infection. However, three crucial requirements need to be fulfilled for the development of disease symptoms: the pathogen should be virulent, the plant host susceptible and the encountered environmental conditions favourable for disease progression [9]. Typical blackleg symptoms comprise water-soaked, blackened stem base in addition to chlorosis and wilting of the leaves [2]. Often the progeny tubers do not develop and in the most severe cases there is a noticeable lack of emergent plants [2]. Regarding soft rot, slimy, water-soaked maceration areas are observable in the inner parenchymatous plant tissue. These zones, if exposed to air, turn brown or black with release of watery exudates [2, 10]. Assessment of the total economic impact of these diseases is demanding as Pectobacterium and Dickeya spp. are present on various plant hosts in diverse geographical regions where miscellaneous seed certification policies remain in force [11].
The pectinolytic bacterial species, which is in focus of this work, belongs to the genus Dickeya. The Dickeya genus was established in 2005 [12] to comprise several former members of at first Erwinia [13] and subsequently Pectobacterium [14] genera. To date, ten species i.e. Dickeya aquatica [15], Dickeya chrysanthemi [12], Dickeya dadantii (including D. dadantii subsp. dadantii and D. dadantii subsp. dieffenbachiae [12, 16]), Dickeya dianthicola [12], Dickeya fangzhongdai [17], Dickeya lacustris [18], Dickeya paradisiaca [12], Dickeya solani [19], D. undicola [20] and Dickeya zeae [12] are classified to the Dickeya genus. It is worth noting that D. solani has drawn a lot of attention ever since its first appearance in Europe in 2004–2005 [19, 21–23]. Outgrouping of uniform isolates belonging to the Dickeya genus was spotted independently, basing on the sequences of 16S rRNA [24], recA [25, 26] or dnaX genes [19, 23], in addition to Repetitive Extragenic Palindromic-PCR (REP-PCR) profiling [23]. Further support for homogeneity of these isolates was provided by whole-cell Matrix-Assisted Laser Desorption Ionization Time-Of-Flight Mass Spectrometry (MALDI-TOF MS), Pulse Field Gel Electrophoresis (PFGE) of total genomic DNA cut with XbaI or I-CeuI restriction enzymes, PCR-based fingerprinting with Enterobacterial Repetitive Intergenic Consensus (ERIC) and BOX primers, comparison of the sequences of intergenic spacer (IGS) in addition to broadening the pool of the analysed housekeeping genes by including dnaN, fusA, gapA, gyrA, purA, rplB and rpoS sequences [19, 27–29]. Even though the observed relatedness in DNA-DNA hybridization (DDH) experiments between the type strains of D. solani and D. dadantii equalled 72%, therefore exceeding the cut-off threshold for species delineation [30], the performed pairwise Average Nucleotide Identity (ANI) calculation with 0.94 value gave contradictory results in favour of separation of these two taxa [19].
Official establishment of D. solani as a distinct clonal species dates back to 2014 [19]. Since then major scientific efforts have been made to provide insight into the occurrence, epidemiology, detection methods, taxonomic position, metabolic profiles, regulation of transcription, genetics and genomics of this phytopathogen [19, 27, 28, 31–39]. The presence of D. solani strains was reported in Europe and beyond, e.g. in the Netherlands [19], Belgium [40], Israel [35], Turkey [41], Finland [28], Norway [42], Portugal [31], Czech Republic [43], Denmark [43], United Kingdom [44], Northern Ireland [45], Greece [46], France [47], Switzerland [48], Spain [49], Slovenia [50], Georgia [51], Russia [52], Germany [32], Brazil [53] and China [54]. Notably, the tested isolates originated from a limited number of plants including potato [27, 28, 35], hyacinth [23] and iris [19], which might be related to previous assumptions on strict linkage between highly specialized pathogens of clonal origin and their host [19, 55]. Remarkable virulence, great devastating potential and easier spread of D. solani strains in contrast to other Dickeya spp. was observed by several research groups [21, 27, 28, 56, 57]. Therefore, there were attempts undertaken to explain foundations of these phenomena on the levels of genomes, transcriptomes and metabolomes [31–33, 38, 39, 47, 58, 59]. It is worth noting though that the majority of genome-oriented research conducted so far benefited from a limited number of whole genome sequences (WGS) [31, 38, 47, 58, 60, 61], impeding broad insight into the intraspecies variation of D. solani. A pangenome-related study is a potent strategy to address comprehensive description of genomic diversity within a bacterial species and to suggest possible genetic determinants for the noted phenotypic differences [31, 62, 63]. ‘Pangenome’ covers all genes detected in a certain bacterial species, while ‘core genome’ comprises the genes present in all the analysed strains, ‘dispensable genome’ encloses the genes observed in two or more strains and ‘unique genome’ consists of the genes detected just in a single bacterial isolate [64]. Undertaking pangenome-based approach allows to state the amount of whole sequenced genomes that would satisfactorily reflect the genetic repertoire of a studied species [31, 63, 65]. If such a number of WGS is reached, the pangenome might be described as closed.
In this study, we aimed at exploiting comparative genomics and pangenome-oriented tools for providing closer insight into biodiversity within the D. solani species. For this purpose, 8 de novo sequenced, assembled and annotated WGS of D. solani strains of diverse origin and year of isolation were acquired. The utilized analytic tools provided insight into extraordinarily high homogeneity among the available 22 D. solani genomes. Importantly, such a number of sequences turned out to be sufficient to report in this work an almost closed status of the pangenome of D. solani species.
Results
D. solani genomic assemblies
The newly sequenced genomes of 8 D. solani strains (Table 1) were assembled into 1–7 scaffolds with no N bases (Table 2) from the PacBio reads with the use of the genome assembly pipeline that we previously described [31]. This method profits solely from PacBio RSII raw reads that are at first filtered from adapters with the use of SMRT Analysis v. 2.3 (Pacific Biosciences, USA) and then corrected, trimmed and assembled with the use of Canu v. 1.5 [66]. Getting consensus and variant calling was achieved with Quiver (SMRT Analysis v. 2.3) [67] and final functional annotation was conducted with Prokka v. 1.12 [68]. The size of these genomes ranged from 4,882,124 bp to 4,934,537 bp, in the case of IFB0487 and IFB0421 D. solani strains, respectively (Table 2). The largest contig of the acquired assemblies varied in size from 4,934,537 bp to 2,394,283 bp regarding either IFB0421 or IFB0311 (Table 2). N50, which refers to the minimum length of contigs in which half of the bases of the assembly are covered, ranged from 755,734 bp to 4,934,537 bp (for IFB0695 or IFB0421; Table 2). L50, describing the number of contigs that comprise half of the genome size, spanned from 1 to 2 (Table 2). The calculated GC content falls within the range of 56.23 to 56.25 (Table 2). None of the contigs from de novo assembled D. solani genomes has been assigned to the sequences of plasmid origin as computed with the use of PlasmidFinder [69]. According to Prokka-based [68] annotation, the newly sequenced genomes of D. solani strains contained in total from 4304 to 4608 genes (in the case of IFB0212 and IFB0417, respectively; Table 1). The number of protein-coding genes varied from 4143 (IFB0212) to 4446 (IFB0417), while the quantities of the annotated rRNA and tRNA amounted to 18–22 and 72–75, respectively (Table 1).
Table 1.
Dickeya solani strains subjected to de novo sequencing in the frames of this study in addition to their genomic contents
| Genome nos /strain nos | Strain | Genome | ||||||
|---|---|---|---|---|---|---|---|---|
| Total number of genes | Number of genes encoding | |||||||
| Country, regiona | Host, year of isolation | Literature reference | Proteins | rRNA | tRNA | tmRNAs | ||
| IFB0167 | Poland, Lower Silesian Voivodeship | Potato cv. Fresco, 2009 | [27] | 4308 | 4146 | 22 | 75 | 1 |
| IFB0212 | Poland, Mazovian Voivodeship | Potato, 2010 | [29] | 4304 | 4143 | 18 | 72 | 1 |
| IFB0231 (VIC-BL-25) | Finland, Liminka | Potato cv. Victoria, 2008 | [28] | 4313 | 4151 | 22 | 75 | 1 |
| IFB0311 | Poland, Pomeranian Voivodeship | Potato cv. Innovator, 2011 | [27] | 4306 | 4144 | 20 | 74 | 1 |
| IFB0417 | Portugal, Santarem | Potato cv. Lady Rosetta, 2012 | This study | 4608 | 4446 | 22 | 75 | 1 |
| IFB0421 | Portugal, Santarem | Potato cv. Lady Rosetta 2012 | This study | 4349 | 4187 | 22 | 75 | 1 |
| IFB0487 | Poland, Podkarpackie Voivodeship | Potato cv. Vineta, 2013 | [27] | 4572 | 4409 | 22 | 75 | 1 |
| IFB0695 | Poland, Kuyavian-Pomeranian | Potato cv. Arielle, 2014 | This study | 4337 | 4172 | 22 | 75 | 1 |
aThe geographical locations of the isolated strains: IFB0167 - Wawrzyszow 50°73′12″ N 17°23′58″ E, IFB0212 - Mlochow 52°02′35.76″ N 20°46′4.01″ E, IFB0231 - High Grade seed potato growing region 64°48′35.46″ N 25°24′55.62″ E, IFB0311 - Lebork 54°32′11.181″ N 17°44′56.144″ E, IFB0417 and IFB0421 39°12′0″ N 8°42′0″ W, IFB0487 - Zdziechowice 50°47′00″ N 22°07′00″ E, IFB0695 - Niwy 53°34′39.443″ N 17°25′49.649″ E. For the origin and the annotated genomic features of the herein included Dickeya solani reference strains see our former study Golanowska et al. (2018) [31]
Table 2.
