

Abstract
Protozoan infections are a serious global health problem1,2. Natural killer (NK) cells and cytolytic T lymphocytes (CTLs) eliminate pathogen-infected cells by releasing cytolytic granule contents—granzyme (Gzm) proteases and the pore-forming perforin (PFN)—into the infected cell3. However, these cytotoxic molecules do not kill intracellular parasites. CD8+ CTLs protect against parasite infections in mice primarily by secreting interferon (IFN)-γ4–10. However, human, but not rodent, cytotoxic granules contain the antimicrobial peptide granulysin (GNLY), which selectively destroys cholesterol-poor microbial membranes11–14, and GNLY, PFN and Gzms rapidly kill intracellular bacteria15. Here we show that GNLY delivers Gzms into three protozoan parasites (Trypanosoma cruzi, Toxoplasma gondii and Leishmania major), in which the Gzms generate superoxide and inactivate oxidative defense enzymes to kill the parasite. PFN delivers GNLY and Gzms into infected cells, and GNLY then delivers Gzms to the intracellular parasites. Killer cell–mediated parasite death, which we term ‘microbe-programmed cell death’ or ‘microptosis’, is caspase independent but resembles mammalian apoptosis, causing mitochondrial swelling, transmembrane potential dissipation, membrane blebbing, phosphatidylserine exposure, DNA damage and chromatin condensation. GNLY-transgenic mice are protected against infection by T. cruzi and T. gondii, and survive infections that are lethal to wild-type mice. Thus, GNLY-, PFN- and Gzm-mediated elimination of intracellular protozoan parasites is an unappreciated immune defense mechanism.
Because GNLY preferentially permeabilizes microbial membranes14, we hypothesized that GNLY could deliver Gzms into parasites. We treated extracellular T. cruzi trypomastigotes, T. gondii tachyzoites or L. major promastigotes that were fluorescently labeled with mCherry or the lipophilic fluorescent dye FM4–64 with Alexa Fluor 488–labeled inactive GzmB (GzmB-488), with or without a sublytic concentration of GNLY (Fig. 1a; Supplementary Figs. 1a and 2a). By using microscopy, we showed that GzmB entered these parasites only in the presence of GNLY. We confirmed these results by analyzing ≥ 300 parasites for every treatment condition and enumerating the number of parasites showing intracellular GzmB (Supplementary Fig. 1b and Supplementary Data).
Figure 1.
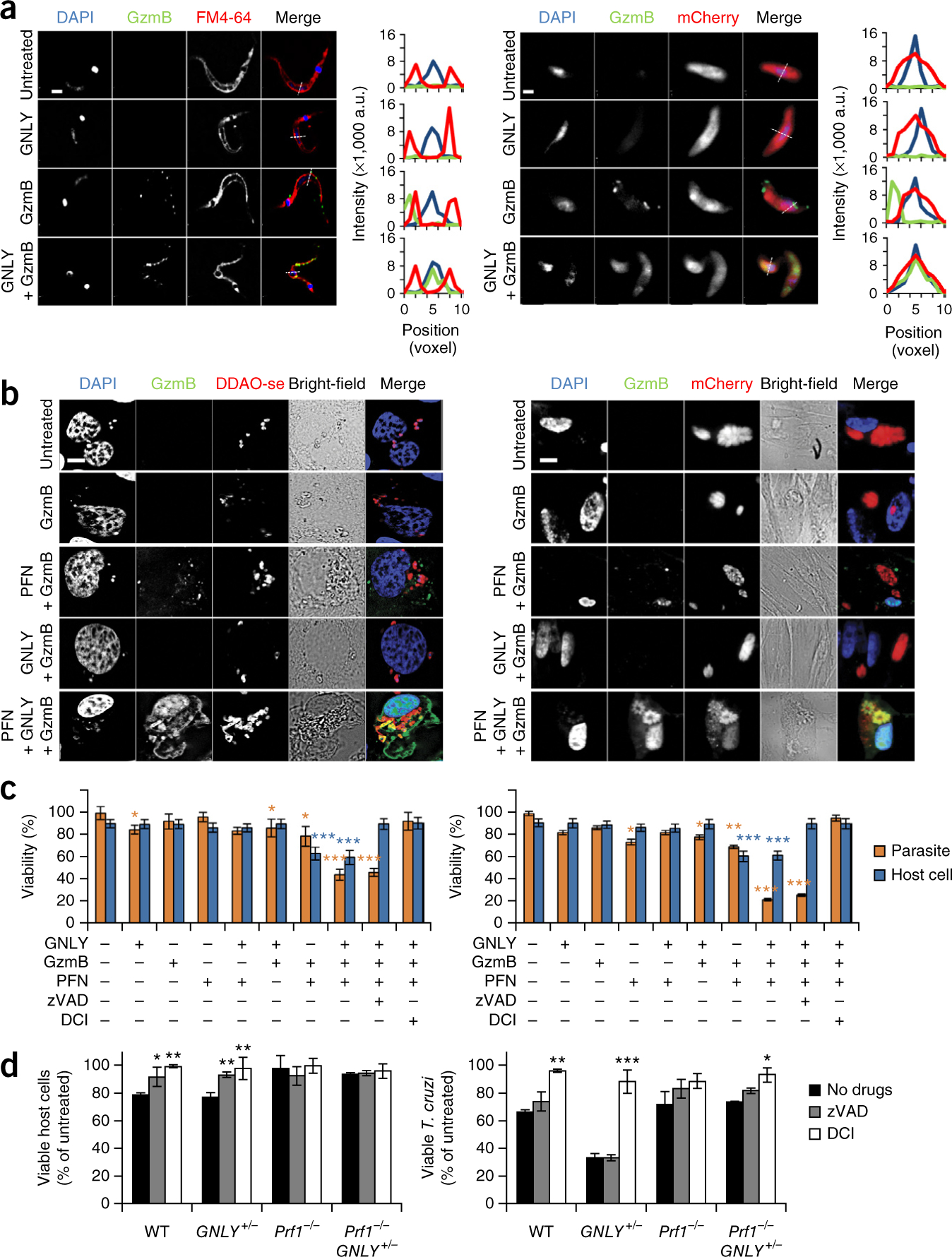
GNLY, PFN and GzmB kill parasites. (a) Representative images (of three experiments) (left) and fluorescence intensity traces along an arbitrary line (right) of FM4–64–labeled T. cruzi trypomastigotes (left) and mCherry-expressing T. gondii (RH strain) tachyzoites (right), that were treated for 30 min with sublethal amounts of GNLY and inactive GzmB-488, and then analyzed by fluorescence microscopy. Nuclei were stained with DAPI. a.u., arbitrary units. Scale bars, 1 μm. (b) Representative images (of three independent experiments) of LLC-MK2 cells infected with T. cruzi labeled with the fluorescent cell-labeling dye DDAO-se (left) and HFF cells infected with T. gondii expressing mCherry (right) that were treated with combinations of PFN, GNLY and GzmB-488. Scale bars, 10 μm. Higher-magnification images are shown in Supplementary Figure 1c. (c) Viability of intracellular T. cruzi (left) and T. gondii (right) and their respective host cells after treatment with combinations of PFN, GzmB and GNLY (parasites were assessed after 1 h of treatment; host cells were assessed after 4 h of treatment). (d) Viability of intracellular T. cruzi in anti-CD3–coated RAW 264.7 cells after a 90-min incubation with WT, GNLY+/−, Prf1−/− or Prf1−/−GNLY+/− splenocytes (effector: target ratio of 5:1). The caspase inhibitor zVAD– fmk or serine protease inhibitor DCI were added as indicated. In c,d, data are mean ± s.e.m. of three independent experiments. ***P < 0.001, **P < 0.01 *P < 0.05; one-way analysis of variance (ANOVA), as compared to untreated cells (c) or to no inhibitor (d). In c, blue asterisks represent P values of blue bars (host cell viability) and orange asterisks represent P values of orange bars (parasite viability).