Basic statistics in addition to the assembly quality metrics for the studied D. solani genomes
| Genome | No. of scaffolds | No. of N bases | Genome size (bp) | Largest contig (bp) | N50 | L50 | %GC | Genbank accession no. | Reference |
|---|---|---|---|---|---|---|---|---|---|
| IFB0167 | 1 | 0 | 4,922,289 | 4,922,289 | 4,922,289 | 1 | 56.25 | CP051457 | This study |
| IFB0212 | 2 | 0 | 4,909,935 | 3,946,010 | 3,946,010 | 1 | 56.25 | JABAON000000000 | This study |
| IFB0231 | 1 | 0 | 4,924,702 | 4,924,702 | 4,924,702 | 1 | 56.24 | CP051458 | This study |
| IFB0311 | 3 | 0 | 4,913,261 | 2,394,283 | 1,850,246 | 2 | 56.24 | JABAOO000000000 | This study |
| IFB0417 | 1 | 0 | 4,924,102 | 4,924,102 | 4,924,102 | 1 | 56.24 | CP051459 | This study |
| IFB0421 | 1 | 0 | 4,934,537 | 4,934,537 | 4,934,537 | 1 | 56.24 | CP051460 | This study |
| IFB0487 | 4 | 0 | 4,882,124 | 3,440,832 | 3,440,832 | 1 | 56.23 | JABAOP000000000 | This study |
| IFB0695 | 7 | 0 | 4,904,769 | 2,442,930 | 755,734 | 2 | 56.25 | JABAOQ000000000 | This study |
| IFB0099 | 1 | 0 | 4,932,920 | 4,932,920 | 4,932,920 | 1 | 56.24 | CP024711 | [31, 76] |
| IFB0158 | 37 | 395 | 4,879,070 | 772,123 | 360,663 | 5 | 56.24 | PENA00000000 | [31] |
| IFB0221 | 38 | 394 | 4,878,255 | 774,432 | 360,663 | 5 | 56.24 | PEMZ00000000 | [31] |
| IFB0223 | 1 | 0 | 4,937,554 | 4,937,554 | 4,937,554 | 1 | 56.24 | CP024710 | [31] |
| IPO 2222 | 1 | 9200 | 4,867,258 | 4,867,258 | 4,867,258 | 1 | 56.22 | AONU01000000 | [44] |
| GBBC 2040 | 1 | 27,548 | 4,860,047 | 4,860,047 | 4,860,047 | 1 | 56.34 | AONX01000000 | [44] |
| MK10 | 3 | 3800 | 4,935,237 | 4,934,019 | 4,934,019 | 1 | 56.21 | AOOP01000000 | [44] |
| MK16 | 3 | 2100 | 4,870,382 | 4,865,372 | 4,865,372 | 1 | 56.23 | AOOQ01000000 | [44] |
| D s0432–1 | 4 | 0 | 4,904,518 | 2,278,175 | 1,562,114 | 2 | 56.20 | AMWE01000000 | [38] |
| PPO 9019 | 24 | 30 | 4,866,823 | 1,553,733 | 485,395 | 3 | 56.25 | JWLS01000000 | [39] |
| PPO 9134 | 22 | 187 | 4,870,830 | 1,553,748 | 485,873 | 3 | 56.24 | JWLT01000000 | [39] |
| RNS 05.1.2A | 37 | 0 | 4,985,571 | 570,255 | 305,078 | 7 | 56.13 | JWMJ01000000 | [39] |
| RNS 07.7.3B | 24 | 325 | 4,871,815 | 688,619 | 485,311 | 4 | 56.24 | JWLR01000000 | [39] |
| RNS 08.23.3.1A | 1 | 12,124 | 4,923,743 | 4,923,743 | 4,923,743 | 1 | 56.25 | AMYI01000000 | [60] |
The genomes depicted in bold have been de novo sequenced and assembled in the frames of this research. The versions of the included reference genomes are the ones downloaded from the Genbank database for Golanowska et al. 2018 [31]
Genomic contents and assembly statistics for the herein reported newly-sequenced D. solani genomes have been juxtaposed to these attributed to 14 reference D. solani sequences (versions of the genomes available in the Genbank database at a time of conducting research have been included; Table 2). The numbers of scaffolds building up the utilized reference genomes are considerably higher (1–38) than the quantities of scaffolds present in 8 de novo sequenced ones (4 are closed, while the remaining ones consist of 2–7 scaffolds; Table 2). Also, the vast majority of reference genomic sequences contain N bases, reaching even the number of 27,548 (GBBC 2040). Other quality metrics of reference assemblies like the largest contig (> 570,255 bp), N50 (> 305,078 bp) or L50 (< 7) are also in favour of the genome assembly pipeline used for the newly sequenced genomes. Moreover, it is worth noting a significantly higher variation (56.13–56.34) in the %GC among the reference genomes than de novo sequenced ones (Table 2).
Interestingly, the stated quantities of tRNA (Table 1) were often lower in the reference genomes, even though the range from 60 to 75 was broader [31]. Regarding rRNA, solely 1 to 4 such genes were annotated for the included versions of the reference genomes of PPO 9019, RNS 05.1.2A, RNS 07.7.3B, IPO 2222, GBBC 2040, MK10, MK16, PPO 9134, IFB0158 and IFB0221 strains [31], in contrast to 18–22 detected in the herein reported de novo sequenced genomes (Table 1).
Taking into consideration that genes coding for 5S, 16S and 23S rRNAs are typically organized into operons encountered in multiple copies, i.e. 1–14 [70], within the bacterial chromosome, such a low number of annotated rRNAs disagrees with the current biological knowledge. Thus, we postulate that the number of the annotated rRNA-encoding genes might be regarded as an informative marker of the achieved quality of de novo assembly of D. solani genomes in view of the fact that highly similar sequences of rRNAs were previously reported to potentially disrupt, due to the occurrence of both highly conserved and variable regions, the assembling process that is typically based on de Bruijn graphs [71]. It should be noted that the genomes possessing a low number of rRNA-coding genes have been assembled from the data generated by Illumina or 454 pyrosequencing platforms with the use of assemblers handling short length reads [31]. For example, the IPO 2222 genome available currently (13.02.20) in the GenBank database was reassembled from both PacBio and Illumina reads and harbours 22 rRNA-encoding genes in contrast to the number of three annotated for the here discussed version [31].
Structural similarities between D. solani genomes
Large scale BLAST comparison of de novo sequenced and reference D. solani genomes, computed with the use of BLAST Ring Image Generator (BRIG) [72], revealed an exceptionally high level of homogeneity among the studied 22 genomes (Fig. 1). The de novo sequenced genomes of D. solani: IFB0167, IFB0212, IFB0231, IFB0311, IFB0417, IFB0421, IFB0487, IFB0695 (Table 1), in addition to IFB0158, IFB0221, IFB0223, RNS 08.23.3.1A and D s0432–1, possess a nearly identical genomic structure to that of IFB0099 (Fig. 1), regardless of the sequencing method used or the closed/draft status of the genome assembly. A notable absence of certain genomic regions is a repeating feature in the case of other D. solani genomes, namely IPO 2222, GBBC 2040, MK10, MK16, PPO 9019 and PPO 9134 (Fig. 1). Some but not all of these sites are likewise not present in the genome of RNS 07.7.3B (Fig. 1). Undoubtedly, the genome of RNS 05.1.2A stands out from the pool of the tested sequences, not only taking into consideration the number, but also the size of the missing regions. It is also worth considering that the genomes of IFB0487 and IFB0695 lack quite large parts of DNA sequences present in the reference IFB0099 genome (Fig. 1). Putatively, it might be associated with the draft character of these genomic assemblies as the number of contigs is reflected in the number of computed synteny blocks. However, the presence of polymorphic sites in these regions cannot be excluded for sure due to the fact that in many cases incompleteness of a bacterial genomic assembly tends to result from the occurrence of repetitive sequences [73].
Fig. 1.

Whole genome comparison for 22 Dickeya solani strains. BLAST Ring Image Generator [72] software was implemented. D. solani IFB0099 was used as a reference. Two first rings correspond to the GC content and GC skew, respectively. Each of the depicted rings refers to one D. solani genome according to the listed coloration. White regions mark dissimilarities. The identities are based on BLAST calculations
Basing genome comparisons on ANI values allows to avoid the bias linked with sequence selections and errors [74]. As this way of genomic distance determination takes advantage of whole-sequence information at high resolution of single nucleotides, three methods of pairwise genome comparisons, i.e. BLAST+ calculation of ANI (ANIb), MUMmer calculation of ANI (ANIm) and computation of the correlation indexes of the tetra-nucleotide signatures (Tetra) were utilized for proving an extraordinarily high similarity level between the analysed 22 D. solani genomes.
In more detail, the vast majority of ANIb values exceeded 99.96, reaching even 100.00 for over a dozen of juxtapositions (Supplementary Table 1). Similarly, in the case of ANIm, 99.98 was often reached, though no 100.00 values were acquired (Supplementary Table 2). It is also worth noticing that a high percentage of all the compared D. solani genomes have been successfully aligned (91.57–99.79 for ANIb and 93.26–100.00 for ANIm; Supplementary Tables 1 and 2). In addition, 1.0 correlation of the tetra-nucleotide signatures was likewise not rarely exhibited by the studied sequences (Supplementary Table 3). Regarding the observed differences, the genome of RNS 05.1.2A strain diverged to the greatest extent from the other sequences studied (Supplementary Tables 1, 2 and 3). ANIb values acquired for comparisons including this genome ranged from 98.55 (vs. PPO 9019) to 98.68 (vs. either RNS 07.7.3B or RNS 08.23.3.1A) (Supplementary Table 1), ANIm varied from 98.71 (towards PPO 9019) to 98.82 (in contrast to RNS 07.7.3B) (Supplementary Table 2), while tetra nucleotide correlation coefficients differed from 0.99976 (vs. either IFB0417 or IFB0487) to 0.99987 (in comparison to MK16) (Supplementary Table 3). ANIb (98.55–99.93) and ANIm (98.71–99.92) calculations also pointed to PPO 9019 and PPO 9134 as the genomes slightly standing out from the others tested (Supplementary Tables 1 and 2), though this deviation was not supported by the correlation coefficients-based method (Supplementary Table 3).
Further insight into the pangenome composition of D. solani
The first glimpse into the structure of D. solani pangenome was provided in our former study [31]. In that work, Mauve-based calculation on 14 (5 closed and 9 draft) D. solani genomes showed that 74.8% (3756 genes) of the gene pool grouped into the core, 11.5% (574 genes) to the accessory and 13.7% (690 genes) to the unique pangenome fraction. In the current research, we significantly enlarged the number of the included D. solani genomes to 22 and applied another software named Bacterial Pan Genome Analysis (BPGA v. 1.3) [75] for handling the computations. The obtained data showed that contribution of the core genome increased to 84.7% (3726 genes) while the accessory and unique pangenome fractions shrank to either 7.2% (318 genes) or 8.1% (356 genes) of the whole D. solani pangenome (4400 genes) as shown in Fig. 2a and Table 3. A reduction in the pool of unique genes was expected due to the larger number of genomic sequences considered. Similarly, the higher quality of genomes used here (as complete genomes) could likely have produced a better assignment of orthologs than in the previous study. However, we cannot a priori exclude a possibility that the use of different software for computing the pangenome between the two studies could have influenced the results.
Fig. 2.
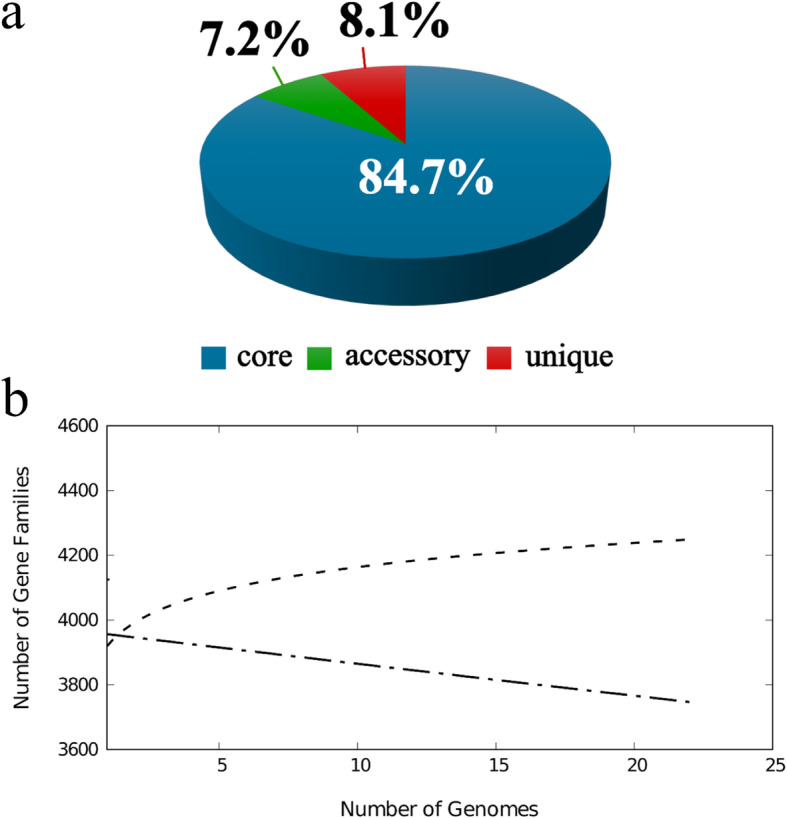
The pangenome profile of Dickeya solani species. BPGA [75] was implemented for the calculations. Abundancy of the core, accessory and unique pangenome fractions within the pangenome of D. solani (a). Total number of distinct gene families referring to the pangenome size (dashed line; power-fit curve equation: f(x) = 3924.52 ∙ x0.0256574) in addition to the number of core gene families (dash-dotted line; exponential curve equation: f1(x) = 3966.10 ∙ e-0.00258611x) are plotted against the number of genomes included (b)
Table 3.
Pangenome statistics for 22 Dickeya solani genomes
| D. solani genome | Pangenome | |||
|---|---|---|---|---|
| Core Genes |
Accessory Genes |
Unique genes |
Absent Genes |
|
| IFB0167 | 3726 | 256 | 0 | 0 |
| IFB0212 | 3726 | 254 | 0 | 0 |
| IFB0231 | 3726 | 256 | 0 | 0 |
| IFB0311 | 3726 | 250 | 0 | 1 |
| IFB0417 | 3726 | 256 | 17 | 5 |
| IFB0421 | 3726 | 255 | 4 | 1 |
| IFB0487 | 3726 | 237 | 20 | 27 |
| IFB0695 | 3726 | 232 | 5 | 19 |
| IFB0099 | 3726 | 255 | 0 | 0 |
| IFB0158 | 3726 | 261 | 1 | 0 |
| IFB0221 | 3726 | 261 | 0 | 0 |
| IFB0223 | 3726 | 249 | 0 | 3 |
| IPO 2222 | 3726 | 271 | 2 | 0 |
| GBBC 2040 | 3726 | 219 | 10 | 24 |
| MK10 | 3726 | 260 | 5 | 2 |
| MK16 | 3726 | 260 | 0 | 0 |
| D s0432–1 | 3726 | 262 | 1 | 0 |
| PPO 9019 | 3726 | 258 | 2 | 0 |
| PPO 9134 | 3726 | 258 | 1 | 0 |
| RNS 05.1.2A | 3726 | 113 | 286 | 107 |
| RNS 07.7.3B | 3726 | 254 | 0 | 0 |
| RNS 08.23.3.1A | 3726 | 255 | 2 | 0 |
Details on the contribution of specific D. solani genomes to the pangenome of this species are depicted in Table 3. The number of accessory genes detected in specific D. solani genomes ranged from 113 (RNS 05.1.2A) to 271 (IPO 2222). Regarding unique genes, there were nine strains deprived of such features (IFB0099, IFB0167, IFB0212, IFB0221, IFB0223, IFB0231, IFB0311, MK16, RNS 07.7.3B), in contrast to RNS 05.1.2A possessing even 286 unique genes (Table 3). Thirteen of the D. solani strains included, i.e. IFB0099, IFB0158, IFB0167, IFB0212, IFB0221, IFB0231, IPO 2222, MK16, D s0432–1, PPO 9019, PPO 9134, RNS 07.7.3B and RNS 08.23.3.1A, did not contain any genes stated as absent, contrary to RNS 05.1.2A strain, which lacked a huge number of 107 genes present in the other genomes analysed (Table 3). Construction and extrapolation of the core- and pan-genome plots (Fig. 2B), calculated with the use of the exponential curve fit model and power-law regression model, respectively, revealed that with the b parameter equalling 0.0256574, the pangenome of D. solani has been almost closed. In other words, the unique gene pool should no longer expand by addition of newly sequenced D. solani genomes.
Functional assignment of the D. solani pangenome fractions
The outcomes of the attribution of the Clusters of Orthologous Groups (COGs) functional categories to the core, accessory and unique gene pools of 22 D. solani strains are depicted in Fig. 3. It might be noted that the core pangenome fraction is most abundantly represented in the general function prediction only (R), followed by amino acid transport and metabolism (E), carbohydrate transport and metabolism (G), transcription (K) and inorganic ion transport and metabolism (P) functional groups (Fig. 3). Regarding the accessory pangenome section, after the genes of general function prediction only (R), the ones involved in transcription (K) were highly represented, next these of function unknown (S), engaged in energy production and conversion (C) in addition to replication, recombination and repair (L), however all these overrepresentations were not statistically significant (Fig. 3). In the case of unique genes, they have been assigned most frequently to general function prediction only (R), function unknown (S), transcription (K), replication, recombination and repair (L) and amino acid transport and metabolism (E) COG categories (Fig. 3). Among the above-mentioned functional groups, just overrepresentations of unique COGs within the function unknown (S) and amino acid transport and metabolism (E) categories were not statistically significant. It is worth to keep in mind that a significant number of general function prediction only (R) and function unknown (S) COG categories attributed to the genes from the unique D. solani pangenome fraction (Supplementary Table 4) by BPGA v. 1.3 belongs now to the X group i.e. mobilome: prophages, transposons. The groups in which both accessory and unique pangenome fractions dominated in contrast to the core section included general function prediction only (R), function unknown (S), transcription (K), replication, recombination and repair (L) and defence mechanisms (V) classifications (Fig. 3).
Fig. 3.
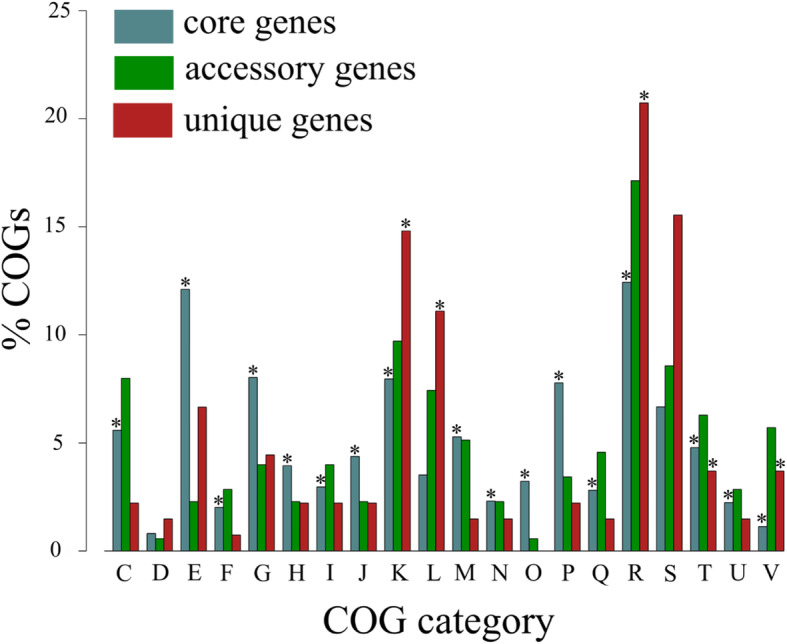
Functional assignment of Dickeya solani pangenome fractions. Comparative functional analysis was performed by mapping of the core (blue), accessory (green) and unique (red) genes to the following COG categories: C - energy production and conversion, D - cell cycle control, cell division, chromosome partitioning, E - amino acid transport and metabolism, F - nucleotide transport and metabolism, G - carbohydrate transport and metabolism, H - coenzyme transport and metabolism, I - lipid transport and metabolism, J - translation, ribosomal structure and biogenesis, K - transcription, L - replication, recombination and repair, M - cell wall/membrane/envelope biogenesis, N - cell motility, O - post-translational modification, protein turnover and chaperones, P - inorganic ion transport and metabolism, Q - secondary metabolites biosynthesis, transport and catabolism, R - general function prediction only, S - function unknown, T - signal transduction mechanisms, U - intracellular trafficking, secretion and vesicular transport and V - defence mechanisms, with the use of BPGA v. 1.3 Pan Genome Functional Analysis module [75]. Overrepresented core, accessory and unique COGs within the attributed functional groups are marked with an asterisk (hypergeometric test; p < 0.05)
It needs to be considered that the number of attributed core COGs was 3300, while the number of accessory and unique COGs equalled 157 and 120, respectively. The largest number of the assigned unique COGs derived from the genome of RNS 05.1.2A (106) followed by IFB0487 (8), IFB0417 (4) IFB0421 (1) and MK10 (1) (Supplementary Table 4). Among the functional roles of the assigned unique D. solani COGs, it is worth noticing, for instance, the genes encoding numerous transcriptional regulators (e.g. AcrR, ArsR, LysR, MarR, RpiR, AlpA, DksA), chemotaxis and adhesion proteins, ABC-type transport system components, proteins engaged in the stress response system (alkylhydroperoxidase, SbcCD, LexA), non-ribosomal peptide synthetase, components of the toxin-antitoxin system (RelBD), efflux permeases (MRS), DNA modification methylases, exo and endonucleases, mobile elements (transposase InsO), in addition to abundant prophage-associated proteins (e.g. tail protein, integrase, portal protein BeeE, primase, protein D, protein U, repressor protein C, protein W, protein X, DNA circulation protein, terminase-like protein, capsid-like protein, YmfQ, head maturation protease, head-tail adaptor) (Supplementary Table 4).
Core-genome-based phylogeny on D. solani strains
To the best of our knowledge, this is the first report on a large-scale genome-wide evolutionary study involving 22 D. solani strains. In the first large clade of the generated neighbour-joining phylogenetic tree computed on concatenated core gene alignments (Fig. 4), two strains obtained from Portugal (IFB0417, IFB0421) grouped in proximity to the ones isolated from hyacinths in the Netherlands (PPO 9019, PPO 9134). The above-listed strains are depicted in a subclade also with D. solani strains isolated in France (RNS 05.1.2A, RNS 07.7.3B, RNS 08.23.3.1A), in addition to IFB0158 strain isolated in Poland that grouped closely to IFB0221 strain from Germany. All before-mentioned strains are hypothesized to share a common ancestor with IFB0311 from Poland that is the last strain included in the first large clade. To start with the second large clade, there is a MK16 strain from Scotland assembled together with the D. solani type strain IPO 2222 from the Netherlands. It is especially intriguing taking into account that Scotland produces 99.5% of its seed potato tubers, while the Netherlands is a potent exporter of this material with strict certification policies [21]. These two D. solani strains share a most recent common ancestor with GBBC 2040 from Belgium, which nicely coincides with the fact that Belgium imports huge amounts of seed potatoes, mainly from the Netherlands, followed by France, Germany and Denmark (https://www.trademap.org/; accessed 18.03.2020). The three above-mentioned strains have the same most recent common ancestor as MK10 from Israel grouped together with IFB0487 isolated in Poland in 2013. These two subclades share a common progenitor with D s0432–1 from Finland, and the previous ancestor with IFB0695 from Poland. The two above-described large clades have most recent common ancestors with the pool of closely related strains: IFB0231 (from Finland), IFB0223 (from Germany), IFB0212 (from Poland), IFB0167 (from Poland) and IFB0099 (from Poland) (Fig. 4). It might be spotted that the trade routes of seed, industrial and table potatoes find some reflection in the computed phylogeny.
Fig. 4.

Phylogenetic analysis of the studied Dickeya solani strains based on concatenated core gene alignments. ‘Species phylogenetic analysis’ functional module of BPGA v.1.3 [75] software was utilized for generation of a neighbour-joining phylogenetic tree. The tree scaling is based on the distance matrix. Flags mark the countries of origin of the included D. solani strains. Also schematic representation of the environmental source (potato tissue, hyacinth tissue, river water or potato rhizosphere) of each D. solani strain is shown
Taking into consideration that the applied BPGA v. 1.3 software extracts protein sequences (excluding paralogs) from 20 random orthologous gene clusters to generate core genome-based phylogeny (Fig. 4), the herein presented visualization might give a hint on the evolutionary relatedness between the studied D. solani strains, but putatively shall not provide conclusive results. Rather, no obvious correlation between the geographical origin of the strains and the computed genome-wide relationships profiting from the core fraction was observed. However, if we have a look at the lengths of the tree branches reflecting the calculated distances, the recognizable divergence of RNS 05.1.2A strain, followed by IFB0487, MK16 and IFB0417 might be spotted (Fig. 4).