To test the hypothesis that GzmB needs both PFN and GNLY to enter intracellular parasites, we added combinations of PFN, GNLY and inactive GzmB-488 to T. cruzi–infected rhesus kidney epithelial cells (LLC-MK2) and T. gondii–infected human foreskin fibroblasts (HFFs) (Fig. 1b and Supplementary Fig. 1c) and found that PFN delivered GzmB into mammalian cells but not into the parasites within these cells. GzmB only entered intracellular parasites in the presence of both PFN and GNLY.
To determine whether combinations of cytotoxic granule proteins kill intracellular parasites, we next compared host-cell viability, as assessed by [51Cr]-release assays, with survival of intracellular T. cruzi or T. gondii, as assessed by the presence of motile parasites or by plaque assays, respectively. We treated T. cruzi–infected LLC–MK2 cells and T. gondii–infected HFF cells with combinations of PFN, GNLY and GzmB (Fig. 1c and Supplementary Fig. 3a) and found that host-cell killing required PFN and GzmB and that this was not enhanced by adding GNLY. Moreover, parasite viability was significantly reduced, but only when all three effector molecules were added. The parasites were killed within 15–30 min, whereas host-cell killing commenced 45–60 min after treatment. Host-cell death was inhibited by pretreatment with the pan-caspase inhibitor zVAD–fmk (which we also refer to as zVAD) and the serine protease inhibitor 3, 4-dichloroisocoumarin (DCI; which blocks Gzm activity), whereas only DCI pretreatment inhibited parasite death. Thus, host-cell and parasite death were independent, and parasite death required a Gzm, PFN and GNLY.
To confirm the role of PFN and GNLY in killer cell–mediated elimination of intracellular parasites, we assessed the killing of anti-CD3–coated T. cruzi–infected RAW 264.7 cells (a mouse macrophage cell line) by interleukin (IL)-15–cultured splenocytes from T. cruzi–infected wild-type (WT), GNLY-transgenic (GNLY+/−)11, PFN-deficient (Prf1−/−) and Prf1−/−GNLY+/− mice (Fig. 1d and Supplementary Fig. 3b). Transgenic GNLY+/− mice were generated using a large bacterial artificial chromosome that contains the 5′ and 3′ regulatory domains of the GNLY gene11,15. As a consequence, these mice express GNLY protein at levels similar to those in humans, but only in NK cells and CTLs. Killing of the infected host cell required PFN, and this was not enhanced by the presence of GNLY; inhibiting Gzms or caspases abrogated host-cell killing (Fig. 1d). Killing of intracellular T. cruzi was greatly enhanced by GNLY and required PFN. The modestly reduced viability of T. cruzi in host cells that were attacked by GNLY-deficient killer cells was probably because dying cells do not fully support parasite growth. DCI treatment, which did not affect target-cell or killer-cell viability on its own (Supplementary Fig. 3c), blocked intracellular parasite death by GNLY+/− killer cells, but zVAD-fmk treatment had no effect. Taken together, these results indicate that Gzms, which are delivered into infected host cells by PFN and then into intracellular parasites by GNLY, kill intracellular parasites in a PFN-, GNLY- and Gzm-dependent, but caspase-independent, manner. Moreover, intracellular parasite killing occurs before, and independently of, host-cell killing, which does not require GNLY.
We next examined the morphology of GNLY- and GzmB-treated T. cruzi and T. gondii cells by using transmission electron microscopy (Fig. 2a,b). Morphological changes, which were completed within 30–60 min after GNLY and GzmB treatment, were similar in both parasites. We first observed mitochondrial swelling and loss of cristae (within 10 and 15 min, respectively). Within 30 min, chromatin condensation, nuclear fragmentation and membrane blebbing—features of mammalian cell apoptosis—were prominent. Because the GNLY- and Gzm-mediated death of bacteria and the caspase-independent CTL-mediated death of mammalian cells are initiated by superoxide-anion generation15,16, we asked whether treatment with a superoxide scavenger (such as Tiron (4,5-dihydroxybenzene-1, 3-disulfonate)) affects T. gondii and T. cruzi death. Treatment with Tiron, which did not affect GzmB, GNLY or PFN activity (Supplementary Fig. 3d–f), abrogated the apoptotic morphological changes, suggesting that parasite death caused by GNLY and GzmB is also mediated by superoxide.
Figure 2.
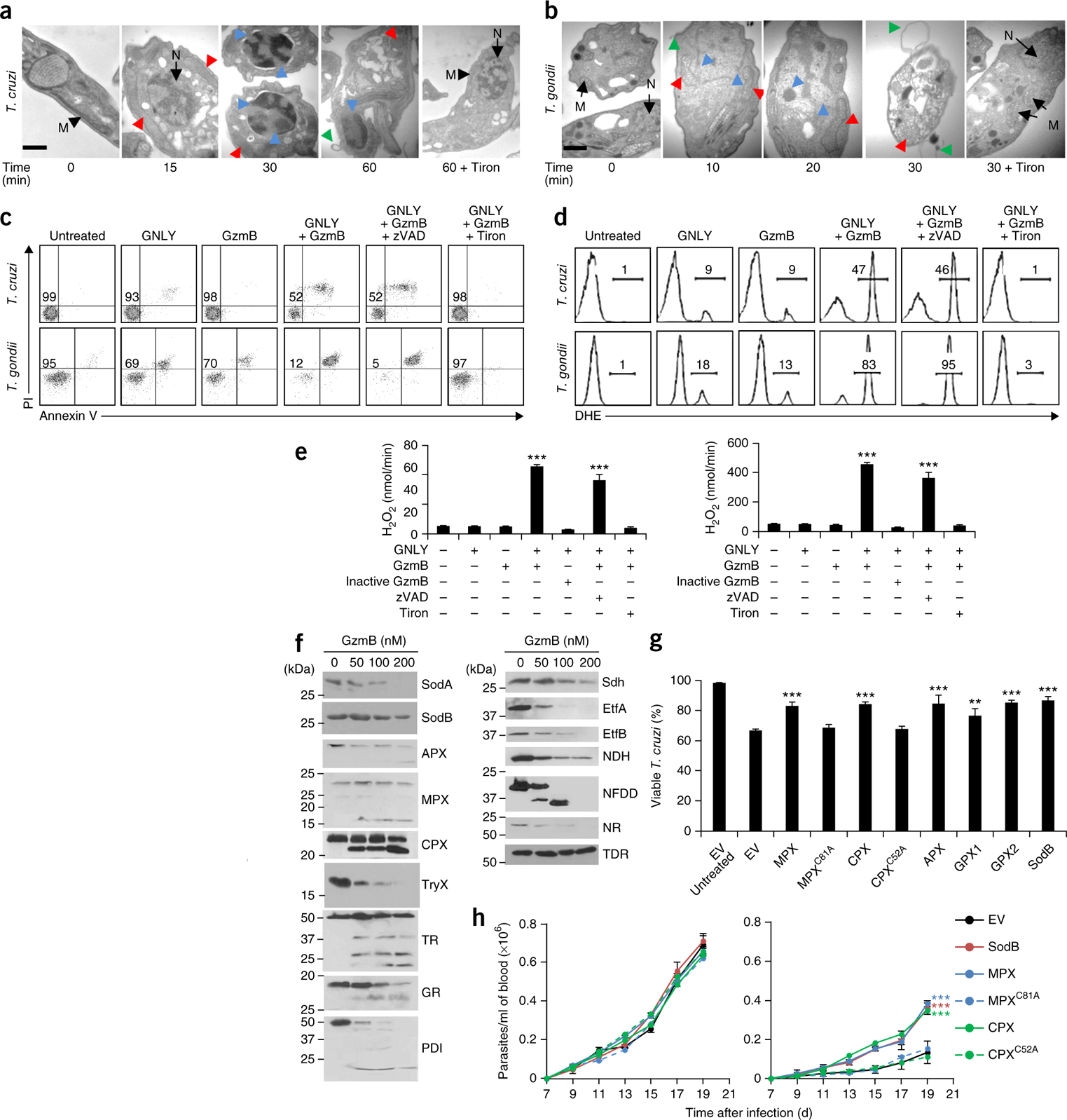
GNLY and GzmB cause oxidative damage–mediated parasite programmed cell death. (a,b) Representative (of three experiments) transmission electron micrographs of extracellular T. cruzi (a) and the T. gondii RH strain (b) after treatment with GzmB and GNLY for the indicated times. Pretreatment of parasites with Tiron is indicated. Black arrows, normal mitochondria (M) and nuclei (N); red arrowheads, swollen mitochondria with abnormal cristae; blue arrowheads, chromatin condensation and fragmented nuclei; green arrowheads, membrane blebs. Scale bars, 100 nm. (c,d) Representative flow cytometry analysis (of three experiments) for annexin V and PI staining (c) or for DHE fluorescence (d) of extracellular T. cruzi (top) and T. gondii (bottom) 30 min after the addition of various combinations of GNLY and GzmB. zVAD–fmk (zVAD) or Tiron were added as indicated. Numbers indicate percentage of viable parasites (c) and percentage of fluorescent cells (d). (e) H2O2 production in treated T. cruzi (left) and T. gondii (right) cells by Amplex Red assays. (f) Representative His6-tag immunoblot analysis (of three experiments) of the cleavage of recombinant T. cruzi proteins 30 min after the addition of the indicated amounts of GzmB. (g) Viable T. cruzi epimastigotes, transfected with an empty vector (EV) or plasmids expressing WT or inactive mutant (MPXC81A and CPXC52A) oxidative defense enzymes, 30 min after the addition of GNLY and GzmB. (h) Parasitemia in WT (left) and GNLY+/− (right) BALB/c mice (n = 5 per group) infected with T. cruzi trypomastigotes that were transfected with an EV or with plasmids expressing WT or mutant oxidative defense enzymes. In e,g, data are mean ± s.e.m. of three independent experiments. In h, data are mean ± s.e.m. of five mice in one experiment. ***P < 0.001, **P < 0.01; by one-way ANOVA relative to untreated cells (e) or to parasites transfected with EV (g,h).