Discussion
Among achievements of this work, one should notice further proofs for high potency of the herein applied genome assembly pipeline described for the first time in our former work [31]. Out of 8 D. solani genomes sequenced de novo in this study, 4 have been closed to a full chromosome and the remaining ones contained just 2–7 scaffolds. It is worth underlining that solely PacBio RSII reads have been used during the assembly process, by these means lowering the required financial effort associated with additional acquisition of MiSeq Illumina reads. Furthermore, all the herein utilized software is open-source, contrary to for instance CLC Genomics Workbench v5 utilized by Pedron et al. [47] for assembling the Illumina HiSeq 2000 reads of D. solani RNS 08.23.3.1A strain into 42 contigs with N50 of 299,659. Interestingly, the sequence of RNS 08.23.3.1A was later on improved by Khayi et al., (2014) [60] into a fully closed chromosome containing the N bases by application of scaffolding, home-made scripts in addition to Sanger sequencing of the PCR amplicons. The herein utilized approach is less laborious and does not require significant bioinformatic skills.
Notably, a remarkable progress has been made on the assembling of D. solani genomic sequences in the recent years. For instance, a draft genome of D. solani IFB0099 reported before [76] consisted of 97 contigs. This sequence was assembled with Celera from 454 pyrosequencing and PacBio reads after trimming with StreamingTrim software [77]. The resulting assembly contained 5,094,121 bp (%GC 56.40), exceeding by 161,201 bp the improved closed circular genome of IFB0099 (%GC 56.24) obtained with the use of the developed genome assembly pipeline [31]. In spite of the same annotation software utilized, the total number of protein-coding genes, i.e. 4365 [76] vs. 4164 [31], in addition to the number of tRNA- or rRNA-encoding sequences, i.e. 129 [76] vs. 97 [31], varied considerably between the above-mentioned versions, which points to the crucial importance of obtaining high quality genomic assemblies prior to undertaking any comparative genomic analyses.
An alternative approach to assembling of D. solani genomes was undertaken by Garlant et al. [38]. The reads for D s0432–1 strain were acquired with Roche 454 GS Flx Titanium chemistry and assembled by using Newbler that generated 98 contigs. Gaps in this assembly were filled in by sequencing PCR or linker-PCR products using an ABI 3730 capillary sequencer. Final gap closing involved the Gap4 program (Staden package). This laborious and costly approach yielded a genome consisting finally of 4 contigs, which discloses obvious benefits of the herein utilized genome assembly pipeline. Another strategy was chosen by Pritchard et al. [44] that assembled 4 D. solani genomes into 23–224 scaffolds by relying on 454 pyrosequencing (3 genomes - MK10, MK16, IPO 2222) or IlluminaGAIIx (1 genome - GBBC 2040) technologies. The genome of IPO 2222 was assembled de novo with the use of 454 Life Sciences Newbler v2.5.3. In the case of MK10 and MK16, meta-assembly of Newbler de novo and reference-guided assemblies to the IPO 2222 reference genome were performed. Regarding a GBBC 2040 genome, for which solely Illumina reads have been acquired, CLC bio assembly module was implemented for mapping the reads to the IPO 2222 reference genome [44]. The N50 values reported for the first released versions of the above-mentioned genomes were much lower (from 40,901 to 485,700, for GBBC 2040 and MK16, respectively [44]) than in the case of the revised versions included here as references. The assemblies reported by Pritchard et al. [44] have been further improved since their release, reaching in the herein utilized versions 1–3 contigs, though the number of the incorporated N bases (2100–27,548) is still quite large. It is worth noticing that the previous assemblies differ significantly from the following ones regarding for instance the genome length. In the work of Pritchard et al. [44], IPO 2222 was reported to possess 4,857,348 bp, the version of this genome included in the here-presented research exhibits 4,867,258 bp, while the length of the one that might be currently (13.02.20) downloaded from the Genbank database equals 4,919,833 bp. This further proves the importance of obtaining high quality assemblies before conducting any genomic comparisons.
One of the reasons behind undertaking search for plasmids in draft genomic sequences of D. solani is that the occurrence of such extrachromosomal molecules might be an explanation for the contig-based status of the assembly. Our data confirmed that up to the present day solely one plasmid sequence has been described in the D. solani species, namely the one harboured by PPO 9019 strain isolated from hyacinth [61]. Notably, this extrachromosomal genetic sequence shared complete identity (100%) with the plasmid of Burkholderia ambifaria AMMD (CP000443.1) [39]. In spite of sharing a common plasmid, there is another argument pointing to the association between D. solani and B. ambifaria, as these two species exhibited notable similarities in the O-polysaccharides (OPS) within their lipopolysaccharide (LPS) structures [78, 79]. In more detail, 6-deoxy-D-altrose that was found in the OPS of D. solani and D. dadantii [78] was up to now reported only as a constituent of a disaccharide repeating unit →4)-α-d-Rhap-(1 → 3)-β-d-6dAltp-(1 → in the OPS of B. ambifaria type strain LMG 19182 [79]. Interestingly, B. ambifaria was noted to possess two diverse OPS molecules, which might be related with the adaptation of these strains to various environmental niches such as plant leaves, roots and rhizospheres, forest soil or even sputum or respiratory tract of patients suffering from cystic fibrosis [80]. Specifically, the B. ambifaria LMG 19182 strain was isolated from the rhizosphere of pea in Wisconsin (USA) in 1985 [81]. As suggested previously, sugar composition of O-antigen follows the availability of monosaccharide substrates [82], therefore the occurrence of D-altrose in the OPS of plant-associated isolates of Dickeya spp. and B. ambifaria, together with the previous proofs for horizontal gene transfer (HGT) resulting from plasmid transmission between these species [39], gives a clue about their coexistence in natural environment.
In this work, 22 whole genomic sequences of D. solani strains were juxtaposed, which revealed extraordinarily high level of homogeneity within this species. Several previous studies also focused on whole genome comparisons that included D. solani chromosomal sequences, but a significantly lesser number of genomes and other bioinformatics software were utilized. Pedron et al. [47] juxtaposed the genome of D. solani 3337 to the one of D. dadantii 3937 with the use of Mauve. In spite of a high level of synteny between these genomes, there were noted two insertions and a notable inversion between two rrs ribosomal RNA-encoding operons [47]. Interestingly, des Essarts et al. [59] spotted two syntenic disruptions and a notable evidence for horizontal gene transfer in the genome of D. solani 3337 in contrast to D. dianthicola RNS04.9. The scale of study has been enlarged in the work of Garlant et al. [38], in which the genomic sequence of D. solani D s0432–1 was compared with a pool of representative genomes of other Dickeya spp. i.e. D. dadantii 3937, D. zeae Ech586, D. paradisiaca Ech703 and D. chrysanthemi Ech1591. The lowest number of rearrangements was observed between D. solani D s0432–1 and D. dadantii 3937 [38]. Subsequently, Khayi et al. [39] reported that the genomes of two D. solani strains, namely 3337 and 0512, exhibit significant syntenic conservation accordingly to Mauve-based visualization. In our previous work [31], the same bioinformatic tool was incorporated to prove the lack of significant chromosomal rearrangements in the closed genomes of 5 D. solani strains. In more detail, the presence of 3 syntenic blocks was revealed in that work with two inversions regarding IFB0099, IFB0223 and RNS 08.23.3.1A strains, contrary to GBBC 2040 and IPO 2222 [31].
Additional evidence for a notable uniformity among the analysed D. solani genomes was provided by the herein computed pairwise comparisons relying on ANIb (98.55–100), ANIm (98.71–99.99) and Tetra correlation (0.99976–1.0) values. All outcomes of the ANI-based calculations easily exceeded the 95–96% species delineation threshold that corresponds to 70% DDH [83]. Previously, Garlant et al. [38] juxtaposed the genome of D. solani D s0432–1 to several other members of the Dickeya genus, i.e. D. dadantii 3937, D. zeae Ech586, D. paradisiaca Ech703 and D. chrysanthemi Ech1591, with the resultant ANI values of 94, 85, 79 and 86%, respectively [38]. The work of des Essarts et al. [59] further supported the closest relationship between D. solani (3337 strain) and D. dadantii (3937 strain) with ANI and DDH values of 94 and 55%. Even though the herein investigated D. solani genomes turned out to be highly homogenous basing on ANI calculations as it was suggested previously [39, 60], the computed values did not always exceed the 99.9 threshold demonstrated before [39, 58, 84]. However, the gathered data are in agreement with our former study [31], in which the ANI values determined for pairwise comparisons among 14 D. solani genomes ranged from 98.60 to 99.99%. It is worth keeping in mind that often various software has been utilized for ANI calculations e.g. nucmer with script calculate_ani.py [84, 85], ChunLab’s online Average Nucleotide Identity Calculator (EzBioCloud) [31, 86] or JSpecies [38, 47], which might be the cause of slight discrepancies in the reported genome-to-genome deviations between D. solani strains.
The fact that the genome of D. solani RNS 05.1.2A clearly stood out from the other analysed both in terms of the genomic structure and the computed pairwise-comparisons is putatively associated with the abundance of unique genes as further proven in the pangenome-related section and suggested in the former studies [31, 39]. Besides, modest dissimilarities in comparison to the included genomic pool were noted for RNS 07.7.3B, PPO 9019 and PPO 9134 sequences, which were also reported to show discrepancies in SNPs/InDels in contrast to other D. solani genomes [58]. Khayi et al. [39] postulated HGT from a closely related habitant of the same ecological niche, namely D. dianthicola, as a possible explanation for this phenomenon. The fact that both PPO 9019 and PPO 9134 strains were acquired from hyacinths and stood out solely in the ANI calculations, in contrast to the computed correlation indexes of the tetra-nucleotide signatures, might be a point in favour of the HGT-based hypothesis.
Moving to the outcomes of pangenome-oriented analyses, the whole gene pool of 22 D. solani strains comprises 4400 genes divided between large core (84.7%) in addition to significantly smaller accessory (7.2%) and unique (8.1%) pangenome fractions. Importantly, with the applied number of whole genomic sequences, the almost closed status of the D. solani pangenome has been reached. Such a feature is regarded as characteristic of the taxa whose representatives either occupy isolated ecological niches or lack efficient mechanisms for gene exchange and recombination [87]. Therefore, D. solani joined the group of real specialized pathogens with closed pangenomes [88] including e.g. Bacillus anthracis [62], Mycobacterium tuberculosis [89], Clostridium difficile [90], Yersinia pestis [91] or Staphylococcus aureus [92]. In contrast to D. solani, we reported that another member of the Pectobacteriaceae family, namely Pectobacterium parmentieri, possesses an open pangenome [93]. Computation with the use of Roary on 15 P. parmentieri genomes disclosed a notably lesser contribution of core (52.8%) and higher of accessory (20.9%) and unique (26.3%) pangenome fractions in comparison to D. solani. We associated the overrepresentation of the dispensable pangenome part with high genomic plasticity of P. parmentieri [93], suggesting a less clonal population structure with respect to that of D. solani [19, 21, 23, 59]. Thus, the closely related P. parmentieri species adhered to the categories of non-specialized species or opportunistic pathogens that often exhibit open pangenomes [88, 94] along with, for instance, Escherichia coli [95], Streptococcus agalactiae [96], Listeria monocytogenes [97], Legionella pneumophila [98] or Salmonella typhi [99]. One should bear in mind that the closed/open pangenome status of a species might have been affected by the number and representativeness of the genomes selected for the analysis [94]. Besides, not without importance is the software utilized for performing the pangenome calculations.
Attribution of the genes originating from diverse D. solani pangenome fractions to functional COG categories showed overrepresentation of phage/mobile elements- and transcription- associated groups in the studied accessory and unique pangenome parts. Notably, the genome of RNS 05.1.2A (followed by the sequences of IFB0487 and IFB0417 strains) had the most significant contribution to this phenomenon. Similarly to this research, our previous study [31] pointed to RNS 05.1.2A genome as the most distant from the others tested, basing on the largest number of unique genes as calculated by Mauve. The MK10 genome was also mentioned in the former study as a divergent one [31], although in the current work it possessed solely 1 unique COG, just as IFB0421.
The herein described high abundance of phage/mobile elements-related COG classifications in the accessory and unique D. solani gene pools is in agreement with our previous study [31] which underlined the importance of prophages in the evolution of D. solani genomes. Out of 35 prophage sequences detected in 14 D. solani genomes, the majority of the strains harbored 2–3 prophages with the exception of RNS 05.1.2A, which showed the presence of 7 such prophage-like elements [31]. Also Khayi et al. [39] reported the RNS 05.1.2A strain to possess unique phage elements and hypothetical or unknown proteins except for some other strain-specific genes coding for two putative ABC transporters, two hypothetical virulence factors and one methyl-accepting chemotaxis protein, similarly to the types of COGs that have been established in the unique pangenome fraction described in the current research. It is also worth noticing that a protein family involved in adhesion has been spotted in the D. solani unique pangenome fraction, which is in accordance with our previous suggestions [31] on the putative involvement of these proteins in the overall D. solani virulence. Furthermore, quite a big number of the observed transcription-associated unique COGs confirms the assumptions of Potrykus et al. [32, 33] on the correlation between the regulation of genes expression and the noted differences in the virulence of various D. solani strains.
In the current study, the first computed core genome-based phylogeny on 22 D. solani strains showed no obvious correlations between the geographical origin of the isolates and their evolutionary relationships. Though, the calculated evolutionary distances pointed to notable divergence of RNS 05.1.2A strain, similarly to what was observed in the other herein conducted comparative genomic analyses. In accordance to our data, an outgrouping of RNS 05.1.2A strain was described previously by Khayi et al. [58] basing on a phylogenetic tree computed on gapA sequences. Also MLST analysis on the concatenated sequences of rpoD, gyrB, recA, rpoS, dnaX, dnaA, gapA, fusA, rplB, purA and gyrA housekeeping genes differentiated RNS 05.1.2A from the other D. solani isolates included [39]. One should bear in mind that the search for phylogenetic relations among D. solani strains is impaired to some degree by high similarity of these isolates and their tendency to group together regardless of the origin and year of isolation. Such a phenomenon was reported before by van der Wolf et al. [19] basing on the phylogenies computed on PFGE profiles, IGS regions, single house-keeping genes, i.e. dnaX, recA, dnaN, fusA, gapA, purA, rpoS, rplB or the concatenated sequences of all the above-listed 8 genes in addition to IGS. In that study, solely the fatty acids fingerprints showed subtle differences between D. solani strains. It seems that phylogenetic relatedness between diverse strains is affected to some extent by the applied marker and bioinformatic method, indicating that the most appropriate approach to be used still has to be revealed.
Conclusions
In view of a high need for extensive comparative genomics studies conducted on the economically important members of the Pectobacteriaceae family [100], at first we decided to enlarge the available pool of D. solani genomes, taking into consideration that this species was pointed to as a significant threat to potato production in Europe [21]. 8 novel D. solani genomes have been sequenced and assembled either to the closed genomes or high-quality draft-status assemblies containing just a few contigs. An exceptionally high level of homogeneity among 22 D. solani genomes was proven in whole-genome comparison, ANIb, ANIm, Tetra and pangenome-oriented analyses. Notably, the genome of D. solani RNS 05.1.2A stood out from the others tested in all the above-mentioned calculations. After the inclusion of 22 D. solani genomes, the pangenome of this species consisting in 84.7% of core, 7.2% of accessory and 8.1% of unique genes, turned out to be almost closed. The assignment of the genes included in the D. solani pangenome fractions to functional COG categories revealed that higher percentages of accessory and unique pangenome parts in contrast to the core section are encountered in phage/mobile elements- and transcription- associated groups, with the genome of RNS 05.1.2A strain having the most significant contribution to this phenomenon. The first large-scale genome-wide phylogenetic study based on concatenated core gene alignments showed rather no obvious correlations between the geographical origin of the strains and the computed evolutionary relationships which might reflect to some point the specificity of the international seed potato market.
Methods
Collection and identification of D. solani strains
Out of 8 D. solani strains subjected to de novo whole-genome sequencing within the frames of this study (Table 1), 7 (IFB0167, IFB0212, IFB0311, IFB0417, IFB0421, IFB0487, IFB0695) have been isolated and identified to the species level by our research group. The implemented methods have been described previously [27, 101]. Briefly, symptomatic potato tissue has been collected from seed potato fields (either in Poland or Portugal; Table 1), homogenized in phosphate buffer, serially-diluted in 0.85% NaCl and plated on semiselective Cristal Violet Pectate (CVP) medium [102]. Post 48 h incubation at 28 °C, the cavity-forming units were collected and purified to reach the axenic culture state by several replating steps on CVP and TSA media. Isolates belonging to the Dickeya genus were identified with the use of PCR either with ADE1 and ADE2 [103] or Df and Dr primers [101, 104]. The isolates have been assigned to D. solani species basing on PCR reactions with SOL-C or SOL-D starters [36] and comparison of the sequences of dnaX housekeeping gene [23]. All strains were subsequently frozen in 40% glycerol and stored in the collection of phytopathogenic bacteria of Intercollegiate Faculty of Biotechnology University of Gdansk and Medical University of Gdansk for subsequent analyses. IFB0231 strain was isolated in Finland (Table 1) and identified to D. solani species as described by Degefu et al. (2013) [28].
De novo sequencing of D. solani genomes
D. solani strains designated for de novo sequencing were selected in such a way as to reflect the highest possible diversity among the already studied isolates [27–29] at our possession (Table 1).
Regarding the firstly analysed four strains (IFB0167, IFB0212, IFB0231 and IFB0311), they have been sent in the form of cell pellets to GATC Biotech (Constance, Germany) for DNA isolation, quality control, libraries preparation and sequencing with the use of two platforms, namely PacBio RSII and Illumina MiSeq. After proposal of the PacBio-based optimal genome assembly pipeline for D. solani [31], DNA of the latter 4 D. solani strains (IFB0417, IFB421, IFB487 and IFB0695) was sequenced at GATC Biotech just on the PacBio RSII platform (Motyka-Pomagruk et al., submitted). Accordingly to the genome assembly pipeline described in our previous work [31], D. solani genomic sequences have been assembled from solely PacBio RSII reads. At first, these raw reads were filtered from adapters with the use of SMRT Analysis software (Pacific Biosciences, USA). The coverage of the filtered reads in terms of IFB0167, IFB0212, IFB0231, IFB0311, IFB0417, IFB421, IFB487 and IFB0695 equalled 274x, 63x, 157x, 57x, 212x, 243x, 211x and 230x, respectively. Then, these reads were corrected, trimmed and assembled with the use of Canu [66]. Getting consensus and variant calling was achieved thanks to Quiver [67], while functional annotation was conducted with Prokka [68] as previously reported [31]. The assembled and annotated de novo sequenced genomes of 8 D. solani strains have been deposited in the National Center for Biotechnology Information (NCBI) Genome database (Bioproject no. PRJNA611911) under the accession numbers listed in Table 2.
Comparative genomics
Beside 8 de novo sequenced genomes (Tables 1 and 2), 14 D. solani reference sequences (Table 2) were included in the conducted comparative genomic analyses: IFB0099 (CP024711 [31]), IFB0158 (PENA00000000 [31]), IFB0221 (PEMZ00000000 [31]), IFB0223 (CP024710 [31]), IPO 2222 (AONU01000000 [44]), GBBC 2040 (AONX01000000 [44]), MK10 (AOOP01000000 [44]), MK16 (AOOQ01000000 [44]), D s0432–1 (AMWE01000000 [38]), PPO 9019 (JWLS01000000 [39]), PPO 9134 (JWLT01000000 [39]), RNS 05.1.2A (JWMJ01000000 [39]), RNS 07.7.3B (JWLR01000000 [39]) and RNS 08.23.3.1A (AMYI01000000 [60]). The above-listed reference sequences have been downloaded from the NCBI Genome database in a FASTA format. To assure the uniformity of the attributed genomic annotations, also the reference D. solani sequences have been processed with Prokka v. 1.12 [68] software as it was the case of de novo assembled sequences.
The number of contigs in the genomes, %GC in addition to N50 and L50 metrics were computed with Quast v. 5.0 [105]. The search for plasmid sequences among the draft D. solani genomic assemblies was accomplished with PlasmidFinder v. 2.0 [69] with the default settings. Whole genome comparison of 22 D. solani sequences has been computed with the use of BRIG v. 0.95 [72]. The included pairwise genome comparisons are based on ANIb, ANIm and computation of the correlation indexes of the tetra-nucleotide signatures by applying JSpecies webserver (accessed 02.2019) [106].
Pangenome analysis
BPGA v. 1.3 [75] was utilized for pangenome studies in addition to the pangenome-based downstream analyses including core genome phylogeny and functional assignments to the COGs categories. Sequence data were pre-processed and clustered with the use of USEARCH (50% cut off) [107]. Further computation of the output led to the generation of a tab delimited gene presence/absence binary matrix (pan-matrix), utilized for pangenome pattern calculations with iterations (20 as a default). For core genome-based phylogeny, BPGA v. 1.3 [75] extracted protein sequences (excluding paralogs) from 20 random orthologous gene clusters. Then MUSCLE [108] was applied for alignment of concatenated core genes resulting in the construction of a neighbour-joining phylogenetic tree. Last but not least, USEARCH [107] was implemented for functional assignments on the basis of the best hits with the reference COG database [109]. COG IDs were attributed to all representative protein sequences from each orthologous protein cluster based on the BLAST algorithm [110]. Percentage occurrences of the assigned COG categories were calculated by BPGA v. 1.3. Overrepresentation of core, accessory and unique COGs within the attributed functional groups was assessed by a hypergeometric test at p < 0.05 in R v. 3.1.3. In addition, the COG IDs assigned to D. solani unique COGs were manually searched against the COG database [109] for stating the up-to-date functions played by the individual protein family clusters.
Supplementary information
Additional file 1: Table S1. ANIb values calculated for the studied Dickeya solani genomes. Description of data: BLAST calculation of ANI (ANIb) was performed with the use of JSpecies [106]. The upper number refers to the ANIb value while the lower depicted in parentheses is the percentage of the aligned sequences.
Additional file 2: Table S2. ANIm values calculated for the studied Dickeya solani genomes. Description of data: MUMmer calculation of ANI (ANIm) was performed with the use of JSpecies [106]. The upper number refers to ANIm value while the lower depicted in parentheses is the percentage of the aligned sequences.
Additional file 3: Table S3. Correlation indexes of the tetra-nucleotide signatures computed for the studied Dickeya solani genomes. Description of data: Computation of the correlation indexes of the tetra-nucleotide signatures was conducted with JSpecies [106].