We next measured superoxide-anion generation by using dihydroethidium (DHE) staining, hydrogen peroxide (H2O2) production by Amplex Red fluorescence, mitochondrial transmembrane potential (Δφ) by DilC1 (hexamethylindodicarbocyanine iodide) and JC-1 (tetraethylbenzimidazolylcarbocyanine iodide) staining, and phosphatidylserine exposure and outer membrane integrity by annexin V and propidium iodide (PI) staining, respectively, in GNLY- and GzmB-treated T. cruzi trypomastigotes, T. gondii tachyzoites and L. major promastigotes (Fig. 2c–e; Supplementary Figs. 2b,c and 4a–c). Without treatment, these obligate parasites survived extracellular conditions for more than the 30- to 60-min incubation times used for these assays, although T. gondii was somewhat more fragile than the other parasites. Addition of GNLY or GzmB on its own had modest effects on production of reactive oxygen species (ROS), annexin V staining or cell membrane integrity. However, addition of GzmB and GNLY together generated superoxide anions within mitochondria (as verified by Mitosox fluorescence) and caused loss of Δφ and of annexin V and PI staining. Superoxide is rapidly converted to peroxide by superoxide dismutases (SODs). We observed that treatment of all three parasites with GNLY and GzmB produced H2O2 and that death required active GzmB (Fig. 2e; Supplementary Figs. 2c and 4a,b). GzmA could substitute for GzmB in these in vitro assays, suggesting that multiple Gzms can kill parasites (Supplementary Fig. 4c and data not shown). Incubation with the ROS scavengers Tiron, MnTBAP, Trolox (TRX), mannitol, N-acetylcysteine (NAC), dimethylthiourea (DMTU) and desferroxamine mesylate (DFO) inhibited T. cruzi death, indicating the importance of ROS in initiating parasite death (Supplementary Fig. 4d).
ROS generation was the earliest event, occurring within 5–10 min of adding GzmB and GNLY to extracellular parasites (Supplementary Fig. 5). It was followed within ~5 min by loss of Δφ. Annexin V and PI staining occurred within 45 min in T. cruzi and L. major. The same order of death-related events occurs during mammalian cell apoptosis. The early detection of superoxide anions coupled with the inhibition of death by a superoxide scavenger (Supplementary Fig. 4c,d) suggests that superoxide has a critical role in parasite killing.
The presence of GzmB and GNLY also activated metacaspases and caspase-like activity, as assessed by the fluorescent indicators Ac-VRPR-AMC and CaspaTag, respectively (Supplementary Fig. 6a,b). Treatment with staurosporine, a kinase inhibitor that induces mammalian cell apoptosis, also activated caspase-like activity in all three parasites. Activation of a caspase sensor was unexpected as the parasites are not known to express caspases. However, treatment with the pan-caspase inhibitor zVAD-fmk had no effect on parasite death.
ROS oxidize DNA to form abasic sites, which can be detected by 8-oxoguanine staining. GzmB- and GNLY-treated T. cruzi and L. major both showed increased levels of 8-oxoguanine by flow cytometry and fluorescence microscopy (Supplementary Fig. 6c,d). However, we did not detect DNA nicking or fragmentation, characteristic features of mammalian cell apoptosis, by TUNEL assays (data not shown), even when the assays were performed 16–24 h after adding GzmB and GNLY. Thus, treatment with GzmB causes oxidative DNA damage, but not DNA fragmentation, in parasites.
T. cruzi’s oxidative defense system uses both evolutionarily conserved enzymes and its own unique enzymes (Supplementary Fig. 7). To identify potential GzmB substrates, we performed differential proteomics analyses of T. cruzi lysates treated with active or inactive GzmB (F.D. and J.L., unpublished data). Proteins involved in oxidative defense or oxidoreduction reactions were overrepresented among the hits. Nanomolar concentrations of GzmB were incubated for 30 min with nine individual purified, recombinant His-tagged anti-oxidant substrate candidates—the superoxide dismutases SodA and SodB, the ascorbate-dependent, mitochondrial and cytoplasmic tryparedoxin peroxidases (APX, MPX, CPX), tryparedoxin (TryX), trypanothione reductase (TR), glutathione reductase (GR) and protein disulfide isomerase (PDI). All nine were cleaved, as assessed by reduced levels of full-length protein or the appearance of a cleavage fragment on immunoblots (Fig. 2f, left). T. cruzi has no well-defined electron transport chain (ETC) complex I. To identify potential sources of superoxide anions, we also assessed the cleavage of seven T. cruzi oxidoreductive enzymes that were hits in our proteomics analysis—succinate dehydrogenase (Sdh), the electron transfer flavoproteins EtfA and EtfB, NAD-FAD–dependent dehydrogenase (NFDD), NADP-dependent oxidoreductase (NDH), nitrate reductase (NR) and thiol-dependent reductase (TDR). Except for TDR, all of the proteins were cleaved (Fig. 2f, right). Thus, GzmB both induces generation of superoxide anions and destroys parasite mechanisms to detoxify it. To assess the importance of crippling oxidative defense in parasite killing, we treated T. cruzi epimastigotes overexpressing SodB, APX, MPX, CPX or a glutathione peroxidase (GPX1 or GPX2)17–20 with GzmB and GNLY and found that overexpression of any of these enzymes, but not their inactive mutants, significantly rescued T. cruzi from death both in vitro (Fig. 2g) and in vivo (Fig. 2h).