Additional file 4: Table S4. The functions of the unique Dickeya solani COGs. Description of data: a The first category was attributed by the BPGA v. 1.3 software. If it differed from the category currently allocated to certain COG IDs in the COG database, the up-to-date assignment after the slash mark is depicted. COG categories: A - RNA processing and modification, B - chromatin structure and dynamics, C - energy production and conversion, D - cell cycle control, cell division, chromosome partitioning, E - amino acid transport and metabolism, F - nucleotide transport and metabolism, G - carbohydrate transport and metabolism, H - coenzyme transport and metabolism, I - lipid transport and metabolism, J - translation, ribosomal structure and biogenesis, K - transcription, L - replication, recombination and repair, M - cell wall/membrane/envelope biogenesis, N - cell motility, O - post-translational modification, protein turnover and chaperones, P - inorganic ion transport and metabolism, Q - secondary metabolites biosynthesis, transport and catabolism, R - general function prediction only, S - function unknown, T - signal transduction mechanisms, U - intracellular trafficking, secretion and vesicular transport, V - defence mechanisms, W - extracellular structures, X - mobilome: prophages, transposons, Y - nuclear structure and Z – cytoskeleton.
Acknowledgements
The authors are highly grateful to Dr. Michal Kabza for his suggestions on the appropriate genome assembling software to use. We also want to acknowledge Dr. Yeshitila Degefu for provision of the VIC-BL-25 (IFB0231) D. solani strain isolated in Finland. The authors are grateful to Dr. Malgorzata Golanowska and Dr. Marta Potrykus for assistance in isolation and identification of several strains characterized in this research.
Abbreviations
- ANI
Average Nucleotide Identity
- ANIb
BLAST+ calculation of ANI
- ANIm
MUMmer calculation of ANI
- BRIG
BLAST Ring Image Generator
- BPGA
Bacterial Pan Genome Analysis
- CVP
Cristal Violet Pectate
- COGs
Clusters of Orthologous Groups
- DDH
DNA-DNA hybridization
- ERIC
Enterobacterial Repetitive Intergenic Consensus
- HGT
Horizontal gene transfer
- IGS
Intergenic spacer
- LPS
Lipopolysaccharide
- MALDI-TOF MS
Matrix-Assisted Laser Desorption Ionization Time-Of-Flight Mass Spectrometry
- NCBI
National Center for Biotechnology Information
- OPS
O-polysaccharides
- PFGE
Pulse Field Gel Electrophoresis
- PCWDEs
Plant cell wall degrading enzymes
- REP-PCR
Repetitive Extragenic Palindromic-PCR
- Tetra
Tetra-nucleotide signatures
- WGS
Whole genome sequences
Authors’ contributions
AM-P performed analyses presented in this work, prepared tables and figures and wrote the first version of this manuscript. SZ and WS isolated, identified to-species and characterized the bacterial strains and discussed the manuscript. AEM provided assistance with the comparative genomics software, calculated overrepresentations of COGs and participated in discussion on the manuscript. EL and AM supervised the whole work and critically revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was funded by National Science Centre in Poland (Narodowe Centrum Nauki) via projects: UMO-2014/14/M/NZ8/00501 attributed to EL and UMO-2016/21/N/NZ1/02783 attributed to AM-P. It was also supported by Ministry of Science and Higher Education in Poland (Ministerstwo Nauki i Szkolnictwa Wyższego) through the Polish-Italian Collaborative Program Canaletto awarded to EL and AM. The funding bodies played no role in the design of the study, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
De novosequenced D. solani genomes.
Data generated in this whole genome sequencing project are available in the Genome database of the National Center for Biotechnology Information (NCBI) under the BioProject PRJNA611911. The analysed D. solani strains have been attributed with the following Biosample nos. SAMN14352303, SAMN14352304, SAMN14352305, SAMN14352306, SAMN14352307, SAMN14352308, SAMN14352309 and SAMN14352310. The assembled and annotated full genomic sequences of the herein de novo sequenced D. solani genomes are available in the NCBI Genome database under the following accession nos: CP051457, JABAON000000000, CP051458, JABAOO000000000, CP051459, CP051460, JABAOP000000000 and JABAOQ000000000 (Table 2).
The datasets generated and/or analyzed during this study will not be publicly available prior to first publication of the herein presented manuscript. After publication, the datasets will be available from the corresponding author on a reasonable request.
The reference D. solani genomes.
The included 14 reference D. solani genomic sequences have been downloaded in a FASTA format from the NCBI Genome database, in which they have been deposited under the following accession nos. CP024711, PENA00000000, PEMZ00000000, CP024710, AONU01000000, AONX01000000, AOOP01000000, AOOQ01000000, AMWE01000000, JWLS01000000, JWLT01000000, JWMJ01000000, JWLR01000000 and AMYI01000000 (Table 2).
Ethics approval and consent to participate
We obtained permissions for the isolation of the studied bacterial isolates from plant samples collected by the National Inspectorates of Plant Health and Seed Inspection Service in Poland. Regarding bacterial strains originating from Portugal, they were isolated from potatoes exported from Poland to Santarem region in Portugal. We have an appropriate agreement with the exporting company concerning the collection of bacterial isolates from this material. All described field studies were conducted in accordance with the policies of the National Inspectorates of Plant Health and Seed Inspection Service in Poland.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-06863-w.
References
- 1.Adeolu M, Alnajar S, Naushad S, Gupta RS. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales Ord. Nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. Nov., Pectobacteriaceae fam. Nov., Yersiniaceae fam. Nov., Hafniaceae fam. Nov., Morganellaceae fam. Nov., and Budviciaceae fam. Nov. Int J Syst Evol Microbiol. 2016;66:5575–5599. doi: 10.1099/ijsem.0.001485. [DOI] [PubMed] [Google Scholar]
- 2.Perombelon MCM, Kelman A. Ecology of the soft rot Erwinias. Annu Rev Phytopathol. 1980;18:361–387. [Google Scholar]
- 3.Hugouvieux-Cotte-Pattat N, Condemine G, Shevchik VE. Bacterial pectate lyases, structural and functional diversity. Environ Microbiol Rep. 2014;6:427–440. doi: 10.1111/1758-2229.12166. [DOI] [PubMed] [Google Scholar]
- 4.Reverchon S, Muskhelisvili G, Nasser W. Virulence program of a bacterial plant pathogen: the Dickeya model. Prog Mol Biol Transl Sci. 2016;142:51–92. doi: 10.1016/bs.pmbts.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Jahn CE, Selimi DA, Barak JD, Charkowski AO. The Dickeya dadantii biofilm matrix consists of cellulose nanofibres, and is an emergent property dependent upon the type III secretion system and the cellulose synthesis operon. Microbiology. 2011;157:2733–2744. doi: 10.1099/mic.0.051003-0. [DOI] [PubMed] [Google Scholar]
- 6.Moleleki LN, Pretorius RG, Tanui CK, Mosina G, Theron J. A quorum sensing-defective mutant of Pectobacterium carotovorum ssp. brasiliense 1692 is attenuated in virulence and unable to occlude xylem tissue of susceptible potato plant stems. Mol Plant Pathol. 2017;18:32–44. doi: 10.1111/mpp.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Expert D, Toussaint A. Bacteriocin-resistant mutants of Erwinia chrysanthemi: possible involvement of iron acquisition in phytopathogenicity. J Bacteriol. 1985;163:221–227. doi: 10.1128/jb.163.1.221-227.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans TJ, Ind A, Komitopoulou E, Salmond GPC. Phage-selected lipopolysaccharide mutants of Pectobacterium atrosepticum exhibit different impacts on virulence. J Appl Microbiol. 2010;109:505–514. doi: 10.1111/j.1365-2672.2010.04669.x. [DOI] [PubMed] [Google Scholar]
- 9.Barrett LG, Kniskern JM, Bodenhausen N, Zhang W, Bergelson J. Continua of specificity and virulence in plant host–pathogen interactions: causes and consequences. New Phytol. 2009;183:513–529. doi: 10.1111/j.1469-8137.2009.02927.x. [DOI] [PubMed] [Google Scholar]
- 10.Perombelon MCM. Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 2002;51:1–12. [Google Scholar]
- 11.Motyka A, Zoledowska S, Sledz W, Lojkowska E. Molecular methods as tools to control plant diseases caused by Dickeya and Pectobacterium spp: a minireview. New Biotechnol. 2017;39:181–189. doi: 10.1016/j.nbt.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Samson R, Legendre JB, Christen R, Fischer-Le Saux M, Achouak W, Gardan L. Transfer of Pectobacterium chrysanthemi (Burkholder et al. 1953) Brenner et al. 1973 and Brenneria paradisiaca to the genus Dickeya gen. nov. as Dickeya chrysanthemi comb. nov. and Dickeya paradisiaca comb. nov. and delineation of four novel species, Dickeya dadantii sp. nov., Dickeya dianthicola sp. nov., Dickeya dieffenbachiae sp. nov. and Dickeya zeae sp. nov. Int J Syst Evol Microbiol. 2005;55(Pt 4):1415–1427. doi: 10.1099/ijs.0.02791-0. [DOI] [PubMed] [Google Scholar]
- 13.Burkholder WR, McFadden LA, Dimock EW. A bacterial blight of Chrysanthemums. Phytopathology. 1953;43:522–526. [Google Scholar]
- 14.Hauben L, Moore ERB, Vauterin L, Steenackers M, Mergaert J, Verdonck L, et al. Phylogenetic position of phytopathogens within the Enterobacteriaceae. Syst Appl Microbiol. 1998;21:384–397. doi: 10.1016/S0723-2020(98)80048-9. [DOI] [PubMed] [Google Scholar]
- 15.Parkinson N, DeVos P, Pirhonen M, Elphinstone J. Dickeya aquatica sp. nov., isolated from waterways. Int J Syst Evol Microbiol. 2014;64:2264–2266. doi: 10.1099/ijs.0.058693-0. [DOI] [PubMed] [Google Scholar]
- 16.Brady CL, Cleenwerck I, Denman S, Venter SN, Rodriguez-Palenzuela P, Coutinho TA, et al. Proposal to reclassify Brenneria quercina (Hildebrand and Schroth 1967) Hauben et al. 1999 into a new genus, Lonsdalea gen. nov., as Lonsdalea quercina comb. nov., descriptions of Lonsdalea quercina subsp. quercina comb. nov., Lonsdalea quercina subsp. iberica subsp. nov. and Lonsdalea quercina subsp. britannica subsp. nov., emendation of the description of the genus Brenneria, reclassification of Dickeya dieffenbachiae as Dickeya dadantii subsp. dieffenbachiae comb. nov., and emendation of the description of Dickeya dadantii. Int J Syst Evol Microbiol. 2012;62:1592–1602. doi: 10.1099/ijs.0.035055-0. [DOI] [PubMed] [Google Scholar]
- 17.Tian Y, Zhao Y, Yuan X, Yi J, Fan J, Xu Z, et al. Dickeya fangzhongdai sp. nov., a plant-pathogenic bacterium isolated from pear trees (Pyrus pyrifolia) Int J Syst Evol Microbiol. 2016;66:2831–2835. doi: 10.1099/ijsem.0.001060. [DOI] [PubMed] [Google Scholar]
- 18.Hugouvieux-Cotte-Pattat N, Jacot-des-Combes C, Briolay J. Dickeya lacustris sp. nov., a water-living pectinolytic bacterium isolated from lakes in France. Int J Syst Evol Microbiol. 2019;69:721–726. doi: 10.1099/ijsem.0.003208. [DOI] [PubMed] [Google Scholar]
- 19.van der Wolf JM, Nijhuis EH, Kowalewska MJ, Saddler GS, Parkinson N, Elphinstone JG, et al. Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum) Int J Syst Evol Microbiol. 2014;64:768–774. doi: 10.1099/ijs.0.052944-0. [DOI] [PubMed] [Google Scholar]
- 20.Oulghazi S, Pedron J, Cigna J, Lau YY, Moumni M, Van Gijsegem F, et al. Dickeya undicola sp. nov., a novel species for pectinolytic isolates from surface waters in Europe and Asia. Int J Syst Evol Microbiol. 2019;69:2440–2444. doi: 10.1099/ijsem.0.003497. [DOI] [PubMed] [Google Scholar]
- 21.Toth IK, van der Wolf JM, Saddler G, Lojkowska E, Helias V, Pirhonen M, et al. Dickeya species: an emerging problem for potato production in Europe. Plant Pathol. 2011;60:385–399. [Google Scholar]
- 22.Czajkowski R, Grabe GJ, van der Wolf JM. Distribution of Dickeya spp. and Pectobacterium carotovorum subsp. carotovorum in naturally infected seed potatoes. Eur J Plant Pathol. 2009;125:263–275. [Google Scholar]
- 23.Slawiak M, van Beckhoven JR, Speksnijder AG, Czajkowski R, Grabe G, van der Wolf JM. Biochemical and genetical analysis reveal a new clade of biovar 3 Dickeya spp. strains isolated from potato in Europe. Eur J Plant Pathol. 2009;125:245–261. [Google Scholar]
- 24.Laurila J, Ahola V, Lehtinen A, Joutsjoki T, Hannukkala A, Rahkonen A, et al. Characterization of Dickeya strains isolated from potato and river water samples in Finland. Eur J Plant Pathol. 2008;122:213–225. [Google Scholar]
- 25.Parkinson N, Stead D, Bew J, Heeney J, Tsror L, Elphinstone J. Dickeya species relatedness and clade structure determined by comparison of recA sequences. Int J Syst Evol Microbiol. 2009;59:2388–2393. doi: 10.1099/ijs.0.009258-0. [DOI] [PubMed] [Google Scholar]
- 26.Waleron M, Waleron K, Podhajska AJ, Łojkowska E. Genotyping of bacteria belonging to the former Erwinia genus by PCR-RFLP analysis of a recA gene fragment. Microbiology. 2002;148:583–595. doi: 10.1099/00221287-148-2-583. [DOI] [PubMed] [Google Scholar]
- 27.Potrykus M, Golanowska M, Sledz W, Zoledowska S, Motyka A, Kolodziejska A, et al. Biodiversity of Dickeya spp. isolated from potato plants and water sources in temperate climate. Plant Dis. 2016;100:408–417. doi: 10.1094/PDIS-04-15-0439-RE. [DOI] [PubMed] [Google Scholar]
- 28.Degefu Y, Potrykus M, Golanowska M, Virtanen E, Lojkowska E. A new clade of Dickeya spp. plays a major role in potato blackleg outbreaks in North Finland. Ann Appl Biol. 2013;162:231–241. [Google Scholar]
- 29.Golanowska M, Kielar J, Lojkowska E. The effect of temperature on the phenotypic features and the maceration ability of Dickeya solani strains isolated in Finland, Israel and Poland. Eur J Plant Pathol. 2017;147:803–817. [Google Scholar]
- 30.Wayne LG. International Committee on Systematic Bacteriology. Report of the ad hoc committee on the reconciliation of approaches to bacterial systematics. ci.nii.ac.jp. 1987;463–4. https://ci.nii.ac.jp/naid/10030638522/. Accessed 11 Feb 2020.
- 31.Golanowska M, Potrykus M, Motyka-Pomagruk A, Kabza M, Bacci G, Galardini M, et al. Comparison of highly and weakly virulent Dickeya solani strains, with a view on the pangenome and panregulon of this species. Front Microbiol. 2018;9:1940. doi: 10.3389/fmicb.2018.01940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potrykus M, Golanowska M, Hugouvieux-Cotte-Pattat N, Lojkowska E. Regulators involved in Dickeya solani virulence, genetic conservation, and functional variability. Mol Plant-Microbe Interact. 2014;27:700–711. doi: 10.1094/MPMI-09-13-0270-R. [DOI] [PubMed] [Google Scholar]
- 33.Potrykus M, Hugouvieux-Cotte-Pattat N, Lojkowska E. Interplay of classic Exp and specific Vfm quorum sensing systems on the phenotypic features of Dickeya solani strains exhibiting different virulence levels. Mol Plant Pathol. 2018;19:1238–1251. doi: 10.1111/mpp.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palacio-Bielsa A, Rodríguez Mosquera ME, Cambra Álvarez MA, Berruete Rodríguez IM, López-Solanilla E, Rodríguez-Palenzuela P. Phenotypic diversity, host range and molecular phylogeny of Dickeya isolates from Spain. Eur J Plant Pathol. 2010;127:311–324. [Google Scholar]
- 35.Tsror L, Ben-Daniel B, Chalupowicz L, van der Wolf J, Lebiush S, Erlich O, et al. Characterization of Dickeya strains isolated from potato grown under hot-climate conditions. Plant Pathol. 2013;62:1097–1105. [Google Scholar]
- 36.Pritchard L, Humphris S, Saddler GS, Parkinson NM, Bertrand V, Elphinstone JG, et al. Detection of phytopathogens of the genus Dickeya using a PCR primer prediction pipeline for draft bacterial genome sequences. Plant Pathol. 2013;62:587–596. [Google Scholar]
- 37.Parkinson N, Pritchard L, Bryant R, Toth I, Elphinstone J. Epidemiology of Dickeya dianthicola and Dickeya solani in ornamental hosts and potato studied using variable number tandem repeat analysis. Eur J Plant Pathol. 2014;141:63–70. [Google Scholar]
- 38.Garlant L, Koskinen P, Rouhiainen L, Laine P, Paulin L, Auvinen P, et al. Genome sequence of Dickeya solani, a new soft rot pathogen of potato, suggests its emergence may be related to a novel combination of non-ribosomal peptide/polyketide synthetase clusters. Diversity. 2013;5:824–842. [Google Scholar]
- 39.Khayi S, Blin P, Pedron J, Chong TM, Chan KG, Moumni M, et al. Population genomics reveals additive and replacing horizontal gene transfers in the emerging pathogen Dickeya solani. BMC Genomics. 2015;16:788–801. doi: 10.1186/s12864-015-1997-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adriaenssens EM, Van Vaerenbergh J, Vandenheuvel D, Dunon V, Ceyssens PJ, De Proft M, et al. T4-related bacteriophage LIMEstone isolates for the control of soft rot on potato caused by ‘Dickeya solani.’. PLoS One. 2012;7:e33227. doi: 10.1371/journal.pone.0033227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozturk M, Aksoy HM. First report of Dickeya solani associated with potato blackleg and soft rot in Turkey. J Plant Pathol. 2017;99:298. [Google Scholar]
- 42.Dees MW, Lebecka R, Perminow JIS, Czajkowski R, Grupa A, Motyka A, et al. Characterization of Dickeya and Pectobacterium strains obtained from diseased potato plants in different climatic conditions of Norway and Poland. Eur J Plant Pathol. 2017;148:839–851. [Google Scholar]
- 43.van der Wolf JM, Vlami MB, van den Boogert PHJF. Dickeya report. In: EUPHRESCO-I. Euphresco phytosanitary Era-net: Wageningen; 2013. [Google Scholar]
- 44.Pritchard L, Humphris S, Baeyen S, Maes M, Van Vaerenbergh J, Elphinstone J, et al. Draft genome sequences of four Dickeya dianthicola and four Dickeya solani strains. Genome Announc. 2013;1:e00087–e00012. doi: 10.1128/genomeA.00087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaczek-Moczydłowska MA, Fleming CC, Young GK, Campbell K, O’Hanlon R. Pectobacterium and Dickeya species detected in vegetables in Northern Ireland. Eur J Plant Pathol. 2019;154:635–647. [Google Scholar]
- 46.Sarris PF, Trantas E, Pagoulatou M, Stavrou D, Ververidis F, Goumas DE. First report of potato blackleg caused by biovar 3 Dickeya sp. (Pectobacterium chrysanthemi) in Greece. New Dis Reports. 2011;24:21. [Google Scholar]
- 47.Pedron J, Mondy S, Van Gijsegem F, Faure D. Genomic and metabolic comparison with Dickeya dadantii 3937 reveals the emerging Dickeya solani potato pathogen to display distinctive metabolic activities and T5SS/T6SS-related toxin repertoire. BMC Genomics. 2014;15:283–296. doi: 10.1186/1471-2164-15-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gill ED, Schaerer S, Dupuis B. Factors impacting blackleg development caused by Dickeya spp. in the field. Eur J Plant Pathol. 2014;140:317–327. [Google Scholar]
- 49.Palacio-Bielsa A, Cambra MA, Lopez MM. Characterisation of potato isolates of Dickeya chrysanthemi in Spain by a microtitre system for biovar determination. Ann Appl Biol. 2006;148:157–164. [Google Scholar]
- 50.Dreo T, Naglić T, Peterka M, Ravnikar M. Zbornik predavanj in referatov 11. slovenskega posvetovanja o varstvu rastlin z mednarodno udeležbo. Ljubljana, Slovenia: Plant Protection Society of Slovenia; 2013. Characterization of Slovenian Pectobacterium and Dickeya isolates from potato; pp. 125–131. [Google Scholar]
- 51.Tsror L, Erlich O, Lebiush S, van der Wolf J, Czajkowski R, Mozes G, et al. First report of potato blackleg caused by a biovar 3 Dickeya sp. in Georgia. New Dis Reports. 2011;23:1. [Google Scholar]
- 52.Kornev K, Ignatov A, Karlov A, Karlov G, Dzhalilov F, Pekhtereva E, et al. Dickeya spp. - emerging pathogen of potato in Russia. Phytopathology. 2012;102:64–65. [Google Scholar]
- 53.Cardoza YF, Duarte V, Lopes CA. First report of blackleg of potato caused by Dickeya solani in Brazil. Plant Dis. 2017;101:243. [Google Scholar]
- 54.Chen XF, Zhang HL, Chen J. First report of Dickeya solani causing soft rot in imported bulbs of Hyacinthus orientalis in China. Plant Dis. 2015;99:155. doi: 10.1094/PDIS-09-14-0916-PDN. [DOI] [PubMed] [Google Scholar]
- 55.Tibayrenc M, Ayala FJ. Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc Natl Acad Sci U S A. 2012;109:E3305–E3313. doi: 10.1073/pnas.1212452109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsror L, Erlich O, Lebiush S, Hazanovsky M, Zig U, Slawiak M, et al. Assessment of recent outbreaks of Dickeya sp. (syn. Erwinia chrysanthemi) slow wilt in potato crops in Israel. Eur J Plant Pathol. 2009;123:311–320. [Google Scholar]
- 57.van der Wolf JM, Kastelein P. The 3rd International Erwinia Workshop on soft rot Enterobacteriaceae and related organisms. 2014. The role of haulm infections in the epidemiology of soft rot Enterobacteriaceae; p. 7. [Google Scholar]
- 58.Khayi S, Blin P, Chong TM, Chan KG, Faure D. Complete genome anatomy of the emerging potato pathogen Dickeya solani type strain IPO 2222T. Stand Genomic Sci. 2016;11:87. doi: 10.1186/s40793-016-0208-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raoul des Essarts Y, Pédron J, Blin P, Van Dijk E, Faure D, Van Gijsegem F. Common and distinctive adaptive traits expressed in Dickeya dianthicola and Dickeya solani pathogens when exploiting potato plant host. Environ Microbiol. 2019;21:1004–1018. doi: 10.1111/1462-2920.14519. [DOI] [PubMed] [Google Scholar]
- 60.Khayi S, Mondy S, Beury-Cirou A, Moumni M, Helias V, Faure D. Genome sequence of the emerging plant pathogen Dickeya solani strain RNS 08.23.3.1A. Genome Announc. 2014;2:e01270–e01213. doi: 10.1128/genomeA.01270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khayi S, Blin P, Chong TM, Chan KG, Faure D. Complete chromosome and plasmid sequences of two plant pathogens, Dickeya solani strains D s0432-1 and PPO 9019. Genome Announc. 2018;6:e00233–e00218. doi: 10.1128/genomeA.00233-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 63.Motyka-Pomagruk A. Genotypic and phenotypic characterization of bacteria from Dickeya solani species and development of novel control methods against phytopathogens. University of Gdańsk, PhD thesis. 2019. [Google Scholar]
- 64.Lapierre P, Gogarten JP. Estimating the size of the bacterial pan-genome. Trends Genet. 2009;25:107–110. doi: 10.1016/j.tig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 65.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 66.Berlin K, Koren S, Chin CS, Drake J, Landolin JM, Phillippy AM. Assembling large genomes with single-molecule sequencing and locality sensitive hashing. Nat Biotechnol. 2015;33:623–630. doi: 10.1101/008003. [DOI] [PubMed] [Google Scholar]
- 67.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 68.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 69.Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rainey FA, Ward-Rainey NL, Janssen PH, Hippe H, Stackebrandt E. Clostridium paradoxum DSM 7308T contains multiple 16S rRNA genes with heterogeneous intervening sequences. Microbiology. 1996;142:2087–2095. doi: 10.1099/13500872-142-8-2087. [DOI] [PubMed] [Google Scholar]
- 71.Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–3352. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuśmirek W, Nowak R. De novo assembly of bacterial genomes with repetitive DNA regions by dnaasm application. BMC Bioinformatics. 2018;19:273. doi: 10.1186/s12859-018-2281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han N, Qiang Y, Zhang W. ANItools web: a web tool for fast genome comparison within multiple bacterial strains. Database (Oxford) 2016;2016:baw084. doi: 10.1093/database/baw084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chaudhari NM, Gupta VK, Dutta C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373. doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golanowska M, Galardini M, Bazzicalupo M, Hugouvieux-Cotte-Pattat N, Mengoni A, Potrykus M, et al. Draft genome sequence of a highly virulent strain of the plant pathogen Dickeya solani, IFB0099. Genome Announc. 2015;3:e00109–e00115. doi: 10.1128/genomeA.00109-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bacci G, Bazzicalupo M, Benedetti A, Mengoni A. StreamingTrim 1.0: a Java software for dynamic trimming of 16S rRNA sequence data from metagenetic studies. Mol Ecol Resour. 2014;14:426–434. doi: 10.1111/1755-0998.12187. [DOI] [PubMed] [Google Scholar]