Although many mammals express a GNLY gene, rodents do not. To assess GNLY’s role in immune protection, we challenged WT or Prf1−/− BALB/c mice, which either expressed or did not express GNLY, with a dose of the CL-Brener T. cruzi strain that causes cardiac infection and inflammation, and kills most WT animals. Although WT mice developed progressive parasitemia, and 9 of 12 (75%) mice died between 21 d and 29 d after infection (Fig. 3a,b), GNLY+/− mice not only had dramatically reduced parasitemia, but also were able to survive infection with the parasite—only 2 of 13 (15%) GNLY+/− mice died. PFN-deficient mice had significantly (P < 0.01) higher levels of parasitemia than WT mice, and all of these mice died within 21 d. We compared cardiac parasite load and inflammation on day 18 and found that the mean cardiac parasite load of WT mice was 70 times higher than that in GNLY+/− mice, which also had less cardiac inflammation and cell necrosis (Fig. 3c,d). PFN-deficient mice, in the presence or absence of GNLY, had about a logfold more parasites in their hearts and more tissue damage. Three WT and 11 GNLY+/− mice that survived to day 40 had no detectable cardiac infection. Thus, both PFN and GNLY contribute to protection from T. cruzi infection, and PFN is essential for GNLY activity in vivo.
Figure 3.
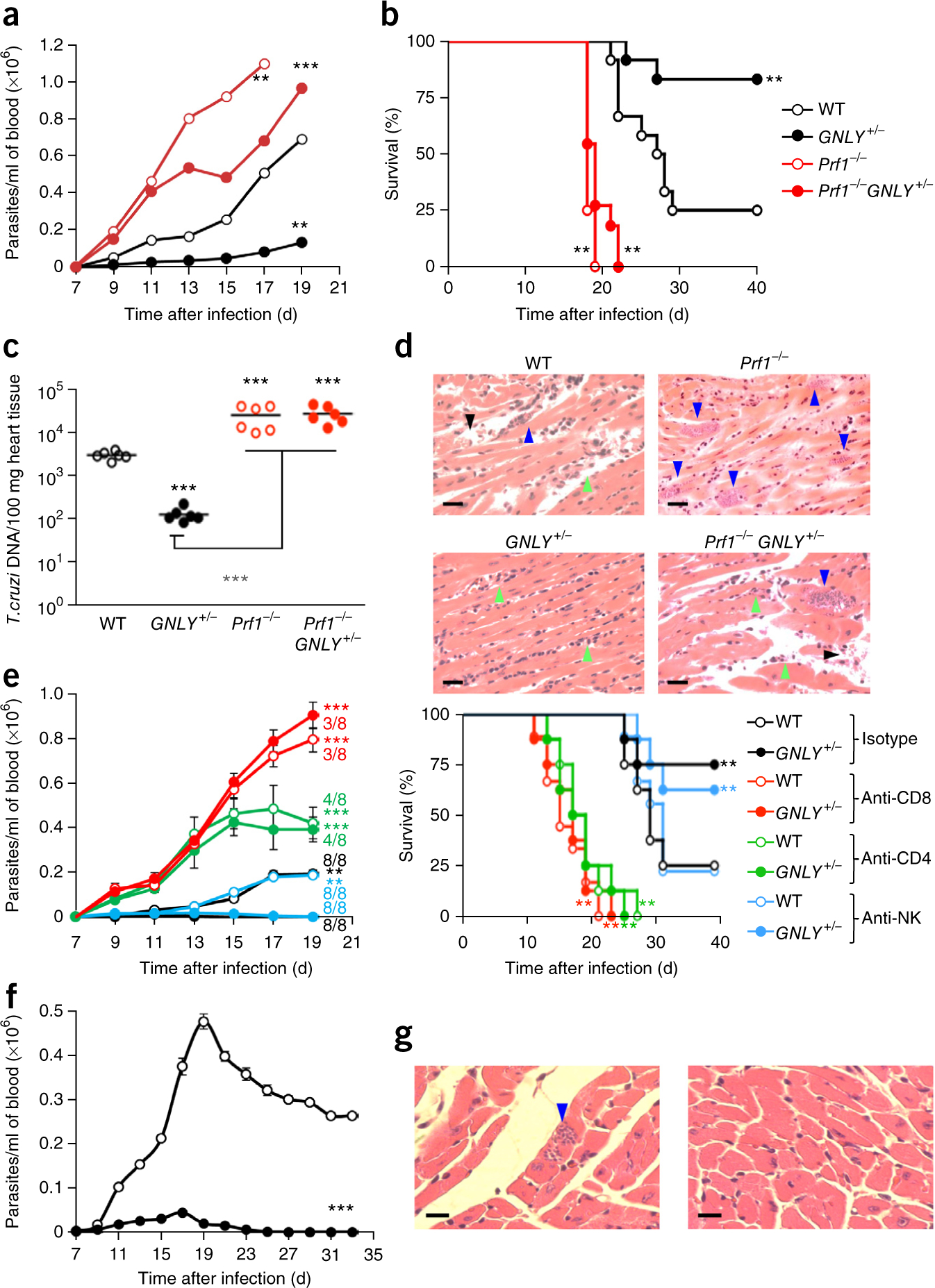
GNLY+/− mice are more resistant to T. cruzi infection than WT mice. (a,b) Parasitemia levels (a) and survival (b) of BALB/c WT (n = 12), GNLY+/− (n = 13), Prf1−/− (n = 12) and Prf1−/−GNLY+/− (n = 12) mice infected with T. cruzi. (c,d) Cardiac parasite load (measured as T. cruzi DNA relative to mouse DNA, using PCR) (c) and representative H&E-stained images of cardiac tissue (d) 18 d after T. cruzi infection of the indicated mice (n = 6 per group). (d) Blue arrowheads, parasite-infested muscle cells; green arrowheads, infiltrating inflammatory cells; black arrowheads, necrotic muscle cells. Scale bars, 20 μm. (e) Parasitemia levels (left) and survival (right) of T. cruzi–infected WT and GNLY+/− mice that had been treated with antibodies to deplete CD8+ T cells, CD4+ T cells or NK cells or with a control antibody (n = 8 per group). Numbers denote the mice that survived in each group 19 d after infection. (f,g) Parasitemia levels (n = 10 per group) (f) and representative images (n = 4 per group) of cardiac parasites by H&E staining at the time of killing (g) of WT and GNLY+/− C57BL/6J mice infected with T. cruzi. A few parasites were seen in cardiac tissue from WT mice (g, left; blue arrow) but none were seen in tissue from GNLY+/− mice (g, right). Scale bars, 20 μm. Error bars represent means ± s.e.m. ***P < 0.001, **P < 0.01; by unpaired Student’s t-test, relative to WT mice (a,e, left; f), or by one-way ANOVA (b,c,e, right).
To examine whether enhanced oxidative defenses would increase T. cruzi’s resistance to treatment with GzmB and GNLY, we compared parasitemia levels in WT and GNLY+/− BALB/c mice infected with parasites that were transfected with an empty vector (EV) or that overexpressed SodB or catalytically active or inactive MPX or CPX (Fig. 2h). WT mice infected with EV-containing T. cruzi had ~7-fold higher parasitemia levels than GNLY+/− mice, confirming that GNLY helps control infection. Overexpression of the active, but not the inactive, oxidative defense enzymes significantly increased parasitemia levels (~4-fold) in GNLY+/− mice but did not alter parasitemia levels in WT mice. Reinforcing the parasite oxidative defenses protected T. cruzi from treatment with GNLY and GzmB, but there was no effect in the absence of GNLY, suggesting that proteolytic disruption of the parasite’s oxidative defenses by GNLY-mediated delivery of granzymes promotes parasite death.