- 78.Ossowska K, Czerwicka M, Sledz W, Zoledowska S, Motyka A, Golanowska M, et al. The uniform structure of O-polysaccharides isolated from Dickeya solani strains of different origin. Carbohydr Res. 2017;445:40–43. doi: 10.1016/j.carres.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 79.De Castro C, Dinischiotu N, Feys B, Lanzetta R, Parrilli M, Molinaro A. Structural identification of the O-antigen fraction from the lipopolysaccharide of the Burkholderia ambifaria strain 19182. Carbohydr Res. 2013;379:95–99. doi: 10.1016/j.carres.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 80.Groenhagen U, Baumgartner R, Bailly A, Gardiner A, Eberl L, Schulz S, et al. Production of bioactive volatiles by different Burkholderia ambifaria strains. J Chem Ecol. 2013;39:892–906. doi: 10.1007/s10886-013-0315-y. [DOI] [PubMed] [Google Scholar]
- 81.Coenye T, Mahenthiralingam E, Henry D, LiPuma JL, Laevens S, Gillis M, et al. Burkholderia ambifaria sp. nov., a novel member of the Burkholderia cepacia complex including biocontrol and cystic fibrosis-related isolates. Int J Syst Evol Microbiol. 2001;51:1481–1490. doi: 10.1099/00207713-51-4-1481. [DOI] [PubMed] [Google Scholar]
- 82.Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant–microbe interactions. FEMS Microbiol Rev. 2002;26:17–47. doi: 10.1111/j.1574-6976.2002.tb00597.x. [DOI] [PubMed] [Google Scholar]
- 83.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Fan Q, Loria R. A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole-genome sequencing data. Syst Appl Microbiol. 2016;39:252–259. doi: 10.1016/j.syapm.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 85.Delcher AL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017;67:1613–1617. doi: 10.1099/ijsem.0.001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mira A, Martín-Cuadrado AB, D’Auria G, Rodríguez-Valera F. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol. 2010;13:45–57. doi: 10.2436/20.1501.01.110. [DOI] [PubMed] [Google Scholar]
- 88.Rouli L, MBengue M, Robert C, Ndiaye M, La Scola B, Raoult D. Genomic analysis of three African strains of Bacillus anthracis demonstrates that they are part of the clonal expansion of an exclusively pathogenic bacterium. New Microbes New Infect. 2014;2:161–169. doi: 10.1002/nmi2.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wozniak M, Wong L, Tiuryn J. CAMBer: an approach to support comparative analysis of multiple bacterial strains. BMC Genomics. 2011;12(Suppl 2):S6. doi: 10.1186/1471-2164-12-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scaria J, Ponnala L, Janvilisri T, Yan W, Mueller LA, Chang YF. Analysis of ultra low genome conservation in Clostridium difficile. PLoS One. 2010;5:e15147. doi: 10.1371/journal.pone.0015147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eppinger M, Worsham PL, Nikolich MP, Riley DR, Sebastian Y, Mou S, et al. Genome sequence of the deep-rooted Yersinia pestis strain Angola reveals new insights into the evolution and pangenome of the plague bacterium. J Bacteriol. 2010;192:1685–1699. doi: 10.1128/JB.01518-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boissy R, Ahmed A, Janto B, Earl J, Hall BG, Hogg JS, et al. Comparative supragenomic analyses among the pathogens Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae using a modification of the finite supragenome model. BMC Genomics. 2011;12:187. doi: 10.1186/1471-2164-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zoledowska S, Motyka-Pomagruk A, Sledz W, Mengoni A, Lojkowska E. High genomic variability in the plant pathogenic bacterium Pectobacterium parmenieri deciphered from de novo assembled complete genomes. BMC Genomics. 2018;19:751. doi: 10.1186/s12864-018-5140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rouli L, Merhej V, Fournier PE, Raoult D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New microbes new Infect. 2015;7:72–85. doi: 10.1016/j.nmni.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rasko DA, Rosovitz MJ, Myers GSA, Mongodin EF, Fricke WF, Gajer P, et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.”. Proc Natl Acad Sci U S A. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deng X, Phillippy AM, Li Z, Salzberg SL, Zhang W. Probing the pan-genome of Listeria monocytogenes: new insights into intraspecific niche expansion and genomic diversification. BMC Genomics. 2010;11:1–21. doi: 10.1186/1471-2164-11-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moliner C, Fournier PE, Raoult D. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev. 2010;34:281–294. doi: 10.1111/j.1574-6976.2010.00209.x. [DOI] [PubMed] [Google Scholar]
- 99.Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, Goodhead I, et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella typhi. Nat Genet. 2008;40:987–993. doi: 10.1038/ng.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bellieny-Rabelo D, Tanui CK, Miguel N, Kwenda S, Shyntum DY, Moleleki LN. Transcriptome and comparative genomics analyses reveal new functional insights on key determinants of pathogenesis and interbacterial competition in Pectobacterium and Dickeya spp. Appl Environ Microbiol. 2019;85:e02050–e02018. doi: 10.1128/AEM.02050-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Potrykus M, Sledz W, Golanowska M, Slawiak M, Binek A, Motyka A, et al. Simultaneous detection of major blackleg and soft rot bacterial pathogens in potato by multiplex polymerase chain reaction. Ann Appl Biol. 2014;165:474–487. doi: 10.1111/aab.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hélias V, Hamon P, Huchet E, van der Wolf JM, Andrivon D. Two new effective semiselective crystal violet pectate media for isolation of Pectobacterium and Dickeya. Plant Pathol. 2012;61:339–345. [Google Scholar]
- 103.Nassar A, Darrasse A, Lemattre M, Kotoujansky A, Dervin C, Vedel R, et al. Characterization of Erwinia chrysanthemi by pectinolytic isozyme polymorphism and restriction fragment length polymorphism analysis of PCR-amplified fragments of pel genes. Appl Environ Microbiol. 1996;62:2228–2235. doi: 10.1128/aem.62.7.2228-2235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Laurila J, Hannukkala A, Nykyri J, Pasanen M, Hélias V, Garlant L, et al. Symptoms and yield reduction caused by Dickeya spp. strains isolated from potato and river water in Finland. Eur J Plant Pathol. 2010;126:249–262. [Google Scholar]
- 105.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–931. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 108.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. ANIb values calculated for the studied Dickeya solani genomes. Description of data: BLAST calculation of ANI (ANIb) was performed with the use of JSpecies [106]. The upper number refers to the ANIb value while the lower depicted in parentheses is the percentage of the aligned sequences.
Additional file 2: Table S2. ANIm values calculated for the studied Dickeya solani genomes. Description of data: MUMmer calculation of ANI (ANIm) was performed with the use of JSpecies [106]. The upper number refers to ANIm value while the lower depicted in parentheses is the percentage of the aligned sequences.
Additional file 3: Table S3. Correlation indexes of the tetra-nucleotide signatures computed for the studied Dickeya solani genomes. Description of data: Computation of the correlation indexes of the tetra-nucleotide signatures was conducted with JSpecies [106].
Additional file 4: Table S4. The functions of the unique Dickeya solani COGs. Description of data: a The first category was attributed by the BPGA v. 1.3 software. If it differed from the category currently allocated to certain COG IDs in the COG database, the up-to-date assignment after the slash mark is depicted. COG categories: A - RNA processing and modification, B - chromatin structure and dynamics, C - energy production and conversion, D - cell cycle control, cell division, chromosome partitioning, E - amino acid transport and metabolism, F - nucleotide transport and metabolism, G - carbohydrate transport and metabolism, H - coenzyme transport and metabolism, I - lipid transport and metabolism, J - translation, ribosomal structure and biogenesis, K - transcription, L - replication, recombination and repair, M - cell wall/membrane/envelope biogenesis, N - cell motility, O - post-translational modification, protein turnover and chaperones, P - inorganic ion transport and metabolism, Q - secondary metabolites biosynthesis, transport and catabolism, R - general function prediction only, S - function unknown, T - signal transduction mechanisms, U - intracellular trafficking, secretion and vesicular transport, V - defence mechanisms, W - extracellular structures, X - mobilome: prophages, transposons, Y - nuclear structure and Z – cytoskeleton.
Data Availability Statement
De novosequenced D. solani genomes.
Data generated in this whole genome sequencing project are available in the Genome database of the National Center for Biotechnology Information (NCBI) under the BioProject PRJNA611911. The analysed D. solani strains have been attributed with the following Biosample nos. SAMN14352303, SAMN14352304, SAMN14352305, SAMN14352306, SAMN14352307, SAMN14352308, SAMN14352309 and SAMN14352310. The assembled and annotated full genomic sequences of the herein de novo sequenced D. solani genomes are available in the NCBI Genome database under the following accession nos: CP051457, JABAON000000000, CP051458, JABAOO000000000, CP051459, CP051460, JABAOP000000000 and JABAOQ000000000 (Table 2).
The datasets generated and/or analyzed during this study will not be publicly available prior to first publication of the herein presented manuscript. After publication, the datasets will be available from the corresponding author on a reasonable request.
The reference D. solani genomes.
The included 14 reference D. solani genomic sequences have been downloaded in a FASTA format from the NCBI Genome database, in which they have been deposited under the following accession nos. CP024711, PENA00000000, PEMZ00000000, CP024710, AONU01000000, AONX01000000, AOOP01000000, AOOQ01000000, AMWE01000000, JWLS01000000, JWLT01000000, JWMJ01000000, JWLR01000000 and AMYI01000000 (Table 2).