To determine which lymphocyte subsets contribute to the enhanced resistance of GNLY+/− mice to parasite infection, we infected WT and GNLY+/− mice after treatment with a control antibody or with antibodies to specifically deplete CD8+ or CD4+ T cells or NK cells (Fig. 3e and Supplementary Fig. 8a). Control antibody–treated GNLY+/− mice had significantly reduced parasitemia levels and showed enhanced survival, as compared to WT mice. NK cell depletion had no significant effect on parasitemia levels or survival, suggesting that NK cells do not contribute significantly to protection. CD8+ T cell–depleted mice fared the worst–they all died within 23 d and their parasitemia levels were as high as those in Prf1−/− mice (Fig. 3a). The parasitemia levels in CD4+ T cell–depleted mice were intermediate to those of WT and CD8+ T cell–depleted mice, and all of the CD4+ T cell–depleted mice died within a few days of the CD8+ T cell–depleted mice. GNLY did not provide a significant survival benefit in either CD4+ T cell– or CD8+ T cell–depleted mice, suggesting that cooperation between CD4+ and CD8+ T cells is needed for GNLY-mediated protection. Cardiac parasite load and histopathology tracked parasitemia levels and survival (Supplementary Fig. 8b,c). In control and NK cell–depleted mice, cardiac infection was reduced by ~60-fold in GNLY+/− mice, as compared to WT mice. NK cell depletion had no significant effect on heart infection. Cardiac infection in CD4+ T cell– or CD8+ T cell–depleted mice resembled that in Prf1−/− mice, and this was not reduced by GNLY. Thus, although CD8+ T cells mostly mediated the protective effect of GNLY, they required CD4+ T cell help.
BALB/c mice are more susceptible to T. cruzi infection than C57BL/6J mice, which develop a stronger IFN-γ response. To verify that the protection afforded by GNLY was not specific to BALB/c mice, we compared T. cruzi infection in WT and GNLY+/− C57BL/6J mice (Fig. 3f,g). As expected, C57BL/6J mice were more resistant to T. cruzi infection than BALB/c mice. Only one of ten WT mice died and no GNLY+/− mice died. Peak parasitemia levels were tenfold higher in WT mice than in GNLY+/− mice. Whereas all of the GNLY+/− mice cleared the parasitemia within 25 d, all of the WT mice had detectable parasitemia at the time of sacrifice on day 33. Thus, GNLY helps to control T. cruzi infection in both backgrounds, although higher levels of IFN-γ may partially compensate for the absence of GNLY in C57BL/6J mice.
To assess the in vivo significance of GNLY against infection by T. gondii, we challenged WT and GNLY+/− mice, in both the BALB/c and C57BL/6J backgrounds, with the type II (Pru strain) of T. gondii (transfected to express luciferase for live-animal imaging), using a parasite dosage that kills most WT animals. The Pru strain killed all of the WT mice by day 16 after infection, whereas all of the GNLY+/− C57BL/6J mice and four of five BALB/c mice survived and cleared the parasitemia, as measured by in vivo imaging and plaque assays (Fig. 4). Thus, GNLY strongly contributes to protection against T. gondii infection in both mouse backgrounds.
Figure 4.
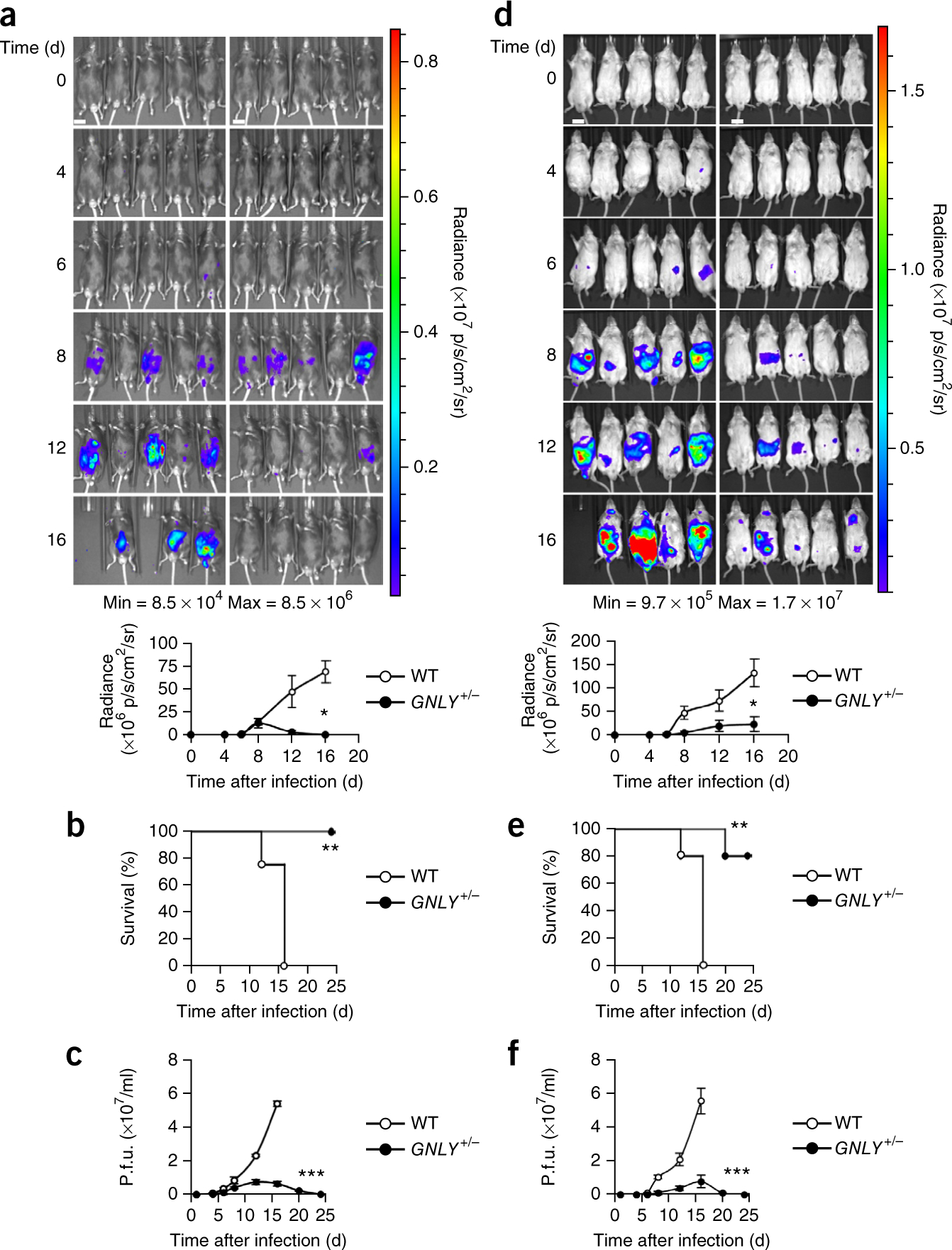
GNLY+/− mice are more resistant to T. gondii infection than WT mice. (a–f) Live-animal imaging analysis (WT mice, left; GNLY+/− mice, right; graph below depicts average whole-animal radiance) (a,d), survival curves (b,e) and quantification of plaque assays (c,f) of WT and GNLY+/− C57BL/6J (a–c) and BALB/c (d–f) mice infected with luciferase-expressing type II T. gondii (Pru strain) (n = 5 per group for each experiment). Scale bars, 1 cm. Error bars in a,c,d,f represent mean ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05; by unpaired Student’s t-test (a,c,d,f) or by one-way ANOVA (b,e).
Here we showed that the cytotoxic granule effectors Gzm, PFN and GNLY work together to kill three protozoan parasites that cause human disease. PFN and GNLY disrupt host cell and parasite membranes, respectively, to deliver the Gzms into intracellular parasites, where they proteolytically act to generate ROS and dismantle parasite oxidative defenses. Scavenging superoxide anions, or overexpressing parasite superoxide dismutase or peroxidases—both in vitro and in vivo—inhibited parasite death, indicating the importance of oxidative damage in parasite killing. Parasite mitochondria generated superoxide anions, a step that was rapidly followed by loss of mitochondrial Δφ and later by the induction of classical features of mammalian cell apoptosis within the parasite. Parasite metacaspase and caspase-like activities were activated but neither was needed for death. Protozoan DNA was oxidized but did not undergo double-stranded breaks. Death of the intracellular parasites occurred independently of, and before, host-cell death, which should limit parasite spreading. A GNLY transgene enabled mice to survive parasite challenges that were lethal to nontransgenic WT mice. Because of the dramatic increase in protection afforded by GNLY, the relevance to humans of previous mouse studies that concluded that IFN-γ secretion, rather than cytotoxicity, is the key protective function of CD8+ T cells in T. cruzi infection needs to be re-evaluated.
On the basis of the broad activity of GNLY seen in cholesterol-poor membranes, killer cell–activated microptosis may be helpful in protection against other protozoan parasites (such as that causing malaria), multicellular parasites (helminths) and fungi. Elucidating the shared and distinct features of microptosis in parasites and other pathogens may identify novel antimicrobial drug targets. In preliminary studies we found that Chagas disease patients but not healthy donors have circulating T. cruzi–specific CD8+ T cells that express GNLY, GzmB and PFN (data not shown), suggesting that this immune response may be important in human disease. However, in patients who are chronically infested with high parasite burdens, develop the severe forms of Chagas disease or are co-infected with HIV, there is a good chance that this protective response is impaired and that anti-parasite killer cells may not express GNLY and PFN. Further studies of the roles of GNLY, GzmB and PFN in defense against human parasite infection should prove interesting.
ONLINE METHODS
Parasite strains and plasmids.
Parasites used were Toxoplasma gondii RH strain (WT or expressing mCherry or YFP), Toxoplasma gondii Pru strain expressing luciferase (provided by M.J. Gubbels, Boston College), Leishmania major (WT or expressing RFP or GFP; provided by K. Okuda, UMass Medical School) and Trypanosoma cruzi CL-Brenner strain. T. cruzi plasmids overexpressing the indicated oxidative stress response genes were obtained from S. Wilkinson (Queen Mary University of London) and J. Kelly (London School of Hygiene & Tropical Medicine). T. gondii was cultured in infected HFF cells (American Type Culture Collection (ATCC)) grown in 1% Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) at 37 °C and tachyzoites were separated from host cells 2–3 d after infection by differential centrifugation as described21. T. cruzi were cultured in LLC-MK2 cells (ATCC) grown in 2% DMEM at 37 °C, and trypomastigotes were collected from the supernatant 5–6 d after infection as described22. T. cruzi epimastigotes were cultured in liver-infused tryptose (LIT) medium (BD Biosciences) at 27 °C. L. major promastigotes were grown in M199 medium (Sigma-Aldrich) at 27 °C as described23.
PFN, GNLY and Gzms.
Native human PFN, GNLY (a mixture of the unprocessed 15-kDa and the processed active 9-kDa isoforms) and GzmB were purified from YT-Indy cells24 (obtained from Z. Brahmi, Indiana University School of Medicine). Recombinant human GzmA and GzmB and inactive serine-to-alanine (S-A) mutants were produced in 293T cells (ATCC) as previously described25. For each batch of purified protein, the sublytic concentration of GNLY or PFN was determined for each parasite or host cell, respectively, as the concentration that led to PI staining of 10–20% of cells after 30–60 min. Because the sublytic concentration varies between batches, it was determined for each enzyme preparation. For PFN, the sublytic concentration ranges from 1–100 nM for 104 mammalian cells; for GNLY the range is 10–100 nM for 105 parasites. GzmB was fluorescently labeled using the Alexa Fluor 488 (AF488) Microscale Protein Labeling Kit (Invitrogen) following the manufacturer’s protocols. Unless otherwise indicated, Gzms were used at a final concentration of 200 nM.
Parasite infection.
RAW 264.7 and LLC-MK2 (ATCC) were used as host cells for T. cruzi, and HFF cells (ATCC) were used as host cells for T. gondii. Host cells were cultured in K10 (DMEM supplemented with 10% heat-inactivated pooled FBS (FBS) (Gemini Bioproducts), 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate, 6 mM HEPES, 1.6 mM l-glutamine, 50 μM β-mercaptoethanol (Sigma-Aldrich)). Cells were seeded at 104–105 cells/well in 96-well plates for in vitro killing experiments or per slide for microscopy and used at ~50% confluence. HFF cells were infected by incubation at 37 °C for 4 h with T. gondii (multiplicity of infection (MOI) = 1) in DMEM containing 1% FBS. RAW 264.7 and LLC-MK2 cells were incubated at 37 °C overnight with T. cruzi (MOI = 30) in DMEM containing 2% FBS. Infected adherent cells were washed three times with medium to remove extracellular parasites.
Imaging GzmB delivery.
For extracellular parasite analyses, T. gondii tachyzoites expressing mCherry, T. cruzi trypomastigotes or L. major promastigotes were treated with 500 nM AF488-labeled inactive GzmB ± sublytic GNLY for 1 h at 37 °C and washed with 10 mM arginine in PBS for 10 min before fixation in 2% formalin in PBS and staining of T. cruzi and L. major cell membranes with FM4–64. For intracellular parasite analyses, HFF cells were infected with mCherry-expressing T. gondii or LLC-MK2 cells with DDAO-se (Invitrogen)–labeled T. cruzi as above. Infected host cells (105 per sample) were treated in 200 μl Hank’s balanced salt solution (HBSS) for 1 h at 37 °C with 500 nM AF488-labeled inactive GzmB ± sublethal amounts of unlabeled GNLY ± sublytic amounts of unlabeled PFN. The slides were washed with 10 mM arginine in PBS for 10 min, fixed in 2% formalin in PBS, and then stained as above. Slides were mounted with VectaShield with DAPI (Vector Labs) and imaged by confocal microscopy using an inverted, fully motorized Axio Observer spinning disk confocal microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) equipped with a cooled electron multiplication charge-coupled device (CCD) camera with 512 × 512 resolution (QuantEM, Photometics, Tuscon, AZ) and a CSU-X1 spinning disc (Yokogawa Electric, Tokyo, Japan) with lasers that were excited at 405, 488, 561 and 640 nm (Coherent, Santa Clara, CA) and emission filter ranges of 452/45, 525/50, 607/36 and 680 long-pass, respectively (Semrock, Rochester, NY). Images were analyzed using SlideBook V5.0 (Intelligent Imaging Inc., Denver, CO) software. Three-dimensional (3D) image stacks were obtained along the z axis using the 63× oil immersion objective by acquiring sequential optical planes spaced 0.25 μm apart. Raw images were deconvolved using SlideBook. To quantify GzmB internalization, 300–500 parasites were assessed per treatment condition.
Parasite treatment with cytotoxic proteins.
Extracellular T. gondii tachyzoites or T. cruzi trypomastigotes and amastigotes, and L. major promastigotes (105) or parasite-infected cells (infected 2–3 d earlier) were incubated with Gzms ± sublytic amounts of GNLY ± sublytic amounts of PFN at 37 °C in 50 μl of 50% PBS diluted in water for the indicated times. Reactions were stopped by diluting with 100–200 μl DMEM with 10% FBS. Host-cell viability was assessed by a 4-h [51Cr]-release assay as described24. Parasite viability was measured 2–3 d after treatment by plaque assays for T. gondii as described21 and by counting motile T. cruzi cells that emerge from infected host cells. For annexin V and PI staining, cells that were incubated with annexin V–FITC for 10 min in the dark at room temperature and then with PI (100 ng/ml) on ice for 5 min were analyzed by flow cytometry.
Mouse strains.
GNLY+/− C57BL/6J mice11 were re-derived at Jackson Laboratory and backcrossed for 14–16 generations into the BALB/c background. These were bred with Prf1−/− mice (Jackson Laboratory). Experiments were performed using 6- to 9-week-old mice that were randomized as to gender in each group with approval of the Animal Care and Use Committees of Boston Children’s Hospital and Harvard Medical School. The numbers of mice used in each experiment varied and are indicated in the figure legends.
Parasite killing by cytotoxic T cells.
To assess killing of intracellular parasites, splenocytes from T. cruzi–infected WT, GNLY+/−, Prf1−/− and Prf1−/−GNLY+/− mice were cultured for 5 d in RPMI1040 medium (GIBCO) that contained 10% FBS and 50 ng/ml IL-15. RAW 264.7 target cells (104/well in 96-well plates), infected with T. cruzi 2–3 d before, were incubated overnight in 2% DMEM before adding 5 μg/ml anti–mouse CD3ε for 15 min (clone 145–2C11, eBioscience). Cultured splenocytes were then added at an effector:target (E:T) ratio of 5:1 for the indicated times. In wells used to measure parasite viability, the splenocytes were washed with 10% FBS DMEM after the indicated times. In some wells splenocytes were preincubated with 250 μM DCI for 30 min at 37 °C and washed with PBS. In other wells, 75 μM zVAD-fmk was added during the killing assay. Host-cell viability was assessed by a 4-h [51Cr]-release assay, and T. cruzi viability was measured by counting motile parasites released 2–3 d after adding splenocytes.
Plaque-forming assay for T. gondii.
6-well plates with confluent HFF cells were inoculated with tenfold serial dilutions of extracellular tachyzoites or blood from infected mice. Uninfected wells or wells infected by a fixed, known number of T. gondii tachyzoites served as controls. The 6-well plates were kept undisturbed for 6–8 d and plaque growth was assessed. Sample wells were washed with PBS, fixed for 5 min in 100% methanol, stained in crystal violet (5× stock: 25 g crystal violet, 250 ml ethanol, 1% ammonium oxalate, 750 ml H2O) and air-dried. Parasite plaques were counted as clear areas against the violet background of confluent HFF cells.
Transmission electron microscopy.
Parasites (T. gondii and T. cruzi) treated with GNLY and GzmB, in the presence or absence of 10 mM Tiron for the indicated times, were fixed for 16–24 h at 4 °C in 2.5% glutaraldehyde, 2% paraformaldehyde in 100 mM cacodylate buffer (pH 7.0) containing 2 mM CaCl2 and 0.2% picric acid. Samples were briefly washed with 200 mM cacodylate buffer (pH 7.0) and then treated for 2 h at 4 °C with 1% osmium tetroxide in 100 mM cacodylate buffer (pH 7.0). After washing with distilled water 3–5 times, samples were stained with 2% aqueous uranyl acetate (Sigma-Aldrich) for 2 h at 4 °C in the dark, dehydrated using increasing ethanol concentrations and then embedded in Epon resin (Sigma-Aldrich). Ultrathin sections of the embedded samples were cut and loaded onto grids and stained further with uranyl acetate for 15–30 min and then with Reynold’s lead citrate (Sigma-Aldrich) for 3–15 min. Grids were dried overnight and observed using a JEOL 1200EX transmission electron microscope equipped with an AMT 2k CCD camera.
ROS and mitochondrial membrane potential assays.
Superoxide-anion generation was monitored by diluting treated parasites 1/50 into PBS containing 2 μM dihydroethidium (DHE) just before flow cytometry. Mitosox (Invitrogen) staining, performed following the manufacturer’s protocol, was used to measure mitochondrial superoxide levels. Mitochondrial membrane potential was measured using DilC1 and JC-1 (Life Technologies) by incubating treated parasites with each dye for 15 min at 37 °C and measuring fluorescence using flow cytometry. H2O2 production was measured as previously described15. Briefly, 105 cells, treated for 5 min, were diluted tenfold in 50% PBS containing 25 μg/ml Amplex UltraRed fluorescent dye (Invitrogen) and 5 U/ml horse-radish peroxidase (Sigma). Control wells contained no parasites. ROS scavengers (manganese(III)-tetrakis(4-benzoic acid)porphyrin (MnTBAP, 0.25 mM, Calbiochem), 4,5-dihydroxybenzene-1,3-disulfonate (Tiron, 10 mM, Sigma), 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox, 0.5 mM, Sigma), mannitol 100 mM, N-acetylcysteine (NAC, 100 mM, Sigma), N,N′-dimethylthiourea (100 mM, Sigma), desferoxamine (1 mM, Sigma)), zVAD-fmk (75 μM) or an equal volume of PBS was added to some wells 5 min before treatment. Stock solutions of the ROS scavengers were buffered to maintain a physiological pH of treated cells as follows: MnTBAP (25 mM in 75 mM NaOH pH 7.5), Tiron (1 M in 1 M Tris pH 7.5), Trolox (10 mM in 5% ethanol, pH adjusted to 7.5 using 75 mM NaOH), mannitol (1 M in ddH2O, pH adjusted to 7.5 using 1 M Tris pH 7.5), N-acetylcysteine (1 M NAC in 1.5 M Tris pH 8.8, which adjusts final pH to 7.5), N,N′-dimethylthiourea (1 M in 1 M Tris pH 7.5), desferoxamine (100 mM in 100 mM Tris pH 7.5). Fluorescence was measured every minute for 1 h using the 540-nm and 620-nm wavelength pair using a Synergy H4 Hybrid Multi-Mode Microplate Reader and calibrated to wells containing H2O2 in final concentrations between 0.1 and 100 μM to calculate the rate of H2O2 production.
Detection of caspase and metacaspase activity.
Parasites were treated with GzmB ± sublytic amounts of GNLY for 30 min at 37 °C before measuring caspase-like activity using the CaspaTag Pan-Caspase In situ Assay Kit (Millipore). Treated parasites were incubated with CaspaTag reagent for 1 h at 25 °C and then fixed and analyzed by confocal microscopy. To detect metacaspase activity, treated parasites were incubated at 37 °C with the fluorogenic metacaspase substrate valine-arginine-proline-arginine–7-amido-4-methylcoumarin (VRPR-AMC) (Bachem) and fluorescence was measured every minute for 2 h at 37 °C using the 360-nm and 460-nm wavelength pair in a Synergy H4 Hybrid Multi-Mode Microplate Reader.
Oxidative DNA damage assays.
Oxidative DNA damage was measured in parasites treated for 1 h at 37 °C with GNLY and/or GzmB, 2 mM arsenite or 0.1 mM H2O2 using the OxyDNA Test kit (EKF Diagnostics) according to the manufacturer’s directions. Briefly, treated parasites were fixed, permeabilized, and incubated in the dark with oxyDNA-FITC conjugate for 1 h at room temperature, and fluorescence was assessed by flow cytometry.
Gzm cleavage.
T. cruzi genes, cloned into pET21a, were expressed with a C-terminal His6 tag in BL21-DE3 Escherichia coli grown in Luria broth after induction with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 1 h. The tagged proteins were purified using Ni-NTA Superflow beads (Qiagen). Purified proteins were diluted 1:100 in 20 mM NaCl, 10 mM Tris, pH 7.5 before adding GzmB at the indicated concentrations. Reactions were stopped after 30 min by boiling in SDS-PAGE loading buffer. Samples were analyzed by immunoblot using anti–6-His mouse monoclonal antibody (Covance, MMS-156P).
Rescue of GzmB-mediated parasite cell death.
Expression plasmids (pTEX backbone) encoding the indicated oxidative defense enzymes or their catalytically inactive mutants, or an empty vector control plasmid was transfected into T. cruzi epimastigotes using Amaxa Human T cell solution and Program U-33, according to the manufacturer’s protocol. Stably transfected parasites, selected for 4–6 weeks in LIT media containing 100 μg/ml G418, were treated with GzmB ± sublytic amounts of GNLY at 37 °C for 30 min in 200 μl of PBS diluted to 50% in water and assessed by annexin V and PI staining.
T. cruzi infection of mice.
T. cruzi were passaged in BALB/c mice. Parasitemia was assessed by counting motile trypomastigotes every 2 d to determine peak parasitemia (14–21 d). Blood parasites were collected in PBS containing 0.5 IU/ml heparin at the time of peak parasitemia. T. cruzi parasites (5,000/mouse) were injected intraperitoneally (i.p.) in 0.5 ml PBS in the indicated mouse strains. Both male and female mice were selected randomly in each group, and the studies were done without blinding the investigators to group allocation. Mice were followed for 40 d for survival and parasitemia, measured every 2 d beginning on day 3, using heparinized blood collected from nicked tails26. In additional experiments mice were sacrificed on day 18 after infection to assess cardiac infection and inflammation. Parasite cardiac load was assessed by PCR for T. cruzi mucin-associated surface protein (MASP) pseudogene DNA (using primers czF: 5′-GCTCTTGCCCACACGGGTGC-3′ and TczR: 5′-CCAAGCAGCGGATAGTTCAGG-3′), normalized to mouse Ifnb1 (encoding interferon (IFN)-β) gene DNA (IFN-beta-F 5′-CTTCTC CACCACAGCCCTCTC-3′ and IFN-beta-R 5′-CCCACGTCAATCTTTCCT CTT-3′). Fixed heart sections were also analyzed by H&E staining and light microscopy. WT and GNLY+/− BALB/c mice were also infected with T. cruzi trypomastigotes with plasmids overexpressing SodB, MPX, CPX or their enzymatically inactive (cysteine-to-alanine) mutants, or with the empty vector (EV). Parasitemia was measured every 2 d beginning on day 3, using heparinized blood collected from nicked tails. An equal number of male and female mice were used in each group of each experiment.
T. gondii infection of mice.
Luciferase-expressing T. gondii type II (Pru strain) cells were passaged in HFF cells. C57BL/6J or BALB/c mice were infected intraperitoneally with 104 Pru parasites. Infection was assessed daily by injecting 1.5 mg d-luciferin intravenously and imaging the mice using the IVIS Spectrum. Mice were assessed for survival and parasitemia, measured daily (by plaque assays, as described above) beginning on day 3, using heparinized blood collected from nicked tails. An equal number of male and female mice were used in each group of each experiment.
Infection of NK cell– and T cell–depleted mice.
WT or GNLY+/− BALB/c mice were injected intraperitoneally with 0.5 mg/mouse of rat anti–mouse CD4 mAb (clone GK 1.5, BioXCell) to deplete CD4 T cells, 0.5 mg/mouse of rat anti–mouse CD8a mAb (clone 2.43, BioXCell) to deplete CD8 T cells, or 0.2 mg/mouse of rabbit anti–mouse asialo-GM1 (Wako Chemicals) to deplete NK cells. Control mice were treated with 0.2 mg/mouse of rat anti-KLH IgG (clone LTF-2, BioXCell). Antibodies were administered by i.p. 3, 2 and 1 d before infection and then every 7 d thereafter. Depletion was verified 1 d before infection.
Statistical analysis.
All values presented are the mean ± s.e.m. of three independent experiments. Statistical analysis was done using Microsoft Excel or Prism 5.0c (GraphPad). One-way analysis of variance (ANOVA) was performed, as specified in the figure legends, in experiments for which multiple comparisons were made; otherwise, unpaired Student’s t-tests were used. P < 0.05 was required for significance. A chi-squared test was used to analyze the data in Supplementary Figure 1b. Data were normally distributed with similar variances between the groups. In all of the experiments, the investigator was not blinded to the group allocation, either while doing the experiment or while assessing the results.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the US National Institutes of Health grant T32HL066987 (F.D.), a Scholar Fellowship from Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) and the David Rockefeller Center for Latin American Studies at Harvard University (R.T.G.), a Science Without Borders Scholar Fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (R.B.P.) and the Brazilian National Institute of Science and Technology for Vaccines grant CNPq/Fapemig/MS 573547/2008-4 (R.T.G.). We thank S. Wilkinson (Queen Mary University, London) and J. Kelly (London School of Hygiene and Tropical Medicine) for T. cruzi overexpression plasmids, K. Okuda (University of Massachusetts Medical School) for L. major, K. Engelberg and M.J. Gubbels (Boston College) for T. gondii, and M. Ericsson and R. Bronson (Harvard Medical School) for technical support.
Footnotes
METHODS
Methods and any associated references are available in the online version of the paper.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Junqueira C et al. The endless race between Trypanosoma cruzi and host immunity: lessons for and beyond Chagas disease. Expert Rev. Mol. Med 12, e29 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Hotez PJ et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl. Trop. Dis 8, e2865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chowdhury D & Lieberman J Death by a thousand cuts: granzyme pathways of programmed cell death. Annu. Rev. Immunol 26, 389–420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrigues MM et al. Importance of CD8 T cell–mediated immune response during intracellular parasitic infections and its implications for the development of effective vaccines. An. Acad. Bras. Cienc 75, 443–468 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Gigley JP, Bhadra R & Khan IA CD8 T cells and Toxoplasma gondii: a new paradigm. J. Parasitol. Res 2011, 243796 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belkaid Y et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol 168, 3992–4000 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Tarleton RL, Koller BH, Latour A & Postan M Susceptibility of β2-microglobulin–deficient mice to Trypanosoma cruzi infection. Nature 356, 338–340 (1992). [DOI] [PubMed] [Google Scholar]
- 8.de Alencar BC et al. Perforin and gamma interferon expression are required for CD4+ and CD8+ T cell–dependent protective immunity against a human parasite, Trypanosoma cruzi, elicited by heterologous plasmid DNA prime–recombinant adenovirus 5 boost vaccination. Infect. Immun 77, 4383–4395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overstreet MG, Cockburn IA, Chen YC & Zavala F Protective CD8 T cells against Plasmodium liver stages: immunobiology of an ‘unnatural’ immune response. Immunol. Rev 225, 272–283 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Braeckel-Budimir N & Harty JT CD8 T cell–mediated protection against liver-stage malaria: lessons from a mouse model. Front. Microbiol 5, 272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang LP, Lyu SC, Clayberger C & Krensky AM Granulysin-mediated tumor rejection in transgenic mice. J. Immunol 178, 77–84 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst WA et al. Granulysin, a T cell product, kills bacteria by altering membrane permeability. J. Immunol 165, 7102–7108 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Stenger S et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282, 121–125 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Barman H et al. Cholesterol in negatively charged lipid bilayers modulates the effect of the antimicrobial protein granulysin. J. Membr. Biol 212, 29–39 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Walch M et al. Cytotoxic cells kill intracellular bacteria through granulysin-mediated delivery of granzymes. Cell 157, 1309–1323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beresford PJ, Xia Z, Greenberg AH & Lieberman J Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 10, 585–594 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Wilkinson SR et al. The Trypanosoma cruzi enzyme TcGPXI is a glycosomal peroxidase and can be linked to trypanothione reduction by glutathione or tryparedoxin. J. Biol. Chem 277, 17062–17071 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Wilkinson SR et al. TcGPXII, a glutathione-dependent Trypanosoma cruzi peroxidase with substrate specificity restricted to fatty acid and phospholipid hydroperoxides, is localized to the endoplasmic reticulum. Biochem. J 364, 787–794 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piacenza L et al. Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage- and endogenously derived peroxynitrite. Biochem. J 410, 359–368 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piacenza L et al. Mitochondrial superoxide radicals mediate programmed cell death in Trypanosoma cruzi: cytoprotective action of mitochondrial iron superoxide dismutase overexpression. Biochem. J 403, 323–334 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfefferkorn ER & Pfefferkorn LC Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp. Parasitol 39, 365–376 (1976). [DOI] [PubMed] [Google Scholar]
- 22.Pires SF et al. Cell culture and animal infection with distinct Trypanosoma cruzi strains expressing red and green fluorescent proteins. Int. J. Parasitol 38, 289–297 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Kapler GM, Coburn CM & Beverley SM Stable transfection of the human parasite Leishmania major delineates a 30-kb region sufficient for extrachromosomal replication and expression. Mol. Cell. Biol 10, 1084–1094 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiery J, Walch M, Jensen DK, Martinvalet D & Lieberman J Isolation of cytotoxic T cell and NK granules and purification of their effector proteins. Curr. Protoc. Cell Biol 47, 3.37.1–3.37.29 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Thiery J et al. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115, 1582–1593 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krettli AU & Brener Z Protective effects of specific antibodies in Trypanosoma cruzi infections. J. Immunol 116, 755–760 (1976). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.